

Abstract
The domestication history of rice remains controversial, with multiple studies reaching different conclusions regarding its origin(s). These studies have generally assumed that populations of living wild rice, O. rufipogon, are descendants of the ancestral population that gave rise to domesticated rice, but relatively little attention has been paid to the origins and history of wild rice itself. Here, we investigate the genetic ancestry of wild rice by analyzing a diverse panel of rice genomes consisting of 203 domesticated and 435 wild rice accessions. We show that most modern wild rice is heavily admixed with domesticated rice through both pollen- and seed-mediated gene flow. In fact, much presumed wild rice may simply represent different stages of feralized domesticated rice. In line with this hypothesis, many presumed wild rice varieties show remnants of the effects of selective sweeps in previously identified domestication genes, as well as evidence of recent selection in flowering genes possibly associated with the feralization process. Furthermore, there is a distinct geographical pattern of gene flow from aus, indica, and japonica varieties into colocated wild rice. We also show that admixture from aus and indica is more recent than gene flow from japonica, possibly consistent with an earlier spread of japonica varieties. We argue that wild rice populations should be considered a hybrid swarm, connected to domesticated rice by continuous and extensive gene flow.
Asian cultivated rice is one of the most ancient and widely consumed staple food crops. Its domestication and cultivation contributed to the rise of agricultural civilization in Asia. Rice is believed to have been domesticated ∼9000 yr ago from one of its sympatric wild species, O. rufipogon (Oka 1988; Fuller et al. 2010). Molecular studies have identified multiple varietal groups in cultivated rice, including two major ones: japonica (keng) and indica (hsien) (Glaszmann 1987; Garris et al. 2005; Sweeney et al. 2007). Indica and japonica are highly differentiated and partially reproductively isolated by a postzygotic barrier (Chang 2003). Despite numerous archaeological and genetic studies on the history of rice domestication, no consensus has been reached on the number of origins of different rice subgroups (Sang and Ge 2007; Huang et al. 2012b; Civáň et al. 2015). Some researchers argue for a single-origin model, which hypothesizes that rice domestication was a single event followed by a post-domestication diversification that created divergent subgroups. This model is supported by molecular research on the domestication genes, sh4 (Li et al. 2006; Lin et al. 2007) and PROG1 (Jin et al. 2008; Tan et al. 2008), which are responsible for two of the most critical domestication traits in rice, nonshattering grains and erect growth, respectively. It has been shown that different varietal groups of cultivated rice share identical sequences at these two domestication genes (Lin et al. 2007; Tan et al. 2008). Additionally, multiple studies that inferred the demographic histories of domesticated rice using independent data sets favor the single-origin model (Gao and Innan 2008; Molina et al. 2011). However, phylogenetic analyses using both nuclear and cytoplasmic DNA markers consistently show that indica and japonica are each associated with different subgroups of O. rufipogon (Cheng et al. 2003; Zhu and Ge 2005; Londo et al. 2006; Rakshit et al. 2007; Huang et al. 2012b; Civáň et al. 2015). Some have used these results to argue that rice domestication occurred more than once, and they attribute the sharing of key domestication loci to gene flow after domestication (Londo et al. 2006; Rakshit et al. 2007; Sang and Ge 2007) or independent selection from standing ancestral variation (Civáň et al. 2015).
Despite these seemingly conflicting viewpoints, it is well accepted that understanding the genetic variation of the primary gene pool, from which rice was domesticated, is critical in studying rice domestication (Vaughan et al. 2008). The primary gene pool is a concept used among plant breeders to define a set of species/subspecies comprised of three components: the cultivated species, its wild ancestor, and in many cases, its weedy counterparts (Harlan and DeWet 1971). Within this gene pool, hybridization occurs easily and hybrid swarms are occasionally formed as a result of crossing between the constituent components (Harlan 1992).
There has been extensive work on the population structure and genetic relatedness of different subgroups within the rice primary gene pool, but incongruent phylogenetic patterns have been observed. Wild rice has an annual ecotype, O. nivara, and its phylogenetic position with the perennial type is inconclusive (Lu et al. 2002; Londo et al. 2006); thus in this study, we will not separate it from O. rufipogon. Early molecular phylogenetic studies using isozymes identified two genetic groups of O. rufipogon, with closer genetic affinity to indica and japonica, respectively (Second 1982). Multiple other DNA studies also identified different genetic subgroups in wild rice populations associated with different domesticated rice subgroups. In addition, they also identified more ancestral genetic groups in wild rice population (Sun et al. 1996; Cheng et al. 2003; Zhu and Ge 2005; Londo et al. 2006). Using genome-wide markers from 48 sequence-tagged sites, Huang et al. (2012a) concluded that there were two distinct groups of wild rice, one genetically related to indica, and one without particular relatedness to any domesticated group. A whole-genome sequencing study (Huang et al. 2012b) categorized wild rice into three groups, two of which cluster with japonica and indica, respectively, in the phylogeny constructed with genome-wide SNP markers. Recently, a genotype-by-sequencing study on 286 diverse O. rufipogon species complex accessions identified six subpopulations and suggested that there was gene flow between O. rufipogon species complex and O. sativa (Kim et al. 2016). To account for the range of genetic variation within the rice primary gene pool, various modeling analyses were also performed (Caicedo et al. 2007; Zhu et al. 2007; Gao and Innan 2008; Molina et al. 2011). They consistently found that domesticated species had suffered severe bottlenecks and that models of nonindependent rice domestication provided better explanation for the pattern of genetic variation within the gene pool (Gao and Innan 2008; Molina et al. 2011). Also, field sampling studies in different regions observed ongoing gene flow among different components of the gene pool (Pusadee et al. 2013, 2016; for summary, see Oka 1988). Numerous concerns were raised regarding the conservation of genetic diversity in wild rice populations, because frequent gene flow from domesticated rice into wild rice populations could cause genetic erosion and diversity loss in wild rice (Oka 1988). It is also well recognized that gene flow between domesticated and wild rice populations is an important factor that might confound phylogenetic studies and demographic history inferences on the rice primary gene pool (Vaughan et al. 2008; Huang et al. 2012a). However, there is no study to date that estimates the amount of gene flow from domesticated rice into natural wild rice populations and/or determine the extent to which gene flow has shaped the genetic landscape of wild rice.
Results
Admixture analysis in the primary gene pool of Asian rice
To investigate population structure and admixture patterns in the primary gene pool of Asian rice, we combined whole-genome sequencing data from 203 cultivated rice varieties (Wang et al. 2016) and 435 accessions of O. rufipogon (Huang et al. 2012b). The cultivated rice accessions were collected from 71 countries and were systematically selected to be representative of rice diversity from more than 18,000 accessions in the USDA rice germplasm seed bank (Agrama et al. 2009). The wild rice samples were collected in situ in wild rice natural habitats (Supplemental Text S1) by scientists from the National Institute of Genetics in Japan (Morishima 2002).
We first estimated ancestry proportions for individuals using NGSadmix (Skotte et al. 2013), which implements a clustering method similar to the one in the popular program ADMIXTURE (Alexander et al. 2009), while incorporating uncertainty in the genotype calls inherent in next generation sequencing (NGS) data. We fit admixture models by varying the number of presumed ancestral populations (K) from 2 to 15 (Supplemental Figs. S1–S3). Generally, the results fit those found in previous studies and those expected from prior knowledge of rice population genetics (Supplemental Text S3). However, accessions of domesticated rice are identified to have a small amount (<5%) of wild rice ancestry, possibly reflecting introgression from wild rice, which was not observed in previous studies (Wang et al. 2016). In the most remarkable case, one domesticated rice accession (GSOR311586) was identified to be of 99% wild ancestry. We conducted field observations, which showed that this accession has shattering grains and black-hull seeds with long awns that are hallmark phenotypes of wild rice (Supplemental Fig. S6). PCR also confirmed that this accession contained a wild allele of sh4. It is very likely that this is, in fact, a wild rice accession that was misidentified as domesticated during germplasm collection.
