

Abstract
Thiol isomerases such as protein-disulfide isomerase (PDI) direct disulfide rearrangements required for proper folding of nascent proteins synthesized in the endoplasmic reticulum. Identifying PDI substrates is challenging because PDI catalyzes conformational changes that cannot be easily monitored (e.g. compared with proteolytic cleavage or amino acid phosphorylation); PDI has multiple substrates; and it can catalyze either oxidation, reduction, or isomerization of substrates. Kinetic-based substrate trapping wherein the active site motif CGHC is modified to CGHA to stabilize a PDI-substrate intermediate is effective in identifying some substrates. A limitation of this approach, however, is that it captures only substrates that are reduced by PDI, whereas many substrates are oxidized by PDI. By manipulating the highly conserved -GH- residues in the CGHC active site of PDI, we created PDI variants with a slowed reaction rate toward substrates. The prolonged intermediate state allowed us to identify protein substrates that have biased affinities for either oxidation or reduction by PDI. Because extracellular PDI is critical for thrombus formation but its extracellular substrates are not known, we evaluated the ability of these bidirectional trapping PDI variants to trap proteins released from platelets and on the platelet surface. Trapped proteins were identified by mass spectroscopy. Of the trapped substrate proteins identified by mass spectroscopy, five proteins, cathepsin G, glutaredoxin-1, thioredoxin, GP1b, and fibrinogen, showed a bias for oxidation, whereas annexin V, heparanase, ERp57, kallekrein-14, serpin B6, tetranectin, and collagen VI showed a bias for reduction. These bidirectional trapping variants will enable more comprehensive identification of thiol isomerase substrates and better elucidation of their cellular functions.
Keywords: disulfide, oxidase, platelet, protein disulfide isomerase, protein-protein interaction, reductase
Introduction
Protein-disulfide isomerase (PDI)2 is the archetypal member of the PDI family, a subgroup of the thioredoxin superfamily, which are proteins that have at least one domain with a thioredoxin-like fold (1–4). PDI is an essential protein folding catalyst and chaperone present at high concentrations in the endoplasmic reticulum (5–7). Although PDI has been shown to have classic chaperone activity toward nascent peptides, its most familiar function is to catalyze disulfide bond formation and rearrangement during the folding process (8–11). PDI consists of four thioredoxin-like domains, two catalytic domains, a and a′, and two non-catalytic domains, b and b′, and has a domain structure of a-b-b′-a′ (12). The two non-catalytic domains are believed to play an important role in the recognition of substrate proteins, whereas each of the catalytic domains contains an independent active site with a highly conserved CXXC motif (1, 4, 8, 13, 14).
The CXXC motif cycles between a reduced state in which both cysteines contain free thiols (-SH groups) and an oxidized state in which both cysteines participate in a disulfide bond. During substrate reduction, the N-terminal cysteine acts as a nucleophile that attacks an oxidized cysteine in the substrate (15, 16). There is a consensus that the local pKa of the N-terminal cysteine allows for a thiolate anion state (-S−). However, several residues have been implicated as required for regulating the pKa (16–19). This attack forms a relatively stable disulfide-linked intermediate between PDI and the substrate protein (20). Resolution of this intermediate is the rate-limiting step for the reaction (21). After the N-terminal cysteine has formed the covalent intermediate with substrate, the C-terminal free thiol attacks its active site partner, forming a new intramolecular disulfide and leaving the substrate protein cysteines in the reduced state (8, 16). The oxidation of substrate proteins is the reverse of this reaction, again transitioning through a covalent disulfide-linked intermediate before being resolved by a third cysteine (5, 15). The reaction mechanism for PDI is diagrammed in Fig. 1.
Figure 1.

PDI reduces and oxidizes substrate proteins. Shown is a reaction scheme diagramming the PDI active site transitioning between a reduced (left) and oxidized (right) state performing disulfide bond reduction (left to right) or oxidation (right to left) on a protein substrate. PDI active site (lower) and protein substrate (upper) are shown.
The thioredoxin superfamily has 20 members with CXXC motifs. The intervening sequences, XX, between the cysteines have been shown to affect the redox potential of the enzyme. PDI has a CGHC sequence for the active site, whereas thioredoxin has a CGPC active site (17). These changes alter the redox potential so that PDI is a better disulfide oxidase and thioredoxin is a better disulfide reductase (4, 22–24). Although the CGHC motif is highly conserved in human thiol isomerases, the substrate recognition domains b and b′ are not well conserved (2, 12, 14). For example, PDI and ERp57, with the closest sequence similarity of the human thiol isomerases, have just under 20% conservation in their b and b′ domains. However, they retain over 50% sequence conservation in their active site, which includes a CGHC motif (4). Despite similarities in active sites, PDI, ERp57, and ERp5, all of which have two thioredoxin domains with a CGHC motif, have different substrate specificities (25, 26).
Previous work has used kinetic substrate trapping to identify substrate proteins of PDI and other thiol isomerases. In these experiments, PDI variants in which the second active site cysteine has been altered to either a serine or an alanine are used. This non-oxidizable variant binds to protein substrates that require reduction but are unable to perform a complete round of catalysis. Instead, the reaction is halted at the intermediate step where substrate and enzyme are covalently linked. This trap-generating reaction mechanism is visually represented in Fig. 2. Free thiol-blocking agents, such as N-ethylmaleimide (NEM), are used to stabilize these “kinetically trapped” complexes, which are subsequently isolated via immunoprecipitation (IP). The trapped substrates are identified by mass spectroscopy.
Figure 2.

Kinetic substrate trapping using either reduced or oxidized PDI variants. Shown are reaction schemes diagramming the kinetic trapping of either the wild-type enzyme, the CXXA variant, or the intervening sequence variants with substrate protein, starting from either the reduced (A) or oxidized (B) state. Bonds that are made or broken during catalysis are shown in red. t0, the initial state; t1 and t2, intermediate states; ttrap, reaction state when stopped with NEM; tfinal, protein-substrate complex, or lack thereof, isolated after IP.
