

Abstract
Ribonucleotide reductases (RNRs) catalyze the conversion of nucleoside diphosphate substrates (S) to deoxynucleotides with allosteric effectors (e) controlling their relative ratios and amounts, crucial for fidelity of DNA replication and repair. Escherichia coli class Ia RNR is composed of α and β subunits that form a transient, active α2β2 complex. The E. coli RNR is rate-limited by S/e-dependent conformational change(s) that trigger the radical initiation step through a pathway of 35 Å across the subunit (α/β) interface. The weak subunit affinity and complex nucleotide-dependent quaternary structures have precluded a molecular understanding of the kinetic gating mechanism(s) of the RNR machinery. Using a docking model of α2β2 created from X-ray structures of α and β and conserved residues from a new subclassification of the E. coli Ia RNR (Iag), we identified and investigated four residues at the α/β interface (Glu350 and Glu52 in β2 and Arg329 and Arg639 in α2) of potential interest in kinetic gating. Mutation of each residue resulted in loss of activity and with the exception of E52Q-β2, weakened subunit affinity. An RNR mutant with 2,3,5-trifluorotyrosine radical (F3Y122•) replacing the stable Tyr122• in WT-β2, a mutation that partly overcomes conformational gating, was placed in the E52Q background. Incubation of this double mutant with His6-α2/S/e resulted in an RNR capable of catalyzing pathway-radical formation (Tyr356•-β2), 0.5 eq of dCDP/F3Y122•, and formation of an α2β2 complex that is isolable in pulldown assays over 2 h. Negative stain EM images with S/e (GDP/TTP) revealed the uniformity of the α2β2 complex formed.
Keywords: conformational change, electron microscopy (EM), electron paramagnetic resonance (EPR), enzyme mutation, glutamate, ribonucleotide reductase, radical transfer, subunit interface
Introduction
Ribonucleotide reductases (RNRs)5 are macromolecular machines that convert nucleoside diphosphates (NDP) to deoxynucleoside diphosphates (dNDP) supplying de novo the pools of monomeric building blocks required for DNA biosynthesis, and controlling in a sophisticated fashion the relative ratios of these pools and their amounts, essential for fidelity of DNA replication and repair (1–3). The class Ia RNRs are found in both humans and Escherichia coli, with the latter serving as the prototype that has been studied for decades. Despite this, the molecular structure of the machine and its gymnastics on binding nucleotides at three distinct sites still remains a mystery. These proteins are composed of two subunits, α and β, which in the case of the E. coli RNR form an active α2β2 complex (3, 4). The NDP substrates, dNTP, and ATP allosteric effectors bind in three sites within α: the catalytic site (C-site), the specificity site (S-site), which controls which NDP is reduced, and the activity site (A-site), which controls the rate of turnover (5–7). The β subunit contains the diferric-tyrosyl radical (Y122•) cofactor essential for nucleotide reduction (8, 9). The initiation of nucleotide reduction requires oxidation of Cys439-α by Tyr122•-β over a distance of 35 Å, utilizing a specific pathway shown in Fig. 1 (3, 4, 10).
Figure 1.

The Uhlin and Eklund docking model for the E. coli active α2β2 complex. The monomers of α2 (PDB code 4R1R) and β2 (PDB code 1RIB) are shown in blue and green and red and yellow, respectively. α2 was crystallized in the presence of GDP (salmon), TTP (purple), and a peptide corresponding to residues 360–375 (pink) of β2. The ATP cone domain housing the activity site is shown in orange. On the left is the RT pathway between Tyr122• in β2 and Cys439 in α2. Trp48 is in brackets as there is currently no evidence for its involvement (18). Note Tyr356 and Glu350 (not shown) are located in the disordered C-terminal tail of β2.
Although studies from the Eklund lab (4, 11) have provided us with atomic resolution structures of α and β, the structure of the active complex has remained a challenge as the subunit interactions are weak (0.2 to 0.4 μm) even in the presence of NDPs and dNTPs and β can act catalytically (12–15). In addition, the C-terminal 30 to 35 residues of all βs are always disordered and this is the region that Sjöberg and co-workers (13) showed was largely responsible for α/β affinity. Within this disordered tail of β reside the conserved residues: Tyr356 and Glu350. Tyr356 is an essential component of the 35-Å pathway involved in the oxidation of Cys439 where NDP reduction occurs (3, 13, 16, 17). Glu350, we have recently shown, plays an essential role in initiation of the conformational gating of this long-range oxidation when NDPs and dNTPs bind to α (18). Briefly, initiation of the radical transfer (RT) process is thought to involve proton transfer from the water on Fe1 in the diferric-Y• cofactor to Tyr122• and electron transfer from Tyr356 forming the Tyr122 phenol (Fig. 1) (18–21). Recent RT studies and new Ia RNR subclassifications (22) have helped us to identify conserved residues that could play an important role at the subunit interface of the RNR in conformational gating. The results of these studies are reported herein.
Currently our thinking about the RNR structure is governed by a docking model of α2β2 generated by Eklund and co-workers (3, 4) using the crystal structures of α2 and β2 and their shape complementarity. Their model is supported by four distance measurements (3) made using pulsed electron electron double resonance (PELDOR) spectroscopy and recent biophysical studies including small angle X-ray scattering and single particle electron microscopy (EM) (23–25). For the most part these methods have taken advantage of RNRs with site-specifically incorporated unnatural amino acids (UAAs) (3). The docking model and the C-terminal 34-amino acid residues of β2 served as the starting point for identifying α/β interface residues.
Two different types of experiments using UAA technology have provided insight about the conformational change(s) effected by binding of S and e to α on the initiation of the rate-limiting conformational gating in β (26, 27). One set of experiments used RNR with 3-aminotyrosine (NH2Y) site-specifically replacing Tyr730 in α. Incubation of NH2Y730-α2, β2 with CDP/ATP resulted in loss of Tyr122• in β, and formation of a new radical at 730 (NH2Y730•) in α2 (24, 26, 28–30). This oxidation, which occurs only upon binding S and e to α, causes an increase in the affinity of the α/β subunits 25-fold relative to the WT RNR and a decrease in the dissociation rate of the subunits by 104, a process formally involving movement of a single hydrogen atom (24)! A second set of experiments investigating the role of proton-coupled electron transfer at Tyr356 and the function of the conserved Glu350 as a proton acceptor of this step (Fig. 1), also provided interesting, unexpected results. Using E350X-β2 (X = Ala, Asp, or Gln) mutants in WT and mutant backgrounds in which Tyr122• was replaced with tyrosine analogs that are hotter oxidants (3-nitrotyrosine, NO2Y122• or 2,3,5-trifluorotyrosine, F3Y122•) (20, 31), we found an inability of E350X-β2 to initiate RT even in the case of the Glu to Asp substitution (18). This result suggested that charged residues might play an important role in gating RT at the interface where the Glu350 residue resides.
This paper focuses on our efforts to identify additional interface residues using mutagenesis and our ability to site-specifically incorporate UAAs into each subunit. Recently using sequence information, the class Ia RNRs (rnrdb.pfitmap.org/) now designated NrdAg and NrdBg were subcharacterized (22). This information and our current structural understanding of α2β2 resulted in the identification and examination of mutations in four conserved residues: Glu52 and Glu350 in β2 and Arg329 and Arg639 in α2 in E. coli RNR. The inactivity of the E. coli mutants established that these residues are essential and the binding studies of α/β interactions established that with the exception of E52Q, the binding affinities decreased 5–20-fold relative to WT-α/β. The tight affinity and inactivity of the E52Q mutant led to further investigation of its properties in the F3Y122• background. Unlike WT-β2, F3Y122•-β2 results in partial uncoupling of the conformational gating that rate limits NDP reduction (20, 32) by rapidly producing the Tyr356• (now detectable). It is likely being reduced to the F3Y122-O− (phenolate) instead of the phenol (18). Despite the inactivity of E52Q-β2, the double mutant, E52Q/F3Y122•-β2, when incubated with α2/S/e (CDP/ATP or GDP/TTP) resulted in formation of 0.5 eq of the Tyr356• intermediate and in the case of CDP, 0.5 eq of dCDP per F3Y122•. Pull-down experiments of the α/β mixture after 5 min and 2 h using a His6-α, gave a high recovery of a 0.6–0.8/1.0 ratio of subunits in the β/α complex. Negative stain electron microscopy (EM) analysis and size exclusion chromatography (SEC) studies revealed that the predominant species is α2β2. The implications of these results on conformational gating and potential structural insight of the active complex are discussed.
Results
Identification of conserved α/β interface residues, their mutation and assay for activity, and subunit binding affinity
Our recent studies investigating the role of Glu350, a conserved residue in the disordered C-terminal tail of β2, suggested that this residue was essential for the conformational gating of the RT initiation process (13, 18). We therefore looked at other conserved charged residues using the α2β2 docking model, to identify those that might reside at the α/β interface. Alignment of 80 sequences in the NrdAg/NrdBg subclass revealed that Glu350 and Glu52 in β, and Arg639 in α were conserved in 80 of 80 sequences, whereas Arg329 in α was conserved in 79 of 80. These residues and additional ones, Arg323 (not conserved) and Arg735 (76/80) in α, became candidates for investigation by mutagenesis.
