

Abstract
Resequencing efforts are uncovering the extent of genetic variation in humans and provide data to study the evolutionary processes shaping our genome. One recurring puzzle in both intra- and inter-species studies is the high frequency of complex mutations comprising multiple nearby base substitutions or insertion-deletions. We devised a generalized mutation model of template switching during replication that extends existing models of genome rearrangement and used this to study the role of template switch events in the origin of short mutation clusters. Applied to the human genome, our model detects thousands of template switch events during the evolution of human and chimp from their common ancestor and hundreds of events between two independently sequenced human genomes. Although many of these are consistent with a template switch mechanism previously proposed for bacteria, our model also identifies new types of mutations that create short inversions, some flanked by paired inverted repeats. The local template switch process can create numerous complex mutation patterns, including hairpin loop structures, and explains multinucleotide mutations and compensatory substitutions without invoking positive selection, speculative mechanisms, or implausible coincidence. Clustered sequence differences are challenging for current mapping and variant calling methods, and we show that many erroneous variant annotations exist in human reference data. Local template switch events may have been neglected as an explanation for complex mutations because of biases in commonly used analyses. Incorporation of our model into reference-based analysis pipelines and comparisons of de novo assembled genomes will lead to improved understanding of genome variation and evolution.
Mutations are not evenly distributed in genome sequences. Base substitutions and short insertions and deletions (“indels,” up to tens of base pairs in length) usually reflect errors in DNA replication and/or repair (Gu et al. 2008) and tend to form clusters (e.g., Averof et al. 2000; Whelan and Goldman 2004; Harris and Nielsen 2014; Sudmant et al. 2015). Explanations for these monogenic point mutation clusters (subsequently referred to as simply “mutation clusters”) vary from an error-prone polymerase (Harris and Nielsen 2014) to indels being mutagenic (Tian et al. 2008; for review, see Ségurel et al. 2014).
In contrast to such short mutation clusters, genomic rearrangements are defined as gross DNA changes, typically thousands to millions of base pairs and covering multiple different genes (Gu et al. 2008). Although difficult to study using traditional genome sequencing methods, they have recently become the focus of intense research (e.g., Pendleton et al. 2015; Sudmant et al. 2015) due to the advent of next-generation sequencing (NGS) techniques and the importance of their effects in both somatic and germ cells, causing cancers and genetic diseases. Earlier mechanisms proposed to explain genomic rearrangements involved recombination, in particular nonallelic homologous recombination (NAHR) and nonhomologous end-joining (NHEJ) (for review, see Gu et al. 2008; Carvalho and Lupski 2016). More recently, replication-based mechanisms such as serial replication slippage (SRS) (Chen et al. 2005a,b,c), break-induced replication (BIR) (Morrow et al. 1997), fork stalling and template switching (FoSTeS) (Lee et al. 2007), and microhomology-mediated break-induced replication (MMBIR) (Hastings et al. 2009a; Sakofsky et al. 2015) have been proposed.
Common to all these mechanisms is that during replication, the 3′ end of the nascent DNA strand dissociates from the original template and invades another (physically close) open replication fork. A segment is incorporated using this new template until the strand dissociates again. Replication may continue through a complex series of such template “switch-and-return” events; eventually, the nascent DNA reassociates with the original template and replication proceeds as normal. Complex examples with multiple switches have been convincingly demonstrated by Chen et al. (2005a) and Lee et al. (2007). Mutations attributed to these replicative repair mechanisms have been associated with long-range template switch events, in which the nascent DNA changes template between distinct replication forks and the inserted segments derive from genomic regions thousands to millions of base pairs distant (Lee et al. 2007) or even from other chromosomes (Chen et al. 2005a,c; Smith et al. 2007), and typically involve major genomic rearrangements (Costantino et al. 2013; Carvalho and Lupski 2016).
Gu et al. (2008) and Carvalho and Lupski (2016) suggest that replication-based genome rearrangement mechanisms could be responsible for both small-scale and large-scale mutations, and their implications for evolution have yet to be investigated. We hypothesized that a generalized model of genome mutation that encompassed the consequences of replication-based mechanisms such as serial replication slippage (SRS), break-induced replication (BIR), fork stalling and template switching (FoSTeS), and microhomology-mediated break-induced replication (MMBIR) might be able to account for the observation of mutation clusters in higher organisms more parsimoniously than invoking a process of successive base substitutions and indels in a small region (i.e., no more than a few tens of base pairs).
Short-range events, involving template switches within the same replication fork and thus with inserted segments deriving from regions nearby in primary sequence, have been considered previously in a limited manner as a possible explanation of mutation clusters. In bacteria, mutations creating perfect inverted repeats occur with high frequency (Dutra and Lovett 2006) and are thought to involve intra-strand template switching, in which the nascent strand is itself used as the template, or inter-strand template switching, in which the strand complementary to the original template is used (Fig. 1A,B; Ripley 1982, 1990; Sinden and Wells 1992). Such template switching is believed to require a preexisting near-perfect inverted repeat, which is converted into a perfect inverted repeat within the nascent strand by the use of complementary sequence for the transient template.
Figure 1.