As seen in Fig. 2A, initially (t0) both the wild-type and variant enzymes are in a reduced state, whereas the target substrate contains a disulfide bond (oxidized). At t1, both the wild-type and variant enzymes have formed a covalent intermediate with the substrate protein. However, at t2, the wild-type enzyme is able to continue catalysis, whereas the variant enzyme is now lagging. When the reaction is halted, ttrap, the wild-type enzyme has finished the reduction of substrate, is no longer covalently linked, and is thus not isolated during immunoprecipitation. But the slower variants are still disulfide-linked to substrate in an intermediate complex. In Fig. 2B, the trapping mechanism begins when PDI is oxidized (t0) and is acting upon a target substrate that requires a disulfide bond (reduced), and it is here where the greatest difference in the CXXA and intervening sequence variant methods is clear. Although kinetic substrate trapping using CXXA-type mutants has elucidated much about thiol isomerase substrates, its major drawback is that this strategy is only able to recognize substrates that are subject to disulfide reduction (20, 23). These variants are unable to identify proteins that require oxidation because the CXXA variants can never exist in an oxidized state as depicted in Fig. 2B. So although the wild-type enzyme and intervening sequence variant interact with protein substrate essentially in the reverse reaction direction of Fig. 2A, there is no interaction with the CXXA variant and substrate protein. To identify substrate proteins that require oxidation using kinetic substrate trapping, a PDI variant must retain both active site cysteine residues but instead be altered to perform the disulfide exchange chemistry much more slowly than wild-type PDI. Addition of alkylating reagents ensures that the disulfide complex remains stable for further isolation and identification (tfinal).
To discover potential PDI substrates that could require either oxidation or reduction, we constructed PDI variants with substitutions only in the intervening sequences of the CXXC motif and then screened these variants for altered substrate kinetics. Seven intervening sequence variants were generated and screened for impaired ability to reduce insulin when compared with wild-type PDI. Of the seven variants, one lost activity altogether (CGDC-PDI) and two had lower activity toward insulin reduction (CGPC-PDI and CGRC-PDI). The two variants with low activity were then used to perform kinetic substrate trapping upon proteins requiring either reduction or oxidation during platelet activation. These complexes were then enriched using 2D gel electrophoresis and identified using mass spectrometry. From these results, several proteins known to function in thrombus formation or the regulation of redox potential were identified as novel substrates for PDI.
Results
The intervening XX amino acids of the PDI active site CXXC are highly conserved throughout the family and were exploited as targets for amino acid substitution in our attempt to make a trapping PDI variant that remains stably complexed with substrate proteins. Amino acid substitutions were made individually to decrease the risk of completely inactivating the enzyme. The mutants included CPHC-PDI, CAHC-PDI, CGAC-PDI, CGDC-PDI, CGFC-PDI, CGPC-PDI, and CGRC-PDI. Substituted amino acids were empirically selected and are listed in Table 1. The electrostatic substitutions were made exclusively at the histidine position, whereas geometry-altering substitutions were made at either the glycine or the histidine position.
Table 1.
List of amino acid substitutions in PDI and hypothesized cause of changes in catalytic activity
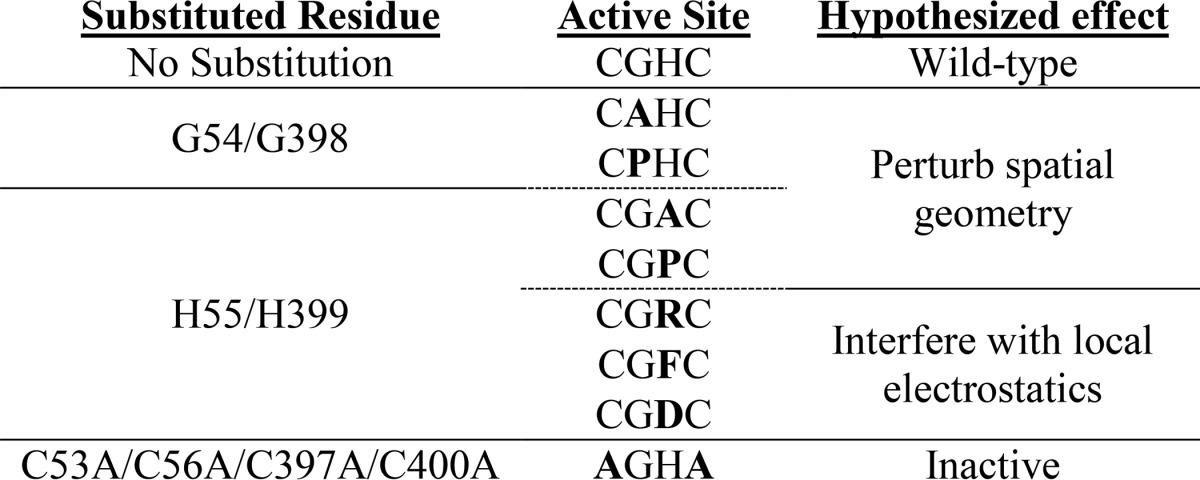
The insulin reduction assay was used to assess the activity of the variants. The insulin reduction assay monitors the aggregation of reduced insulin through turbidity measurements. PDI reduces insulin when added to the reaction mixture. Reduced insulin aggregates, and the resulting increase in turbidity is monitored spectrophotometrically (27). The goal of our amino acid substitutions was to make a PDI variant that has a more stable, covalently bound intermediate, effectively slowing the overall reaction due to a prolonged transition state. In the context of the insulin assay, a more stable PDI-insulin intermediate would cause a lag in the time it takes to generate sufficient free insulin to form aggregates. Both wild-type PDI (CGHC) and the catalytically inactive variant AGHA-PDI were used as controls. Of the seven variants, CGRC-PDI, CGPC-PDI, and CGDC-PDI showed a significant increase in lag time when compared with wild-type PDI (Fig. 3A). CGPC-PDI showed an approximate 4-fold increase in lag time, CGRC-PDI showed an increase of ∼5-fold, and CGDC-PDI never showed any reduction of insulin whatsoever, strongly resembling the effect caused by the catalytically inactive variant AGHA-PDI. To confirm that the inability of these variants to reduce insulin was not due to complete inactivation from the amino acid substitution, activities of the functionally compromised variants (CGRC, CGPC, and CGDC) were measured using the di-eosin glutathione disulfide (di-eosin-GSSG) assay (Fig. 3B). CGPC-PDI and CGRC-PDI were both still capable of reducing the small molecule substrate, showing that these amino acid substitutions were not completely inactivating. Like insulin, the CGDC variant was incapable of reducing the fluorescent substrate di-eosin-GSSG, indicating that this amino acid substitution more than likely inactivates the enzyme completely and for that reason was not further evaluated.
Figure 3.

Effect on reductase activity of intervening sequence substitutions in the CXXC motif of PDI.