In the case of the glutamates, each residue was changed to Ala, Gln, and Asp, whereas in the case of the arginines, each was changed to Ala, Gln, and Lys. The proteins were expressed and purified to homogeneity based on SDS-PAGE analysis using the WT protocols (supplemental Fig. S1). In the case of the β2 mutants, the diferric-Y122• was self-assembled to give a cofactor with a Y122• content similar to WT-β2 (Table 1). All mutants were assayed for activity and a Kd for each α/β interaction was determined (Table 1). The E52X-β2 (X = Ala, Asp, or Gln) mutants have activity ∼0.15% of WT-β2, within the levels typically observed for endogenously copurifying WT-β2. The Kd measurements revealed that the Ala and Asp mutants are 5- and 10-fold higher than WT, whereas Gln is similar to WT (Fig. 2A). These studies suggest that Glu52 plays an important role in catalysis.
Table 1.
Specific activity and Kd for E52X (X = Ala, Asp, or Gln)-β2
| β2 | Y122•/β2 | Specific activitya | Wild-type activityb | Kda |
|---|---|---|---|---|
| nmol/min/mg | % | μm | ||
| WT | 1.2 | 7000 | 100.0 | 0.18b |
| E52A | 1.0 | 13.4c | 0.2 | 0.96 |
| E52D | 1.2 | 10.2c | 0.2 | 2.33 |
| E52Q | 1.1 | 8.6c | 0.1 | 0.12 |
| F3Y122• | 0.7 | 686 | 9.8 | |
| E52Q/F3Y122• | 0.9 | 5.8c | 0.1 | <4 nmd |
a Specific activity was determined by the radioactive assay (49) and Kd was determined by the competitive binding method (12). All data are representative of at least two independent experiments.
b Previously reported (12).
c The wild-type NrdB that co-purifies with mutants may cause the low activity.
d Upper limit for Kd of E52Q/F3Y122•.
Figure 2.
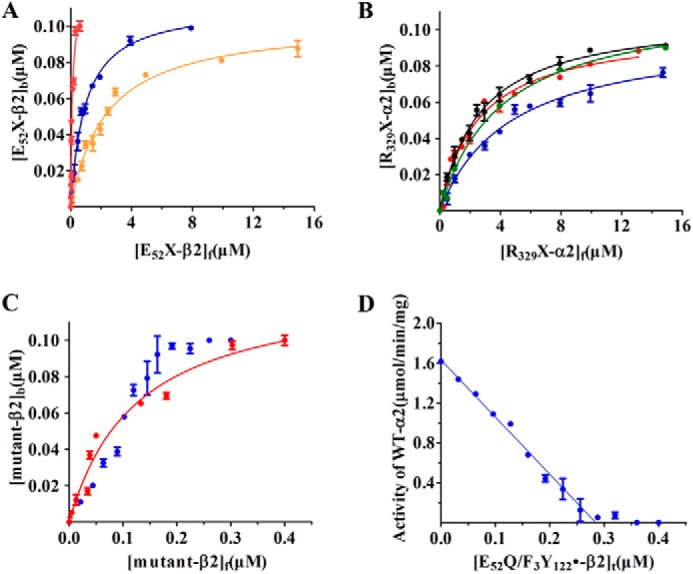
Kd between α2 and β2 in the presence of CDP/ATP determined by the competitive inhibition spectrophotometric assay (12). The data were fit (solid line) to Equation 1. All data are representative of two independent experiments and are expressed as mean ± S.D. Subscript b, f, and t are the bound, free, and total protein concentrations, respectively. A, Kd for α2/E52X-β2 (X = Ala, Asp, or Gln). E52A (blue), E52D (orange), E52Q (red) are shown. B, Kd for mutant-α2/β2: R329A (blue), R329K (red), R329Q (black), and R639Q (green). C, binding for α2/E52Q/F3Y122•-β2 shows a stoichiometric titration under standard assay conditions (blue) and an expanded version of α2/E52Q-β2 shown in A (red). D, analysis of activity with increasing concentrations of E52Q/F3Y122• (0.7 F3Y122•, see text).
N3CDP as a probe of E52X-β2 (X = Ala, Asp, or Gln)
Because RNR is essential, the issue of endogenous WT-RNR co-purifying with the mutants always hinders determination of a lower level of enzymatic activity. An alternative way to assess activity has been to use the mechanism-based inhibitor 2′-azido-2′-deoxycytidine diphosphate (N3CDP) (13, 33). This NDP analog binds in the active site and is enzymatically converted to a nitrogen-centered nucleotide radical (N•), that becomes covalently bound to a cysteine in the active site. The inactivation is stoichiometric with the WT-β2, with complete loss of activity resulting from 1 Tyr122•/β2 being converted to 0.5 eq of N•, leaving 0.5 eq of the Tyr122• remaining (34–36). This unusual stoichiometry is associated with the half-sites reactivity of all class I RNRs. The N• has been extensively characterized by isotopic labeling and EPR methods. With mutant β2s the rate of formation of N• is often slow and the radical is quenched slowly with time; the kinetics often preclude N• detection, thus analysis of total radical loss as a function of time is monitored (13). The results of experiments in which E52X-β2/α2/N3CDP/TTP (X = Ala, Asp, and Gln) were incubated and analyzed by EPR over 120 min are summarized in Fig. 3A (33). No N• is observed and the total Tyr122• varies no more than 10% over the 2-h time period. With WT-β2, 0.5 eq of N• is formed within 30 s. Thus, no activity of E52X mutants is apparent by this method either.
Figure 3.
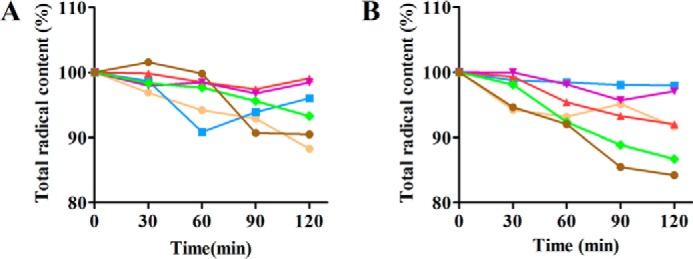
Time-dependent inactivation of RNR mutants in the presence of N3CDP at 25 °C. A, time-dependent radical loss of E52X-β2 (X = Ala, Asp, or Gln), WT-α2, and TTP in the absence (orange, red, or green) and presence (blue, purple, or brown) of N3CDP. B, time-dependent radical loss of R329X-α2 (X = Ala, Lys, or Gln), WT-β2, and TTP in the absence (orange, red, or green) and presence (blue, purple, or brown) of N3CDP. Each point represents the average of two independent trials.
A third method to assess RNR activity is to place E52X into a different background: specifically one in which the Tyr122• is replaced with F3Y122•. The F3Y122•-β2 mutant when incubated with α2/CDP/ATP has been studied in detail and shown to generate dCDP and the pathway Tyr356• (Fig. 1) at 25 s−1 in the first turnover and then reoxidize the putative F3Y122-O− to the F3Y122• in the rate-limiting step in the steady-state (20). This mutant is a hotter oxidant than Tyr122• and disrupts conformational gating of the RT process (20, 32, 37). The E52Q mutant in this background has 0.1% WT activity (Table 1), likely associated with endogenous levels of co-purifying WT-β2. Thus all assays pointed to inactivity of E52Q-β2.
Efforts to determine the Kd for subunit interactions with this double mutant gave data distinct from the single E52Q mutant (Fig. 2C) and the other mutants (Figs. 2A). The sharp break suggests a “stoichiometric” titration. Reanalysis of these data in which activity is monitored with increasing concentrations of E52Q/F3Y122•-β2 reveal that for 0.1 μm α2β2 complex, 0.28 μm of the double mutant was required for complete inactivation (Fig. 2D). Given that the mutant protein used in this experiment has 0.7 F3Y122•/β2 with the radical equally distributed between the two β monomers and assuming that the diferric-cluster without radical binds much more weakly, then one would predict the requirement for 0.29 μm mutant, very similar to the experimental observation.