Classic template switch mechanism and the new four-point model. (A,B) The classic template switch mechanism creates perfect inverted repeats. (A) DNA replication (blue arrow) exchanges template and converts a nearly perfect inverted repeat (dashed red arrows) into a perfect one (solid red arrows), causing a cluster of differences (bulge, bottom); this can happen by an intra-strand (left) or an inter-strand (right) switch. (B) An inter-strand switch may invert the spacer of the repeat (black dots). (C,D) Our new four-point model generalizes the template switch mutation process while remaining compatible with the classic model proposed by Ripley (1982): C describes both cases of A, and D is consistent with B. Template exchanges are described with four switch points (labeled ①–④) projected onto a reference sequence (R). The points define three sequence fragments (F1–F3) which, when concatenated, create a mutated output. F1 and F3 are copied from R; F2 is copied complementary to either F1 (intra-strand switch) or R (inter-strand switch). (E,F) Examples of mutation clusters compatible with the new model. The template switches (top) can perfectly explain complex mutations observed in real data (bottom; mismatches shown in lower case in the human sequence). (E) Event “3-2-1-4,” named for the order of the switch points along R, creates an inverted repeat (bottom; red arrows) (Link to the original data: http://grch37.ensembl.org/Homo_sapiens/Location/Compara_Alignments?align=548;r=11:133333935-133333985). (F) Event “3-1-2-4” creates an inverted repeat (red arrows) separated by an inverted spacer (dotted line) (Link to original data: http://grch37.ensembl.org/Homo_sapiens/Location/Compara_Alignments?align=548;r=12:74744810-74744853). (G,H) Predicted secondary structures generated by the inverted repeats created in the human sequences, E and F, respectively.
Under this model, both intra- and inter-strand template switch types can cause sequence changes within the repeat (Fig. 1A), and the latter can additionally invert the “spacer” sequence (the region between the repeat fragments) (Fig. 1B). Although these changes can appear as clusters of differences (Dutra and Lovett 2006) and have been detected in genes implicated in human genetic disease (Chen et al. 2005b), this bacterial-style mechanism has not been considered significant in the evolution of higher organisms (Ladoukakis and Eyre-Walker 2008). These conclusions were based on limited data, however, and on an assumption that the mechanism necessarily creates perfect inverted repeats. We compared human and chimp genomes and observed mutation clusters that create novel inverted repeats consistent with the bacterial mechanism. Many clusters could only partially be explained by the creation of an inverted repeat, however, and novel repeats were often flanked by indels or dissimilar sequence, inconsistent with the classical model.
Even with the underlying biological mechanism uncertain, we realized that the existence and properties of a template switch mutation process, capable of creating inverted repeats, could be studied using pairs of closely related genome sequences. In this study, we devised the “four-point model” of template switching and implemented a computational tool for genome-wide searches for mutational patterns consistent with this model. We applied this to alignments of human and chimp and to multiple human genome sequences in order to see whether template switch events have a role in the origin of short mutation clusters and whether current reference-based NGS mapping strategies for population resequencing data give an unbiased picture of clustered mutations. Our analysis detects many such template switch events and calls into question the accuracy of current resequencing strategies.
Results
Four-point model of template switching
Any single template switch-and-return event can be described by a model that projects four sequence positions onto a reference sequence and then constructs a replication copy from the three fragments defined by these points. For convenience, we describe the process as involving the nascent leading strand; the model equally well describes events corresponding to the lagging strand. We have implemented the model with the assumption that template switches are short-range (i.e., use the same replication fork) and involve “jumps” in the replication process to use a template strand other than the original one (“replication slippage in trans” in the terminology of Chen et al. 2005b). This can be the nascent DNA strand itself (intra-strand switching) or the lagging strand (inter-strand switching). We do not attempt to use the model to explain long-range template switches or multiple successive rounds of template switching. Although the four-point model could in principle be extended to cover these possibilities, including all of the outcomes that may arise from the SRS, BIR, FoSTeS, and MMBIR mechanisms, it would be computationally intractable and unlikely to find compelling examples given that essentially the entire genome would be available as the possible explanation of a relatively small number of nearby base substitutions and indels.
Our four-point model of template switching, based on short-range switch-and-return events, is illustrated in Figure 1C–F. Assuming that replication proceeds from left (Ⓛ) to right (®), points ① and ② indicate the location of the first switch event with the nascent strand dissociating from the leading strand at location ① and continuing at ② (lagging strand, or equivalent location on the nascent strand). Similarly, the second (return) switch event comprises a second dissociation taking place at ③ and reassociation with the leading strand at ④. The replication copy then consists of fragments Ⓛ→①, ②→③ (complemented, note), ④→® (Fig. 1E,F). If fragment ②→③ overlaps with fragment Ⓛ→① or ④→®, the mutation creates a novel inverted repeat that then may be capable of forming a RNA secondary structure (Fig. 1G,H).
Modeling the template switch process like this has two major advantages. First, it allows for a formal analysis of mutation events and their evaluation in comparison to alternative explanations. Second, our description of the process is general and has few a priori constraints for the template exchanges. We make no assumptions about the causes or mechanisms of template switching, and our projection of switch points onto a reference is impartial regarding the type of the switch event, either intra- or inter-strand: the model only requires that the ②→③ fragment is copied in reverse-complement orientation. The possible outcomes under the four-point model are defined by the relative order and distance of the switch points, and the classical mechanism proposed to explain inverted repeats in bacteria (Ripley 1982, 1990) is a special case of our generalized model (Fig. 1A–D). Supplemental Figure S1 illustrates all the possible cases under the model, covering the scenarios described before (Fig. 1A,B) as well as several others, including creation of inverted and direct repeats flanked by dissimilar sequence and one case causing inversion of a sequence fragment only. For creation of mutation clusters, an important characteristic of the model is that replacement of the ①→④ fragment with the reverse-complement of the ②→③ fragment by a single switch event can generate changes that, when viewed in a linear alignment, will appear as multiple nearby substitutions and indels.
Application of the four-point model
To test whether biological data support the proposed model as an explanation of mutation clusters in the human genome, we implemented a computational tool based on a custom dynamic programming (dp) algorithm. The tool identifies clusters of differences between two aligned genomic sequences and then searches for an explanation of the region of dissimilarity in one sequence (replicate output) by copying a fragment from the other sequence (reference) in reverse-complement orientation, as achieved in the four-point model. With two closely related sequences, parallel mutation will be rare, and we arbitrarily designate one sequence as the reference and assume that it represents the ancestral form around each mutation event in the replicate lineage. For full details of the dp algorithm used to determine the optimal four-point model explanation for each mutation cluster, see Methods, Supplemental Figure S2, and Supplemental Algorithm S1. The tool is computationally tractable for genome-wide searches.