A, PDI variants were tested for enzymatic activity using the insulin reductase assay. PDI was added to insulin in the presence of DTT, and turbidity was monitored at 650 nm for 1 h. B, variants that showed slower kinetics in the insulin reductase assay were tested for enzymatic activity using the di-eosin-GSSG assay. PDI was added to di-eosin-GSSG in the presence of DTT, and fluorescence was monitored for 10 min after excitation at 525 nm and recording emission at 545 nm. Wild-type PDI and the catalytically inactive AGHA PDI variant were used as positive and negative controls, respectively. Assays were performed in triplicate, and error bars represent standard deviation. ♦, wild-type PDI; ♢, AGHA-PDI; ▴, CAHC-PDI; ▵, CPHC-PDI; ●, CGPC-PDI; ○, CGRC-PDI; ■, CGAC-PDI; □, CGFC-PDI;  , CGDC-PDI. RFU, relative fluorescence units.
, CGDC-PDI. RFU, relative fluorescence units.
Because our goal was to create PDI variants capable of forming stable, covalently bound intermediates with substrate proteins requiring either reduction or oxidation, we determined whether our PDI variants could be both prereduced and preoxidized PDI variants. The reduction and oxidation of the variants was monitored with Ellman's reagent (DTNB) (Fig. 4A). DTNB reacts with free thiols and detects changes in the number of free thiols after variants are reduced or oxidized by exposure to DTT or GSSG. Using wild-type PDI and the catalytically inactive AGHA-PDI as controls, we found that the CGRC-PDI and CGPC-PDI variants showed similar levels of DTNB reactivity to wild type when all of the proteins were in the reduced state and that in the oxidized state each of the proteins showed similar reactivity to the AGHA variant.
Figure 4.

PDI variants retain activity following reversible oxidization and reduction. A, DTNB was used to measure free thiols on the PDI variants after reduction with DTT (gray bars) and oxidation with GSSG (white bars). Samples are normalized to the reduced wild-type PDI. B, PDI variants were tested for enzymatic activity after reduction (gray bars) and oxidation (white bars) using the di-eosin-GSSG assay. The catalytically inactive AGHA was used as a negative control. Assays were performed in triplicate, and error bars represent 3σ.
The results from the insulin assay show that the enzyme variants did retain some level of activity, but there remained the possibility that the slow reaction rate was caused by an inability of these variants to perform more than one round of catalysis. Using the di-eosin-GSSG method to measure thiol isomerase activity (28), aliquots of DTT-reduced variants were assayed, and the remainder of the samples were oxidized with GSSG and assayed again (Fig. 4B). The ability of these variants to reduce the di-eosin-GSSG substrate was unaffected by the cycle of reduction and oxidation. The di-eosin-GSSG assay is performed in the presence of DTT, so the similar reductase activity after GSSG oxidation could only be achieved if the enzyme was first reduced by the assay environment. This result combined with the DTNB confirmation shows that these variants are capable of cycling through multiple rounds of catalysis, moving from a reduced state to an oxidized state and back again.
Although it is known that PDI and other thiol isomerases play a pivotal role in thrombus formation, the substrate proteins that require PDI are not well defined (29–31). Vitronectin has been the best characterized of the plasma PDI substrates in thrombus formation that have been identified by using trapping mutants after mutation of the active site cysteines (32). Given the slow rate of turnover of the CGPC-PDI and CGRC-PDI variants, we hypothesized that we could capture variant-substrate intermediates using the thiol-blocking reagent NEM to halt any further catalysis. This leaves the PDI variants in an intermediate state of turnover, trapped in stable covalent disulfide linkages with substrate proteins. To this end, platelets were activated with thrombin in the presence of Ca+2, Mn+2, and one of the PDI variants in either a prereduced or preoxidized state. The reaction was terminated with the addition of hirudin, and the platelet lysate and releasate fractions were separated. PDI-substrate complexes, stabilized by the addition of NEM, were immunoprecipitated using anti-FLAG antibody-coupled beads. Because only the exogenous PDI variants contain FLAG, complexes involving endogenous PDI were not immunoprecipitated. After SDS gel electrophoresis under non-reducing conditions, the immunoprecipitate was immunoblotted for FLAG antigen using anti-FLAG antibodies (Fig. 5). SDS gels were purposely overloaded to ensure visualization of substrates with low prevalence. The catalytically inactive PDI variant AGHA was used as a negative control. Both variants, whether prereduced or preoxidized, formed high-molecular-weight complexes in both the lysate and releasate fractions, whereas the inactive variant showed no complex formation.
Figure 5.
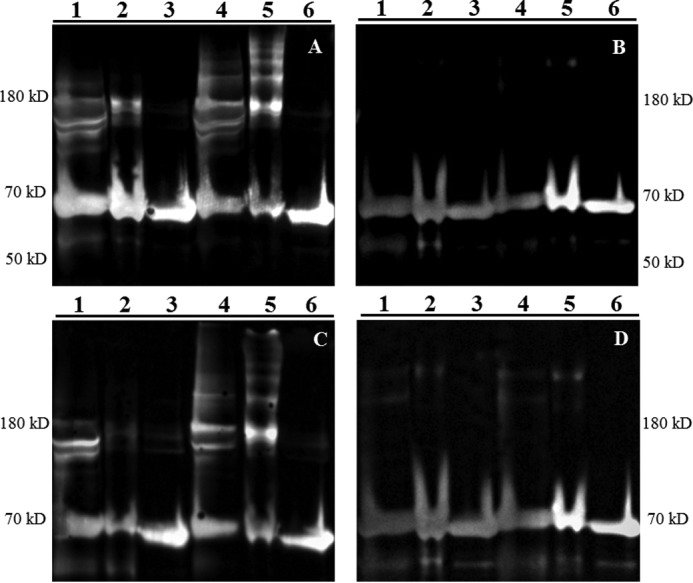
Prereduced and preoxidized CGPC and CGRC form reduction-dependent complexes with substrates from activated washed platelets. Platelet releasates (A and B) and lysates (C and D) were prepared as described under “Experimental procedures.” Samples were then immunoprecipitated with anti-FLAG antibody, purposefully overloaded to detect PDI-substrate complexes, and separated by SDS-PAGE in either non-reducing (A and C) or reducing (B and D) conditions followed by immunoblotting using anti-FLAG antibody. The catalytically inactive AGHA variant was used as a control. Lane 1, prereduced CGPC-PDI; lane 2, prereduced CGRC-PDI; lane 3, prereduced AGHA-PDI; lane 4, preoxidized CGPC-PDI; lane 5, preoxidized CGRC-PDI; lane 6, preoxidized AGHA-PDI. Molecular mass standards are shown on the left and right.