CDP/ATP, GDP/TTP, and N3CDP/TTP to probe E52Q/F3Y122•-β2 activity by EPR methods
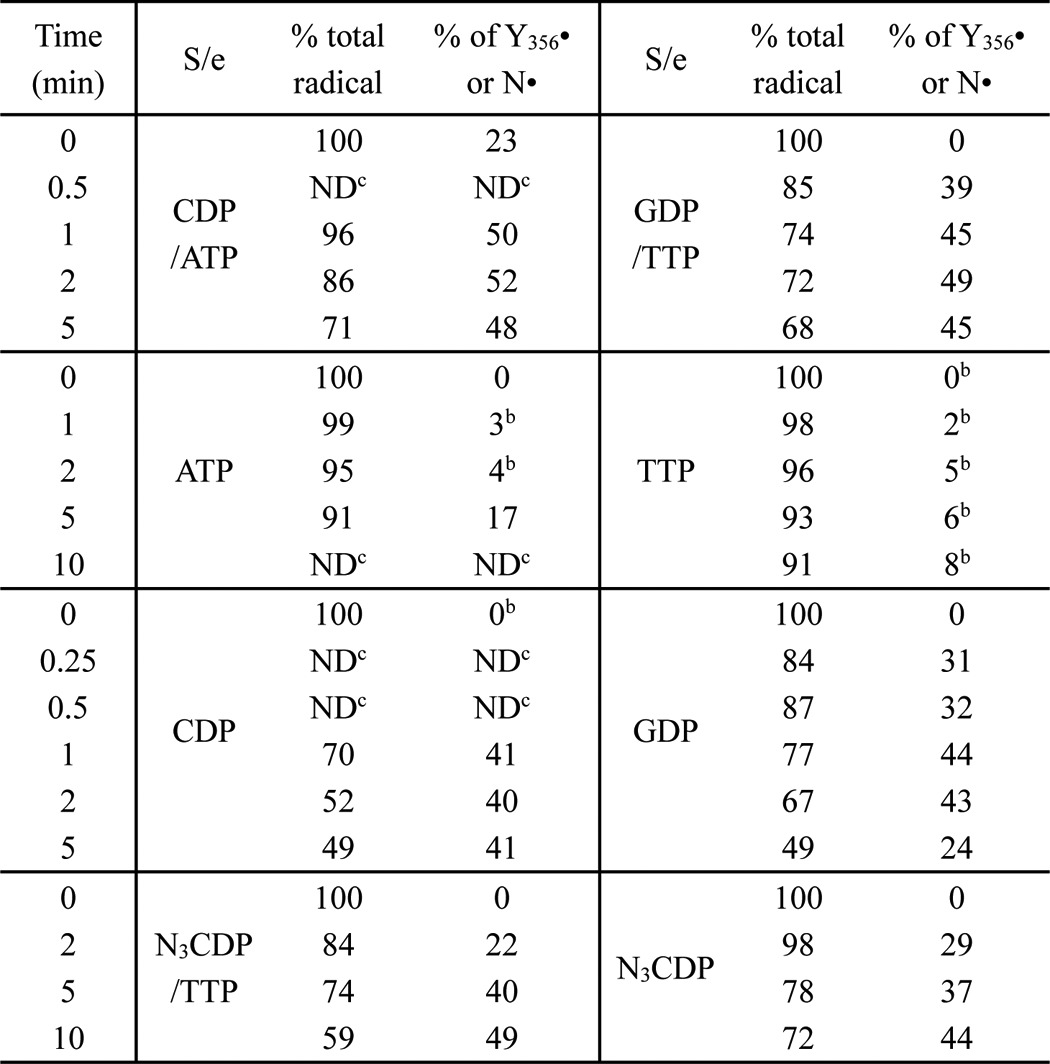
Although no activity of E52Q-β2 or E52Q/F3Y122•-β2 was observed under steady-state conditions, additional experiments were performed on E52Q/F3Y122•-β2 to determine whether chemistry could be observed in the first turnover. As noted above, addition of CDP/ATP/α2 to the single mutant, F3Y122•-β2, results in formation of Tyr356• and a burst of dCDP (0.5 eq/F3Y122•). The double mutant, E52Q/F3Y122•-β2 was incubated with CDP/ATP and analyzed by EPR spectroscopy for production of the Tyr356•. The results are shown in Fig. 4A and are summarized in Table 2. The data reveal that only 4% of the total radical is lost within 1 min and that it increases to 30% by 5 min. Also within the 1-min time frame, 0.50 eq of Tyr356• is formed. The rate of loss of the total radical is substantially reduced when CDP is omitted. When ATP is omitted, however, the total radical is reduced to 50% by 5 min and the amount of Tyr356• is increased to 40% of the total radical by 1 min and remains unchanged at 5 min. Thus CDP is the predominant driver of Tyr356• formation and the effector (ATP) appears to stabilize the F3Y122• radical in β2 when no substrate is present for reduction.
Figure 4.
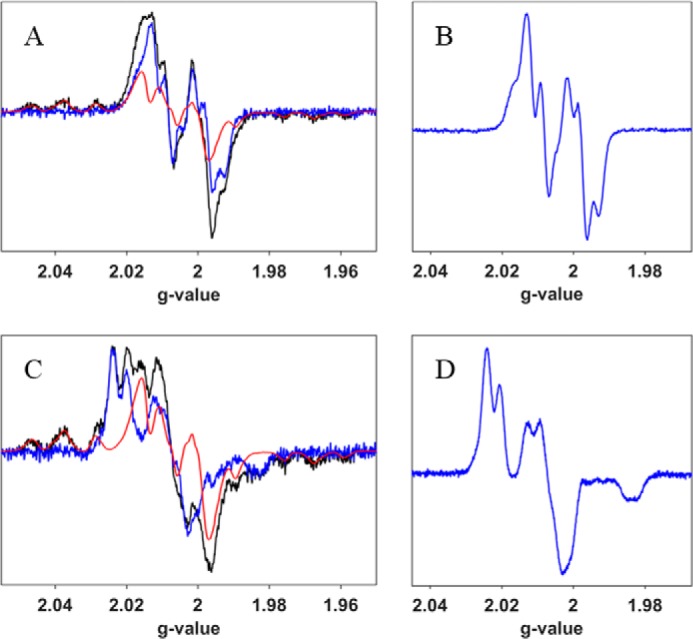
Reaction of E52Q/F3Y122•-β2, WT-α2 with CDP/ATP (A) or N3CDP/TTP (C) monitored by EPR spectroscopy. A, subtraction of the F3Y• spectrum (red) from the composite spectrum from the reaction of E52Q/F3Y122•-β2, WT-α2, CDP, and ATP at 1 min (black) reveals the spectrum in blue. B, spectrum of Tyr356• observed in the reaction of F3Y122•-β2, WT-α2, CDP, and ATP as a reference (20). C, subtraction of F3Y122• (red) from the composite spectrum at 10 min (black) from the reaction of E52Q/F3Y122•-β2, WT-α2, N3CDP, and TTP reveals the spectrum in blue. D, spectrum of N• observed in the reaction of WT-β2, WT-α2, N3CDP, and TTP as a reference (33).
Table 2.
Reaction of E52Q/F3Y122•-β2a and WT-α2a with either ATP/CDP or TTP/GDP or TTP/N3CDP analyzed by EPR spectroscopy
a The concentration, 15 to 50 μm of 1:1 E52Q/F3Y122•-β2 and WT-α2.
b The spectrum after subtraction was similar to background.
c ND, not determined.
An identical set of experiments carried out with purine substrates and effectors have the same phenotypes. The results are summarized in Table 2. With GDP/TTP, by 5 min 30% of the total radical is lost, whereas 0.5 eq of Tyr356• is formed within 2 min. The effector TTP stabilizes total radical and limits Tyr356• formation, whereas GDP is the predominant driver of Tyr356•. What is most amazing about these results is that under steady-state conditions where neither E52Q nor E52Q/F3Y122•-β2 make dCDP, E52Q/F3Y122•-β2 can initiate RT subsequent to S/e binding.
As noted above, a second way to look for activity, uses N3CDP or N3CDP/TTP. The results of this set of experiments are shown in Fig. 4C and summarized in Table 2. In contrast to the results with the single mutant (E52Q), N• is formed and accounts for 49% (N3CDP/TTP) versus 43% (N3CDP) of the total radical at 10 min (compare Fig. 4, C with D, an authentic standard for N•). Thus these data also support the activity of the double mutant, E52Q/F3Y122•-β2, at least on the first turnover.
E52Q/F3Y122•-β2 with pre-reduced α2, CDP, and ATP can produce dCDP
The above observation that the double mutant, E52Q/F3Y122•-β2, is capable of RT to the α2 catalytic site suggests that this protein may be able to make dCDP, even though no (or very low) activity is observed in the steady-state. To test for dCDP formation, an assay was carried out with a 1:1 ratio of subunits at 20 μm in the presence of CDP alone (blue), CDP/ATP (red), and CDP/ATP with reductant TR/TRR/NADPH (green) and the reaction was monitored as a function of time (Fig. 5). The amount of the Tyr356• (0.5 eq) observed (Table 2) is likely formed during reverse RT and suggested that 0.5 eq of dCDP would be generated. The results shown in Fig. 5 suggest that this is the case. There is a burst of dCDP formation and it is independent of the presence of reductant. The size of the burst in all three experiments is similar to the amount of Tyr356• formed, consistent with half-sites reactivity and one turnover. In all experiments, the burst phase is followed by a slow phase that occurs from 0.2 to 0.6% (1.6, 3.4, and 4.4 nmol/min/mg in Fig. 5, blue, red, and green, respectively) of that observed with the single mutant, F3Y122-β2 (686 nmol/min/mg). The rate is fastest with TR/TRR/NADPH/CDP/ATP > CDP/ATP > CDP. A number of explanations are possible for this slow phase observed in all experiments. In the absence of reductant (red and blue, Fig. 5) the slow phase could be associated with endogenous β2 acting catalytically, with very slow completion of the catalytic cycle in which Tyr356• must reoxidize the F3Y-O− or with slow release of cytosine catalyzed by the oxidized form of RNR. This issue remains unresolved. However, the interesting result is that E52Q/F3Y122•-β2 is able to carry out one turnover! Thus, although the steady-state assays do not reveal significant activity (0.1% WT, Table 1), the double mutant is capable of the radical-based reactions that result in dCDP formation.
Figure 5.