In the wild rice population, however, we identified six subgroups (Fig. 1A), which we denote as Or-A, Or-B, Or-C, Or-D, Or-E, and Or-F, respectively, according to the order of emergence when increasing K in the admixture analyses (Fig. 1A; Supplemental Fig. S1). We also find good correspondence between subpopulations assigned here and previously described genetic subgroups (Huang et al. 2012b) based on phylogenetic analyses (Supplemental Table S1; Supplemental Fig. S7). Notably, a large proportion (42%) of wild rice individuals seems to be substantially admixed and thus could not be assigned to a single ancestry group, suggesting a complicated history of hybridization and differentiation among wild rice. Among the identified clusters, four components (Or-A, Or-B, Or-C, and Or-D) are unique to wild rice. The Or-A component is the first to emerge in wild rice when we increase K from 2 to 3. This component has a broad geographic distribution, with highest ancestry proportions concentrated in the oceanic regions and lower ancestry proportions in West India and Sri Lanka (Fig. 1B). Or-B emerged when five ancestral populations were included in the model. Geographically, Or-B is found almost exclusively in China, and it has been hypothesized that Or-B may represent the wild ancestor of both indica and japonica since this population harbors ancestral alleles at domestication-related loci shared by indica and japonica (Huang et al. 2012b). Adding one additional ancestral population to the model (K = 6) results in the emergence of Or-C, which is found mostly in South and Southeast Asia and comprises the majority of the wild rice genomes in the West India and Sri Lanka populations. Or-D is found almost exclusively in the Indochina Peninsula, Bangladesh, and East India (Fig. 1B). Intriguingly, for the last two subgroups (Or-E and Or-F), the major genetic components are shared with aus and indica, respectively. To further characterize the genetic relationships among subgroups in this gene pool, we carried out a principal component analysis (PCA) (Fig. 1C; Supplemental Fig. S8). In the PCA space constructed with the first two PCs, japonica forms an isolated cluster, whereas indica and wild rice form a separate, more diffuse cluster. Or-E and Or-F colocalize with aus and indica in the PCA plot. PC3 separates indica and aus, each forming a cluster. However, Or-E and Or-F still cluster with aus and indica, respectively, and the clustering pattern persists even at higher dimensions of the PCA space (Supplemental Fig. S8). This suggests a very high degree of genetic relatedness between wild rice subgroups Or-E/Or-F and the domesticated rice subgroups aus/indica, respectively.
Figure 1.

Population structure of the rice primary gene pool, including O. sativa and O. rufipogon. (A) Clustering using NGSadmix assuming K = 2 and K = 9. At K = 2, the samples are divided into indica and japonica components. At K = 9, five subgroups of domesticated rice are recovered, and four unique components of wild rice are identified. The color bars beneath the clusters denote the subgroup assignments. The red arrow points to the misidentified domesticated accession (GSOR311586), which was confirmed to have wild rice ancestry. The abbreviations of subgroups in cultivated rice are as follows: (ADM) admixture; (IND) indica; (AUS) aus; (ARO) aromatic; (TRJ) tropical japonica; (TEJ) temperate japonica. (B) Geographic distribution of rice samples. South and Southeast Asia, which are the major habitats for wild rice and also major rice cultivation areas, are shown on the map. The area was divided into five regions: (1) South Asia; (2) Ganges Basin; (3) Indochina Peninsula; (4) China; (5) Archipelago countries. The color code of the bar beneath the clustering plot indicates cultivated (green) and wild (black) rice. The abbreviations at the bottom left are as follows: (AS) Asia; all rice samples from Asia but not shown on the map are included in this category; (AM) America; (AF) Africa; (EU) Europe. (C) PCA of the combined population with wild (triangle) and cultivated (dot) rice samples. The abbreviation codes are the same as those in A.
Gene flow between O. rufipogon and O. sativa
The exceptional genetic similarity between Or-E/Or-F and the corresponding domesticated subgroups revealed by PCA and admixture analyses can be explained by two possible hypotheses. First, Or-E and Or-F could be extant representatives of the ancestral source population used in the domestication process, and the genetic affinity with aus and indica could result from standing ancestral polymorphism segregating in these domesticated subgroups. Second, it could be caused by gene flow between domesticated rice and the corresponding wild subgroups. To test these hypotheses, we first conducted a correlation analysis between geographic distance and genetic distance in all sativa–rufipogon pairs. We find a highly significant correlation (ρ = 0.15, P < 2.2 × 10−16) (Supplemental Fig. S9), indicating that geographically close sativa–rufipogon sample pairs tend to be more genetically related than expected. One possible explanation for the correlation could be that the correlation is driven by shared ancestral polymorphism between two species, but this is only tenable when there are multiple geographic sites where rice was domesticated independently. Moreover, the correlation is also present within smaller regions, such as India (ρ = 0.18, P = 1.1 × 10−12) and Bangladesh (ρ = 0.27, P = 3.1 × 10−3) (Supplemental Fig. S10). An explanation of the correlation based solely on multiple independent domestications would further require multiple such domestication events within each country, with local variability and structure preserved since the time of domestication—a very unlikely scenario. A more tenable hypothesis is substantial local gene flow between domesticated and wild rice in these regions.
To further examine the hypothesis of gene flow between domesticated and wild rice populations, we analyzed the local ancestry at two known domestication-related genes, sh4 and PROG1, and asked whether the domesticated alleles are found in the wild population or vice versa. These two genes were previously shown to be responsible for key morphological transitions from wild to domesticated rice: a mutation (G → T) in the coding sequence of sh4 causes reduced shattering of rice grains (Li et al. 2006; Lin et al. 2007), and genetic variants in PROG1 contribute to the transition from prostrate to erect growth in domesticated rice (Jin et al. 2008; Tan et al. 2008). To our knowledge, these are the only two genes in the rice genome that control critical traits distinguishing wild and domesticated rice; meanwhile, all domesticated rice share identical domesticated alleles at these loci (Lin et al. 2007; Tan et al. 2008), despite enormous allelic diversity commonly observed at other genomic loci among subgroups of domesticated rice. The domestication alleles confer traits strongly preferred by humans, but they are presumably highly deleterious in the wild: The nonshattering phenotype will increase the probability of herbivory of rice seeds, and erect growth will make rice plant more easily spotted and grazed by herbivores (Tan et al. 2008). We first examined the haplotype content at the sh4 locus using a clustering approach (Methods; Supplemental Fig. S11). Despite varying K from 2 to 5, all domesticated rice accessions except the “misidentified” GSOR311586 remain assigned to a single component (Fig. 2A), suggesting they harbor closely related haplotypes, as previously argued (Tan et al. 2008). Surprisingly, 94 samples (21.6% of all wild samples) from the wild rice population are also consistently assigned to the domesticated cluster, suggesting that they have the domesticated allele at sh4. Using a PCR assay, we confirmed that all the assayed samples contained the derived allele (T) at the functional SNP position, supporting the local ancestry assignment method as an effective approach in discerning alleles (Supplemental Text S6; Supplemental Table S1). Since we adopted the 95% ancestry cutoff for identifying domesticated allele (Methods), the result suggests that 94 may represent a conservative estimate of the number of wild samples harboring the domesticated allele at sh4. This estimate is consistent with a previous study which determined that ∼27% of wild rice contain the nonshattering allele at sh4 (Zhu et al. 2012).