The high-molecular-weight complexes formed by each variant are only of interest if the individual proteins are complexed via disulfide bonds. Detection of a disulfide-linked (i.e. the reduction-sensitive complex) demonstrates that the target protein was enzymatically altered by PDI when the reaction was stopped by the addition of NEM. To separate out the proteins that were covalently disulfide-bonded to PDI, 2D electrophoresis was performed. First, the immunoprecipitate was separated by SDS-PAGE under non-reducing conditions. The gel lane was then excised and incubated with DTT to reduce any disulfide-linked protein complexes in the gel. The lane was then stained with Coomassie Blue and separated orthogonally in another dimension of SDS-PAGE under reducing conditions. After the reducing dimension, the entire gel was visualized using silver stain (Fig. 6). Any protein that traveled the same distance in both directions, indicating that it was not affected by DTT reduction, would travel along the diagonal line observed in each gel. Those proteins that were affected would travel further in the reducing conditions as their apparent molecular mass would have changed when the disulfide-linked complex was reduced. The protein spots that appeared below the unaffected protein diagonal were excised and subjected to mass spectrometry. The same analysis was performed using the catalytically inactive AGHA variant, and any proteins identified in those samples were excluded from the list of potential targets.
Figure 6.

Reduction-dependent complexes are confirmed via separation with 2D electrophoresis. Platelet releasates and lysates were prepared as described. Proteins were first separated by SDS-PAGE under non-reducing conditions (left to right) and then separated again by SDS-PAGE under reducing conditions (top to bottom). Spots circled in red or green identify proteins specific for the CGRC variant. Spots boxed in blue or black squares identify proteins specific for the oxidized condition.
Potential substrates for PDI during platelet activation were identified through mass spectrometry, and of those potential substrates there were several proteins known to play a role in thrombus formation. The proteins that we chose to evaluate and the fractions in which they were identified are listed in Table 2. To confirm that these proteins formed reduction-sensitive complexes with PDI, immunoblotting was performed on the immunoprecipitate samples under either reducing or non-reducing conditions. As examples, immunoblots under both reducing and non-reducing conditions are shown for cathepsin G, glutaredoxin-1, fibrin, and heparanase (Fig. 7). If the target protein is trapped in a high-molecular-weight disulfide-linked complex, then the band will shift lower in the reduced samples when compared with the non-reduced samples.
Table 2.
Proteins identified as potential PDI substrates

Figure 7.

Immunoblotting confirms complexed substrates identified via mass spectrometry. Platelet lysates and releasates were prepared as described. Samples were then immunoprecipitated with anti-FLAG antibody, purposely overloaded to detect PDI-substrate complexes, and separated by SDS-PAGE under either non-reducing (lanes 1–6) or reducing (lanes 7–12) conditions followed by immunoblotting onto PVDF membranes. Samples were visualized using both primary anti-FLAG antibody and primary antibody to either cathepsin G (A), heparanase (B), glutaredoxin-1 (C), or fibrin (D). Anti-FLAG signal is shown in green, target protein signal is displayed in red, and co-localization is displayed in yellow. The catalytically inactive AGHA variant was used as a control. Lanes 1 and 7, prereduced CGPC-PDI; lanes 2 and 8, prereduced CGRC-PDI; lanes 3 and 9, prereduced AGHA-PDI; lanes 4 and 10, preoxidized CGPC-PDI; lanes 5 and 11, preoxidized CGRC-PDI; lanes 6 and 12, preoxidized AGHA-PDI.
During the confirmation of these target proteins, we observed that although some proteins were identified by mass spectrometry in only the prereduced or the preoxidized samples others were identified in both. The immunoblotting studies confirmed a bias among target proteins for either prereduced or preoxidized PDI variants (Fig. 8). We assessed this bias by first using densitometry to quantitate the band intensities of both the fluorescent PDI signal and the target protein signal for each immunoblot lane (as seen in Fig. 7) to generate a target protein:total PDI ratio. This serves to normalize for any variation in the amount of total PDI immunoprecipitated from each oxidation or reduction condition. Then, using the target protein:total protein ratios for both the reduced and oxidized conditions, we can generate a bias for the target protein and either the reduced or oxidized form of the variant in question. When performing substrate trapping using the CGPC variant, four of our target proteins were biased toward the preoxidized form of the variant: thioredoxin, glutaredoxin, cathepsin G, and GP1b. All of the other target proteins seemed to favor the prereduced form of CGPC. The CGRC variant showed the same four proteins favoring the preoxidized state but also showed fibrinogen preferring the preoxidized form to the reduced CGRC.
Figure 8.

The bias of the CGPC and CGRC PDI variants for forming complexes in their prereduced or preoxidized state. Shown is a graphical representation of the ratio of densitometry quantification of the target proteins identified for the CGPC variant (A) or the CGRC variant (B) in the prereduced or preoxidized samples. First, a target protein:PDI ratio was generated using the band intensities of both proteins from the oxidized or reduced samples (target:PDI variant). Then, a reduced:oxidized bias was generated using those values. Target proteins whose bias was toward the preoxidized state are oriented toward the left, and target proteins whose bias was toward the prereduced state are oriented toward the right. The black vertical bar represents the point of no bias, and the length of the bar represents the strength of the bias. Cath, cathepsin; Coll, collagen; Grx, glutaredoxin; Trx, thioredoxin; Fbgn, fibrinogen; KK, kallekrein.
After confirming the target proteins via immunoblotting, the effects of oxidized or reduced PDI on cathepsin G activity were studied in activated platelet releasate (Fig. 9). Cathepsin G is a protease that, by cleaving several substrate proteins, serves to regulate thrombosis (33, 34). After activating platelets in the presence of either preoxidized or prereduced forms of both wild-type PDI and the trapping variants, an increase in cathepsin G activity was observed in the presence of oxidized PDI (Fig. 9). Although cathepsin G activity is easily measured through the cleavage of a colorimetric substrate, the presence of biological substrates can serve to inhibit the colorimetric development because of their greater affinity for the enzyme. One such protein substrate found in abundance in activated platelet releasate is thrombospondin-1. Thrombospondin-1 is cleaved by cathepsin G, and one of the cleavage products then serves to inhibit further cathepsin G activity (35).
Figure 9.