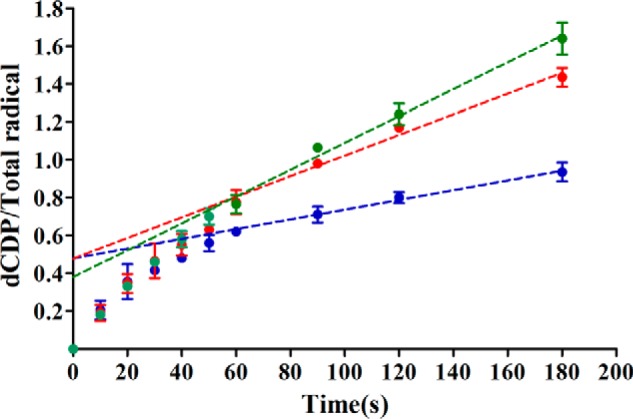
dCDP formation by WT-α2 and E52Q/F3Y122•-β2 (0.91 Y122•/β2) in the presence of CDP (blue), CDP/ATP (red), or CDP/ATP and reductant TR/TRR/NADPH (green). The reaction mixture contained 20 μm of 1:1 subunits in 30 μl. In these experiments α2 was pre-reduced. Each point represents the average of two independent trials.
Interaction of His6-α2 and E52Q/F3Y122•-β2 using pulldown assays and SDS-PAGE analysis
Our previous studies showed that incubation of His6-NH2Y730-α2 with β2, CDP, and ATP resulted in formation of NH2Y730• concomitant with Tyr122• loss. Rapid purification of His6-NH2Y730-α2 from this mixture using a Ni-NTA affinity resin by centrifugation followed by SDS-PAGE analysis showed that α and β co-purified (24).
Given these results and the apparent stoichiometric titration of E52Q/F3Y122•-β2 with α2 in our binding assay (Fig. 2, C and D), similar pulldown experiments with His6-α2/E52Q/F3Y122•-β2/ATP/CDP using a Ni-NTA resin were undertaken. Purification was carried out by centrifugation (Fig. 6A) (24) or column gravity workup (Fig. 6B) with quantitation by SDS-PAGE and densitometry using α and β standards (Fig. 6A, left).
Figure 6.
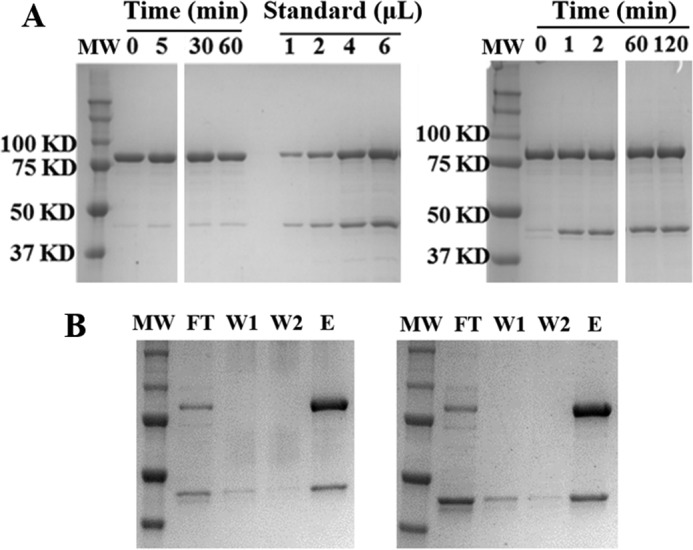
Pulldown assays of different β2s by His6-WT-α2 analyzed by 10% SDS-PAGE. A, elution fractions of a time course from WT-β2 (left) and E52Q/F3Y122•-β2 (right) by His6-WT-α2 in the presence of CDP/ATP using the centrifugation assay. Standards for quantification (1 μm His6-WT-α2 and 1 μm WT-β2) loaded in different amounts are indicated in the left panel. B, pulldown assays with 1:1 (left) or 1:2 (right), α2:E52Q/F3Y122•-β2 with CDP/ATP by gravity with a Ni-affinity column showing flow through (FT), washes (W1 and W2), and elution (E).
Centrifugation analysis monitoring supernatants from time 0 to 120 min incubation prior to workup revealed that when no CDP was present (time 0), no E52Q/F3Y122•-β2 was pulled down, but within 1 min of its addition, the pulldown was maximized and remained unchanged (Fig. 6A, right). The majority of the pulldown experiments were carried out using a column gravity workup (Fig. 6B), as it typically gave higher recoveries of His6-α2 (>80%). A variety of experiments were carried out in which the S (CDP or GDP), e (ATP or TTP), or S/e pairs and the incubation times, 5 or 30 min, were varied. In addition, controls with β2, E52Q/Y122•-β2, and F3Y122•-β2, or E52Q/Y122•-β2 without S/e were also examined. The results summarized in Table 3 reveal that with S alone or S/e that a β2/α2 ratio of 0.5–0.8 was observed, where with e alone, the ratio was lower at 5 min, but increased by 30 min (experiments 8 and 12). The data together suggest that the appropriate S/e pair form “tight” complexes rapidly and that tight complex remains at 30 min. These conclusions are supported by the controls (Table 3, 1–4) that all have low β2/α2 ratios, 0.0–0.2, in the pulldowns. These studies suggest the F3Y122•, a conformational uncoupler that generates the Tyr356 pathway radical in combination with the E52Q mutation are important for successful α2β2 complex formation.
Table 3.
Pulldowns with E52Q/F3Y122•-β2, WT-(His)6-α2, S/e, S, e, and controls
| β2 | Y122•/β2 | S/e | β2/α2 ratio (5 min) | β2/α2 ratio (30 min) | |
|---|---|---|---|---|---|
| 1 | WT | 1.2 | CDP/ATP | 0.01 | 0.02 |
| 2 | E52Q | 1.1 | CDP/ATP | 0.19 | 0.13 |
| 3 | F3Y | 0.8 | CDP/ATP | 0.22 | 0.17 |
| 4 | E52Q/F3Y | 0.9 | –/– | 0.05 | 0.03 |
| 5a | E52Q/F3Y | 0.9 | CDP/ATP | 0.58 | 0.62 |
| 5b | E52Q/F3Y | 0.9 | CDP/ATP | 0.59 | 0.64 |
| 6 | E52Q/F3Y (Y731F-α2)a | 0.9 | CDP/ATP | 0.44 | 0.59 |
| 7 | E52Q/F3Y | 0.9 | CDP/– | 0.76 | 0.76 |
| 8 | E52Q/F3Y | 0.9 | –/ATP | 0.30 | 0.50 |
| 9 | E52Q/F3Y | 0.9 | GDP/TTP | 0.59 | 0.43 |
| 11 | E52Q/F3Y | 0.9 | GDP/– | 0.54 | 0.53 |
| 12 | E52Q/F3Y | 0.9 | –/TTP | 0.15 | 0.45 |
| 13 | E52Q/F3Y(1x)b | 0.9 | CDP/ATP | 0.63 | – |
| 14 | E52Q/F3Y(2x)b | 0.9 | CDP/ATP | 0.80 | – |
| 15 | E52Q/F3Y(1x)b | 0.9 | GDP/TTP | 0.62 | – |
| 16 | E52Q/F3Y(2x)b | 0.9 | GDP/TTP | 0.79 | – |
a In 6, the RT pathway block Y731F-α2 was used.
b In 13–16, E52Q/F3Y-β2 was at a 1:1 (×1) or 2:1 (×2) ratio with α2.
Characterization of the reaction mixture by SEC and negative stain EM
Two additional types of experiments were carried out to support an α2β2 complex structure and the tightness of the complex. In one set of experiments the reaction of E52Q/F3Y122•-β2 was incubated with 0.5 eq of α2 (1:2, α2:β2 subunit ratio), GDP, and TTP and loaded on a Superdex 200 SEC column and then eluted with assay buffer containing 50 μm GDP and 10 μm TTP. The results shown in Fig. 7A reveal a peak eluting at 12.1 ml and a broad peak at 13.7 ml. Comparison with molecular weight standards in Fig. 7B suggests that the former is α2β2 and the latter is β2 and the ratio is 1:1 based on a comparison of the relative peak areas as expected from experimental design (Fig. 7, red). When the FPLC experiment was carried out in the absence of nucleotides in the elution buffer, peaks were observed at very similar elution volumes (Fig. 7, black), but the ratio of the peak intensities suggest only ∼40% α2β2 complexation. In a control with F3Y122•-β2/α2/GDP/TTP, no α2β2 complex was observed (Fig. 7A, blue). Control experiments with E52Q/Y122•-β2 in place of E52Q/F3Y122•-β2 showed α2β2 complex formation with GDP/TTP in the elution buffer, whereas no α2β2 was observed without GDP/TTP (not shown). In these experiments, the peaks corresponding to α2β2 at 12 ml eluted 25 min after reaction initiation with GDP and TTP. Thus although the pulldown experiments allow isolation of α2β2 with very high recovery and no GDP/TTP in the elution buffer, the SEC data tell us that on the 30-min time scale of the SEC analysis, the two subunits come apart in the absence of nucleotides during chromatography.
Figure 7.
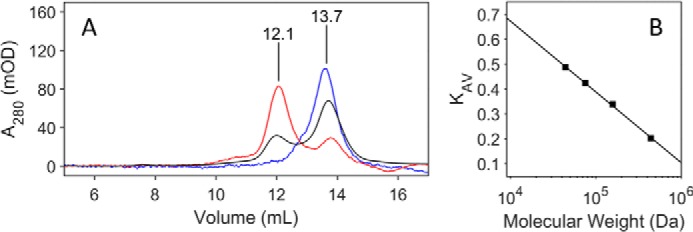
SEC of E52Q/F3Y122•-β2 with α2, GDP, and TTP in the presence (red) or absence (black) of nucleotides in the elution buffer and a control experiment with F3Y122•-β2, α2, GDP, and TTP in the presence of GDP/TTP in eluent (blue). A, the peak eluting at 12.1 min has a molecular weight consistent with α2β2, whereas the broad peak at 13.7 min is likely uncomplexed β and α. The experiment was carried out under the same conditions as the negative stain EM images. A 1:2 ratio of α2:β2 was used to maximize complex formation. B, molecular mass standards are ferritin (440 kDa), aldolase (158 kDa), conalbumin (75 kDa), and ovalbumin (44 kDa).