Figure 2.

sh4 haplotypes in wild and domesticated rice populations. (A) Local ancestry inference at sh4 locus for K = 5. The bar at the bottom denotes O. sativa (green) and O. rufipogon (black) accessions, respectively. (B) Diversity reduction at the selective sweep region of sh4. The y-axis shows the ratio of pairwise differences estimator (π) of nucleotide diversity among populations. (C) dXY values between populations at the selective sweep region of sh4. The gray line indicates the sh4 gene region in B and C.
The observation that these “wild” accessions contain the domestication allele at this key domestication gene can be explained by two hypotheses: introgression from domesticated rice or shared ancestral variation. In the first scenario, we would expect that these individuals might share the signal of the domestication-related selective sweep at the sh4 locus and show a reduction in genetic distance to domesticated rice relative to the distance between other wild rice and domesticated rice at this locus. However, if these varieties harbor the domestication allele simply due to shared ancestry from pre-domestication, they should not show the signal of a domestication-related selective sweep. To test this hypothesis, we examined local diversity at this locus on wild rice carrying the domesticated allele of sh4 (hereafter, WRDS) and found a fourfold reduction in relative nucleotide diversity across the 200-kb region that perfectly coincides with a similar diversity reduction in domesticated rice (Fig. 2B; Supplemental Fig. S12). Also, Tajima's D (Tajima 1989) is −2.63 in this region (Supplemental Fig. S15), indicating an excess of rare alleles relative to equilibrium expectations, which is also consistent with the scenario of a recent selective sweep. At the sweep region, the genetic divergence between WRDS and domesticated rice drops to 0 (Fig. 2C), indicating they share nearly identical haplotypes. However, the divergence between WRDS-wild and cultivated-wild population is consistently high and resemble background genomic levels (Fig. 2C; Supplemental Fig. S13). Taken together, these results show that the genetic similarity between WRDS and domesticated rice at sh4 is caused by sharing of the same domestication allele transferred by gene flow from domesticated into wild rice populations. The fact that nominal wild rice has the shattering phenotype (Zhu et al. 2012), even when carrying the domesticated sh4 haplotype, suggests that one or more compensatory mechanisms have evolved in wild rice populations in order to compensate for the extremely high influx of the domesticated sh4 allele through continuous gene flow from domesticated rice.
When applying the same analysis to the PROG1 locus, we identified 113 wild rice accessions (26.0% of all wild rice samples) carrying the domestication allele prog1 (Methods; Supplemental Fig. S14); in these, the nucleotide diversity is reduced and Tajima's D is −2.42 (Supplemental Figs. S15–S18), similar to the pattern observed for sh4. A significant excess of these accessions (n = 66; P < 0.01, χ2 test) also carry the domestication allele at sh4. In total, 23 of 25 accessions in subgroup Or-E carry prog1, and 20 accessions carry the domesticated sh4 allele. In the Or-F subgroup, 11 of the 12 accessions carry the prog1 allele, and all of them harbor the domestication allele of sh4. When combined with the genome-wide admixture inferences, these results strongly argue that the Or-E and Or-F subgroups either emerged as a result of feralization of domesticated rice or have received very high levels of gene flow, most likely from the aus and indica varieties, respectively. Therefore, the shared ancestry of Or-E/Or-F with domesticated subgroups observed under the K = 9 should be interpreted as a consequence of extensive gene flow from domesticated rice. Moreover, it is noteworthy that 104 accessions of other subgroups of wild rice harbor the domesticated allele at either PROG1 or sh4, resulting in a total 32% of annotated wild rice accessions carrying domestication alleles, suggesting that gene flow/feralization is substantial and not limited to only a subset of the wild rice subgroups (Supplemental Fig. S19).
Morphologically, domesticated rice has closed floret, making cross pollination difficult and keeping them largely self-fertilized. Wild rice, however, typically has open floret with exerted stigma, resulting in a higher rate of outcrossing, and this is mirrored by lower inbreeding coefficient estimates when compared with domesticated rice (t-test, P ≪ 0.01) (Supplemental Fig. S20). Thus, morphological differences predict an asymmetric pattern of gene flow, with its dominant direction from domesticated into wild populations. Moreover, the census sizes of domesticated rice populations are much larger relative to wild rice populations, which also suggests that gene flow will predominantly be from domesticated to wild rice. Consistent with these expectations, we find 207 domestication alleles at sh4/PROG1 in wild rice populations, whereas the wild alleles in domesticated accessions are rarely observed (n = 3). Genome-wide admixture analyses are also consistent with this hypothesis: varying K from 2 to 9, we consistently observe domestication components in wild rice populations, but very little wild ancestry in domesticated rice (Suplpemental Fig. S1). For the K = 9 model, 50% of wild rice have >10% domesticated ancestry (Supplemental Fig. S21). Interestingly, wild rice populations are enriched with accessions containing 50%–60% or 90%–100% domesticated ancestry (Supplemental Fig. S21), possibly due to very recent gene flow.
Geographic pattern of gene flow
Monitoring the geographic pattern of the gene flow is important and may help guide the protection of wild rice germplasm. Using introgression of sh4 and PROG1 as an indicator, we found a significantly biased geographic distribution and could reject the hypothesis of a uniform amount of gene flow in all regions (P < 0.01, χ2 test) (Fig. 3A; Supplemental Table S2). In Bangladesh, 75% of wild accessions have domesticated alleles at one of the loci, and 45% have domestication alleles at both loci; in East India, we find 60.4% have one domesticated allele, and 41.5% have both. These numbers are much higher than the average level of 32.4% and 15.2%. In contrast, the northeast ranges of wild rice habitat show little or no introgression at either locus, e.g., only 17.5% of wild rice in China and 6.7% in Laos harbored domesticated alleles. In Indonesia, none of the rice accessions show evidence of introgression. Estimates of domesticated ancestry proportions (K = 9) in the genome of wild rice show a pattern of gene flow similar to that inferred using the two domestication loci (Fig. 3B; Supplemental Fig. S22). Varieties from Bangladesh have the highest proportion of domesticated rice ancestry among wild rice populations, with an estimate of 60% (Supplemental Fig. S22). The high level of gene flow in this region is consistent with field observations arguing that wild rice collected in this region may be heavily admixed (Morishima 2002). The neighboring regions of East India and Malaysia also have high estimates of 50% and 43% domesticated ancestry, respectively. In contrast, wild accessions from regions such as China, Laos, and Indonesia are relatively unadmixed, with domesticated admixture proportions as low as 8%, 10%, and 15%, respectively (Supplemental Fig. S22).
Figure 3.
