

Abstract
Background
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a lethal inherited arrhythmia syndrome characterized by adrenergically stimulated ventricular tachycardia. Mutations in the cardiac ryanodine receptor gene (RYR2) cause an autosomal dominant form of CPVT, while mutations in the cardiac calsequestrin 2 gene (CASQ2) cause an autosomal recessive form.
Objective
The aim of this study was to clinically and genetically evaluate a large family with severe autosomal dominant CPVT.
Methods
Clinical evaluation of family members was performed, including detailed history, physical examination, electrocardiogram, exercise stress test, and autopsy review of decedents. We performed genome-wide linkage analysis in 12 family members and exome sequencing in 2 affected family members. In silico models of mouse and rabbit myocyte electrophysiology were used to predict potential disease mechanisms.
Results
Severe CPVT with dominant inheritance in 6 members was diagnosed in a large family with 2 sudden deaths, 2 resuscitated cardiac arrests, and multiple appropriate implantable cardioverter-defibrillator shocks. A comprehensive analysis of cardiac arrhythmia genes did not reveal a pathogenic variant. Exome sequencing identified a novel heterozygous missense variant in CASQ2 (Lys180Arg) affecting a highly conserved residue, which cosegregated with disease and was absent in unaffected family members. Genome-wide linkage analysis confirmed a single linkage peak at the CASQ2 locus (logarithm of odds ratio score 3.01; θ = 0). Computer simulations predicted that haploinsufficiency was unlikely to cause the severe CPVT phenotype and suggested a dominant negative mechanism.
Conclusion
We show for the first time that a variant in CASQ2 causes autosomal dominant CPVT. Genetic testing in dominant CPVT should include screening for heterozygous CASQ2 variants.
Keywords: CPVT, Cardiac calsequestrin, Heterozygous, Autosomal dominant
Introduction
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a highly lethal, rare inherited arrhythmia syndrome characterized by ventricular tachycardia (VT), which is adrenergically stimulated, usually by physical exertion.1,2 The prevalence of CPVT is 1 in 10,000 among Europeans.3,4 The diagnostic criteria include a structurally normal heart and normal baseline electrocardiogram (ECG) with the development of polymorphic VT or bidirectional VT after exercise, usually in a patient younger than 40 years.3 CPVT can also be diagnosed in patients with a known pathogenic gene variant and in family members of an index case who develop premature ventricular contractions during exercise.
The pathophysiology of CPVT involves abnormal intra-cellular calcium regulation with subsequent abnormal excitation-contraction coupling and delayed afterdepolariza-tions.5 The most common form of CPVT is due to autosomal dominant variants in the cardiac ryanodine receptor gene (RYR2).6,7 A rare form of autosomal recessive CPVT results from homozygous or compound heterozygous variants in the cardiac calsequestrin 2 gene (CASQ2).8–11 Two reports of small families have proposed that autosomal dominant CASQ2 mutations may cause CPVT.12,13 Additional genes implicated in CPVT include the potassium channel inwardly rectifying subfamily J2 (KCNJ2), ankyrin B (ANK2), triadin (TRDN), and calmodulin 1 (CALM1).14–17
Pathogenic variants in RYR2 and CASQ2 account for up to 65% of CPVT.18 In families where the genetic cause remains elusive after screening of known CPVT genes, next generation sequencing approaches provide a more comprehensive platform for novel gene discovery.19 We show for the first time a heterozygous CASQ2 variant causing autosomal dominant CPVT in a large family with a severe phenotype.
Methods
Clinical evaluation of family members
Clinical evaluation of the family included a detailed history, physical examination, 12-lead ECG, echocardiogram, exercise stress test (EST), medical record review, and postmortem report review of decedents. Children younger than 5 years underwent adrenaline challenge testing at the discretion of the treating pediatric electrophysiologist. All studies were conducted with approval and in strict accordance with the Sydney Local Health District Ethics Review Committee, Australia.
Cardiac arrhythmia gene analyses
Full RYR2 sequencing, including multiplex ligation–dependent probe amplification, was performed by a commercial testing laboratory (Regional Genetics Laboratory, Manchester, UK). CALM1/2/3 sequencing (Aarhus University, Aarhus, Denmark), a 33-gene cardiac arrhythmia panel (Online Supplemental Appendix 1), and mitochondrial genome sequencing were performed (MNG Services, Atlanta, GA). Variants in known CPVT genes were assessed for pathogenicity using American College of Medical Genetics (ACMG) criteria.20
Exome sequencing, variant filtering, and validation
After genomic DNA extraction from fresh or frozen postmortem blood, whole exome sequencing (WES) was performed as previously reported by our group (Online Supplemental Appendix 2).21,22 Variants were excluded if they had a minor allele frequency (MAF) of > 0.1% in the 1000 Genomes Project (1KG) data (http://www.1000genomes.org/) or Exome Sequencing Project (ESP) data (http://evs.gs.washington.edu/EVS/) or occurred in 410% of the 96 in-house exome sequences. Data from Illumina′s Human BodyMap 2.0 project were used to estimate the level of gene expression in cardiac tissue, as previously reported. The in silico predicted effect of variants on protein function and conservation across species was assessed using SIFT (Sorting Intolerant From Intolerant; http://sift-dna.org/), Polyphen-2 (http://genetics.bwh.harvard.edu/pph2/ ), GERP (Genomic Evolutionary Rate Profiling), Mutation Taster (www.mutationtaster.org), and combined annotation-dependent depletion (CADD). After identifying candidate genes, the frequency of the variant in the Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org) was reviewed. Rare variants for validation were polymerase chain reaction amplified (Online Supplemental Appendix 3). Sanger sequencing was performed, and electropherograms were manually inspected using Sequencher version 5.1 (Gene Codes Corp, Ann Arbor, MI)
Linkage analysis
Genome-wide linkage analysis was performed in 12 family members using Illumina HumanCytoSNP-12 genotyping array and Multipoint Engine for Rapid Likelihood Inference linkage analysis software.23 The genotype data were formatted for Multipoint Engine for Rapid Likelihood Inference using LINKDATAGEN and included a check for Mendelian errors to exclude large deletions.24 Linkage analysis was performed assuming autosomal dominant inheritance, 99% penetrance, and a disease allele frequency of 0.0001. All genotyped affected and unaffected family members were included in the calculation.