In a second set of experiments, α2β2 complex formation was examined by negative stain EM. Our previous studies on the reaction of NH2Y730-α2, β2, CDP, and ATP reported our first efforts to look for the “active” α2β2 complex by this method (24). The resulting low resolution (∼32 Å) model revealed a subunit arrangement that was consistent with the α2β2 docking model (Fig. 1). Interestingly, when WT-α2 and WT-β2 were mixed and observed on an EM grid with negative stain, almost all observed particles were of free α2 and almost no α2β2 complex was observed. Free β2 is too small (87 kDa) to be visualized. NH2Y730-α2 with WT-β2 gave rise to ∼70% α2β2 particles (24).
Here, negative stain EM experiments with WT-α2 and E52Q/F3Y122•-β2 with GDP/TTP were carried out under similar conditions to the SEC (Fig. 7) and pulldown (Table 3) experiments. What is immediately striking is the large number of α2β2 complexes that are present (Fig. 8), estimated to be 90%. The ratio of 1:2 for α2:β2 was chosen to maximize the chemistry (Tyr356• formation) as typically there are ∼0.8 E52Q/F3Y122•-β2. Taken together, the pulldown studies, EM, and SEC analysis reveals α2β2 complexes that are supported by biochemical analysis that shows active RT and dCDP formation. The SEC data reveal that further work, such as our stopped flow fluorescence studies on NH2Y730-α2, will be informative in determining a quantitative assessment of the subunit affinity in the complex observed.
Figure 8.

Negative stain EM grid of a reaction mixture of the E52Q/F3Y122•-β2/α2 (2:1) ratio incubated with 1 mm GDP and 0.2 mm TTP for 15 min reveals predominantly α2β2. Representative α2β2 and α2 particles are indicated by red and blue squares, respectively.
Activity and subunit binding affinity of additional α and β mutants (Table 4)
Table 4.
Specific activities for mutant-α2s with 5-fold WT-β2 or 10-fold F3Y122•-β2 determined by the radioactive assay (49) and Kd for mutant-α2/WT-β2 interaction determined by the competitive inhibition assay (12)
All data are representative of at least two independent experiments.
| α2 | WT-β2 |
F3Y122•-β2 |
Kd | ||
|---|---|---|---|---|---|
| Specific activity | Wild-type activity | Specific activity | Wild-type activity | ||
| nmol/min/mg | % | nmol/min/mg | % | μm | |
| WT | 2428 | 100.00 | 805 | 100.0 | 0.18a |
| R329A | 0b | 0.00 | 5.2 | 0.6 | 4.65 |
| R329K | 2.8 | 0.12 | 22.3 | 2.8 | 2.56 |
| R329Q | 0b | 0.00 | 8.0 | 1.0 | 2.79 |
| R323K | 837 | 34.47 | NDc | ND | ND |
| R639Q | 0b | 0.00 | 2.8 | 0.4 | 3.30 |
| R735Q | 1492 | 61.45 | ND | ND | ND |
a Previously reported by Climent et al. (12).
b The counts were the same as the background control.
c ND, not determined.
In addition to Glu52, our search for charged interface residues suggested that Arg329 and Arg639 in α were also of interest. Arg329 is located in loop 3 of α and is adjacent to a second Arg at 323 that is not conserved (see supplemental Fig. S2). Mutants of Arg329 (Ala, Lys, and Gln) were made and assayed for activity and binding to WT-β2. The results of mutation of Arg329 to Ala, Lys, and Gln and a control of Arg323 to Lys are shown in Table 4. R329A and R329Q have no detectable activity, whereas R329K has 0.12% activity of WT-α2. Binding studies (12) revealed that all three mutants exhibit 10–20-fold weaker binding than WT, similar to the phenotypes of the E52X mutants with the exception of Gln (Fig. 2, A and B). Given the weak Kd values, the mutants were assayed at higher protein concentration and still found to have no activity (Table 4). In the case of R329K, an additional experiment was carried out with N3CDP to look for N• formation and total radical loss. These results (Fig. 3B) also indicate that this mutant is inactive and hence important. The control, a lysine mutant of Arg323 has 34% the activity of the WT-α2 and was thus not considered further.
The R329X-α mutants were also studied with F3Y122•-β2. Activity of 1 to 3% WT was observed with the Lys mutant having the highest level (Table 4). These studies also suggest that Arg329 plays an important role in the RNR catalysis. Finally, studies with an R639Q-α mutant revealed that it is inactive, whereas mutations of the non-conserved Arg735, also proposed to be at the interface, results in active enzyme.
Discussion
In the past decade using technology to site-specifically incorporate UAA coupled to time-resolved kinetic measurements to study the consequences of the incorporation, much has been learned about the long distance RT process required to initiate nucleotide reduction in RNR (3, 26, 38). The UAAs have been one of the crucial perturbants to allow uncoupling of the rate-limiting conformational gating that masks the RT and the nucleotide reduction chemistry in the WT enzyme (20).
Binding of the appropriate S and e pairs to α2 followed by binding β2 has long been known to trigger the essential conformational change(s) that occurs over the 37 Å (C-site) or 39 Å (S-site) in α2 to the RT initiation site in β2 (Fe1 to S of Cys439 or 2-O of TTP, respectively) (19, 32). The binding of CDP/ATP or CDP (or GDP/TTP or GDP) changes the loop 2 structure in α2 and also induces a closure of the barrel structure around the catalytic site on α2 (39). These changes must be transmitted across the α/β subunit interface, likely through a conserved network of residues to position the water bound to the Fe1 in the diferric cluster, so that the proton can be efficiently delivered to Tyr122• concomitant with its reduction (Fig. 1).
Our recent studies on the conserved Glu350 in β2, suggested that it likely plays a very important role in conformational gating (3, 18). We thus decided to investigate the possible role of other conserved interface residues including Glu52-β2, Arg329-α2, and Arg639-α2.
The results with the E52Q in the WT and F3Y122• backgrounds are most striking. In the WT background it is unable to make dNDPs and is inactive in the formation of the N• from N3CDP. E52Q-β2, in contrast to the Glu and Asp mutants, binds similarly to α2 as WT-β2 (Table 1, Fig. 2A) and for E52Q/F3Y122•-β2 the binding to α2 is stoichiometric (Fig. 2, C and D). Additionally, although the E52Q/F3Y122•-β2 is inactive in the steady-state assay, it is able to make N• from N3CDP, Tyr356• in the presence of CDP/ATP, and catalyze 1/2 turnover (one CDP/two F3Y122•), consistent with the half-site reactivity of RNR (20, 23). It is likely that the reoxidation of F3Y122-O− to the F3Y122• by Tyr356• is too slow to compete with loss of the total radical (Table 2) (18, 20, 37), potentially explaining the lack of activity under steady-state conditions. This reoxidation is also slow for F3Y122-O− in the WT background, but the E52Q mutation appears to result in an even slower process.
To investigate α/β binding, we used several pulldown approaches. The experiment with His6-α2, S/e (where S is CDP or GDP and e is ATP or TTP), and E52Q/F3Y122•-β2 allowed isolation of a complex by Ni-NTA affinity chromatography with a β2/α2 subunit ratio of ∼0.6. In contrast, the ratio of 0.01 was observed with the WT control after a 5-min incubation (Table 3).
Interestingly, the double mutant complex has a longer lifetime than the pathway (Tyr356•) radical in the pulldown assays. The total amount of radical (F3Y122• and Tyr356•) decreases 30 to 50% over 5 min (Table 2), yet the complex can be isolated over 2 h (Fig. 6A, right, and time course data not shown with the other experiments in Table 3). Thus, the conformation of the α2β2 complex that allowed its isolation appears to have a “kinetic” memory, that is, it remains in an altered conformation after much of the pathway radical has decayed. This observation of a kinetic memory is strikingly similar to our recent studies with α on the human RNR. This subunit forms a hexameric structure, α6, in the presence of dATP or the phosphorylated drugs clofarabine di- or triphosphate (ClFDP or ClFTP) (40–42). When dATP dissociates from α6, the hexamer returns to a monomeric state. However, when ClFDP or ClFTP dissociate, the hexameric structure remains. The molecular basis for the continued tight binding of α2β2 in the case of the E. coli RNR double mutant and α6 in the hRNR remain unknown. However, it is intriguing in the case of the E. coli RNR that a conservative chemical substitution Gln for Glu in the F3Y122•-β2 has such a dramatic effect on α/β interactions in pulldown assays.