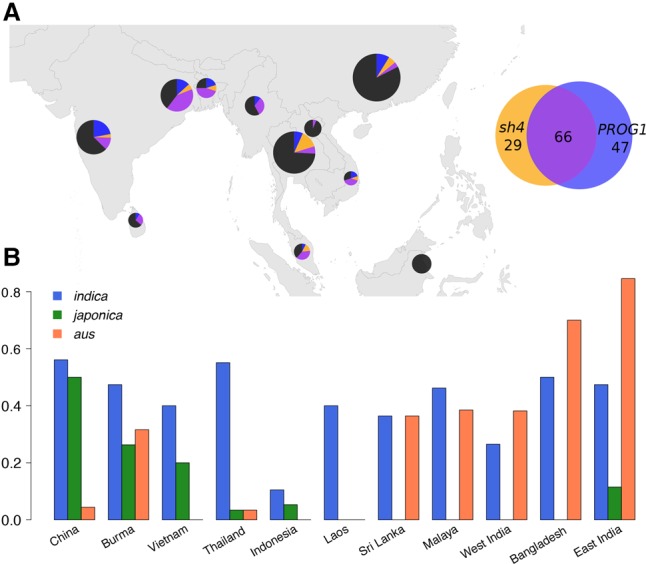
Geographic and subspecific pattern of gene flow. (A) Geographic distribution of domesticated alleles introgression at PROG1 and sh4 loci. Each pie chart represents the wild rice population of a region, and the area is proportional to its sample size. Each chart was divided into four categories according to the haplotype information at the two domestication loci: the domestication sh4 allele only (yellow), the wild prog1 allele only (blue), domestication alleles for both sh4 and PROG1 (purple), and wild sh4 and PROG1 alleles (black). Regions with less than 10 samples are not shown. (B) Geographic distribution of gene flow from indica, japonica, and aus. The proportions of admixed wild accessions with >5% ancestry of a certain subspecies in different regions are plotted. Wild accessions with indica have a pandemic distribution across rice cultivation regions, whereas accessions with japonica ancestry are endemic to regions including China, Burma, and Vietnam. Accessions with aus ancestry are mainly found in the Ganges Basin region, Sri Lanka, and Malaysia.
When examining gene flow from the perspective of the donors, we find a great difference in contribution from different domesticated rice subgroups, with 50% indica, 46% aus, and only 4% japonica. There are several factors likely contributing to this pattern. First, indica and aus varieties more readily shatter than japonica varieties (Konishi et al. 2006; Vaughan et al. 2008), so they are more likely to contribute to feralization. Second, wild rice is more likely to be sampled from areas in which varieties from the indica and aus subgroups are cultivated. Japonica varieties are mainly cultivated in the north, including North China, Korea, and Japan (G Khush, pers. comm.), where wild rice is rare and hence has not been included in wild rice sampling efforts. The extensive overlap of indica/aus planting area with wild rice habitat provides more opportunity for gene flow. In countries such as Laos, Vietnam, and Thailand, where gene flow mainly comes from the indica subgroup (100%, 94%, and 93%, respectively). However, in Bangladesh and India, gene flow is mostly contributed by the aus subgroup (75% and 55%, respectively). Interestingly, consistent with the broad distribution of indica cultivation, gene flow from the indica subgroup is present in wild populations from most geographic regions with an average of 50% of admixed samples carrying >5% indica ancestry (Fig. 3B). In contrast, aus and japonica are planted in more restricted geographic regions, and the distribution of gene flow into wild populations reflects these geographic biases (Fig. 3B). The proportion of wild accessions with >5% aus ancestry is high in Bangladesh and India (86% and 61%, respectively), which coincides well with the traditional planting area of aus varieties (Glaszmann 1987; Khush 1997). A considerable proportion of wild accessions from Malaysia and Sri Lanka (38% and 36%, respectively) also carry substantial aus ancestry. Wild accessions with >5% japonica ancestry are found in high proportions specifically in regions such as China, Burma, and Vietnam, representing the northeast range of wild populations where the planting region of japonica varieties and wild rice populations overlap.
To determine whether gene flow from each domesticated subgroup occurred during the same or different time periods, we used local ancestry inference in admixed wild rice to identify introgressed domesticated chromosomal segments. Since the introgressed segments are broken into smaller segments by recombination over time, the distribution of introgressed tract lengths is informative about the age of admixture (Pool and Nielsen 2009; Moreno Estrada et al. 2013). The results of the local ancestry inference are consistent with our global ancestry inferences (Supplemental Fig. S23) and further support geographic biases in domesticated sources of gene flow (Fig. 4A). We summarized the length distribution of introgressed tracts from each domesticated subgroup and found that the length distribution of japonica haplotypes is enriched for smaller segments with an average of 8 centimorgan (cM) (Fig. 4B). The distribution of japonica haplotypes is significantly shorter than that of both indica (t-test, P < 1 × 10−8) and aus (t-test, P < 1 × 10−8), which have average haplotype length of 27 cM and 18 cM, respectively. This result indicates that the gene flow from japonica to wild rice is older than that of aus and indica.
Figure 4.

Distribution of chromosomal segments with domesticated ancestry in wild rice. (A) Ancestry assignment for wild rice from different regions. Each row represents a chromosome of one individual. Data for Chromosome 7 is presented. Bar colors indicate ancestry as follows: (gray) wild rice; (coral) aus; (blue) indica; (green) japonica. Introgression in Bangladesh is dominated by aus; Thailand is dominated by indica. Chinese wild rice harbors tracts of both indica and japonica ancestry. (B) Cumulative distribution of ancestry tract lengths from different subgroups of domesticated rice.
Feralization plays an important role in gene flow
The gene flow from O. sativa to O. rufipogon may follow two different evolutionary pathways: pollen dissemination or seed dispersal. If seed spillage were involved, we would expect to find cytoplasmic genomes with domesticated rice haplotype in wild populations. In this study, we took advantage of the high copy number of the chloroplast genome, providing an average of 200× sequencing coverage for each accession in the sequencing data (Methods), to obtain highly accurate haplotype information. We first estimated a maximum likelihood (ML) phylogenetic tree of the chloroplast haplotypes (Supplemental Fig. S24). The domesticated rice samples were found in two clusters, corresponding to the indica and japonica subgroups. Interestingly, many wild rice chloroplast genomes were nested within the domesticated rice clusters. To further quantify the number of wild rice accessions that are closely related to domesticated rice chloroplast haplotype, we constructed a haplotype network using common polymorphic sites across rice chloroplast genomes (Supplemental Text S4), which summarizes all major chloroplast haplotypes in the primary gene pool of rice and the phylogeny among them (Fig. 5). Surprisingly, we found 98 accessions (28.8% of 340) of wild rice with identical chloroplast haplotypes to those of domesticated rice. For both Or-E and Or-F, which we have shown to carry domesticated nuclear ancestry, an excess of accessions harbor domesticated chloroplast haplotypes as well (17 of 24 for Or-E, P = 0.01; 8 of 12 for Or-F, P = 0.06, χ2 test). This further supports that these accessions in fact are established by seed dispersal, i.e., feral rice. These results suggest an evolutionary scenario that includes ancient feralization events followed by subsequent backcrossing with wild rice populations. In line with the analysis at domestication loci, gene flow from domesticated rice is not limited to just Or-E and Or-F subgroups, because domesticated chloroplast genomes are carried by other groups of wild rice as well (Supplemental Fig. S25).
Figure 5.