In silico simulations
The CPVT phenotype was modeled as described in our recent study25 using computationally reduced CASQ2 by modifying the Soltis-Saucerman (rabbit) model26 and the Morotti-Grandi (mouse) cardiac cell model.27 We reduced the CASQ2 total protein concentration by 25%, 50%, and 75% and performed virtual myocyte pacing experiments in the presence of 1 uM isoprenaline (Online Supplemental Methods).
Results
Clinical evaluation of family members
Family BFC was identified through the Australian Genetic Heart Disease Registry. The clinical characteristics and pedigree are presented in Table 1 and Figure 1A. The proband (II-2) had a history of unexplained syncope during physical activity. She was initially diagnosed with “unexplained VT” at the age of 23 after a resuscitated VT arrest. She was subsequently diagnosed with CPVT in 2005 through EST (Figure 1B), commenced on β-blocker therapy, and received an implantable cardioverter-defibrillator (ICD). Despite good compliance with therapy, she has experienced an appropriate ICD shock.
Table 1. Clinical and genetic characteristics of family BFC.
| Code | Sex | Current age (y) | Diagnosis | Age at CPVT diagnosis | Genotype | Diagnostic criterion | SCD/RCA/VT | Syncope | ICD | App ICD Therapy | β-Blocker |
|---|---|---|---|---|---|---|---|---|---|---|---|
| I-2 | F | 92 | Normal | NA | −/− | NA | No | No | No | NA | No |
| II-2 | F | 64 | CPVT | 23 | +/− | Bidirectional VT on EST | Yes | Yes | Yes | Yes | Yes |
| II-3 | M | Deceased aged 16 | MVA | NA | NA | Unknown | NA | No | NA | NA | NA |
| II-4 | M | 62 | Normal | NA | −/− | No | No | No | No | NA | No |
| II-7 | F | 61 | CPVT | 51 | +/− | Polymorphic ectopy on EST | Yes | Yes | Yes | No | Yes |
| II-9 | F | 58 | Normal | NA | −/− | No | No | No | No | No | No |
| III-1 | F | 41 | Normal | NA | −/− | No | No | No | Yes | No | No |
| III-3 | F | 38 | CPVT | 31 | +/− | Polymorphic ectopy on EST | Yes | Yes | Yes | Yes | Yes |
| III-5 | M | 34 | Normal | NA | −/− | No | No | No | No | NA | No |
| III-11 | M | 33 | Normal | NA | −/− | No | No | No | No | NA | No |
| III-13 | F | 32 | CPVT | 28 | +/− | Polymorphic ectopy on EST | No | No | Yes | No | Yes |
| IV-1 | F | Deceased aged 5 | CPVT/SCD | 5 | +/− | Unexplained sudden death in CPVT family | Yes | No | No | No | No |
| IV-2 | F | 4 | CPVT | 3 | +/− | Polymorphic ectopy on adrenaline challenge | No | No | No | NA | Yes |
| IV-6 | F | 1.5 | Possible CPVT | NA | NA | Atrial arrhythmias on adrenaline challenge | No | No | No | NA | Yes |
−/− = no variant identified; +/− = presence of Lysl80Arg variant; App ICD therapy = appropriate implantable cardioverter-defibrillator therapy; CPVT= catecholaminergic polymorphic ventricular tachycardia; EST= exercise stress test; F= female; ICD =implantable cardioverter-defibrillator; M = male; MVA = motor vehicle accident; NA = not applicable; RCA = resuscitated cardiac arrest; SCD = sudden cardiac death;
Figure 1.