From the many β2 structures available, we know that Glu52 located on the surface of β is conformationally flexible with “out,” “in,” and “intermediate” conformations (supplemental Fig. S2, B and C). Its “in” conformation connects through waters to a conserved residue, Arg236, within β. Arg236 has connectivity to Trp48 that in turn connects to Asp237, which connects to His118, a ligand to Fe1 of the cofactor (supplemental Fig. S2C). It is the water on Fe1 that is proposed to deliver the proton to Tyr122• upon Tyr122• reduction (supplemental Fig. S2C) (11, 19, 43). Also shown in supplemental Fig. S2 is the location of the “out” conformation of Glu52 relative to the conserved Arg329 in loop 3 of α in the α2β2 docking model. Supporting the importance of Arg329, mutants (Gln, Lys, and Ala) show weak binding to β2, with Kd values elevated 10-fold relative to WT, similar to the results with Glu350 and Glu52 mutants. The inactivity of Glu52 and Arg329 mutants might result from their altered conformations in this region of α2. The studies with E52Q/F3Y122•-β2 and the requirement for S/e suggest its importance in conformational triggering of RT across α/β. The unexpected observation of the high percentage of the α2β2 complex formed in the double mutant may provide the opportunity to gain insight into the structure of this complex based on our negative stain EM images (Fig. 8).
Finally, the least well studied mutant, Arg639-α has very low activity and has weakened binding to β. Recent structures from the Drennan lab (39) show that in the presence of the correct S/e pairs, loop 2 (yellow, supplemental Fig. S3A) becomes ordered, the barrel clamps around the catalytic site, and the β-hairpin (supplemental Fig. S3B, blue to orange) moves to potentially protect the active site. Arg639, which is adjacent to this hairpin may play a role in stabilizing the differential hairpin conformations. Interestingly, this β-hairpin is conserved in the class II RNRs and is observed to move when the adenosylcoblamin cofactor, the radical initiator, binds to initiate nucleotide reduction via formation of a thiyl radical (44).
Conclusions
The reversible long distance RT between α and β continues to be a fascinating feature of the class I RNRs. RT is gated subsequent to binding the appropriate S and e pairs on the α subunit, requiring communication across the subunits over a distance of 35 to 40 Å. The transient nature of the α and β interactions in the E. coli RNR, the flexibility of its α and β tails both essential in catalysis, the complexity and number of nucleotide-binding sites, have all made an understanding of the molecular mechanism of conformational gating and a structure of an active RNR elusive. Here we have identified conserved residues likely to control conformational gating at the α/β interface. The most intriguing results are that the double mutant of E52Q/F3Y122•-β2 when incubated with α2, S, and e, potentially forms the “tightest” complex thus far reported based on pulldown assays, SEC, and negative stain EM studies. The conservative mutation of Glu52 to Asp, on the other hand, weakens subunit affinity compared with WT. Clearly the design of the subunit interface is intricate, providing the exquisite control that is needed for the RT chemistry mediated by S/e in this essential enzyme.
Experimental procedures
Materials
All primers and plasmids utilized in this study are shown in supplemental Table S1. All primers were provided by Integrated DNA Technologies. Site-directed mutagenesis was performed using the Stratagene QuikChange kit and all constructs were confirmed by sequencing at QuintaraBio (Boston). WT-β2 (7000 nmol/min/mg) and E52X-β2 (X = Ala, Asp, or Gln) were isolated as previously reported with typical yields of ∼20–30 mg/g of cell paste (45). WT-α2 (2428 nmol/min/mg) and mutant-α2s were purified following the published protocol with typical yields of ∼20–30 mg/g of cell paste (29). Thioredoxin (TR, 40 units/mg) and thioredoxin reductase (TRR, 1400 units/mg) were purified following the standard protocols (46, 47).
F3Y was enzymatically synthesized from the corresponding phenol using tyrosine phenol lyase TPL (27). The pBAD-nrdB-TAG122 and pEVOL-FnYRS-E3 plasmids were generated and isolated as described (20). Apo-F3Y122-β2 and apo-E52Q/F3Y122-β2 were expressed, purified, and reconstituted as previously reported (20). Typical yields were ∼8–10 mg/g of cell paste.
[5′-3H]CDP was purchased from ViTrax (Placentia, CA). Roche Applied Science provided the calf alkaline phosphatase (20 units/μl). Sigma provided Hepes, MgSO4, EDTA, LB, 2× YT microbial medium, ampicillin, chloramphenicol (Cm), hydroxyurea, ATP, CDP, TTP, GDP, deoxycytidine (dC), and NADPH. Isopropyl β-d-1-thiogalactopyranoside and dithiothreitol (DTT) were obtained from Promega. N3CTP (2′-Azido-2′-deoxycytidine 5′-triphosphate) was purchased from TriLink Biotechnologies and converted to the diphosphate (N3CDP) as previously described (48). Assay buffer consisted of 50 mm Hepes (pH 7.6), 15 mm MgSO4, 1 mm EDTA. The temperature was controlled using a Lauda circulating water bath for all experiments: at 25 °C for E. coli RNR. All E. coli α2 and β2 concentrations are reported per dimer.
RNR activity assays
The activity of E. coli WT-α2 or WT-β2 (0.15 μm) was determined in the presence of 5-fold excess of the second subunit, β2 or α2 (0.75 μm). E52X-β2 (X = Ala, Asp, or Gln, 0.5, 1.0, and 2 μm), F3Y122•-β2 (0.5 μm), E52Q/F3Y122•-β2 (2 μm), or mutant-α2s (2 μm) were also assayed in a 5-fold excess of WT-α2 or WT-β2. A typical assay mixture of 140 μl contained RNR, TR (30 μm), TRR (0.5 μm), and ATP (3 mm) in assay buffer and the reaction mixture was initiation with [5-3H]CDP (1 mm, 3769 cpm/nmol). The method of Steeper and Stuart (49) was used for analysis.
Time-dependent inactivation of RNR mutants in the presence of N3CDP
A 250-μl reaction mixture contained: protein (30 μm WT-α2 with 30 μm E52X-β2 (X = Ala, Asp, or Gln) or 30 μm R329X-α2 (X = Ala, Lys, or Gln) with 30 μm WT-β2], 0.2 mm TTP, 50 mm Hepes (pH 7.6), 1 mm EDTA, 15 mm MgSO4 and was incubated at 25 °C for 1 min. The time 0 sample was frozen in liquid nitrogen and the EPR spectrum was recorded. The sample was then thawed and the reaction started by addition of 0.25 mm N3CDP. The control had no N3CDP. Each sample was warmed to 25 °C and used for a complete time course study by repeated freeze-thaw cycles (50). The amount of radicals were quantitated as previously described (28).
Kd measurements for the interaction between α2 and β2 mutants
The interaction between E52X-β2 (X = Ala, Asp, or Gln) for α2 and R329X-α2 (X = Ala, Lys, Gln, or Glu) for β2 were determined in assay buffer at 25 °C by the competitive inhibition assay (12). A typical assay mixture in a final volume of 310 μl contained variable amounts of mutant-β2, 0.15 μm WT-α2, and 0.3 μm WT-β2 or variable amounts of mutant-α2, 0.30 μm WT-α2, and 0.15 μm WT-β2, along with 50 μm TR, 1 μm TRR, 1 mm CDP, 1.6 mm ATP, 0.2 mm NADPH. The concentration of E52X-β2 and R329X-α2 were varied from 0.1 to 15 and 0.01 to 0.5 μm for E52Q-β2. Nucleotide reduction activity was determined by monitoring the A340 nm change and the data were then fit to Equation 1.
| (Eq. 1) |
Efforts to determine the affinity of E52Q/F3Y122•-β2 for α2 were carried out by the same procedure. However, the binding curves could only be fit under the assumption that the subpopulation of E52Q/F3Y122-β2 lacking F3Y122• does not competitively inhibit in the concentration range of the experiment (Fig. 2C). An upper limit of the Kd was estimated from this model (Table 1). Efforts to develop an alternative method for Kd determination are ongoing.
dCDP formation with pre-reduced α2 and E52Q/F3Y122•-β2 monitored by chemical quench
α2 was treated with hydroxyurea to inactivate the small amount of β2 that is always present in α2 samples (32). The protein was then desalted on a Sephadex G-25 column (1.5 × 20 cm) equilibrated with 50 mm Tris (pH 7.6) and 5% glycerol. The reaction mixture contained 20 μm pre-reduced WT-α2, 20 μm E52Q/F3Y122•-β2 (0.91 Y122•/β2), ±3 mm ATP, ± 40 μm TR, 1.6 μm TRR, 1 mm NADPH. The reaction was initiated by [5-3H]CDP (1 mm, 9982 cpm/nmol) at 25 °C. RNR activity was quenched with 2.0% HClO4 and the dCDP was measured by the method of Steeper and Stuart (49).
Reaction of E52Q/F3Y122•-β2 with WT-α2 monitored by EPR spectroscopy
In a final volume of 250 μl the reaction mixture contained WT-α2 (15 to 50 μm), with 1 eq of β2, substrate (CDP (1 mm) or GDP (1 mm) or N3CDP (0.25 mm)), ± effector (ATP (3.0 mm) or TTP (0.2 mm)) in assay buffer. Samples were incubated for a specified time in a circulating water bath at 25 °C and quenched for EPR analysis in liquid nitrogen. EPR spectra were recorded at 77 K in the Department of Chemistry Instrumentation Facility on a Bruker ESP-300 X-band spectrometer equipped with a quartz finger Dewar filled with liquid nitrogen. Typical EPR parameters were as follows: microwave frequency = 9.45 GHz, power = 32 μW, modulation amplitude = 1.5 G, modulation frequency = 100 kHz, time constant = 40.96 ms, scan time = 41.9 s. Analysis of the resulting spectra was carried out using WinEPR (Bruker) and an in-house written program in Matlab R2016a (50). EPR spin quantitation was carried out using CuII as standard.
