

Abstract
Inactivation of the tumor suppressor protein p53 by mutagenesis, chemical modification, protein-protein interaction, or aggregation has been associated with different human cancers. Although DNA is the typical substrate of p53, numerous studies have reported p53 interactions with RNA. Here, we have examined the effects of RNA of varied sequence, length, and origin on the mechanism of aggregation of the core domain of p53 (p53C) using light scattering, intrinsic fluorescence, transmission electron microscopy, thioflavin-T binding, seeding, and immunoblot assays. Our results are the first to demonstrate that RNA can modulate the aggregation of p53C and full-length p53. We found bimodal behavior of RNA in p53C aggregation. A low RNA:protein ratio (∼1:50) facilitates the accumulation of large amorphous aggregates of p53C. By contrast, at a high RNA:protein ratio (≥1:8), the amorphous aggregation of p53C is clearly suppressed. Instead, amyloid p53C oligomers are formed that can act as seeds nucleating de novo aggregation of p53C. We propose that structured RNAs prevent p53C aggregation through surface interaction and play a significant role in the regulation of the tumor suppressor protein.
Keywords: amyloid, fluorescence, kinetics, p53, prion, protein aggregation, protein folding, RNA, domain V of 23S rRNA, p53C
Introduction
In 1989, 10 years after the identification of its gene, p532 was classified as a tumor suppressor protein. Since then thousands of studies have been dedicated to understanding p53's function, or rather the loss of such, which has been directly linked to more than half of all human cancers and >95% of all lung cancers (1). In about half of all p53-related tumors, loss of p53 function is the direct consequence of specific mutations in the p53 gene. In all other cases the tumor suppressor protein is inactivated through various interactions; some viral and others of host cell origin (2, 3).
A flexible phosphoprotein composed of 393 amino acids, p53 is only functional in the form of a homo-tetramer. The N terminus, also known as the transactivation domain (Fig. 1), is intrinsically disordered and separated from the remainder of the protein by a proline-rich region. It is the preferred site of interaction for many proteins, such as the human double minute 2 homolog (MDM2). Residues 94–312 encompass the region responsible for the primary function of p53; that is, sequence-specific DNA binding. It is referred to as the central core domain (CCD) and is commonly designated as p53C. This region of p53 has the highest concentration of cancer-related mutations (4) and is characterized by a high propensity to aggregate in an amyloid-like fashion (5). p53C also shows considerable overlap with disease-causing p53 isoforms described below. All of these properties make p53C a suitable model system for p53 research, which has been used extensively in earlier studies. The p53C domain is followed by a tetramerization domain and finally a regulatory domain, also shown to bind DNA but nonspecifically (6).
Figure 1.
Schematic representation of p53 and its functional regions. Starting from the N terminus, the transactivation domain (TAD) (red) is followed by the proline-rich region (PRR) (yellow), central core domain (p53C) (green), tetramerization domain (TD) (blue), and regulatory domain (RD) (light brown).
A major component of a complex network of molecular interactions and cellular quality control processes, p53 has been speculated to perform a multitude of biochemical functions. It is only activated when cells are stressed or damaged. In such situations p53 interacts with specific DNA sequences and induces the transcription of adjacent genes, the products of which lead to cell cycle arrest or apoptosis (3). p53 abundance and transcriptional activity are the subjects of multifactorial regulation. The human gene TP53 expresses 12 different p53 isoforms with varied sequences and properties. The p53C is the predominant constituent of the isoforms Δ133p53β, Δ133p53γ, Δ160p53β, and Δ160p53γ (7), which play significant roles in the regulation of full-length p53 expression (8) and behavior in cancerous and normal cells (9, 10). Their relative mRNA expression varies in different tissues and has been correlated to stress response and p53 activation (11). Additionally, various chemical modifications, such as acetylation, phosphorylation, glycosylation, and ubiquitination, promote key conformational changes in p53, thereby stabilizing the protein, labeling it for degradation or altering its affinity toward DNA and its functional partners (3, 12). Apart from modifications, the stability and activity of p53 also depend on its interaction with other proteins. Some of them, like MDM2, deactivate p53 so that the cells do not remain arrested indefinitely. Alternatively, p53 interaction with the oncoproteins of some DNA viruses renders them inactive or marked for degradation (2). Being a eukaryotic transcription factor, p53 naturally contains nuclear-export and -import signals essential for its cellular localization. This is yet another avenue for p53 regulation in the cell (13).