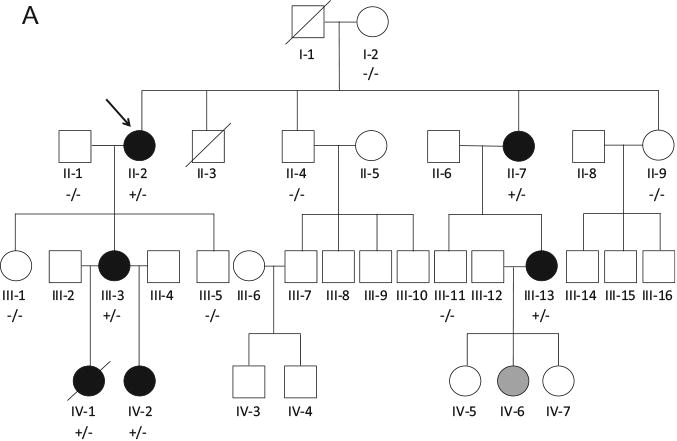
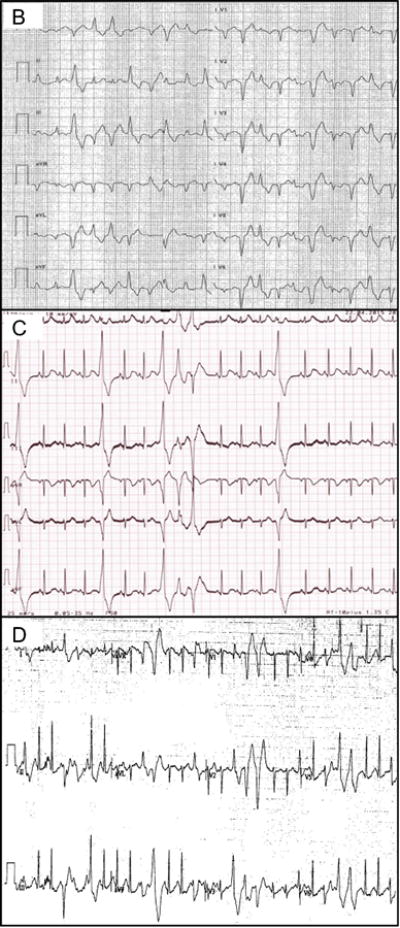
A: Family BFC pedigree. Squares indicate males; circles, females; line through a symbol, deceased individual; solid black symbols, clinically affected individuals; open symbols, clinically unaffected individuals; solid gray symbol, clinically borderline individual; arrow, proband; +/ −, presence of heterozygous Lys180Arg; and −/ −, no variant identified. B: Exercise stress test from proband (II-2) showing exercise-induced bidirectional ventricular tachycardia. C: Adrenaline challenge test from (IV-2) showing polymorphic ventricular ectopy. D: Exercise stress test from other family member (III-13) who underwent whole exome sequencing showing polymorphic ventricular ectopy.
The proband's daughter (III-3) experienced an unexplained resuscitated cardiac arrest while swimming at the age of 14. She was later diagnosed with CPVT at the age of 31 after an EST arranged as part of family screening, commenced β-blockers, and received an ICD. She has experienced 2 appropriate shocks and numerous self-terminating episodes of VT. Her daughter (IV-1) died suddenly at the age of 5 while swimming; the cause of death was unexplained after postmortem examination, and blood was stored. She underwent normal clinical evaluation with an ECG and echocardiogram 2 weeks before her death and was awaiting EST. Her sister (IV-2) was diagnosed with CPVT at the age of 3 after polymorphic ectopy was induced on adrenaline challenge testing (Figure 1C) and has commenced β-blocker therapy.
The proband's sister (II-7) was diagnosed with CPVT on EST on the background of unexplained syncope; VT has been documented while under general anesthesia. Her daughter (III-13) was diagnosed with CPVT on EST performed as part of family screening (Figure 1D). She has a history of presyncope and palpitations but no syncope. Her daughter (IV-6) was noted to have atrial arrhythmias on adrenaline challenge testing, an abnormal response, but nondiagnostic for CPVT; and β-blockers were recommended. The proband's brother (II-3) died in a motorbike accident at the age of 16. The remaining family members have all undergone regular cardiovascular review with EST every 3 years, all of which have been negative for CPVT.
Cardiac arrhythmia gene analysis
Commercial genetic testing had been performed in the family as part of routine clinical management. The proband (II-2) had no pathogenic variant identified by complete RYR2 sequencing. After this, her niece (III-13) underwent repeat RYR2 sequencing (along with multiplex ligationdependent probe amplification), sequencing of CALM1/2/3 and the mitochondrial genome, and a 33-gene cardiac arrhythmia panel. Three variants of uncertain significance were found: JUP (Thr249Met), KCNE1 (Asp85Asn), and DSG2 (Ile293Val). Recently, the KCNE1 and DSG2 variants have been reclassified as benign because of their high frequency in the ExAC database. The JUP variant did not cosegregate with disease and was reclassified as benign.
WES
Since no pathogenic variant was found after a focused analysis of arrhythmia genes, the proband (II-2) and her niece (III-13) underwent research-based WES, as they were the most distantly related, clinically affected family members and were alive. The average coverage of target sequences was 98-fold for III-13 and 92-fold for II-2, with 91% of the target sites having 20-fold coverage. A total of 54 shared rare variants (1KG/ESP MAF < 0.1%) were identified (48 single nucleotide variants, 6 insertions and/or deletions) (Appendix 4). The pipeline used for rare variant stratification is shown in Figure 2A. To identify candidate variants for autosomal dominant CPVT, we first searched for variants in genes involved in CPVT or calcium regulation. A novel missense variant in CASQ2 (Lys180Arg) was identified, which had not been previously reported, as well as a 19 bp frameshift deletion in EF-hand calcium binding domain 4B (EFCAB4B). We then prioritized variants in genes known to affect cardiac function in humans or in animal models, those predicted to alter the protein sequence (ie, nonsense, frameshift, and splice site variants), and those with high cardiac expression, high conservation across species, and in silico prediction of pathogenicity. Four additional candidate variants were identified (Table 2).
Figure 2.