Pulldown assays
A final volume of 100 μl contained untagged-β2s (10 μm), His6-WT-α2 (10 μm), ATP (3 mm), or TTP (0.2 mm) in assay buffer at 25 °C. CDP (1 mm) or GDP (1 mm) or alternatively mutant β2 was added to initiate the reaction. The reaction mixture was incubated for 1 to 120 min at 25 °C and then combined with a nickel-nitrilotriacetic acid resin (∼60 or 300 μl, from Qiagen) suspended in the EDTA-free assay buffer and rotated by hand at room temperature for 1 min. The sample was then centrifuged (30 s, 3,000 × g, 4 °C) and the supernatant was removed. Alternatively, the NTA resin (300 μl) was placed in a small column and eluted by gravity. In the former case, the resin “pellet” was rapidly resuspended in 600 μl of wash buffer (EDTA-free assay buffer with 300 mm NaCl and 15 mm imidazole (pH 7.6)) and centrifuged (30 s, 3,000 × g, 4 °C). This wash step was repeated a second time. Resin-bound protein was then eluted by resuspending it in elution buffer (100 μl, EDTA-free assay buffer with 250 mm imidazole (pH 7.6)), followed by centrifugation (30 s, 3,000 × g, 4 °C). The procedure (flow through, washes (W1 and W2), and elution (E)) took 5 min. The recovery of α is typically 40 to 50%.
In the latter case, gravity elution, the procedure (loading, washes, and elution) is the same except that the procedure takes 2 to 3 min and the recovery of α is typically ∼90%. The contents of each fraction were assessed by SDS-PAGE (10%) and compared with the fractions obtained in a control experiment with standards made from stock solutions: 1 μm His6-WT-α2 and 1 μm WT-β2.
Negative stain EM on α2 with E52Q/F3Y122•-β2
A reaction mixture was prepared with 5 μm α2, 10 μm E52Q/F3Y122•-β2, 1 mm GDP, and 0.2 mm TTP in assay buffer (50 mm Hepes, pH 7.6, 15 mm MgSO4, and 1 mm EDTA) where β2 was added last to initiate the reaction. The mixture was incubated 3 min at 25 °C and then diluted 130-fold in assay buffer containing 1 mm GDP and 0.2 mm TTP giving final protein concentrations of 40 nm α2 and 80 nm E52Q/F3Y122•-β2. The solution was applied to a 300-mesh continuous carbon grid (EMS) and stained three times with a 1% uranyl acetate solution. The total time between reaction initiation and application onto the grid was ∼15 min.
Data collection
All images were collected at the W. M. Keck Institute for Cellular Visualization at Brandeis University. The grids were imaged at 200 kV on a Tecnai F20 electron microscope (FEI) equipped with an UltraScan 4000 CCD camera (Gatan) using SerialEM operated in manual low-dose mode at a magnification of 62,000 with a pixel size of 1.79 Å at the specimen level.
Size exclusion chromatography
A reaction of 300 μl contained 50 mm Hepes (pH 7.6), 15 mm MgSO4, 2 μm α2, 4 μm E52Q/F3Y122•-β2, 1 mm GDP, and 0.2 mm TTP. The reaction mixture was loaded into a 200-μl loop and injected onto a Superdex 200 10/300 GL preequilibrated in 50 mm Hepes (pH 7.6), 15 mm MgSO4, 50 μm GDP, 10 μm TTP, and 150 mm NaCl. The flow rate was 0.5 ml/min. Ferritin (440 kDa), aldolase (158 kDa), conalbumin (75 kDa), and ovalbumin (44 kDa) were used as the protein standards to generate a standard curve for molecular weight estimation.
Author contributions
Q. L., M. J. P., and J. S. designed the study and wrote the paper. Q. L. made the mutants and performed activity assays, EPR experiments, and pulldown assays. M. J. P. identified the interface residues and proposed the structural models for their role. A. T. T. analyzed EPR data and performed pulldown assays and SEC experiments. K. R. played an important intellectual role in experimental design. A. K. performed assays related to Arg639 and Arg735 in α. G. H. K., A. T. T., and C. L. D. carried out the negative stain EM experiments and analysis and provided structural insight into the function of the mutants. All authors approved the final version of the manuscript.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant GM29595 (to J. S.), National Natural Science Foundation of China Grant 81372138 and Foundation of Zhejiang University for International Cooperation and Exchanges (to J. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Table S1 and Figs. S1–S3.
- RNR
- ribonucleotide reductases
- NDP
- nucleoside diphosphates
- dNDP
- deoxynucleoside diphosphates
- RT
- radical transfer
- UAA
- unnatural amino acid
- SEC
- size exclusion chromatography
- N3CDP
- 2′-azido-2′-deoxycytidine diphosphate
- F3Y122•
- 2,3,5-trifluorotyrosine radical
- Ni-NTA
- nickel-nitrilotriacetic acid
- TR
- thioredoxin
- TRR
- thioredoxin reductase
- NO2Y122•
- 3-nitrotyrosine radical.
References
- 1. Hofer A., Crona M., Logan D. T., and Sjöberg B. M. (2012) DNA building blocks: keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 47, 50–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nordlund P., and Reichard P. (2006) Ribonucleotide reductases. Annu. Rev. Biochem. 75, 681–706 [DOI] [PubMed] [Google Scholar]
- 3. Minnihan E. C., Nocera D. G., and Stubbe J. (2013) Reversible, long-range radical transfer in E. coli class Ia ribonucleotide reductase. Acc. Chem. Res. 46, 2524–2535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Uhlin U., and Eklund H. (1994) Structure of ribonucleotide reductase protein R1. Nature 370, 533–539 [DOI] [PubMed] [Google Scholar]
- 5. Brown N. C., and Reichard P. (1969) Role of effector binding in allosteric control of ribonucleoside diphosphate reductase. J. Mol. Biol. 46, 39–55 [DOI] [PubMed] [Google Scholar]
- 6. Rofougaran R., Crona M., Vodnala M., Sjöberg B. M., and Hofer A. (2008) Oligomerization status directs overall activity regulation of the Escherichia coli class Ia ribonucleotide reductase. J. Biol. Chem. 283, 35310–35318 [DOI] [PubMed] [Google Scholar]
- 7. von Döbeln U., and Reichard P. (1976) Binding of substrates to Escherichia coli ribonucleotide reductase. J. Biol. Chem. 251, 3616–3622 [PubMed] [Google Scholar]
- 8. Sjöberg B. M., and Reichard P. (1977) Nature of the free radical in ribonucleotide reductase from Escherichia coli. J. Biol. Chem. 252, 536–541 [PubMed] [Google Scholar]
- 9. Sjöberg B. M., Reichard P., Gräslund A., and Ehrenberg A. (1978) Tyrosine free-radical in ribonucleotide reductase from Escherichia coli. J. Biol. Chem. 253, 6863–6865 [PubMed] [Google Scholar]
- 10. Stubbe J., and van Der Donk W. A. (1998) Protein radicals in enzyme catalysis. Chem. Rev. 98, 705–762 [DOI] [PubMed] [Google Scholar]
- 11. Nordlund P., Sjöberg B. M., and Eklund H. (1990) Three-dimensional structure of the free radical protein of ribonucleotide reductase. Nature 345, 593–598 [DOI] [PubMed] [Google Scholar]
- 12. Climent I., Sjöberg B. M., and Huang C. Y. (1991) Carboxyl-terminal peptides as probes for Escherichia coli ribonucleotide reductase subunit interaction: kinetic analysis of inhibition studies. Biochemistry 30, 5164–5171 [DOI] [PubMed] [Google Scholar]
- 13. Climent I., Sjöberg B. M., and Huang C. Y. (1992) Site-directed mutagenesis and deletion of the carboxyl terminus of Escherichia coli ribonucleotide reductase protein R2: effects on catalytic activity and subunit interaction. Biochemistry 31, 4801–4807 [DOI] [PubMed] [Google Scholar]
- 14. Hassan A. Q., and Stubbe J. (2008) Mapping the subunit interface of ribonucleotide reductase (RNR) using photo cross-linking. Bioorg. Med. Chem. Lett. 18, 5923–5925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ravichandran K. R. (2016) Mechanistic investigations of the radical transport pathway in fluorotyrosine-substituted class Ia ribonucleotide reductases. Ph.D. thesis, Massachusetts Institute of Technology, Massachusetts Institute of Technology, Cambridge, MA [Google Scholar]
- 16. Stubbe J., Nocera D. G., Yee C. S., and Chang M. C. (2003) Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem. Rev. 103, 2167–2201 [DOI] [PubMed] [Google Scholar]