Already in 1992 the transmission of the dominant-negative effect of p53 through hetero-oligomerization was suggested by Lane (14). According to this model the presence of mutant or destabilized p53 monomers reduces the affinity of the tetramer for consensus DNA, making it less stable at physiological temperatures (15, 16), thereby increasing the population of partially unfolded, intermediate species prone to aggregation (17). These metastable conformers closely resemble misfolded p53 mutants identified as key contributors to tumor development (18). In comparison to the major prion protein PrP, p53 mutants and p53C display high propensity for aggregation (19). The ability of misfolded p53C to aggregate in an amyloid fashion (5, 20) and to penetrate eukaryotic cells via micropinocytosis and sequester intracellular native p53 (21) is consistent with a prion-like mechanism of propagation, which was proposed to have evolved as an additional control mechanism of p53 proteostasis (20, 22). Which cellular factors contribute to the prion-like behavior of p53 is unclear (22); however, studies on canonical members of the prion family, such as PrP, point toward the contribution of nucleic acids in this process (23).
Functional prions as well as disease-associated prion-like proteins have long been known to associate with nucleic acids, particularly RNA (24). After p53 was isolated in a complex with ribosomal RNA (25) and was shown to bind and repress the translation of its own mRNA (26), p53-RNA interactions have drawn substantial attention for scientific investigations (27, 28). Although the majority of RNA binding appears to involve at least 30 C-terminal residues of the protein, p53 lacking the regulatory domain has also shown affinity for RNA even at high salt concentrations (29). These observations led to several hypotheses on the functional aspects of the interaction. Among those, latent p53 reactivation, mRNA-linked nuclear export, competition with consensus DNA sequences, and RNA-p53-MDM2 ternary complex formation seem to bear the hallmark of a mechanism akin to the intricate p53 regulatory network.
Here we have studied the role of RNA of various origin and sequences on the aggregation of wild-type human p53C using Rayleigh light scattering (LS), fluorescence, immunoblotting, and cryo-transmission electron microscopy (TEM) assays. To understand the biological relevance of our observations, we have also studied the aggregation of full-length p53 (FLp53) with LS and dynamic light scattering (DLS). Our results suggest that RNA plays dual roles in p53C aggregation depending on the RNA:protein ratio. It induces large amorphous aggregate formation at a low (1:50) RNA:protein ratio. In contrast, at a higher ratio (1:8 or higher), RNA shows a distinct inhibitory effect on the aggregation of p53C. We, therefore, speculate that RNA has a profound influence on the abundance and function of the tumor suppressor protein in the cell.
Results
Aggregation of p53C under different conditions
The ability of p53C to form aggregates both in vivo and in vitro has been demonstrated previously (5, 17, 18, 30–33). The kinetics of p53C aggregation without agitation was primarily studied by monitoring the increase in light scattering at 400 nm (LS) as a function of time. The LS versus time curves exhibited classical nucleation-growth kinetics (Fig. 2A). Initially, a slow lag phase was observed where the unfolded/misfolded proteins accumulate until sizable seeds are formed. This is then followed by an exponential phase associated with a fast increase of the LS signal, indicating rapid particle growth. When the aggregates approached optimal size the LS signal plateaued. Prolonged incubation (>2000 s) of higher protein concentrations led to a gradual decrease in intensity, indicating the precipitation of the larger aggregates (data not shown).
Figure 2.

Kinetics of p53C aggregation followed by Rayleigh light scattering at 400 nm. A, time course of p53C aggregation at various protein concentrations at 37 °C. B, time course of p53C (4.5 μm) aggregation at different temperatures. Kinetics of p53C (4.5 μm) aggregation in the presence of various concentrations of dom V rRNA (C) and DHFR mRNA (D). Effect of NaCl (E) and KCl (F) on p53C aggregation, studied by LS after 2 h of incubation at 37 °C. AU, arbitrary units.