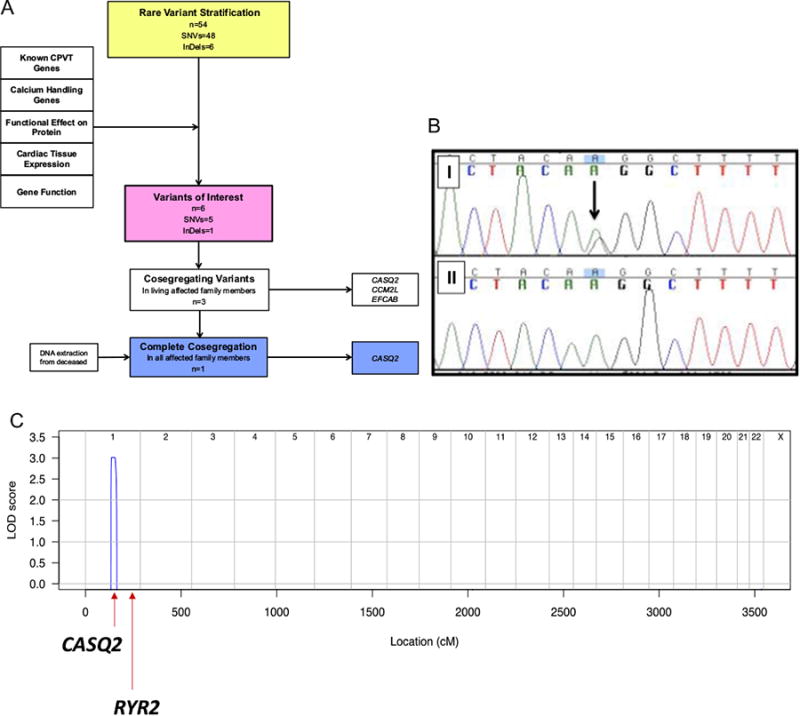
A: Genetic analysis of proband and family with pipeline for the stratification of rare variants. B: Sanger sequencing in decedent (B-I) and unaffected family member (B-II). C: Genome-wide linkage analysis showing single peak for disease linkage at chromosome 1 at the site of CASQ2. CASQ2 = cardiac calsequestrin 2 gene; CCM2L = cerebral cavernous malformation 2-like gene; CPVT = catecholaminergic polymorphic ventricular tachycardia; EFCAB = EF-hand calcium binding domain gene; InDel = insertion and/or deletion; LOD = logarithm of odds ratio; RYR2 = ryanodine receptor gene; SNV = single nucleotide variant.
Table 2. Top 6 ranked variants identified using exome sequencing.
| Gene symbol | Gene name | Cardiacexpression(fkpm) | cDNA change | Amino acid change | CADD score | 1KG MAF | ESP count | ExAC count |
|---|---|---|---|---|---|---|---|---|
| CASQ2 | Cardiac calsequestrin 2 | 399 | c.539A> G | Lys180Arg | 23 | 0 | 0 | 0 |
| CCM2L | Cerebral cavernous malformation 2-like | Null | c.235G> C | Glu85Gln | 34 | 0 | 2 | 17 |
| GTF2E2 | General transcription factor IIE | 5.5 | c.53C> T | Ser18Phe | 27 | 0 | 0 | 0 |
| KCTD19 | Potassium channel tetramerization domain containing 19 | Null | c.814G> A | Gly272Arg | 18 | 0 | 2 | 18 |
| PKP3 | Plakophilin 3b | Null | c.830G> C (homozygous) | Arg277Pro | 13 | 0 | 0 | 2712 |
| EFCAB4B | EF-hand calcium binding domain 4B | 1.2 | c.1389_1407del | Tyr464Glufs*22 | Null | 0 | 0 | 0 |
1KG= 1000 Genomes Project; CADD = combined annotation-dependent depletion; cDNA = complementary DNA; ESP = Exome Sequencing Project; ExAC = Exome Aggregation Consortium; MAF = minor allele frequency.
Genotyping and cosegregation studies
Segregation of the 6 candidate variants was performed in affected living family members and 3 segregated with disease: Lys180Arg in CASQ2, Glu85Gln in CCM2L, and Tyr464Glufs*22 in EFCAB4B. Only the Lys180Arg CASQ2 variant segregated in the decedent (IV-1, Figure 2B). Further genotyping revealed no unaffected family members with the Lys180Arg CASQ2 variant (Figure 1A and Table 1).
To exclude the possibility of compound heterozygous CASQ2 variants, all common and rare CASQ2 variants in the 2 exomes were reviewed. There were 7 shared CASQ2 variants; the Lys180Arg variant, 2 variants in the 3′ untranslated region (UTR), 1 variant in the 5′UTR, 2 intronic variants and a synonymous variant (Asp395Asp), all of which were excluded because of high frequencies in population databases (MAF ≥0.43).
Linkage analysis
Genome-wide linkage analysis revealed a single linkage peak in family BFC covering a 47.1 Mb region of chromosome 1 (1:106161201-153283429; logarithm of odds ratio [LOD] score 3.01; θ = 0) (Figure 2C). At this locus, there were 3 single nucleotide variants identified in the exomes of II-2 and III-13: the novel Lys180Arg CASQ2 variant and a variant in PDE4DIP and MRPS21, both of which were considered as too frequent to cause CPVT (European MAF 0.008 and 0.0007, respectively). There was absence of linkage at the RYR2 locus (Figure 2C).