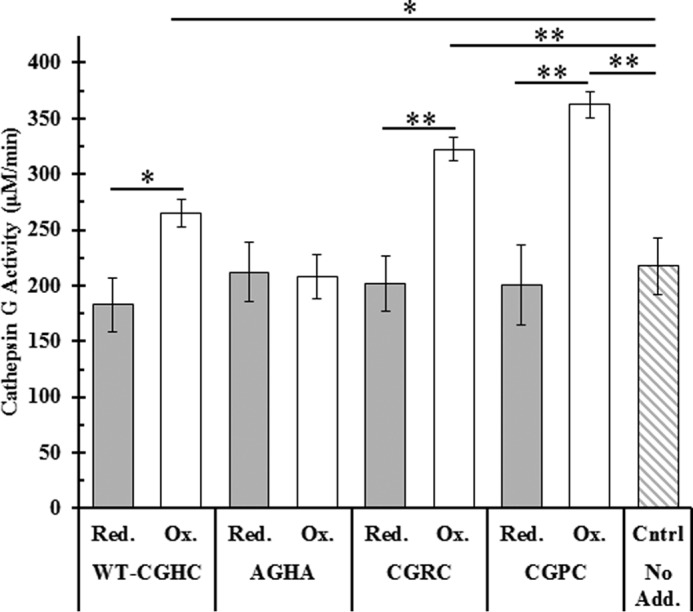
Effect of the oxidation state of PDI on cathepsin G activity in washed platelet releasate. Washed platelets were first incubated with either prereduced (gray) or preoxidized PDI (white) and then activated with 0.1 unit/ml thrombin. Platelet releasates were clarified with centrifugation and assayed for cathepsin G activity using a colorimetric probe monitored at 410 nm. Cathepsin G activities are shown as averages of triplicate experiments; error bars represent S.E. Paired t tests were used to determine significance: *, p < 0.05; **, p < 0.01. Red., reduced; Ox., oxidized; Cntrl, control; No Add., no additions.
Discussion
Kinetic substrate trapping has been successfully used to understand the kinetics and substrate specificity of thiol isomerases. However, the variant isomerases used for this technique have been limited. To form the most stable form of substrate traps, the second active site cysteine is modified, forming a CXXA or CXXS motif in the active site. Because of the loss of that second “resolving” cysteine (Fig. 1), the disulfide-linked protein complexes are much more stable. However, these complexes can only be formed with proteins requiring disulfide reduction (Fig. 2). This limitation potentially removes from the substrate pool those substrate proteins that require isomerization and those that require oxidation. Our results demonstrate the utility of an alternative approach. By using PDI variants with intervening sequence substitutions, we can perform kinetic substrate trapping to identify substrate proteins that require either reduction or oxidation by PDI.
The enzymatic function of a thiol isomerase family member does not perfectly correlate with the four active site residues. Several groups have studied PDI structure and function by making “PDI-like” active site motifs in thioredoxin by substituting a histidine for the proline in the CGPC active site motif of thioredoxin (12, 19, 26). Substitution of that proline to a histidine does change the redox potential of thioredoxin to closely resemble that of PDI, but the altered potential is still significantly different from wild-type PDI (24). Similar work has attempted to bestow thioredoxin kinetics to the viral T4 thioredoxin (25). The viral T4 thioredoxin contains a CVYC motif. This study made use of both the double mutant and the individual mutant proteins, creating the full CGPC motif as well as CVPC and CGYC. When the three of these variants were compared with wild-type thioredoxin, the substitution of the tyrosine, and not the substitution of the valine, was found to contribute the vast majority of the conferred thioredoxin-like properties.
Direct substitution of amino acids does not seem to confer additional function or stability from one family member to another, but the wide variety of attempts at this within the thiol isomerase family has uncovered some correlations. The better the active site residues are able to stabilize the thiolate anion in the active site, the more oxidative the enzyme behaves (19, 22–24, 26). Furthermore, the more stable the thiolate anion is in the active site, the less stable the protein is when in its oxidized disulfide-bound state (16, 22–25, 36). In PDI, the active site histidine residue plays a substantial role in stabilizing a thiolate anion on the N-terminal cysteine (N-Cys). Typically the pKa of a cysteine thiol is 8.3, but in wild-type PDI the pKa of the N-Cys is 4.5. Upon substitution of the histidine with proline, however, the N-Cys pKa rises to 6.6, showing the dramatic effect of the neighboring histidine (24). The N-Cys in thioredoxin has a much higher pKa and thus a less stable thiolate anion. This instability renders thioredoxin a better reductase because the oxidized form is more stable. Thioredoxin is therefore a better disulfide bond acceptor than PDI or other thiol isomerases with stable thiolates. Like PDI, DsbA and GRX1 also have a drastically lowered pKa on their N-Cys (16, 22, 24, 25, 36). DsbA also has a histidine in its active site, but GRX1 possesses a CPYC motif. The pKa of tyrosine is over 10, and although it is most likely lowered by the nearby thiolate it does not achieve the same thiolate stability that a more ionizable residue like histidine does. Although the histidine in PDI is responsible for lowering the pKa substantially, the pKa of free cysteine is 8.3, 2 orders of magnitude higher than the observed pKa value of 6.6. This additional thiolate stabilization is attributed to positive charges residing on the local protein backbone, indicating that overall structure of the protein can influence stability, not simply the active site residues (24). A study examining thioredoxin showed that when certain amino acids outside the active site are mutated to alanines, D26A and P76A, drastic changes to redox potential and stability are observed that are attributed to a cascading disruption in charge distribution across the enzyme (17).
To fully study the wide range of substrate proteins for PDI, we generated variants with amino acid substitutions in the intervening sequences while retaining the active site cysteines. Of several single amino acid substitutions to the intervening sequence, there were two variants, CGPC and CGRC, that were still catalytically active, albeit with reduced reductase capability in the presence of insulin. The intervening sequence substitutions that formed stable oxidative traps were both substitutions at the histidine residue of the CGHC motif in PDI, which helps regulate the stability of the thiolate anion in the active site. The substitution of this histidine to either proline or arginine created a PDI variant that could form stable complexes when performing either reducing or oxidizing disulfide chemistry. Alternatively, substituting an aspartic acid residue for histidine completely inactivated the enzyme. This addition of more negative charge into the active site renders the thiolate anion extremely unstable, so much so that it resulted in the complete inactivation of the enzyme. The His → Arg substitution has not been previously studied, but we utilized the previous work analyzing substrate kinetics and functions when the histidine is substituted for a proline. Although the loss of the histidine certainly destabilizes the N-Cys thiolate anion, the pKa remains lower than physiological pH, indicating that the thiolate anion is still favored and the enzyme is still active. The less favorable rate of transition from a reduced to oxidized state has been observed using a CGPC-PDI variant (24). The increase in N-Cys pKa renders the glutathione-induced transition between oxidized and reduced states almost 2 orders of magnitude less favorable. This altered kinetics of oxidation and reduction could explain why we were able to utilize a His → Pro substitution in PDI to form more stable substrate traps than we would with wild-type PDI.