We focused on the complex and unique regions of human and chimp genomes and, for every mutation cluster of two or more nonidentical bases within a 10-bp window, compared the solution involving a template switch to the original linear sequence alignment. Due to the low complexity of four-base DNA sequences, short local matches might be found for any region containing a cluster of base substitutions and indels, creating the appearance of a template switch event. To assess the rate of such false positives, we computed the best solutions explaining the dissimilar sequence regions with the fragment ②→③ copied in reverse (i.e., not reverse-complement) orientation. The underlying assumption of this false positive model is that there is no biological mechanism that copies DNA in a reverse manner and any sequence fragment appearing as such must have been generated by random substitutions. The prevalence of such “false copy-events” is a proxy for reverse-complement copy events appearing by chance, i.e., false positives. Supplemental Algorithm S1 also describes the modification of our dp algorithm to compute this control.
Based on the sequence context of each candidate event (Methods), we filtered a set of high-confidence template switch events for a more detailed analysis.
Discovery of four-point mutation events from human-chimp data
We first applied our model to genome-wide Ensembl EPO alignments (v.71, six primates) of human and chimp (Paten et al. 2008; Flicek et al. 2013), considering the chimp sequence the reference and the human sequence the mutated copy. The portion of human–chimp alignment data not masked as repeats or low-complexity sequence (48.5% of total length) (Methods) contains 14.51 × 106 base differences and 1.19 × 106 indels. Of these, 3.84 × 106 base differences (26.4%) and 0.76 × 106 indels (63.9%) are within mutation clusters consisting of multiple nearby base differences or alignment gaps. Using our computational tool, we found 4778 candidate four-point mutation events, spread across all human chromosomes, overlapping with 11,723 base differences and 1288 indels, or 0.31% and 0.17% of total clustered unmasked events, respectively. We considered the possibility that some events might be false positives caused by errors in the assembly of the human or chimp genomes. However, every case we inspected in subsequent analyses of human resequencing data (see below) was confirmed as reliable assembly.
Some candidate events were consistent with the original mechanism proposed for bacteria (Ripley 1982, 1990) and convert a near-perfect inverted repeat into a perfect one (for example, see Fig. 2A,B), but the majority were associated with large sequence changes, causing multiple base differences and indels in linear alignments (Fig. 2C,D). Although any complex mutation could be generated by a combination of simple, “traditional,” mutations, Occam's razor suggests that a four-point model template switch mutation is a better explanation than multiple substitutions and indels occurring in such a cluster. However, we also noticed that matches shorter than 12–13 bp are often found by chance (Supplemental Figs. S3, S4) and, despite strict filtering (Methods), our list of candidate events might still contain false positives. To get an unbiased picture of the process, we removed events with ②→③ fragment shorter than 14 bp. This was done to improve the signal to noise ratio and does not mean that short template switch events could not happen; in contrast, many cases with a short ②→③ fragment appear highly convincing (Fig. 1E).
Figure 2.

Example events detected in human. (A) A near-perfect inverted repeat in chimp (dashed black arrows, the one mismatch indicated with asterisks) has been converted into a perfect inverted repeat (red arrows) in human (top). The cluster of six additional dissimilarities (dotted line) in fact represents perfect inversion of the 6-bp spacer sequence and makes the template switch (bottom) a likely explanation (Original data: http://grch37.ensembl.org/Homo_sapiens/Location/Compara_Alignments?align=548;r=2:85464419-85464489). (B) Predicted DNA secondary structure before (chimp; bottom) and after (human; top) the template switch event. The dotted arrows indicate the reverse-complemented spacer region, which the four-point model explains with a single event. (C,D) Additional complex mutation patterns (mismatches in lower case) that can be explained by a single template switch event. (C) Event “1-3-2-4” only converts the spacer sequence (http://grch37.ensembl.org/Homo_sapiens/Location/Compara_Alignments?align=548;r=9:113151972-113152067). (D) Event “3-1-4-2” converts the spacer sequence and creates two inverted repeats (red and magenta arrows) (http://grch37.ensembl.org/Homo_sapiens/Location/Compara_Alignments?align=548;r=2:135684492-135684613).
After this filtering, we assigned the 794 remaining candidate events to specific event types based on the relative positions of the switch points and computed their frequencies. We found that, of the 12 possible conformations of switch points, only six are present (Table 1, human versus chimp comparison). Of these, two event pairs are “mirror cases” indistinguishable from one another if both leading and lagging strand replication are considered (Supplemental Fig. S1), and the six conformations observed therefore define four distinct switch event types. Type “1-4-3-2” (with its mirror case “3-2-1-4”) (Supplemental Fig. S1a; Fig. 1F) creates an inverted repeat and accounts for 31% of the high-confidence events detected in the chimp–human comparison. Type “1-3-4-2” (and mirror case “3-1-2-4” ) (Supplemental Fig. S1a; Fig. 1F) creates an inverted repeat separated by an inverted spacer sequence and accounts for 23% of events. The remaining two types are novel and only achievable under our four-point model: type “1-3-2-4,” accounting for 45% of events, only inverts a sequence fragment and creates no repeat (Fig. 2C), and type “3-1-4-2” creates two inverted repeats separated by an inverted spacer (Fig. 2D) and accounts for 1% of events.
Table 1.