Functional domain of variant
The CASQ2 Lys180 residue is found within the sarco-plasmic reticulum (SR) junctional face membrane inter action domain (Figure 3A) and is highly conserved across species (Figure 3B).4 A number of other pathogenic variants in CASQ2 are found in this region (Figure 3A). In silico prediction tools CADD, SIFT, Mutation Taster, and GERP demonstrate the variant to be in a conserved domain with a predicted deleterious effect on protein function; Polyphen-2 predicts the variant to be benign.
Figure 3.
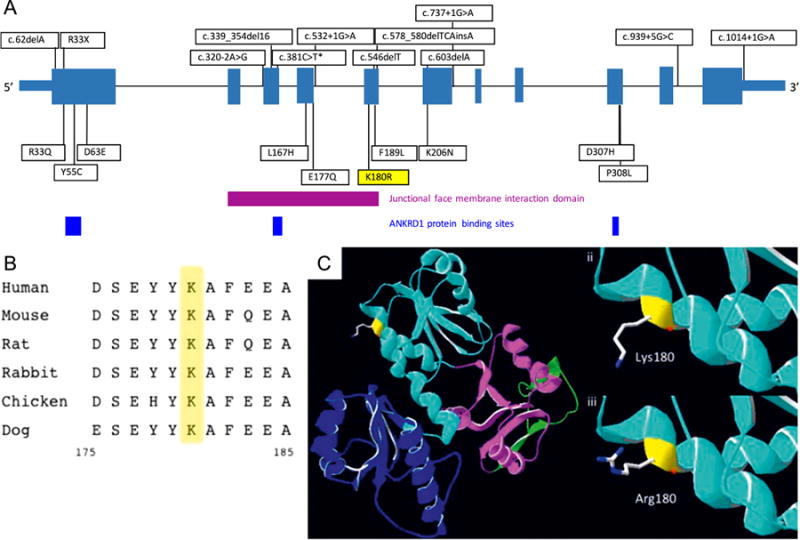
A: Overall CASQ2 protein structure with documented pathogenic or likely pathogenic loss-of-function mutations (above) and missense mutations (*c381C4> T synonymous mutation demonstrated to affect messenger RNA splicing) (below). The Lys180Arg mutation is demonstrated in exon 5 within junctional face membrane interaction domain (purple), and cardiac ankyrin repeat domain 1 protein binding sites are also shown. Modified from Leenhardt et al.4 B: Lysine residue at position 180 is highly conserved among species. C: Predictive protein modeling (using DeepView/Swiss PDB Viewer) showing (i) overall CASQ2 structure, (ii) lysine at position 180, and (iii) change in structure at the junctional face membrane domain interaction with the Lys180Arg mutation. ANKRD1 = ankyrin repeat domain 1; CASQ2 = cardiac calsequestrin 2.
In silico disease modeling
In the rabbit myocyte electrophysiology model, >50% reduction in CASQ2 was required to disrupt voltage and calcium transient dynamics (Figure 4). In the mouse myocyte electrophysiology model, 50% reduction in CASQ2 produced intracellular calcium alternans and 75% reduction resulted in spontaneous action potential triggers (Figure 5A). A 1-dimensional coupled rabbit tissue model harboring 75% reduction in CASQ2 demonstrated evidence of spontaneous delayed afterdepolarizations and triggered action potentials with corresponding changes in calcium transients (Figure 5B). Collectively, these simulations suggest haploinsufficiency to be unlikely to cause the CPVT phenotype and a dominant negative mechanism to be more likely, with 450% reduction in CASQ2 reproducing the CPVT phenotype.
Figure 4.
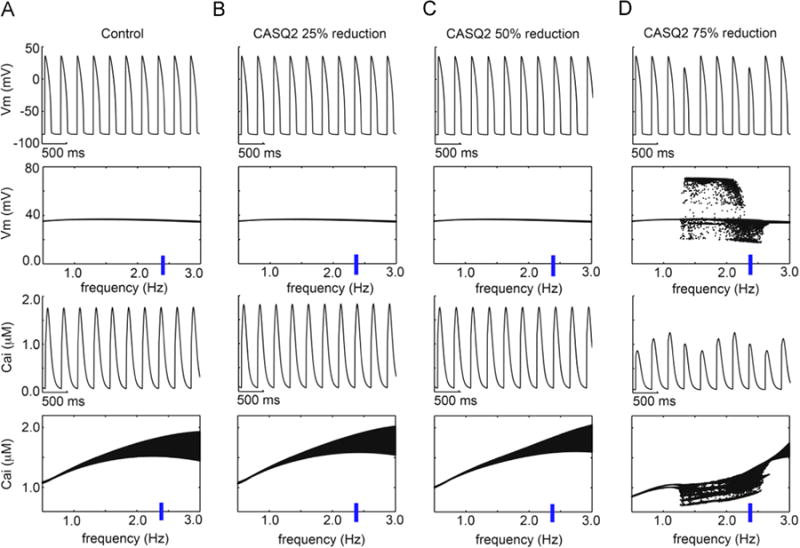
Simulated rabbit model prediction of CASQ2 reductions with 1 µM simulated isoproterenol. The time course of action potentials is in the top rows, and calcium transients are in the third row at 2.35 Hz. Voltage and Ca2+ transient dynamics over a range of physiological frequencies is shown below each simulated time course at 2.35 Hz: the voltage maxima are binned for each pacing frequency between 0.5 and 3 Hz during the second half of a 2-minute simulation time course. (A) Control, (B) CASQ2 25% reduction, (C) 50% reduction, and (D) 75% reduction. Cai = intracellular calcium concentration, CASQ2 = cardiac calsequestrin 2, Vm= membrane voltage.