How did substitutions of histidine in the active site enable mutants that could capture either oxidizing or reducing reactions? We can infer from previous work that the substitutions of the histidine residue altered the pKa of the nucleophilic cysteines, thus making the transition from reduced to oxidized and back again less favorable than in wild-type PDI. The variants could still transition from a reduced or oxidized state but at a rate that allowed us to perform kinetic substrate trapping with substrates requiring either reduction or oxidation. Although we are able to begin the kinetic trapping reaction with PDI in either a prereduced or preoxidized state, it is quite possible that the state of the enzyme can change before interacting with the substrate that is kinetically trapped. An outline of this premise is shown in Fig. 10. Because our intervening sequence variants are slower but fully functional, the possibility exists that, before we quench the thiol exchange with NEM to trap our hypothetical substrate Protein X, our variants are able to fully complete the required disulfide chemistry on Protein X and the complex we isolate is in fact the second substrate, Protein Y. Isolation of complexed platelet proteins identified five proteins, cathepsin G, glutaredoxin-1, thioredoxin, fibrinogen, and GP1b, which appear to be biased for interacting with oxidized PDI. An effect of oxidized PDI on cathepsin G was observed using washed platelet releasate in an ex vivo assay, and other published work had identified GP1b as a PDI substrate that requires oxidation as well (29). The CGHC motif is present in many members of the thiol isomerase family, including all catalytically active vascular thiol isomerases (29–31, 37, 38). The use of thiol isomerase variants that are fully functional but with a prolonged intermediate state will not only elucidate the relationships between structure and function of these enzymes but will also help evaluate their roles in biological systems.
Figure 10.

Kinetic substrate trapping using described PDI variants has potential to identify both reduction and oxidation substrates. Shown are reaction schemes diagramming the ideal reaction during kinetic trapping as well as a two-cycle kinetic trapping reaction. Wild type is shown for comparison. Green diagrams denote the hypothetical Protein X, blue diagrams denote the hypothetical Protein Y, and bonds that are made or broken during catalysis are shown in red. t0, the initial state; t1 and t2, intermediate states; ttrap, reaction state when stopped with NEM; tfinal, protein-substrate complex, or lack thereof, isolated after IP.
Experimental procedures
PDI variant generation and purification
The cDNA encoding for human PDI was inserted into the pT7-FLAG-SBP-1 vector purchased from Sigma. Point mutations in the PDI sequence were made in subsequent rounds using the QuikChange site-directed mutagenesis kit available from New England Biolabs. PDI was expressed in the BL21 strain of Escherichia coli, first grown in LB medium at 37 °C until induced overnight at 25 °C with 1 mm isopropyl 1-thio-β-d-galactopyranoside. Cells were lysed using the BPER bacterial protein extraction kit from Thermo Fisher. Clarified lysates were incubated with high capacity streptavidin-coated agarose beads overnight at 4 °C, washed for at least 10 column volumes with 1× TBS containing 0.1% Tween 20 (TBST), and then eluted with the same buffer containing 2 mm biotin. Eluates were dialyzed three times against 1× PBS, concentrated, and stored frozen at −80 °C. An SDS-polyacrylamide gel showing expression levels and final purity of each PDI construct is shown in supplemental Fig. S1.
Platelet preparation
Resting platelets were isolated by centrifuging whole blood at 300 × g for 20 min in the presence of 10% citrate. Platelet-rich plasma was removed, incubated at 37 °C for 30 min, and then centrifuged at 1000 × g for 10 min in the presence of 20% acid-citrate-dextrose, 0.05 unit/ml a-pyrase, and 1 μm prostaglandin E1. Platelets were resuspended thrice in HEPES-Tyrode buffer (134 mm sodium phosphate, 2.9 mm KCl, 12 mm sodium bicarbonate, 20 mm HEPES, 1 mm Mn2+, 5 mm glucose, pH 7.3) and incubated at 37 °C for 30 min.
Insulin reduction assay
PDI reductase was assayed by measuring the turbidity of reduced insulin (27). PDI (12.5 mg/ml) was added to 0.35 mg/ml insulin in the presence of 300 μm DTT. The reaction was carried out in 100 mm potassium phosphate, pH 7.0, with 2 mm DTT. The turbidity of reduced aggregated insulin was monitored by absorbance at 650 nm.
Di-eosin-GSSG assay
Preparation of di-eosin-GSSG was performed as described (28). PDI activity was determined by adding 50 nm PDI to 150 nm di-eosin-GSSG probe and 5 μm DTT in 1× PBS. Eosin fluorescence was monitored by excitation of eosin-GSH at 525 nm and emission at 545 nm.
DTNB assay
PDI proteins were diluted to a concentration of 1 mg/ml and then reduced or oxidized by incubating with either 20 mm DTT or 20 mm GSSG for 20 min on ice. The GSSG/DTT was then removed using a size-exclusion desalting column as described (Zeba, Thermo Fisher). Samples were then divided and either assayed for activity as described above, reincubated with the opposing redox compound to assess reversibility, or diluted 10-fold into 100 mm Tris buffer, pH 8.3, in the presence of DTNB (100 mm) to assess free thiol oxidation. For DTNB measurements, samples were incubated for 5 min at room temperature and then measured for absorbance at 412 nm.
Kinetic trapping
Purified PDI variants, wild-type PDI, or inactive PDI was added to washed platelets (1–3 × 109) in the presence or absence of 1 mm CaCl2, 10 mm MnCl2, and 1.0 unit/ml thrombin and allowed to incubate at room temperature for 3 min. Hirudin (20 units/ml) was added and incubated for 1 min, and then 20 mm NEM was added and allowed to incubate at room temperature in the dark for 30 min. The platelet suspension was then spun down at 1000 × g for 10 min and separated into releasate and pellet fractions. The releasate fraction was spun for an addition 5 min at 2800 × g. The pellet fraction was resuspended in 1 ml of cold 20 mm Tris, pH 7.4, 2% Triton X-100, 20 mm EGTA, 20 mm MgCl2, 2 mm ATP, 20 mm NEM, 0.025 mg/ml cytochalasin D, 0.8 mm PMSF, and 1× protein inhibitor mixture (Thermo Fisher) and incubated on ice for 30 min with regular inversion. The platelet lysate was then mixed with 1 ml of cold 2 m Tris, pH 7.0, 0.5 mm ATP, 0.2 mm MgCl2, and 20 mm NEM and incubated on ice for 30 min with regular inversion. The lysate was then centrifuged at 13,100 × g for 10 min at 4 °C. PDI-substrate complexes were captured with anti-FLAG magnetic beads (Sigma) from clarified lysate and clarified releasate overnight at 4 °C away from light with inversion. The magnetic beads were separated and rinsed three times with 20 mm HEPES, pH 7.4, 5% Triton X-100, and 500 mm NaCl. Complexes were eluted by heating at 95 °C for 15 min in Laemmli buffer.