Proportion of event types
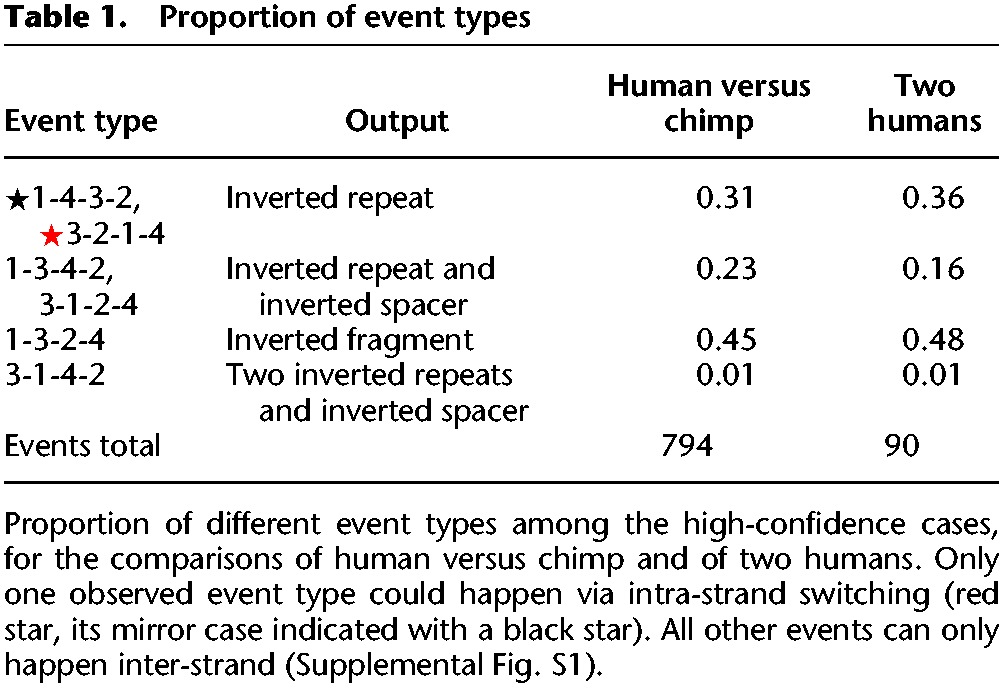
The unifying feature of the event types theoretically possible under the model but not observed in real sequence data is that in the ordering of the switch points, ④ precedes ①. This would be the hallmark of an event in which the second (return) template switch requires the opening of the newly synthesized DNA double helix (Supplemental Fig. S1). In addition, we observe numerous cases of inversion of spacer sequences; this cannot occur when ② precedes ①, a prerequisite of intra-strand switches. These discoveries suggest that template switches occur inter-strand, that is, the fragment ②→③ is copied from the opposite strand (Fig. 1A,B).
Although inversions of spacer sequences have been observed in bacteria (Ripley 1982, 1990), the intra-strand mechanism has been the dominant hypothesis (Dutra and Lovett 2006). It appears that this is not correct, at least for evolution since the human–chimp divergence. We also find that the relative frequencies of different event types are very different. In part this may be determined by factors such as the length distribution of the copied fragment (Supplemental Fig. S3) and type “3-1-4-2” requiring that the fragment ②→③ overlaps with both ① and ④. However, the frequencies of different event types may also reflect the properties of the mutation process, e.g., template switching benefiting from the proximity of the DNA strands, or the chance of the new mutation escaping error correction (Sinden and Wells 1992).
Identification of polymorphic mutations in human data
To understand whether template switch events are actively shaping human genomes, we analyzed human resequencing data and searched for polymorphic loci. We first aligned the human reference genome GRCh37 (International Human Genome Sequencing Consortium 2004) to that of a male of predominantly European ancestry (denoted HuRef) (Levy et al. 2007), both based on classical capillary sequencing and assembled independently. We then considered HuRef as the reference and identified clusters of mutations in GRCh37 that were consistent with different types of four-point model template switch events. Using the same approach as in the human–chimp comparisons, we identified 267 candidate events in the unmasked portion of the human genome and then selected a smaller set of high-confidence cases for a more detailed analysis (Methods). For these 90 events, the proportions of different event types were similar to those found in human–chimp comparisons. Again, only the six types not requiring opening of the new helix were found, and the majority of events require inter-strand switches (Table 1: two humans comparison).
Still focusing on these 90 candidate events, we manually studied the HuRef sequence data mapped onto the reference genome (Li and Durbin 2011). We could resolve the genotype of HuRef for 76 (84%) of the candidate events and found 40 of them heterozygous, i.e., the sequence data contain reads consistent with both HuRef and GRCh37 alleles (Fig. 3A,B; Supplemental Table S1; Supplemental Data S1). In two cases, the read data revealed that the mutations forming the cluster are not linked and are the result of two independent mutation events (Supplemental Fig. S5), and in the remaining 14 cases, mapping of HuRef sequence reads against GRCh37 was inconsistent with the alignment of genome assemblies; we did not consider these ambiguous mutations any further.
Figure 3.

A template switch mutation event with variable allele frequencies in human populations. (A) Four-point model explanation of a complex mutation between the human reference GRCh37 and HuRef. Notation is as in Figure 1. (B) A subset of the original sequencing reads from HuRef (top) and the 1KG individual NA12878 (bottom). Dots and commas indicate the read matching to the reference on the forward and reverse strand, uppercase and lowercase characters denote the corresponding mismatches, and asterisks mark the alignment gaps. These reads reveal heterozygosity at the locus. (C) The EPO alignment for primates reveals that GRCh37 is the ancestral form. As all other primates resemble the reference allele, the most parsimonious explanation is that the mutation (HuRef) happened in the human lineage since its divergence from the human–chimp ancestor. (D) 1KG variation data explain this event as a cluster of seven single-nucleotide polymorphisms and four indels. The phased genotypes for NA12878 (1|0) indicate that the variant alleles are linked and all in the same haplotype. The single origin of the whole cluster is further supported by the uniform derived allele frequencies across the sites within all 1KG data (AF) and within each superpopulation (AFR, AMR, EAS, EUR, SAS).
We then looked at the same loci in the 1000 Genomes (1KG) data (The 1000 Genomes Project Consortium 2015) and studied the alignment data for individual NA12878. We found that NA12878 has a nonreference allele at 47 of the 76 resolved loci (62%) and, with the exception of the two cases mentioned, all the changes are found within the same sequence reads. This finding has two implications. First, with two different sequencing technologies (capillary and Illumina) and analysis pipelines showing the same mutation patterns, we can reject the possibility that the observed events could be technical artifacts. Second, the agreement of short-read data with assembly based on long capillary reads suggests that the template switch mutation model can be studied using modern resequencing data.