Figure 5.
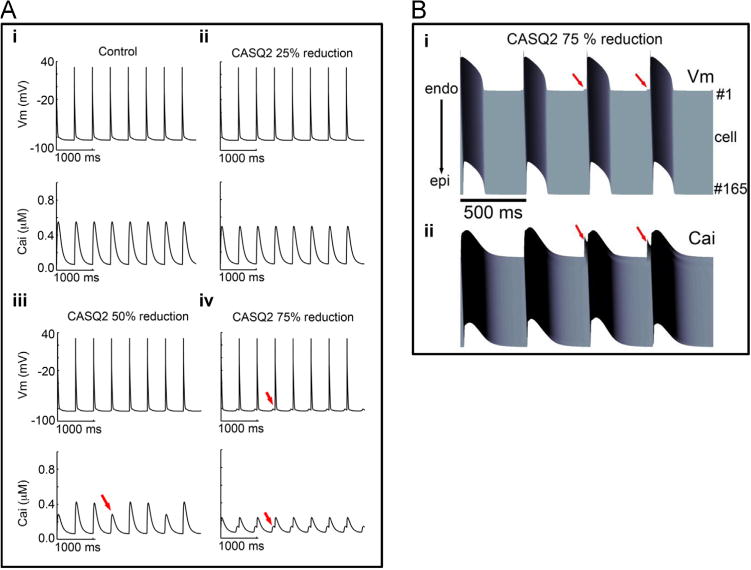
A: Simulated CASQ2 reduction in virtual mouse model membrane voltage (top) and intracellular Ca2+ transients (bottom) are shown during 2 Hz pacing. (i) Control with 1 µM simulated isoprenaline, (ii) CASQ2 25% reduction, (iii) 50% reduction, and (iv) 75% reduction. Red arrows indicate spontaneous triggers. Cells were paced for 2 minutes. Last 4 seconds are shown in the figure. (B: Space-time representation generated from the 1-dimensional coupled rabbit tissue model harboring 75% reduction in CASQ2 at a frequency of 2 Hz. (i) Voltage time course and (ii) corresponding calcium transient. Red arrows indicate spontaneous delayed afterdepolarizations, triggered action potentials, and Ca2+ transients. Cai = intracellular calcium concentration; CASQ2 = cardiac calsequestrin 2; Vm= membrane voltage.
Discussion
This study demonstrates the first large autosomal dominant CPVT family with linkage to a novel heterozygous missense variant in CASQ2 (LOD 3.01; θ = 0). The family shows autosomal dominant inheritance with 6 affected members, including 2 affected half-siblings of an affected mother. Complete disease penetrance is observed, with a severe disease phenotype including sudden deaths, resuscitated cardiac arrests, documented VT/ventricular fibrillation, appropriate ICD shocks, and syncope. The key clinical implication of this study is that genetic testing for heterozygous CASQ2 variants should be considered in all patients with CPVT.
The genetic investigation of this family highlights the utility of genome-wide linkage analysis working in synchrony with WES in the identification of disease-causing mutations. Exome sequencing in family “BFC” identified a Lys180Arg variant in CASQ2. The family had previously undergone extensive genetic testing of cardiac arrhythmia genes with no result. Given that CASQ2 is a known arrhythmia gene and was included on the 33-gene panel, the variant should have been identified previously. It is possible that the variant was missed owing to being in an area of low sequence coverage. However, even if this variant had been successfully identified by panel testing, the dominant inheritance pattern may have led to this variant being ignored. Subsequent genome-wide linkage analysis confirmed a single disease linkage peak outside the RYR2 locus. By using WES and genome-wide linkage analysis, we have taken a nonbiased approach to identify a novel pathogenic variant in this family.
Our study is the first to clearly identify a dominantly inherited heterozygous mutation in CASQ2 that is disease causing in CPVT. CASQ2 maps to chromosome 1p13.3-p11 and encodes 399 amino acids across 11 exons.8,9 CASQ2 is a major intracellular calcium storage protein modulating the Ca2+ release from the SR.5,29 Mutations in this gene are well described in CPVT disease pathogenesis due to disruption in SR calcium release.5 However, all pathogenic variants reported to date are autosomal recessive, that is, either homozygous or compound heterozygous.8–12 Postma et al12 identified a CASQ2 nonsense variant, Arg33*, in a patient with CPVT and 2 family members who exhibited episodes of bigeminy and premature ventricular contractions (PVCs) on exercise rather than premature VT. However, 3 relatives also carried this variant with no CPVT phenotype. Kirchhefer et al13 reported a novel CASQ2 missense variant, Lys206Asn, in a patient with CPVT signs including supra-ventricular and ventricular extrasystoles and couplets during EST. Her son, who died of cardiac arrest at the age of 13, had a history characteristic of CPVT, though she never had an exercise test performed and DNA was not available for genotyping. A second son who carried the Lys206Asn variant was asymptomatic at the age of 28, suggesting reduced penetrance.13 In our family, the pedigree is large, the phenotype is severe, and, in those in whom segregation analyses have been performed, there is 100% penetrance.