2D electrophoresis
Immunoprecipitates were overloaded on the gel and separated by SDS-PAGE under non-reducing conditions with an acrylamide concentration of 5%. Bands were excised, washed twice with H2O, and then incubated for 30 min in 2× Laemmli buffer containing 125 mm DTT. The bands were again washed twice with H2O and then incubated for 20 min in 2× Laemmli buffer containing 190 mm NEM. Bands were again washed twice with H2O and placed orthogonally on a 5% acrylamide gel. Proteins were separated in the second dimension by SDS-PAGE under reducing conditions and visualized by silver staining.
Mass spectroscopy
Peptide sequences were determined with tandem mass spectroscopy using an Orbitrap Elite MS system (Thermo Fisher), and those sequences were matched to proteins of interest using BLAST.
Antibodies
All primary antibodies were directed toward human antigens except for FLAG antibodies. All antibodies were used at manufacturer-recommended dilutions. Anti-fibrin 59D8 (39), rabbit anti-serpin B6 (Novus Biologicals, NBP1-86648), and rabbit anti-heparanase (LifeSpan Biosciences, LS-C178439) were obtained from the indicated sources. All remaining antibodies were obtained from Abcam: rabbit anti-ERp57 (ab10287), rabbit anti-GP1b/CD42c (ab96565), rabbit anti-thioredoxin (ab86255), rabbit anti-glutaredoxin (ab45953), rabbit anti-cathepsin G (ab131407), rabbit anti-collagen VI (ab6588), rabbit anti-fibrinogen γ chain (ab96532), rabbit anti-annexin V (ab30941), mouse anti-tetranectin (ab167667), mouse anti-kallekrein14 (ab167234), rabbit anti-FLAG (ab1162), and goat anti-FLAG (ab1257).
Immunoblotting
Immunoblotting was performed using the Mini Trans-Blot Cell system (Bio-Rad) at 4 °C at either 100 V for 1 h or 30 V overnight. Proteins were transferred onto low-fluorescence PVDF membranes (Bio-Rad), and membranes were blocked in 5% milk in TBST for either 1 h at room temperature or overnight at 4 °C. Membranes were then incubated with primary antibodies overnight at 4 °C, washed three times for15 min with TBST, incubated with secondary antibodies for 1 h at room temperature, and washed five times for 5 min with TBST before visualization. Visualization was performed using an ImageQuant LAS 4000 gel imaging system (GE Healthcare), and ImageQuant TL software was used for all quantification and analysis. For HRP-conjugated secondaries, an ECL Western blotting kit (Abcam, ab133406) was used according to the manufacturer's instruction
Cathepsin G activity
For the platelet releasates, washed platelets were generated as described above. Platelets were diluted to a concentration of 400,000/μl and then activated in the presence of prereduced or preoxidized PDI (0.5 mg/ml), 1 mm CaCl2, 10 mm MnCl2, and 0.1 unit/ml thrombin and allowed to incubate at room temperature for 3 min. Hirudin (2 units/ml) was added and incubated for 1 min. Platelet releasate was generated by centrifugation at 1000 × g for 10 min. Colorimetric cathepsin G substrate (N-succinyl-Ala-Ala-Pro-Phe p-nitroanilide; Sigma) was diluted in 100 mm Tris, pH 8.0, at a final concentration of 10 mm and then diluted 10-fold into the platelet releasate sample for measurement. Activity was measured via absorbance at 410 nm over time.
Author contributions
B. F. conceived and coordinated the study. K. M. B. performed the experiments shown in Fig. 4. S. P. G. assisted in designing and optimizing the cathepsin G experiments. J. D. S. designed, performed, and analyzed the remainder of the experiments shown. J. D. S., R. F., and B. F. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
This work was supported by National Institutes of Health Grants U54HL112302 and T32HL07917. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Fig. S1.
- PDI
- protein-disulfide isomerase
- NEM
- N-ethylmaleimide
- GSSG
- glutathione disulfide
- IP
- immunoprecipitation
- DTNB
- 5,5′-dithio-bis(2-nitrobenzoic acid).
References
- 1. Ellgaard L., and Ruddock L. W. (2005) The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 6, 28–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Freedman R. B., Klappa P., and Ruddock L. W. (2002) Protein disulfide isomerases exploit synergy between catalytic and specific binding domains. EMBO Rep. 3, 136–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ferrari D. M., and Söling H. D. (1999) The protein disulphide-isomerase family: unravelling a string of folds. Biochem. J. 339, 1–10 [PMC free article] [PubMed] [Google Scholar]
- 4. Appenzeller-Herzog C., and Ellgaard L. (2008) The human PDI family: versatility packed into a single fold. Biochim. Biophys. Acta 1783, 535–548 [DOI] [PubMed] [Google Scholar]
- 5. Lyles M. M., and Gilbert H. F. (1991) Catalysis of the oxidative folding of ribonuclease A by protein disulfide isomerase: dependence of the rate on the composition of the redox buffer. Biochemistry 30, 613–619 [DOI] [PubMed] [Google Scholar]
- 6. Ohba H., Harano T., and Omura T. (1981) Intracellular and intramembranous localization of a protein disulfide isomerase in rat liver. J. Biochem. 89, 889–900 [DOI] [PubMed] [Google Scholar]
- 7. LaMantia M. L., and Lennarz W. J. (1993) The essential function of yeast protein disulfide isomerase does not reside in its isomerase activity. Cell 74, 899–908 [DOI] [PubMed] [Google Scholar]
- 8. Wilkinson B., and Gilbert H. F. (2004) Protein disulfide isomerase. Biochim. Biophys. Acta 1699, 35–44 [DOI] [PubMed] [Google Scholar]
- 9. Laboissiere M. C., Sturley S. L., and Raines R. T. (1995) The essential function of protein-disulfide isomerase is to unscramble non-native disulfide bonds. J. Biol. Chem. 270, 28006–28009 [DOI] [PubMed] [Google Scholar]
- 10. Gilbert H. F. (1998) Protein disulfide isomerase. Methods Enzymol. 290, 26–50 [DOI] [PubMed] [Google Scholar]
- 11. Freedman R. B., Dunn A. D., and Ruddock L. W. (1998) Protein folding: a missing redox link in the endoplasmic reticulum. Curr. Biol. 8, R468–R470 [DOI] [PubMed] [Google Scholar]