We followed the kinetics of p53C aggregation at different protein concentrations and temperatures. In agreement with previous data (34), increasing protein concentrations facilitated p53C aggregation, which was associated with gradual shortening of the lag phase and an increased rate and amplitude of the exponential phase (Fig. 2A). Following previous reports on the kinetic instability of p53C at physiological conditions (16), we then tested the aggregation profile of p53C in a temperature range between 16 and 40 °C (Fig. 2B). No significant increase in LS was observed up to 30 °C, even after extended periods of incubation. At 34 °C, a small increase in LS was observed after 80 min of incubation. However, a significant gain in LS was recorded at 37 °C, reaching a maximum amplitude after approximately 1 h. At 40 °C, the aggregation of p53C was even greater, with a higher rate and a shorter lag phase.
Effect of RNA on p53C aggregation followed by light scattering
To study the role of RNA in p53C aggregation, soluble p53C was incubated at 37 °C for 90 min in the absence and presence of the RNAs of different sequences and origin in varied concentrations. The level of aggregation was estimated by recording LS at the end of the incubation period. The RNAs tested were the in vitro transcribed domains V, IV, and II of the 23S ribosomal RNA of Escherichia coli (referred to as dom V/IV/II rRNA), the central-loop region of domain V of Bacillus subtilis 23S rRNA (referred to as RNA1), and the mRNAs encoding E. coli dihydrofolate reductase (DHFR mRNA), green fluorescent protein + (GFP+ mRNA), and human carbonic anhydrase I (HCA mRNA). Bulk-tRNAs isolated from E. coli MRE600 were also used, and as a control, we tested free ribonucleoside triphosphates (rNTPs), double-stranded DNAs encoding p53C and GFP+, and an 82-nucleotide-long single-stranded DNA.
Almost all the RNAs tested here affected p53C aggregation in a concentration-dependent manner (Fig. 3A). The addition of RNAs of low concentration (with a final RNA:protein ratio of 1:50) resulted in a 1.5–2-fold increase in the LS intensity compared with the no-RNA sample (Fig. 3A). Conversely, with an increasing RNA:protein ratio, all RNAs gradually suppressed p53C aggregation below the level of p53C alone, evidenced by the decrease in LS intensity. The efficacy of the suppression of p53C aggregation varied among the RNAs. Although dom V of the 23S rRNA was the most effective suppressor of p53C aggregation, bulk-tRNAs had the least impact. It appeared that comparatively higher concentrations of the shorter RNAs were necessary to achieve the same level of suppression as that with the longer RNAs. Free ribonucleotides, however, did not show any influence on p53C aggregation, even at much higher concentrations (Fig. 3B). Furthermore, the addition of double-stranded DNA (dsDNA) did not suppress, but rather stimulated p53C aggregation (Fig. 3B). Single-stranded DNA (ssDNA), of length comparable to tRNA, also led to increase in p53C aggregation (Fig. 3B). However, at concentrations equal to the highest tested concentration for tRNA, ssDNA only reduced p53C aggregation to its native level. These results altogether indicate that the suppression of p53C aggregation is an activity specific to RNA. Finally, the suppressive effect of the RNAs could be completely reversed by RNase A treatment before p53C addition (Fig. 3C). Considering that the secondary structure modeling of our single-stranded sequences predicted a mixture of duplex and single-stranded regions for all RNAs (Fig. 4), our results suggest that only structured RNA can effectively reduce p53C aggregation.
Figure 3.

p53C aggregation in the presence of nucleic acids and rNTPs at 37 °C followed by LS at 400 nm. A, relative aggregation of p53C (4.5 μm) in the absence/presence of different RNAs after 1.5 h of incubation. B, relative aggregation of p53C in the absence/presence of single/double-stranded DNA and rNTPs after 1.5 h of incubation. C, reversal of the RNA effect on p53C aggregation by RNase treatment (5 μm RNase A) before incubation with p53C. All measurements were performed at least in triplicate, and the error bars indicate S.D. AU, arbitrary units.
Figure 4.