Elimination of mutation accumulation hypothesis
In principle, perfect linkage of adjacent sequence changes in two unrelated individuals could also be explained by mutations being accumulated over a long period of time in complete absence of recombination. To rule that out, we assessed the maximum age of the mutation clusters using phylogenetic information (Fig. 3C). The EPO alignments contain data from at least two additional primate species for 73 loci. The two alleles detected between the two humans GRCh37 and HuRef segregate among the primate species in only one of these loci; in all 72 other cases, all primate sequences resemble one of the two human alleles, whereas the second human allele is unique (Fig. 3C; Supplemental Data S2). Although some loci could be polymorphic in nonhuman primates, the result suggests that a great majority of the events are young, and the adjacent changes within the mutation clusters result from single mutation events.
Mutation clusters in 1000 Genomes variation data
NA12878 is only one individual and, to understand how large a proportion of the template switch mutations are polymorphic in humans, we investigated whether the 76 candidate loci could be detected in population resequencing data. Using the 1KG variant calls (The 1000 Genomes Project Consortium 2015), we found that this is indeed the case: of the 76 confirmed events between GRCh37 and HuRef, the mutation pattern created by the event is completely explained by combinations of the 1KG variants (separate calls of indels and single-nucleotide polymorphisms) at 35 loci, and partially explained at 16 loci. In most of these 51 cases, the mutations at a locus have uniform allele frequencies within human populations and are in near-perfect linkage disequilibrium (standard deviations of allele frequencies predominantly <0.02, D′ values predominantly >0.99) (Supplemental Table S2), further demonstrating the single origin for the full mutation cluster (Fig. 3D; Supplemental Data S1). The variation data confirm the two earlier cases as combinations of independent mutations (Supplemental Fig. S5), but for all other inconsistencies between inferred template switch mutations and the 1KG variation data, the underlying alignment data show the incomplete mutation patterns and the nonuniform allele frequencies to be artifacts from erroneous mapping and variant calling.
For example, Figure 4 shows a locus where HuRef is heterozygous for a template switch mutation that explains an apparent cluster of 22 substitutions and one 17-bp deletion within a 69-bp region. NGS reads from 1KG individuals NA12872 (homozygous for the mutation) and NA12873 (heterozygous) illustrate how this locus has been miscalled in the 1KG analyses, with many sequence differences undetected and the linkage of the detected differences inferred incorrectly (Fig. 4B,D). The underlying reason for these errors is the inability of current mapping software to align short reads containing multiple differences to the reference sequence. This is visible in the mean sequencing coverage across the locus: although individuals matching the reference show even coverage, heterozygotes and homozygotes for the variant allele have drastically lower coverage for the mutation sites (Fig. 4C). Such errors in short read mapping and subsequent variant calling demonstrate the difficulty of correctly detecting complex mutations using current reference-based analysis methods.
Figure 4.

Complex mutation partially called in 1KG data. (A) A mutation pattern explained by a template switch event. (B) Sequence reads from HuRef and 1KG individuals NA12872 and NA12873. Well-aligned capillary reads reveal HuRef as a heterozygote. In the 1KG individuals, the short NGS reads for the variant allele mostly fail to map: based on two indicative sites (blue box), NA12872 is a homozygote and NA12873 is a heterozygote. (C) Mean sequencing depth of all 2504 1KG (phase 3) low-coverage individuals, grouped according to their genotype for the two indicative sites. (D) 1KG variation data, with genotypes for NA12872 (left) and NA12873 (right). Adjacent variants created by a template switch event are transmitted together and are expected to have identical genotypes, giving uniform allele frequencies in population data. This is not the case here, because fewer than half of the differences (colored boxes, corresponding to those in B) are called, and of those, the terminal ones are called with a higher certainty, the mutation pattern is incomplete, and the variant alleles at adjacent positions appear to occur at different frequencies.
Despite highly uniform allele frequencies, the 1KG variant calls consider the template switch events that we identified to be clusters of independent mutations events—the largest clusters consisting of more than 10 apparently independent mutation events (Fig. 4; Supplemental Fig. S6)—and thus seriously exaggerate the estimates of local mutation rate. On the other hand, uniform allele frequencies at adjacent positions indicate a shared history for a mutation cluster and potentially allow computational detection of events. To test this, we investigated whether any of the events found between human and chimp are still polymorphic in humans and associated with a cluster of SNP positions with uniform allele frequencies (Methods). We found several such events, the frequencies of the two haplotypes varying from close to 0 to nearly 1, and the frequencies differing significantly between populations (Supplemental Fig. S7). This finding demonstrates two things: first, a greater number of loci than were detected by a comparison of two human individuals are polymorphic and segregate among human populations; second, if the read mapping and variant calling were perfect, variation data combined with variant sequence reconstruction could be used for de novo computational detection of template switch mutations. As a proof of concept, we applied the method to the parent-offspring data of Besenbacher et al. (2016) and could explain several complex de novo mutations as template switch events (Supplemental Fig. S8).
Discussion
Our generalized template switch model can explain a large number of complex mutation patterns—clusters of apparent base substitutions and indels—with a single mutation event. Although only explaining 0.3% of base differences and 0.2% of indels within mutation clusters (i.e., regions with two or more nonidentical bases within a 10-bp window) in the human–chimp comparison, this is nevertheless a large number of individual events and far exceeds the numbers previously found in higher organisms. Note that our inferences are likely underestimates, because of our strict criteria and filtering. The model is compatible with, and significantly extends, the one previously proposed for bacteria (Ripley 1982, 1990) and described replication-based mechanisms for genome rearrangements such as BIR, SRS, FoSTeS and MMBIR. Unlike previous models for short-range template switching, significant preexisting repeats or sequence similarity are not required and the process can thus create completely novel repeats (Fig. 2; Supplemental Fig. S9). This is consistent with the reported cases of major genomic rearrangements in which identity of only two or three bases is observed at the switch points (Lee et al. 2007; Hastings et al. 2009a; Costantino et al. 2013). We also found no evidence of the intra-strand events of the bacterial model, possibly because they would require breaking of the bonds between the leading strand template and the newly synthesized DNA. On the other hand, the most common event type that we detected, which only inverts a sequence fragment, can only be found by our generalized model.