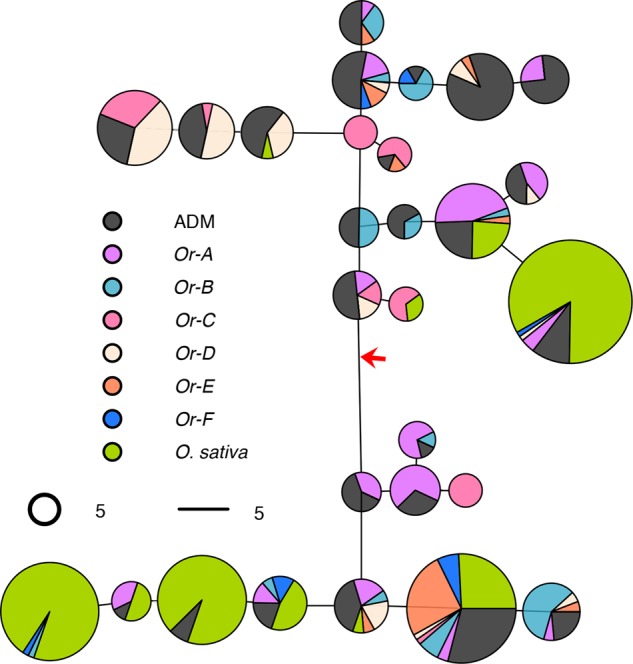
Chloroplast haplotype network among the 28 common haplotypes in rice primary gene pool. The haplotypes were defined using 74 common SNPs from the rice chloroplast genome. Each pie chart represents one haplotype, and it was further divided according to the subgroup information of the samples. All domesticated rice samples were colored in green, and wild rice samples were divided into seven subgroups. The root of the haplotype network was inferred using the chloroplast genome of O. meridionalis and is indicated by a red arrow. The length of lines connecting pie charts is proportional to the pairwise distance between haplotypes. The areas of the pie charts are proportional to the number of samples with the haplotype.
Selection and adaptation in feral rice
The exceptional relatedness of both nuclear and chloroplast genomes between Or-E and aus indicates that Or-E might have arisen from aus varieties in the very recent past and then diverged during adaptation to the local wild environments. Thus, a comparison of Or-E and aus genomes provides a unique opportunity to investigate the genetic basis of plant feralization. In order to identify loci that might have been differentially selected between domesticated and feral rice, we first scanned the genome using FST to identify highly differentiated genes between Or-E and aus. We performed gene ontology (GO) enrichment analysis on genes with FST values ranking in the top 5% of the empirical distribution. The top enriched GO terms are mostly high-hierarchy terms that are too general to provide any specific biological hints (Supplemental Table S3). However, among the top enriched GO terms that refer to explicit biological functions, abiotic and biotic resistance terms, including response to fungus (P = 8.1 × 10−7), bacterium (P = 1.5 × 10−9), salt (P = 8.4 × 10−11), cold (P = 4.8 × 10−6), and wounding (P = 3.6 × 10−8), are prominently enriched. This suggests that rice might have faced different biotic and abiotic selection pressures under domestic and wild conditions. Interestingly, the GO term “long-day photoperiodism” is also enriched, an enrichment which persists even if the GO analysis is limited to genes with the top 1% FST values, indicating that genes underlying flowering time in long-day condition are among the most differentiated genes between Or-E and aus. We subsequently identified genes under selection in Or-E that may have been targeted by natural selection during the feralization process. Interestingly, HD1, a gene underlying major quantitative trait locus (QTL) for photoperiod-dependent flowering (Yano et al. 2000), is among those with the most dramatic diversity reduction across Or-E rice genomes, ranking in the top 0.3% of diversity-reduction genes across Or-E rice genomes, suggesting strong selection on this locus in the Or-E population. A comparison of the haplotypes of Or-E and aus at this gene identified the most differentiated SNP as a nonsynonymous polymorphism (G/A, G387S) that is fixed for G in Or-E but has low allele frequency in aus (13.3%), a potential candidate causal mutation. It is likely that HD1 is a target of selection for rice feralization and that the nonsynonymous mutation has contributed to the flowering time adaptation of rice in the wild habitat.
Implications for rice domestication
The high level of gene flow between wild and domesticated rice has consequences for our understanding of the process of rice domestication. To illustrate this, we estimated admixture graphs of geographically defined wild rice and major groups of domesticated rice using TreeMix (Pickrell and Pritchard 2012), which uses a maximum likelihood (ML) method based on a Gaussian model of allele frequency change. We divided wild rice into five regional populations based on geographic characteristics of the wild rice area and potential boundaries between subgroups (Methods; Fig. 1B). Four major subgroups of domesticated rice were also included. Although the topology of the ML trees changes depending on the number of migration events (m) allowed in the model (Supplemental Fig. S26), certain patterns persist and are robust toward assumptions regarding m. First, the domesticated rice subgroups consistently show evidence of more genetic drift, likely because they underwent strong bottlenecks caused by the domestication process and by artificial selection. The japonica subgroups have exceptionally long branches consistent with the previously reported much stronger bottleneck in their domestication history (Caicedo et al. 2007; Zhu et al. 2007; Gao and Innan 2008). Wild rice populations in the Ganges Basin (GBW) consistently form a clade with indica and aus (Supplemental Fig. S26). Two hypotheses could explain this pattern: (1) indica and aus were domesticated from the GBW very recently, or (2) as suggested by the previous analyses in this manuscript, the GBW populations are a product of feralization from domesticated aus and indica rice. Similarly, temperate and tropical japonica forms a clade with Chinese rice when assuming no migration.
Allowing just one migration event (m = 1) (Supplemental Fig. S26), we observe an admixture from indica into the Indochina wild rice population (ICW) contributing 46% of the DNA in Indochina. This is consistent with the results that a substantial amount of indica ancestry is observed in ICW (Figs. 1B, 4A). Allowing two admixture events (m = 2) (Supplemental Fig. S26), a substantial amount of gene flow from Indochina to China is observed. This is possibly a consequence of Chinese wild rice being admixed between original wild rice and domesticated rice. This is supported by the fact that when m = 3, wild Chinese rice groups cluster with wild rice in Indochina and the Archipelago, but with substantial gene flow (49%) from the ancestor of japonica (Fig. 6). Likely, the true wild ancestor of japonica rice is not represented in the sample by any current wild descendant population. The Or-B component found in China may not be an “authentic” wild component, but rather it is a product of admixture between wild rice and ancient japonica. The wild rice ancestral to the domesticated japonica may be, in fact, already extinct. For models with m = 3, we observed an admixture event, with a proportion of 19%, from aus to tropical japonica (Fig. 6), indicating substantial genetic ancestry shared between these two subgroups. We consistently observe japonica sharing high residuals with aus/indica (Supplemental Fig. S26), which likely reflects that they share many genomic components caused by hybridization in their domestication and breeding history.
Figure 6.
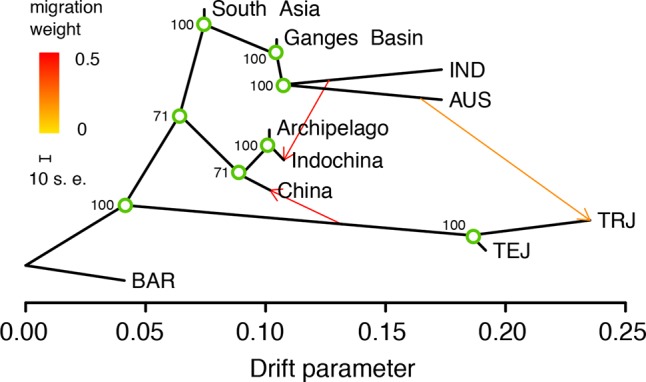
Maximum-likelihood admixture graph on the primary gene pool of Asian domesticated rice. The wild rice (O. rufipogon) population was divided into five geographic populations (Methods). The abbreviations for the major domesticated rice subgroups are the same as in Figure 1. African wild rice, O. barthii (BAR), was used to root the tree. The bootstrap values on the tree are based on 1000 replicates. Arrows on the graph represent admixture events among different rice populations.