The mechanism underpinning how the Lys180Arg variant in CASQ2 leads to severe CPVT is intriguing. The Lys180Arg variant is located within a highly conserved large alpha helix at the junctional face membrane interaction domain. Disruption of CASQ2 protein at this position may affect its assembly within the SR.4 Predictive protein modeling demonstrated how the substitution of a larger arginine residue for the lysine at position 180 might alter the binding affinity of the CASQ2 protein with altered polarity of the residues at the junctional face membrane interface (Figure 3C).30
We undertook a modeling and simulation approach to predict a plausible mechanism for the phenotype-genotype correlation. In the simulations, we titrated the defective CASQ2 effect into the model and predicted the functional effect. If all of the mutant CASQ2 were defective, then we would expect haploinsufficiency to reproduce the severe clinically observed phenotype. However, the predictions suggest that even a 50% reduction in CASQ2 is insufficient to cause the severity of phenotype observed in the family.
Our previous study included extensive validation for the computational model.25 Although the model predictions cannot be used to definitively prove that the Lys180Arg CASQ2 mutation is autosomal-dominant, they suggest that 450% reduction in CASQ2 is necessary for the phenotype. This could be achieved by a dominant negative mechanism of the mutant allele on the wild-type allele, as observed in other inherited arrhythmias.31,32 This finding would also be consistent with the observation that in the family described by Postma et al,12 the Arg33* nonsense variant was tolerated with no evidence of CPVT in multiple family members, presumably because of the normal allele acting independently of the truncated mutant. Finally, the computational simulations are consistent with the mouse model described by Chopra et al in 2007.33 They also demonstrated that even modest reductions in CASQ2 are sufficient to lead to changes in SR Ca2+ release and CPVT phenotype in vivo. Therefore, although our computational model does not assess the particular variant in question, the model provides important insights into the potential mechanism of heterozygous CASQ2 variants. It also challenges the notion that almost complete absence of CASQ2 is required to lead to a CPVT phenotype. The particular functional mechanism of our Lys180Arg variant will be the subject of our future functional studies.
Conclusion
We report the first large family with clear autosomal dominant inherited CPVT and genome-wide linkage (LOD 3.01) to a heterozygous missense variant in CASQ2. This family demonstrates strong cosegregation of the variant in affected and unaffected relatives with 100% penetrance, a high LOD score, excellent genotype-phenotype concordance, and supportive evidence that the variant is located in a key functional domain of the protein. This result sheds further light on the mechanism of CPVT because of CASQ2 mutations. The findings may also provide insights for families who may harbor pathogenic heterozygous CASQ2 variants that may previously have been overlooked because of the inheritance pattern not being recessive. Genetic testing in autosomal dominant CPVT families should include screening for heterozygous CASQ2 variants.
Supplementary Material
Footnotes
Appendix: Supplementary data Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.hrthm. 2016.05.004
References
- 1.Leenhardt A, Lucet V, Denjoy I, Grau F, Ngoc DD, Coumel P. Catecholaminergic polymorphic ventricular tachycardia in children: a 7-year follow-up of 21 patients. Circulation. 1995;91:1512–1519. doi: 10.1161/01.cir.91.5.1512. [DOI] [PubMed] [Google Scholar]
- 2.Coumel PFJ, Lucet V, Attuel P, Bouvrain Y. Catecholamine-induced severe ventricular arrhythmias with Adams-Stokes syndrome in children: report of four cases. Br Heart J. 1978;40:28–37. [Google Scholar]
- 3.Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm. 2013;10:1932–1963. doi: 10.1016/j.hrthm.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 4.Leenhardt A, Denjoy I, Guicheney P. Catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. 2012;5:1044–1052. doi: 10.1161/CIRCEP.111.962027. [DOI] [PubMed] [Google Scholar]
- 5.Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011;108:871–883. doi: 10.1161/CIRCRESAHA.110.226845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laitinen PJ, Brown KM, Piippo K, et al. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103:485–490. doi: 10.1161/01.cir.103.4.485. [DOI] [PubMed] [Google Scholar]
- 7.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 8.Lahat H, Eldar M, Levy-Nissenbaum E, Bahan T, Friedman E, Khoury A, Lorber A, Kastner DL, Goldman B, Pras E. Autosomal recessive catecholamine-or exercise-induced polymorphic ventricular tachycardia: clinical features and assignment of the disease gene to chromosome 1p13-21. Circulation. 2001;103:2822–2827. doi: 10.1161/01.cir.103.23.2822. [DOI] [PubMed] [Google Scholar]
- 9.Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, Levy-Nissenbaum E, Khoury A, Lorber A, Goldman B, Lancet D, Eldar M. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet. 2001;69:1378–1384. doi: 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.di Barletta MR, Viatchenko-Karpinski S, Nori A, Memmi M, Terentyev D, Turcato F, Valle G, Rizzi N, Napolitano C, Gyorke S, Volpe P, Priori SG. Clinical phenotype and functional characterization of CASQ2 mutations associated with catecholami-nergic polymorphic ventricular tachycardia. Circulation. 2006;114:1012–1019. doi: 10.1161/CIRCULATIONAHA.106.623793. [DOI] [PubMed] [Google Scholar]