Mutation frequency is known to vary significantly across genomes (Ségurel et al. 2014). Involvement of DNA polymerase zeta (Pol ζ), an error-prone translesion polymerase, has been suggested to explain regions of elevated rate (Harris and Nielsen 2014), and substitutions at adjacent sites have been taken as evidence of positive selection (Bazykin et al. 2004). When the template switch event does not involve loss or gain of sequence, the mutation pattern that it creates appears as a multinucleotide substitution. Although sequence context, replication timing and gene expression may affect propensity for template switching, the contribution of consequent mutations to the large-scale spatial variation in the mutation rate is likely to be small. Our results demonstrate, however, that local template switch mutation has a significant role in the de novo creation of clusters of adjacent substitutions (Supplemental Fig. S8, cf. Besenbacher et al. 2016) and can explain them without the involvement of selection. Moreover, many template switch events are associated with indels in the alignment (Supplemental Fig. S10), and the process we have identified provides an alternative to the proposition of indels being mutagenic and triggering nearby base substitutions (Tian et al. 2008).
The proposed four-point model has consequences for our understanding of genome evolution and the methods used for studying it. Although template switching is known to have a role in genomic rearrangements (Gu et al. 2008; Hastings et al. 2009b; Costantino et al. 2013; Carvalho and Lupski 2016), our analyses demonstrate that it can also take place in a local context. As such, it provides a one-step mechanism for the generation of hairpin loops and, in combination with other mutations, provides a pathway to more complex secondary structures (Ding et al. 2014; Rouskin et al. 2014; Wan et al. 2014). The model also provides a mechanism for the evolution of existing DNA secondary structures and provides an explanation for the long-standing dilemma of exceptionally high rates for compensatory substitutions (Dixon and Hillis 1993; Tillier and Collins 1998; Meer et al. 2010). Interestingly, the mechanism may also maintain apparent DNA secondary structures without selective force. A number of human disease mutations (Chen et al. 2005a,b,c; Lee et al. 2007; Hastings et al. 2009a; Zhang et al. 2009) have been attributed to events that can be described by our template switch model. We also note that key mutations implicated in the de novo origin of the putative human protein coding gene DNAH10OS (Ensembl ID ENSG00000204626) (Fig. 4 of Knowles and McLysaght 2009, enabling 10-bp insertion CCTCATTCCT and G→A substitution 2 bp downstream; Xie et al. 2012) can be explained by the four-point model.
A probable reason why template switch mutations have not received greater attention may be bias in commonly used analysis methods. Tight clusters of differences, the typical signature of the process, make read mapping and subsequent variant calling challenging. This is demonstrated by phase 3 of the 1KG Project (The 1000 Genomes Project Consortium 2015), which provides significant improvements in comparison to earlier releases but, as we have shown (e.g., Fig. 4), still contains errors and inconsistencies around the regions we have studied. High quality sequence and assembly has been vital to improving understanding of structural variation of genomes (Pendleton et al. 2015; Sudmant et al. 2015). We have shown that improving genome assemblies to the level of individual bases and short indels relative to reference sequences is needed in order to permit correct interpretation of the causes of population-level differences and of the information most commonly used to study intra- and inter-species evolution. Mapping methods that simultaneously consider multiple references (Schneeberger et al. 2009; Maciuca et al. 2016) and improved algorithms for local assembly (Wala et al. 2017) are beginning to become available, and the new mutation model we propose could be modeled and considered in future analyses. With a rapidly growing number of high quality de novo-assembled genomes and improved algorithms for local assembly, the full extent of local template switch events among the mutation processes acting on genomes can be uncovered.
Methods
Discovery of four-point mutations
We downloaded the Ensembl (v.71) EPO alignments (Paten et al. 2008; Flicek et al. 2013) of six primates and included all blocks containing only one human and chimp sequence, covering in total 2.648 Gb of the human sequence and 94.8% of the EPO alignment regions. Keeping only human and chimp sequences, we identified alignment regions where two or more nonidentical bases (mismatches or indels) occur within a 10-bp window. For each such mutation cluster, we considered the surrounding sequence (for human and chimp, respectively, 100 and 200 bp upstream of and downstream from the cluster boundaries), and in accordance with our four-point model attempted to reconstruct the human query from the chimp reference with imperfect copying (allowing for mismatches and indels) of the forward strand and two freely placed template switch events. Candidate switch events were required to have high sequence similarity and, within the ②→③ fragment, only mismatches were allowed. If exact positions of switch events could not be determined (Supplemental Fig. S11), our approach maximized the length of ②→③ fragment and reported this upper limit of the strand-switch event length. For comparison, we reconstructed the human query from the chimp reference with imperfect copying of the forward strand only (i.e., linear alignment) using the same scoring. A custom dynamic programming algorithm to determine the optimal four-point model explanation for each mutation cluster is described in Supplemental Figure S2 and Supplemental Algorithm S1.