Discussion
Elucidating the pattern of gene flow among wild and domesticated rice is important for understanding the history of rice domestication. Multiple studies argued for the independent domestication of rice based on reciprocal monophyly of indica and japonica when using different nuclear DNA markers in independent rice collections (Cheng et al. 2003; Zhu and Ge 2005; Rakshit et al. 2007; Civáň et al. 2015). In contrast, treating O. rufipogon as a single homogenous group in an analysis of divergence times and population trees, Molina et al. (2011) argued for a single domestication event. Based on comparisons to wild rice samples, Huang et al. (2012b) similarly argued that rice domestication originated in South China. Recently, Civáň et al. (2015) argued that the aus group had been independently domesticated in the Ganges Basin area. In this study, we showed that there is extensive, continuous gene flow from domesticated rice into wild rice populations. It suggests that the patterns described in previous studies are likely caused by gene flow from domesticated rice into wild rice populations after rice domestication. We show that wild rice in the Ganges Basin is likely feral rice, recently diverged from domesticated rice, and Chinese wild rice has received extensive gene flow from an ancient japonica population. Furthermore, the indica and aus groups are always sister groups, suggesting a single domestication event for these two groups. TreeMix results are largely compatible with a dual origin of domestication given the deep divergence observed between indica and japonica subgroups. The divergence even spans the diversity of present-day Asian wild rice, but we caution that current wild rice samples may be biased due to incomplete sampling or loss of “authentic” wild rice samples in germplasm centers during preservation. We cannot exclude the possibility of a single domestication hypothesis because the deep divergence could also be caused by substantial independent gene flow from other wild rice species into different domesticated rice subgroups, which is practiced in rice breeding (Brar and Khush 1997). The single domestication hypothesis would require either (1) extensive gene flow from wild rice into the indica/aus subgroups so that their genomes now are dominated by gene flow from wild rice, combined with a subsequent loss (or lack of representation) of true “ancestral” wild rice; or (2) a single domestication hypothesis could also be compatible with the data if all wild rice populations represented in the panel are dominated by gene flow from local domesticated rice occurring continuously over the past ∼9,000 yr. However, the dual domestication model is arguably a simpler scenario.
Rice was introduced into the United States <400 yr ago, and rice cultivation was not widely expanded until the 1750s (Dethloff 2003). However, weedy rice is now common in rice growing regions in the United States and is one of the major weeds limiting rice production (Ziska et al. 2015). Genetic analysis has shown that American weedy rice population arose independently from indica and aus varieties (Londo and Schaal 2007). These observations indicate that rice could frequently revert to the wild state in domestication traits. Weedy rice is a conspecific form of cultivated rice, while displaying distinguishing features including shattering grains and strong seed dormancy typical of wild rice (Ferrero 2003; Song et al. 2014). The shattering and seed dormancy phenotypes acquired in weedy rice are presumably adaptive in wild conditions, potentially further facilitating feralization. A crop–weed–wild complex is found throughout regions where wild and cultivated rice overlap, and gene flow among components within the species complex is frequently observed (Ellstrand et al. 2013; Pusadee et al. 2013, 2016; Song et al. 2014). In Asia, rice cultivation has been performed for thousands of years, and rice feralization has likely happened throughout this period as well (Vaughan et al. 2005). In fact, much presumed wild rice in many parts of Asia may possibly be descendants of ancient feralization/hybridization events. Wild and weedy rice found all over the world might simply represent different stages of the feralization process. It is even possible that what we today characterize as wild rice in Asia, may largely be feral rice that has undergone thousands of generations of natural selection in the wild, and the original species from which O. sativa was domesticated is either extinct or has been almost entirely overwhelmed by the massive amounts of gene flow from domesticated rice. O. rufipogon may then represent a nominal species created by human domestication and subsequent feralization/hybridization.
Methods
Genomic data acquisition
The genomic data of wild rice was downloaded from the European Nucleotide Archive under the accession number ERP001143 (Supplemental Text S2). Domesticated rice data was downloaded from the NCBI BioProject Repository (project number: PRJNA301661).
Short-read mapping
Reads were mapped to the rice genome (IRGSP-1.0) (Kawahara et al. 2013) with BWA (version 0.7.0) (Li and Durbin 2009), and the mapping was further improved with Stampy (version 1.0.20) (Lunter and Goodson 2011). PCR duplicates were removed by “rmdup” in SAMtools (version 0.17) (Li et al. 2009). We realigned reads at gapped regions with GATK (version 2.6) (DePristo et al. 2011).
Population structure, phylogeny, network, and TreeMix analyses
We estimated genotype likelihoods of populations with the “-GL” option in ANGSD (version 0.542) (Korneliussen et al. 2014). Inbreeding coefficients for each individual were calculated using a probabilistic framework implemented in ngsF (Vieira et al. 2013). The variability and allele frequency of each genomic site was estimated by ANGSD using the “-doMaf” command. Variable sites were extracted and used for further analyses. A genotype likelihoods-based method, implemented in NGSadmix (Skotte et al. 2013), was used for global ancestry inference. The analysis was conducted on the combined population, including 203 domesticated and 435 wild rice accessions. We randomly picked one variable site for every 5-kb genomic region from variable sites to reduce effects of linkage disequilibrium. In total, 60,722 evenly distributed markers were used. With these markers, we successively tested 14 clustering models in the population with K (presumed cluster number) ranging from 2 to 15. For each K, we ran 200 independent replicate optimizations, picked the clustering model with the highest log likelihood value, and the corresponding log likelihoods are shown in Supplemental Figure S3. PCA was performed with the same genotype likelihoods data set using ngsCovar from the ngsTools package (Fumagalli et al. 2014). All plots were generated with R (version 3.0.2) (R Core Team 2016). We estimated admixture trees, phylogenies, and haplotype networks using standard methods explained in Supplemental Text S4 and S5.
Introgression analyses at two domestication loci
To identify domestication haplotypes at the sh4 locus in wild rice, we inferred local ancestry in a 10-kb region centered on sh4. Using genotype likelihoods, we ran NGSadmix for varying values of K, and domesticated rice accessions were consistently assigned to one component from K = 2 to K = 5 except for the misidentified sample, GSOR311586. At K = 6, the domesticated rice population splits into two major components, which conflicted with prior knowledge that there is one haplotype at this locus in the domesticated rice population (Li et al. 2006; Lin et al. 2007), suggesting that K = 6 model is overfitting. To further investigate this issue, we randomly sampled three samples from each domesticated rice population assigned to different ancestries under the K = 6 model and PCR amplified the sh4 locus in these accessions. They turned out in all cases to harbor the domesticated allele at the causal variant site. Consequently, we proceeded to use the K = 5 model for allele identification of the domestication haplotype. Wild rice samples with at least 95% domesticated ancestry at the locus were inferred to carry the domesticated allele. For the PROG1 locus, there is also a strong selective sweep (He et al. 2011), and all domesticated rice share identical haplotypes in this region (Tan et al. 2008). We thus applied the same procedures to identify introgression at this locus. For both genes, we confirmed that the domesticated haplotypes identified from the wild rice population contained the domesticated allele at the functional SNP site through PCR amplification (Supplemental Text S6; Supplemental Table S1).
Tajima's D and θπ statistics were calculated under a probabilistic framework designed for low-coverage data (Korneliussen et al. 2013). The methods are implemented in ANGSD and can be invoked by parameter “-doThetas.” dXY between populations was calculated in 10-kb nonoverlapping windows. For each window, dXY values were calculated for all paired polymorphic sites and then averaged over sites. For each polymorphic site, the allele frequencies in population X and Y are denoted as pX and qX, and pY and qY, respectively, and dXY for a site is calculated as dXY = pXqY + pYqX.