- 11.de la Fuente S, Van Langen IM, Postma AV, Bikker H, Meijer A. A case of catecholaminergic polymorphic ventricular tachycardia caused by two calseques-trin 2 mutations. Pacing Clin Electrophysiol. 2008;31:916–919. doi: 10.1111/j.1540-8159.2008.01111.x. [DOI] [PubMed] [Google Scholar]
- 12.Postma AV, Denjoy I, Hoorntje TM, Lupoglazoff JM, Da Costa A, Sebillon P, Mannens MM, Wilde AA, Guicheney P. Absence of calsequestrin 2 causes severe forms of catecholaminergic polymorphic ventricular tachycardia. Circ Res. 2002;91:e21–e26. doi: 10.1161/01.res.0000038886.18992.6b. [DOI] [PubMed] [Google Scholar]
- 13.Kirchhefer U, Wehrmeister D, Postma AV, Pohlentz G, Mormann M, Kucerova D, Muller FU, Schmitz W, Schulze-Bahr E, Wilde AA, Neumann J. The human CASQ2 mutation K206N is associated with hyperglycosylation and altered cellular calcium handling. J Mol Cell Cardiol. 2010;49:95–105. doi: 10.1016/j.yjmcc.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Vega AL, Tester DJ, Ackerman MJ, Makielski JC. Protein kinase A-dependent biophysical phenotype for V227F-KCNJ2 mutation in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. 2009;2:540–547. doi: 10.1161/CIRCEP.109.872309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K, Priori SG, Keating MT, Bennett V. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natll Acad Sci U S A. 2004;101:9137–9142. doi: 10.1073/pnas.0402546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nyegaard M, Overgaard MT, Sondergaard MT, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet. 2012;91:703–712. doi: 10.1016/j.ajhg.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roux-Buisson N, Cacheux M, Fourest-Lieuvin A, et al. Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human. Hum Mol Genet. 2012;21:2759–2767. doi: 10.1093/hmg/dds104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyo-pathies. Heart Rhythm. 2011;8:1308–1339. doi: 10.1016/j.hrthm.2011.05.020. [DOI] [PubMed] [Google Scholar]
- 19.Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med. 2013;369:1502–1511. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bagnall RD, Das KJ, Duflou J, Semsarian C. Exome analysis-based molecular autopsy in cases of sudden unexplained death in the young. Heart Rhythm. 2014;11:655–662. doi: 10.1016/j.hrthm.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 22.Bagnall RD, Molloy LK, Kalman JM, Semsarian C. Exome sequencing identifies a mutation in the ACTN2 gene in a family with idiopathic ventricular fibrillation, left ventricular noncompaction, and sudden death. BMC Med Genet. 2014;15:99. doi: 10.1186/s12881-014-0099-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. MERLIN—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 24.Bahlo M, Bromhead CJ. Generating linkage mapping files from Affymetrix SNP chip data. Bioinformatics. 2009;25:1961–1962. doi: 10.1093/bioinformatics/btp313. [DOI] [PubMed] [Google Scholar]
- 25.Yang PC, Moreno JD, Miyake CY, Vaughn-Behrens SB, Jeng MT, Grandi E, Wehrens XH, Noskov SY, Clancy CE. In silico prediction of drug therapy in catecholaminergic polymorphic ventricular tachycardia. J Physiol. 2016;594:567–593. doi: 10.1113/JP271282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soltis AR, Saucerman JJ. Synergy between CaMKII substrates and beta-adrenergic signaling in regulation of cardiac myocyte Ca(2+) handling. Biophys J. 2010;99:2038–2047. doi: 10.1016/j.bpj.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morotti S, Edwards AG, McCulloch AD, Bers DM, Grandi E. A novel computational model of mouse myocyte electrophysiology to assess the synergy between Na+ loading and CaMKII. J Physiol. 2014;592:1181–1197. doi: 10.1113/jphysiol.2013.266676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ingles J, Semsarian C. The Australian Genetic Heart Disease Registry. Int J Cardiol. 2013;168:e127–e128. doi: 10.1016/j.ijcard.2013.08.036. [DOI] [PubMed] [Google Scholar]
- 29.Franzini-Armstrong C, Protasi F, Tijskens P. The assembly of calcium release units in cardiac muscle. Ann N Y Acad Sci. 2005;1047:76–85. doi: 10.1196/annals.1341.007. [DOI] [PubMed] [Google Scholar]
- 30.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kagan A, Yu Z, Fishman GI, McDonald TV. The dominant negative LQT2 mutation A561V reduces wild-type HERG expression. J Biol Chem. 2000;275:11241–11248. doi: 10.1074/jbc.275.15.11241. [DOI] [PubMed] [Google Scholar]
- 32.Wang Z, Tristani-Firouzi M, Xu Q, Lin M, Keating MT, Sanguinetti MC. Functional effects of mutations in KvLQT1 that cause long QT syndrome. J Cardiovasc Electrophysiol. 1999;10:817–826. doi: 10.1111/j.1540-8167.1999.tb00262.x. [DOI] [PubMed] [Google Scholar]
- 33.Chopra N, Kannankeril PJ, Yang T, Hlaing T, Holinstat I, Ettensohn K, Pfeifer K, Akin B, Jones LR, Franzini-Armstrong C, Knollmann BC. Modest reductions of cardiac calsequestrin increase sarcoplasmic reticulum Ca2+ leak independent of luminal Ca2+ and trigger ventricular arrhythmias in mice. Circ Res. 2007;101:617–626. doi: 10.1161/CIRCRESAHA.107.157552. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.