Filtering of events
For each mutation cluster, we recorded the coordinates of the inferred template switch events and computed similarity measures for the different parts of the template switch and forward alignments as well as the differences in the inferred numbers of mutations between the two solutions; we also recorded whether the regions include repeatmasked (Smit et al. 2013–2015) or dustmasked (Morgulis et al. 2006) sites, as well as the number of different bases included in the ②→③ fragments. We then selected a set of events as high-confidence candidates using the following criteria: (1) the switch points ① and ④ are at most 30 bp upstream and downstream, respectively, from the cluster boundaries; (2) the ②→③ fragment is at least 10 bp long; (3) the ②→③ fragment as well as 40-bp flanking regions upstream and downstream show at least 95% identity between the sequences; (4) the forward alignment indicates at least two differences (of which at least one a mismatch) more than the template switch alignment (which may also contain up to 5% mismatches); and (5) the ②→③ fragment is not repeatmasked or dustmasked and contains all four bases. As a control to assist in assessing the occurrence of false positives, we repeated the analysis without complementing the ②→③ fragment: no biological function is known for reverse repeats, and we consider them a proxy for the probability of observing a repeat of particular length by chance.
Identification of polymorphic mutations
For comparisons between humans, we use the GRCh37 reference sequence and coordinates. Major differences between GRCh37 and the most recently released reference relate to mtDNA and centromeres, coordinate changes, and alternative haplotypes. These changes have negligible effect on our results. Much of the additional nonreference data that we use (e.g., 1KG) is computed against GRCh37; aligning to this maintains continuity with other papers’ original notation.
The GRCh37 and HuRef sequences were aligned using LASTZ (Harris 2007) and following the UCSC analysis pipeline (Kent et al. 2002). The four-point mutation events were identified using the same approach as with human–chimp data. The 1KG variation data from ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502/ were analyzed using BCFtools (Li 2011), and selected regions of resequencing data from ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data were visualized using SAMtools (Li et al. 2009). Mutation clusters with uniform allele frequencies were identified as follows: (1) 1KG variant calls were extracted for the mutation cluster plus 10 bp of flanking region; (2) for each locus, runs of adjacent positions with <10% difference in global allele frequency (AF) were recorded; and (3) the runs of selected length (e.g., 3) with AF between 0.01 and 0.99 were outputted. The 1KG variant alleles were reconstructed using GATK (McKenna et al. 2010). Short-read alignment data, 1KG variant calls, and primate sequence alignments for the candidate template switch event loci are shown in Supplemental Data S1 and S2.
Reconstruction of de novo mutations
The complex de novo mutations identified in comparison of parent–offspring data are provided only in a spreadsheet format by Besenbacher et al. (2016). We wrote a custom script (Supplemental Methods) that reads the data in this nonstandard tabular format and locates the mutations on NCBI36, the reference sequence used in the original study. We applied this script to clusters that were less than 100 bp in size and reconstructed the mutated copies with 100 bp of flanking sequence. These de novo mutations were then compared to the unaltered NCBI36 reference sequence, and the best explanations involving a template switch event were determined using our dp algorithm.
Other computational analyses
DNA secondary structures were predicted with the ViennaRNA package (Lorenz et al. 2011), using the command “RNAfold --paramFile=dna_mathews2004.par --noconv --noGU”. The length distribution (Supplemental Fig. S3) and the allele frequencies (Fig. 3D) were visualized with R (R Core Team 2014).
Data access
The short-read alignment data and the 1KG variant calls for the candidate template switch event loci detected between two humans are available in Supplemental Data S1, and the primate sequence alignments for the same loci in Supplemental Data S2. The computational tool used for the analyses is available as Supplemental Algorithm S1 and at https://github.com/ariloytynoja/fpa.
Supplementary Material
Acknowledgments
We thank Martin Taylor for help and comments in early stages of the study, Aylwyn Scally and an anonymous reviewer for their constructive feedback on the manuscript, and CSC–IT Center for Science, Finland, for computational resources.
Author contributions: N.G. devised the extended four-point model. A.L. implemented the method and performed the analyses. N.G. and A.L. designed the study, discussed the results, and wrote the manuscript. Both authors read and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.214973.116.
References
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averof M, Rokas A, Wolfe KH, Sharp PM. 2000. Evidence for a high frequency of simultaneous double-nucleotide substitutions. Science 287: 1283–1286. [DOI] [PubMed] [Google Scholar]
- Bazykin GA, Kondrashov FA, Ogurtsov AY, Sunyaev S, Kondrashov AS. 2004. Positive selection at sites of multiple amino acid replacements since rat-mouse divergence. Nature 429: 558–562. [DOI] [PubMed] [Google Scholar]
- Besenbacher S, Sulem P, Helgason A, Helgason H, Kristjansson H, Jonasdottir A, Jonasdottir A, Magnusson OT, Thorsteinsdottir U, Masson G, et al. 2016. Multi-nucleotide de novo mutations in humans. PLoS Genet 12: e1006315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho CM, Lupski JR. 2016. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17: 224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JM, Chuzhanova N, Stenson PD, Férec C, Cooper DN. 2005a. Complex gene rearrangements caused by serial replication slippage. Hum Mutat 26: 125–134. [DOI] [PubMed] [Google Scholar]
- Chen JM, Chuzhanova N, Stenson PD, Férec C, Cooper DN. 2005b. Intrachromosomal serial replication slippage in trans gives rise to diverse genomic rearrangements involving inversions. Hum Mutat 26: 362–373. [DOI] [PubMed] [Google Scholar]
- Chen JM, Chuzhanova N, Stenson PD, Férec C, Cooper DN. 2005c. Meta-analysis of gross insertions causing human genetic disease: novel mutational mechanisms and the role of replication slippage. Hum Mutat 25: 207–221. [DOI] [PubMed] [Google Scholar]
- Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, Haber JE, Iliakis G, Kallioniemi OP, Halazonetis TD. 2013. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343: 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, Assmann SM. 2014. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505: 696–700. [DOI] [PubMed] [Google Scholar]