Genotype calling and local ancestry inference
To infer the local ancestry in admixed wild rice genomes, we first set up reference wild, temperate japonica, aus and indica panels. Under the K = 9 admixture model, wild rice individuals whose ancestry were inferred to be ≥80% from one of four wild rice specific components, and which contained neither prog1 nor domesticated allele of sh4, were used in the reference wild rice panel. Domesticated rice with ≥80% inferred ancestry from one of indica, aus, or temperate japonica was used as reference indica, aus, or temperate japonica panel, respectively. Wild rice samples with combined ancestry of indica, aus, and temperate japonica ≥20% were included as admixed accessions. Local ancestry assignment was performed on admixed rice genomes with RFMix (Maples et al. 2013). Since this algorithm uses haplotypes as input, we called genotypes in both admixed and reference panel samples with ANGSD (Korneliussen et al. 2014). Imputation and phasing was further performed on the data sets with BEAGLE (version 3.3.2) (Browning and Browning 2007).
Selection detection in feral rice
We calculated FST between the Or-E and aus populations for all rice genes using ngsTools (Fumagalli et al. 2014), which calculates FST using genotype likelihoods, taking genotyping uncertainty into account. We performed GO analysis on the ranked gene list based on the FST values: GO annotation of all rice genes was downloaded from Gramene (http://www.gramene.org/; release 49), the enrichment of each GO was tested using Fisher's exact test, corrected for multiple tests using a Bonferroni correction. The significantly enriched GO terms for the top 5% FST genes can be found on Supplemental Table S3. Nucleotide diversity reduction in both aus and Or-E genomes was estimated by comparing with diversity in wild rice populations. The diversity for each population was estimated using the “-doThetas” command in ANGSD (Korneliussen et al. 2014).
Data access
PCR-amplified sequences have been submitted to NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) under accession numbers KY701787-KY701861 (for sh4) and KY701862-KY701970 (for PROG1).
Supplementary Material
Acknowledgments
This work was supported by grants from the Chinese Academy of Sciences (XDA08010401) and National Natural Science Foundation of China (31430063). J.C. was supported by a National Institutes of Health Ruth L. Kirschstein National Research Service Award. We thank Qi Feng and Bin Han from National Center for Gene Research, Chinese Academy of Sciences for providing wild rice DNA for PCR assay. We thank Yunhua Xiao for suggestions on PCR conditions and primer design.
Author contributions: R.N. and C.C. supervised the project. H.W., F.G.V., and J.C. performed the analysis. H.W. and R.N. wrote the manuscript with critical input from all authors. All authors read and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.204800.116.
Freely available online through the Genome Research Open Access option.
References
- Agrama H, Yan W, Lee F, Fjellstrom R, Chen MH, Jia M, McClung A. 2009. Genetic assessment of a mini-core subset developed from the USDA rice genebank. Crop Sci 49: 1336–1346. [Google Scholar]
- Alexander DH, Novembre J, Lange K. 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19: 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar DS, Khush GS. 1997. Alien introgression in rice. In Oryza: from molecule to plant (ed. Sasaki T, Moore G), pp. 35–47. Springer, Dordrecht. [PubMed] [Google Scholar]
- Browning SR, Browning BL. 2007. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet 81: 1084–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caicedo AL, Williamson SH, Hernandez RD, Boyko A, Fledel-Alon A, York TL, Polato NR, Olsen KM, Nielsen R, McCouch SR, et al. 2007. Genome-wide patterns of nucleotide polymorphism in domesticated rice. PLoS Genet 3: 1745–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TT. 2003. Origin, domestication, and diversification. In Rice: origin, history, technology, and production (ed. Smith CW, Dilday RH), pp. 3–25. Wiley, Hoboken, NJ. [Google Scholar]
- Cheng C, Motohashi R, Tsuchimoto S, Fukuta Y, Ohtsubo H, Ohtsubo E. 2003. Polyphyletic origin of cultivated rice: based on the interspersion pattern of SINEs. Mol Biol Evol 20: 67–75. [DOI] [PubMed] [Google Scholar]
- Civáň P, Craig H, Cox CJ, Brown TA. 2015. Three geographically separate domestications of Asian rice. Nat Plants 1: 15164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethloff HC. 2003. American rice industry: historical overview of production and marketing. In Rice: origin, history, technology, and production (ed. Smith CW, Dilday RH), pp. 67–86. Wiley, Hoboken, NJ. [Google Scholar]
- Ellstrand NC, Meirmans P, Rong J, Bartsch D, Ghosh A, de Jong TJ, Haccou P, Lu BR, Snow AA, Stewart CN, et al. 2013. Introgression of crop alleles into wild or weedy populations. Annu Rev Ecol Evol Syst 44: 325–345. [Google Scholar]
- Ferrero A. 2003. Weedy rice, biological features and control. In Weed management for developing countries. FAO Plant Production and Protection Paper, pp. 89–107. FAO, Rome, Italy. [Google Scholar]
- Fuller DQ, Sato YI, Castillo C, Qin L, Weisskopf AR, Kingwell Banham EJ, Song J, Ahn SM, Van Etten J. 2010. Consilience of genetics and archaeobotany in the entangled history of rice. Archaeol Anthropol Sci 2: 115–131. [Google Scholar]
- Fumagalli M, Vieira FG, Linderoth T, Nielsen R. 2014. ngsTools: methods for population genetics analyses from next-generation sequencing data. Bioinformatics 30: 1486–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao LZ, Innan H. 2008. Nonindependent domestication of the two rice subspecies, Oryza sativa ssp. indica and ssp. japonica, demonstrated by multilocus microsatellites. Genetics 179: 965–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris AJ, Tai TH, Coburn J, Kresovich S, McCouch S. 2005. Genetic structure and diversity in Oryza sativa L. Genetics 169: 1631–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaszmann JC. 1987. Isozymes and classification of Asian rice varieties. Theor Appl Genet 74: 21–30. [DOI] [PubMed] [Google Scholar]
- Harlan JR. 1992. Crop and man. American Society of Agronomy, Crop Science Society of America, Madison, WI. [Google Scholar]
- Harlan JR, De Wet JM. 1971. Toward a rational classification of cultivated plants. Taxon 20: 509–517. [Google Scholar]
- He Z, Zhai W, Wen H, Tang T, Wang Y, Lu X, Greenberg AJ, Hudson RR, Wu CI, Shi S. 2011. Two evolutionary histories in the genome of rice: the roles of domestication genes. PLoS Genet 7: e1002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Molina J, Flowers JM, Rubinstein S, Jackson SA, Purugganan MD, Schaal BA. 2012a. Phylogeography of Asian wild rice, Oryza rufipogon: a genome-wide view. Mol Ecol 21: 4593–4604. [DOI] [PubMed] [Google Scholar]
- Huang X, Kurata N, Wei X, Wang ZX, Wang A, Zhao Q, Zhao Y, Liu K, Lu H, Li W, et al. 2012b. A map of rice genome variation reveals the origin of cultivated rice. Nature 490: 497–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin J, Huang W, Gao JP, Yang J, Shi M, Zhu MZ, Luo D, Lin HX. 2008. Genetic control of rice plant architecture under domestication. Nat Genet 40: 1365–1369. [DOI] [PubMed] [Google Scholar]
- Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, Tanaka T, Wu J, Zhou S, et al. 2013. Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khush GS. 1997. Origin, dispersal, cultivation and variation of rice. In Oryza: from molecule to plant, pp. 25–34. Springer, NY. [PubMed] [Google Scholar]