- 17. Stubbe J. (1998) Ribonucleotide reductases in the twenty-first century. Proc. Natl. Acad. Sci. U.S.A. 95, 2723–2724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ravichandran K., Minnihan E. C., Lin Q., Yokoyama K., Taguchi A. T., Shao J., Nocera D. G., and Stubbe J. (2017) Glutamate 350 plays an essential role in conformational gating of long-range radical transport in Escherichia coli class Ia ribonucleotide reductase. Biochemistry 56, 856–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wörsdörfer B., Conner D. A., Yokoyama K., Livada J., Seyedsayamdost M., Jiang W., Silakov A., Stubbe J., Bollinger J. M. Jr., and Krebs C. (2013) Function of the diiron cluster of Escherichia coli class Ia ribonucleotide reductase in proton-coupled electron transfer. J. Am. Chem. Soc. 135, 8585–8593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ravichandran K. R., Minnihan E. C., Wei Y., Nocera D. G., and Stubbe J. (2015) Reverse electron transfer completes the catalytic cycle in a 2,3,5-trifluorotyrosine-substituted ribonucleotide reductase. J. Am. Chem. Soc. 137, 14387–14395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yokoyama K., Uhlin U., and Stubbe J. (2010) A hot oxidant, 3-NO2Y122 radical, unmasks conformational gating in ribonucleotide reductase. J. Am. Chem. Soc. 132, 15368–15379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lundin D., Torrents E., Poole A. M., and Sjöberg B. M. (2009) RNRdb, a curated database of the universal enzyme family ribonucleotide reductase, reveals a high level of misannotation in sequences deposited to Genbank. BMC Genomics 10, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seyedsayamdost M. R., Chan C. T., Mugnaini V., Stubbe J., and Bennati M. (2007) PELDOR spectroscopy with DOPA-β2 and NH2Y-α2s: distance measurements between residues involved in the radical propagation pathway of E. coli ribonucleotide reductase. J. Am. Chem. Soc. 129, 15748–15749 [DOI] [PubMed] [Google Scholar]
- 24. Minnihan E. C., Ando N., Brignole E. J., Olshansky L., Chittuluru J., Asturias F. J., Drennan C. L., Nocera D. G., and Stubbe J. (2013) Generation of a stable, aminotyrosyl radical-induced α2β2 complex of Escherichia coli class Ia ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A. 110, 3835–3840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ando N., Brignole E. J., Zimanyi C. M., Funk M. A., Yokoyama K., Asturias F. J., Stubbe J., and Drennan C. L. (2011) Structural interconversions modulate activity of Escherichia coli ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A. 108, 21046–21051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Seyedsayamdost M. R., and Stubbe J. (2009) Replacement of Y730 and Y731 in the α2 subunit of Escherichia coli ribonucleotide reductase with 3-aminotyrosine using an evolved suppressor tRNA/tRNA-synthetase pair. Methods Enzymol. 462, 45–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Seyedsayamdost M. R., Yee C. S., and Stubbe J. (2007) Site-specific incorporation of fluorotyrosines into the R2 subunit of E. coli ribonucleotide reductase by expressed protein ligation. Nat. Protoc. 2, 1225–1235 [DOI] [PubMed] [Google Scholar]
- 28. Seyedsayamdost M. R., Xie J., Chan C. T., Schultz P. G., and Stubbe J. (2007) Site-specific insertion of 3-aminotyrosine into subunit α2 of E. coli ribonucleotide reductase: direct evidence for involvement of Y730 and Y731 in radical propagation. J. Am. Chem. Soc. 129, 15060–15071 [DOI] [PubMed] [Google Scholar]
- 29. Minnihan E. C., Seyedsayamdost M. R., Uhlin U., and Stubbe J. (2011) Kinetics of radical intermediate formation and deoxynucleotide production in 3-aminotyrosine-substituted Escherichia coli ribonucleotide reductases. J. Am. Chem. Soc. 133, 9430–9440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nick T. U., Lee W., Kossmann S., Neese F., Stubbe J., and Bennati M. (2015) Hydrogen bond network between amino acid radical intermediates on the proton-coupled electron transfer pathway of E. coli α2 ribonucleotide reductase. J. Am. Chem. Soc. 137, 289–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yokoyama K., Smith A. A., Corzilius B., Griffin R. G., and Stubbe J. (2011) Equilibration of tyrosyl radicals (Y356•, Y731•, Y730•) in the radical propagation pathway of the Escherichia coli class Ia ribonucleotide reductase. J. Am. Chem. Soc. 133, 18420–18432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ge J., Yu G., Ator M. A., and Stubbe J. (2003) Pre-steady-state and steady-state kinetic analysis of E. coli class I ribonucleotide reductase. Biochemistry 42, 10071–10083 [DOI] [PubMed] [Google Scholar]
- 33. Sjöberg B. M., Gräslund A., and Eckstein F. (1983) A substrate radical intermediate in the reaction between ribonucleotide reductase from Escherichia coli and 2′-azido-2′-deoxynucleoside diphosphates. J. Biol. Chem. 258, 8060–8067 [PubMed] [Google Scholar]
- 34. Salowe S. P., Ator M. A., and Stubbe J. (1987) Products of the inactivation of ribonucleoside diphosphate reductase from Escherichia coli with 2′-azido-2′-deoxyuridine 5′-diphosphate. Biochemistry 26, 3408–3416 [DOI] [PubMed] [Google Scholar]
- 35. Salowe S., Bollinger J. M. Jr., Ator M., Stubbe J., McCraken J., Peisach J., Samano M. C., and Robins M. J. (1993) Alternative model for mechanism-based inhibition of Escherichia coli ribonucleotide reductase by 2′-azido-2′-deoxyuridine 5′-diphosphate. Biochemistry 32, 12749–12760 [DOI] [PubMed] [Google Scholar]
- 36. van Der Donk W. A., Stubbe J., Gerfen G. J., Bellew B. F., and Griffin R. G. (1995) EPR investigations of the inactivation of E. coli ribonucleotide reductase with 2′-azido-2′-deoxyuridine-5′-diphosphate: evidence for the involvement of the thiyl radical of C225-R1. J. Am. Chem. Soc. 117, 8908–8916 [Google Scholar]
- 37. Ravichandran K. R., Zong A. B., Taguchi A. T., Nocera D. G., Stubbe J., and Tommos C. (2017) Formal reduction potentials of difluorotyrosine and trifluorotyrosine protein residues: defining the thermodynamics of multistep radical transfer. J. Am. Chem. Soc. 139, 2994–3004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Minnihan E. C., Young D. D., Schultz P. G., and Stubbe J. (2011) Incorporation of fluorotyrosines into ribonucleotide reductase using an evolved, polyspecific aminoacyl-tRNA synthetase. J. Am. Chem. Soc. 133, 15942–15945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zimanyi C. M., Chen P. Y., Kang G., Funk M. A., and Drennan C. L. (2016) Molecular basis for allosteric specificity regulation in class Ia ribonucleotide reductase from Escherichia coli. eLIFE 5, 10.7554/eLife.07141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ando N., Li H., Brignole E. J., Thompson S., McLaughlin M. I., Page J. E., Asturias F. J., Stubbe J., and Drennan C. L. (2016) Allosteric inhibition of human ribonucleotide reductase by dATP entails the stabilization of a hexamer. Biochemistry 55, 373–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aye Y., and Stubbe J. (2011) Clofarabine 5′-di and -triphosphates inhibit human ribonucleotide reductase by altering the quaternary structure of its large subunit. Proc. Natl. Acad. Sci. U.S.A. 108, 9815–9820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aye Y., Brignole E. J., Long M. J., Chittuluru J., Drennan C. L., Asturias F. J., and Stubbe J. (2012) Clofarabine targets the large subunit (α) of human ribonucleotide reductase in live cells by assembly into persistent hexamers. Chem. Biol. 19, 799–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ekberg M., Pötsch S., Sandin E., Thunnissen M., Nordlund P., Sahlin M., and Sjöberg B. M. (1998) Preserved catalytic activity in an engineered ribonucleotide reductase R2 protein with a nonphysiological radical transfer pathway: the importance of hydrogen bond connections between the participating residues. J. Biol. Chem. 273, 21003–21008 [DOI] [PubMed] [Google Scholar]
- 44. Sintchak M. D., Arjara G., Kellogg B. A., Stubbe J., and Drennan C. L. (2002) The crystal structure of class II ribonucleotide reductase reveals how an allosterically regulated monomer mimics a dimer. Nat. Struct. Biol. 9, 293–300 [DOI] [PubMed] [Google Scholar]
- 45. Yokoyama K., Uhlin U., and Stubbe J. (2010) Site-specific incorporation of 3-nitrotyrosine as a probe of pKa perturbation of redox-active tyrosines in ribonucleotide reductase. J. Am. Chem. Soc. 132, 8385–8397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chivers P. T., Prehoda K. E., Volkman B. F., Kim B. M., Markley J. L., and Raines R. T. (1997) Microscopic pKa values of Escherichia coli thioredoxin. Biochemistry 36, 14985–14991 [DOI] [PubMed] [Google Scholar]
- 47. Russel M., and Model P. (1985) Direct cloning of the trxB gene that encodes thioredoxin reductase. J. Bacteriol. 163, 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Persson A. L., Eriksson M., Katterle B., Pötsch S., Sahlin M., and Sjöberg B. M. (1997) A new mechanism-based radical intermediate in a mutant R1 protein affecting the catalytically essential Glu-441 in Escherichia coli ribonucleotide reductase. J. Biol. Chem. 272, 31533–31541 [DOI] [PubMed] [Google Scholar]
- 49. Steeper J. R., and Steuart C. D. (1970) A rapid assay for CDP reductase activity in mammalian cell extracts. Anal. Biochem. 34, 123–130 [DOI] [PubMed] [Google Scholar]
- 50. Ravichandran K. R., Taguchi A. T., Wei Y., Tommos C., Nocera D. G., and Stubbe J. (2016) A >200 meV uphill thermodynamic landscape for radical transport in E. coli ribonucleotide reductase determined using fluorotyrosine-substituted enzymes. J. Am. Chem. Soc. 138, 13706–13716 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.