- Dixon MT, Hillis DM. 1993. Ribosomal RNA secondary structure: compensatory mutations and implications for phylogenetic analysis. Mol Biol Evol 10: 256–267. [DOI] [PubMed] [Google Scholar]
- Dutra BE, Lovett ST. 2006. Cis and trans-acting effects on a mutational hotspot involving a replication template switch. J Mol Biol 356: 300–311. [DOI] [PubMed] [Google Scholar]
- Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, et al. 2013. Ensembl 2013. Nucleic Acids Res 41: D48–D55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Zhang F, Lupski JR. 2008. Mechanisms for human genomic rearrangements. PathoGenetics 1: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris R. 2007. “Improved pairwise alignment of genomic DNA.” PhD thesis, Pennsylvania State University, State College, PA. [Google Scholar]
- Harris K, Nielsen R. 2014. Error-prone polymerase activity causes multinucleotide mutations in humans. Genome Res 24: 1445–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR. 2009a. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5: e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Lupski JR, Rosenberg SM, Ira G. 2009b. Mechanisms of change in gene copy number. Nat Rev Genet 10: 551–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium. 2004. Finishing the euchromatic sequence of the human genome. Nature 431: 931–945. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12: 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles DG, McLysaght A. 2009. Recent de novo origin of human protein-coding genes. Genome Res 19: 1752–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladoukakis ED, Eyre-Walker A. 2008. The excess of small inverted repeats in prokaryotes. J Mol Evol 67: 291–300. [DOI] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR. 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131: 1235–1247. [DOI] [PubMed] [Google Scholar]
- Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G, et al. 2007. The diploid genome sequence of an individual human. PLoS Biol 5: e254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2011. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27: 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2011. Inference of human population history from individual whole-genome sequences. Nature 475: 493–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. 2011. ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciuca S, del Ojo Elias C, McVean G, Iqbal Z. 2016. A natural encoding of genetic variation in a Burrows-Wheeler Transform to enable mapping and genome inference. bioRxiv 10.1101/059170. [DOI] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meer MV, Kondrashov AS, Artzy-Randrup Y, Kondrashov FA. 2010. Compensatory evolution in mitochondrial tRNAs navigates valleys of low fitness. Nature 464: 279–282. [DOI] [PubMed] [Google Scholar]
- Morgulis A, Gertz EM, Schäffer AA, Agarwala R. 2006. A fast and symmetric DUST implementation to mask low-complexity DNA sequences. J Comput Biol 13: 1028–1040. [DOI] [PubMed] [Google Scholar]
- Morrow DM, Connelly C, Hieter P. 1997. “Break copy” duplication: a model for chromosome fragment formation in Saccharomyces cerevisiae. Genetics 147: 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paten B, Herrero J, Fitzgerald S, Beal K, Flicek P, Holmes I, Birney E. 2008. Genome-wide nucleotide-level mammalian ancestor reconstruction. Genome Res 18: 1829–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton M, Sebra R, Pang AWC, Ummat A, Franzen O, Rausch T, Stutz AM, Stedman W, Anantharaman T, Hastie A, et al. 2015. Assembly and diploid architecture of an individual human genome via single-molecule technologies. Nat Methods 12: 780–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. [Google Scholar]
- Ripley LS. 1982. Model for the participation of quasi-palindromic DNA sequences in frameshift mutation. Proc Natl Acad Sci 79: 4128–4132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripley LS. 1990. Frameshift mutation: determinants of specificity. Annu Rev Genet 24: 189–213. [DOI] [PubMed] [Google Scholar]
- Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. 2014. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505: 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakofsky CJ, Ayyar S, Deem AK, Chung WH, Ira G, Malkova A. 2015. Translesion polymerases drive microhomology-mediated break-induced replication leading to complex chromosomal rearrangements. Mol Cell 60: 860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger K, Hagmann J, Ossowski S, Warthmann N, Gesing S, Kohlbacher O, Weigel D. 2009. Simultaneous alignment of short reads against multiple genomes. Genome Biol 10: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ségurel L, Wyman MJ, Przeworski M. 2014. Determinants of mutation rate variation in the human germline. Annu Rev Genomics Hum Genet 15: 47–70. [DOI] [PubMed] [Google Scholar]
- Sinden RR, Wells RD. 1992. DNA structure, mutations, and human genetic disease. Curr Opin Biotechnol 3: 612–622. [DOI] [PubMed] [Google Scholar]
- Smit AF, Hubley R, Green P. 2013–2015. RepeatMasker Open-4.0. http://www.repeatmasker.org/. [Google Scholar]
- Smith CE, Llorente B, Symington LS. 2007. Template switching during break-induced replication. Nature 447: 102–105. [DOI] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Hsi-Yang Fritz M, et al. 2015. An integrated map of structural variation in 2,504 human genomes. Nature 526: 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Wang Q, Zhang P, Araki H, Yang S, Kreitman M, Nagylaki T, Hudson R, Bergelson J, Chen JQ. 2008. Single-nucleotide mutation rate increases close to insertions/deletions in eukaryotes. Nature 455: 105–108. [DOI] [PubMed] [Google Scholar]
- Tillier ER, Collins RA. 1998. High apparent rate of simultaneous compensatory base-pair substitutions in ribosomal RNA. Genetics 148: 1993–2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wala J, Bandopadhayay P, Greenwald N, O'Rourke R, Sharpe T, Stewart C, Schumacher SE, Li Y, Weischenfeldt J, Yao X, et al. 2017. Genome-wide detection of structural variants and indels by local assembly. bioRxiv 10.1101/105080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, Zhang J, Spitale RC, Snyder MP, Segal E, et al. 2014. Landscape and variation of RNA secondary structure across the human transcriptome. Nature 505: 706–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan S, Goldman N. 2004. Estimating the frequency of events that cause multiple-nucleotide changes. Genetics 167: 2027–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C, Zhang YE, Chen JY, Liu CJ, Zhou WZ, Li Y, Zhang M, Zhang R, Wei L, Li CY. 2012. Hominoid-specific de novo protein-coding genes originating from long non-coding RNAs. PLoS Genet 8: e1002942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. 2009. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nat Genet 41: 849–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.