- Kim H, Jung J, Singh N, Greenberg A, Doyle JJ, Tyagi W, Chung JW, Kimball J, Hamilton RS, McCouch SR. 2016. Population dynamics among six major groups of the Oryza rufipogon species complex, wild relative of cultivated Asian rice. Rice 9: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi S, Izawa T, Lin SY, Ebana K, Fukuta Y, Sasaki T, Yano M. 2006. An SNP caused loss of seed shattering during rice domestication. Science 312: 1392–1396. [DOI] [PubMed] [Google Scholar]
- Korneliussen TS, Moltke I, Albrechtsen A, Nielsen R. 2013. Calculation of Tajima's D and other neutrality test statistics from low depth next-generation sequencing data. BMC Bioinformatics 14: 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korneliussen TS, Albrechtsen A, Nielsen R. 2014. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15: 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Zhou A, Sang T. 2006. Rice domestication by reducing shattering. Science 311: 1936–1939. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Griffith ME, Li X, Zhu Z, Tan L, Fu Y, Zhang W, Wang X, Xie D, Sun C. 2007. Origin of seed shattering in rice (Oryza sativa L.). Planta 226: 11–20. [DOI] [PubMed] [Google Scholar]
- Londo J, Schaal B. 2007. Origins and population genetics of weedy red rice in the USA. Mol Ecol 16: 4523–4535. [DOI] [PubMed] [Google Scholar]
- Londo JP, Chiang YC, Hung KH, Chiang TY, Schaal BA. 2006. Phylogeography of Asian wild rice, Oryza rufipogon, reveals multiple independent domestications of cultivated rice, Oryza sativa. Proc Natl Acad Sci 103: 9578–9583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu BR, Zheng K, Qian H, Zhuang J. 2002. Genetic differentiation of wild relatives of rice as assessed by RFLP analysis. Theor Appl Genet 106: 101–106. [DOI] [PubMed] [Google Scholar]
- Lunter G, Goodson M. 2011. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res 21: 936–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maples BK, Gravel S, Kenny EE, Bustamante CD. 2013. RFMix: a discriminative modeling approach for rapid and robust local-ancestry inference. Am J Hum Genet 93: 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina J, Sikora M, Garud N, Flowers JM, Rubinstein S, Reynolds A, Huang P, Jackson S, Schaal BA, Bustamante CD, et al. 2011. Molecular evidence for a single evolutionary origin of domesticated rice. Proc Natl Acad Sci 108: 8351–8356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno Estrada A, Gravel S, Zakharia F, McCauley JL, Byrnes JK, Gignoux CR, Ortiz Tello PA, Martínez RJ, Hedges DJ, Morris RW, et al. 2013. Reconstructing the population genetic history of the Caribbean. PLoS Genet 9: e1003925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima H. 2002. Reports of the study-tours for investigation of wild and cultivated rice species. Part II, pp. 196–199. https://shigen.nig.ac.jp/rice/oryzabase/ricereport/. [Google Scholar]
- Oka HI. 1988. Origin of cultivated rice. Elsevier, New York. [Google Scholar]
- Pickrell JK, Pritchard JK. 2012. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet 8: e1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pool JE, Nielsen R. 2009. Inference of historical changes in migration rate from the lengths of migrant tracts. Genetics 181: 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusadee T, Schaal BA, Rerkasem B, Jamjod S. 2013. Population structure of the primary gene pool of Oryza sativa in Thailand. Genet Resour Crop Evol 60: 335–353. [Google Scholar]
- Pusadee T, Jamjod S, Rerkasem B, Schaal B. 2016. Life-history traits and geographical divergence in wild rice (Oryza rufipogon) gene pool in Indochina Peninsula region. Ann Appl Biol 168: 52–65. [Google Scholar]
- Rakshit S, Rakshit A, Matsumura H, Takahashi Y, Hasegawa Y, Ito A, Ishii T, Miyashita NT, Terauchi R. 2007. Large-scale DNA polymorphism study of Oryza sativa and O. rufipogon reveals the origin and divergence of Asian rice. Theor Appl Genet 114: 731–743. [DOI] [PubMed] [Google Scholar]
- R Core Team. 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. [Google Scholar]
- Sang T, Ge S. 2007. The puzzle of rice domestication. J Integr Plant Biol 49: 760–768. [Google Scholar]
- Second G. 1982. Origin of the genic diversity of cultivated rice (Oryza spp.): study of the polymorphism scored at 40 isozyme locil). Jpn J Genet 57: 25257. [Google Scholar]
- Skotte L, Korneliussen TS, Albrechtsen A. 2013. Estimating individual admixture proportions from next generation sequencing data. Genetics 195: 693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song BK, Chuah TS, Tam SM, Olsen KM. 2014. Malaysian weedy rice shows its true stripes: wild Oryza and elite rice cultivars shape agricultural weed evolution in Southeast Asia. Mol Ecol 23: 5003–5017. [DOI] [PubMed] [Google Scholar]
- Sun C, Wang X, Atsushi Y, Kazuyki D, Nobuo I. 1996. RFLP analysis of nuclear DNA in common wild rice (O. rufipogon Griff.) and cultivated rice (O. sativa L.). Scientia Agricultura Sinica 30: 37–44. [Google Scholar]
- Sweeney MT, Thomson MJ, Cho YG, Park YJ, Williamson SH, Bustamante CD, McCouch SR. 2007. Global dissemination of a single mutation conferring white pericarp in rice. PLoS Genet 3: e133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L, Li X, Liu F, Sun X, Li C, Zhu Z, Fu Y, Cai H, Wang X, Xie D, et al. 2008. Control of a key transition from prostrate to erect growth in rice domestication. Nat Genet 40: 1360–1364. [DOI] [PubMed] [Google Scholar]
- Vaughan DA, Sanchez PL, Ushiki J, Kaga A, Tomooka N. 2005. Asian rice and weedy rice evolutionary perspectives. In Crop ferality and volunteerism (ed. Gressel J), pp. 257–277. CRC, Boca Raton, FL. [Google Scholar]
- Vaughan DA, Lu BR, Tomooka N. 2008. The evolving story of rice evolution. Plant Sci 174: 394–408. [Google Scholar]
- Vieira FG, Fumagalli M, Albrechtsen A, Nielsen R. 2013. Estimating inbreeding coefficients from NGS data: impact on genotype calling and allele frequency estimation. Genome Res 23: 1852–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Xu X, Vieira FG, Xiao Y, Li Z, Wang J, Nielsen R, Chu C. 2016. The power of inbreeding: NGS based GWAS of rice reveals convergent evolution during rice domestication. Mol Plant 9: 975–985. [DOI] [PubMed] [Google Scholar]
- Yano M, Katayose Y, Ashikari M, Yamanouchi U, Monna L, Fuse T, Baba T, Yamamoto K, Umehara Y, Nagamura Y, et al. 2000. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 12: 2473–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Ge S. 2005. Phylogenetic relationships among A-genome species of the genus Oryza revealed by intron sequences of four nuclear genes. New Phytol 167: 249–265. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Zheng X, Luo J, Gaut BS, Ge S. 2007. Multilocus analysis of nucleotide variation of Oryza sativa and its wild relatives: severe bottleneck during domestication of rice. Mol Biol Evol 24: 875–888. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Ellstrand NC, Lu BR. 2012. Sequence polymorphisms in wild, weedy, and cultivated rice suggest seed-shattering locus sh4 played a minor role in Asian rice domestication. Ecol Evol 2: 2106–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziska LH, Gealy DR, Burgos N, Caicedo AL, Gressel J, Lawton Rauh AL, Avila LA, Theisen G, Norsworthy J, Ferrero A, et al. 2015. Weedy (red) rice: an emerging constraint to global rice production. Adv Agron 129: 181–228. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.