- 12. Darby N. J., Kemmink J., and Creighton T. E. (1996) Identifying and characterizing a structural domain of protein disulfide isomerase. Biochemistry 35, 10517–10528 [DOI] [PubMed] [Google Scholar]
- 13. Kemmink J., Darby N. J., Dijkstra K., Nilges M., and Creighton T. E. (1997) The folding catalyst protein disulfide isomerase is constructed of active and inactive thioredoxin modules. Curr. Biol. 7, 239–245 [DOI] [PubMed] [Google Scholar]
- 14. Klappa P., Ruddock L. W., Darby N. J., and Freedman R. B. (1998) The b′ domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J. 17, 927–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Walker K. W., Lyles M. M., and Gilbert H. F. (1996) Catalysis of oxidative protein folding by mutants of protein disulfide isomerase with a single active-site cysteine. Biochemistry 35, 1972–1980 [DOI] [PubMed] [Google Scholar]
- 16. Darby N. J., Freedman R. B., and Creighton T. E. (1994) Dissecting the mechanism of protein disulfide isomerase: catalysis of disulfide bond formation in a model peptide. Biochemistry 33, 7937–7947 [DOI] [PubMed] [Google Scholar]
- 17. Gleason F. K. (1992) Mutation of conserved residues in Escherichia coli thioredoxin: effects on stability and function. Protein Sci. 1, 609–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chivers P. T., Prehoda K. E., and Raines R. T. (1997) The CXXC motif: a rheostat in the active site. Biochemistry 36, 4061–4066 [DOI] [PubMed] [Google Scholar]
- 19. Lundström J., Krause G., and Holmgren A. (1992) A Pro to His mutation in active site of thioredoxin increases its disulfide-isomerase activity 10-fold. New refolding systems for reduced or randomly oxidized ribonuclease. J. Biol. Chem. 267, 9047–9052 [PubMed] [Google Scholar]
- 20. Walker K. W., and Gilbert H. F. (1995) Oxidation of kinetically trapped thiols by protein disulfide isomerase. Biochemistry 34, 13642–13650 [DOI] [PubMed] [Google Scholar]
- 21. Walker K. W., and Gilbert H. F. (1997) Scanning and escape during protein-disulfide isomerase-assisted protein folding. J. Biol. Chem. 272, 8845–8848 [DOI] [PubMed] [Google Scholar]
- 22. Creighton T. E., and Freedman R. B. (1993) A model catalyst of protein disulphide bond formation. Curr. Biol. 3, 790–793 [DOI] [PubMed] [Google Scholar]
- 23. Darby N. J., and Creighton T. E. (1995) Characterization of the active site cysteine residues of the thioredoxin-like domains of protein disulfide isomerase. Biochemistry 34, 16770–16780 [DOI] [PubMed] [Google Scholar]
- 24. Kortemme T., Darby N. J., and Creighton T. E. (1996) Electrostatic interactions in the active site of the N-terminal thioredoxin-like domain of protein disulfide isomerase. Biochemistry 35, 14503–14511 [DOI] [PubMed] [Google Scholar]
- 25. Joelson T., Sjöberg B. M., and Eklund H. (1990) Modifications of the active center of T4 thioredoxin by site-directed mutagenesis. J. Biol. Chem. 265, 3183–3188 [PubMed] [Google Scholar]
- 26. Krause G., Lundström J., Barea J. L., Pueyo de la Cuesta C., and Holmgren A. (1991) Mimicking the active site of protein disulfide-isomerase by substitution of proline 34 in Escherichia coli thioredoxin. J. Biol. Chem. 266, 9494–9500 [PubMed] [Google Scholar]
- 27. Holmgren A. (1979) Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J. Biol. Chem. 254, 9627–9632 [PubMed] [Google Scholar]
- 28. Raturi A., and Mutus B. (2007) Characterization of redox state and reductase activity of protein disulfide isomerase under different redox environments using a sensitive fluorescent assay. Free Radic. Biol. Med. 43, 62–70 [DOI] [PubMed] [Google Scholar]
- 29. Chiu J., Passam F., Butera D., and Hogg P. J. (2015) Protein disulfide isomerase in thrombosis. Semin. Thromb. Hemost. 41, 765–773 [DOI] [PubMed] [Google Scholar]
- 30. Furie B., and Flaumenhaft R. (2014) Thiol isomerases in thrombus formation. Circ. Res. 114, 1162–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cho J., Furie B. C., Coughlin S. R., and Furie B. (2008) A critical role for extracellular protein disulfide isomerase during thrombus formation in mice. J. Clin. Investig. 118, 1123–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bowley S. R., and Fang C., Merrill-Skoloff G., Furie B. C., and Furie B. (2017) Protein disulfide isomerase secretion following vascular injury initiates a regulatory pathway for thrombus formation. Nat. Commun. 8, 14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Korkmaz B., Moreau T., and Gauthier F. (2008) Neutrophil elastase, proteinase 3 and cathepsin G: physicochemical properties, activity and physiopathological functions. Biochimie 90, 227–242 [DOI] [PubMed] [Google Scholar]
- 34. Sambrano G. R., Huang W., Faruqi T., Mahrus S., Craik C., and Coughlin S. R. (2000) Cathepsin G activates protease-activated receptor-4 in human platelets. J. Biol. Chem. 275, 6819–6823 [DOI] [PubMed] [Google Scholar]
- 35. Hotchkiss K. A., Chesterman C. N., and Hogg P. J. (1996) Catalysis of disulfide isomerization in thrombospondin 1 by protein disulfide isomerase. Biochemistry 35, 9761–9767 [DOI] [PubMed] [Google Scholar]
- 36. Wunderlich M., Jaenicke R., and Glockshuber R. (1993) The redox properties of protein disulfide isomerase (DsbA) of Escherichia coli result from a tense conformation of its oxidized form. J. Mol. Biol. 233, 559–566 [DOI] [PubMed] [Google Scholar]
- 37. Jordan P. A., Stevens J. M., Hubbard G. P., Barrett N. E., Sage T., Authi K. S., and Gibbins J. M. (2005) A role for the thiol isomerase protein ERP5 in platelet function. Blood 105, 1500–1507 [DOI] [PubMed] [Google Scholar]
- 38. Holbrook L.-M., Sasikumar P., Stanley R. G., Simmonds A. D., Bicknell A. B., and Gibbins J. M. (2012) The platelet-surface thiol isomerase enzyme ERp57 modulates platelet function. J. Thromb. Haemost. 10, 278–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hui K. Y., Haber E., and Matsueda G. R. (1983) Monoclonal antibodies to a synthetic fibrin-like peptide bind to human fibrin but not fibrinogen. Science 222, 1129–1132 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.