Secondary structure predictions of the single-stranded RNA and DNA sequences. A, tRNA-bulk. E. coli (∼ 75 nt). B, ssDNA (82 nt). C, dom IV of E. coli 23S rRNA (361 nt). D, RNA1, central loop region of dom V of B. subtilis 23S rRNA (337 nt, the stem loop part of dom V has been replaced with an artificial, 8-nucleotides-long oligo; uppermost region) (59). E, DHFR mRNA, E. coli (516 nt). F, dom V of E. coli 23S rRNA (660 nt). G, dom II of E. coli 23S rRNA (682 nt). H, GFP+ mRNA (714 nt). I, HCA mRNA, Homo sapiens (783 nt). rRNA secondary structure maps were obtained from the rRNA secondary Structures (RibosomeGallery) database (see Ref. 70). All other secondary structure predictions were performed using the Vienna RNA Websuite (71).
The effect of RNA on p53C aggregation was further investigated by following the kinetics of p53C aggregation in the presence of dom V rRNA and DHFR mRNA, where the increase in LS was followed over time. As shown in Fig. 2, C and D, at a higher RNA:protein ratio (≥1:8), both RNAs led to a substantial decrease in the rate and amplitude of aggregation, with the final amplitude being ∼16% of that for p53C incubated without RNA. On the other hand, when p53C was incubated with a six times lower RNA concentration (RNA:p53C = 1:50), the extent of p53C aggregation increased, and the initial lag phase was reduced to a large extent.
We also investigated the effect of salt on the aggregation of p53C in the presence and absence of RNA (Fig. 2, E and D). For both KCl and NaCl, the increase in salt concentration coincided with a decrease in protein aggregation in the absence of nucleic acids. When RNA was present, an increase in aggregation was observed up to 60 mm salt, which was then followed by a significant decrease.
Effect of RNA on p53C aggregation followed by intrinsic fluorescence
Protein p53C contains a single tryptophan and eight tyrosine residues, all of which contribute to the intrinsic fluorescence of the protein. Changes in the polarity of the environment of these aromatic residues alter the fluorescence emission spectrum of the protein (35). Probing for those spectral changes is an approach often used as a measure of conformational rearrangements. We recorded the fluorescence spectra of p53C in the native, denatured, and aggregated states (Fig. 5D). The denatured state was achieved by incubating p53C overnight in 8 m urea at 37 °C. The aggregated state was produced by incubating p53C at 37 °C for 3 h.
Figure 5.

p53C aggregation at 37 °C followed by intrinsic fluorescence (F340). A–C, time course of p53C (4.5 μm) aggregation in the presence of dom V rRNA (A), DHFR mRNA (B), and GFP+ mRNA (C). D, fluorescence emission spectra of native (blue), aggregated (black) and denatured (magenta) states of p53C, prepared as described under “Experimental Procedures.” The spectrum for the native state was recorded at room temperature. AU, arbitrary units.
Similar to the full-length protein (36), native p53C exhibited a tyrosine-dominated emission spectrum with an emission maximum at 305 nm (excitation: 280 nm) (Fig. 5D). The tryptophan contribution to the spectrum was at that stage undetectable and only appeared with the progression of denaturation. The tyrosine contribution to the spectrum decreased moderately in the aggregated state and significantly in the denatured state (Fig. 5D). Aggregated p53C displayed a pronounced, tryptophan-dominated peak at 335 nm (5, 18, 37). In comparison, the fluorescence peak for the denatured state was of reduced intensity and significantly red-shifted, with an emission maximum at 356 nm. The distinctive spectral features of the three p53C states allowed for the application of intrinsic fluorescence as a secondary probe to study the kinetics of p53C aggregation (Fig. 5, A–C). As suggested by Fersht and co-workers (36), fluorescence emission at 340 nm (F340) is characteristic of high-order molecular interactions between p53C species and can be used as a quantitative tool for aggregation.
To complement our LS data, the time course of p53C aggregation was studied by following F340 after excitation at 280 nm. Soluble p53C was incubated at 37 °C alone or with two concentrations (0.1 μm (low) and 0.6 μm (high)) of three different RNAs: dom V rRNA, DHFR, and GFP+ mRNAs. Similar to our LS data, F340 increased exponentially after a short lag phase and, for the no-RNA samples, reached a plateau at ∼2000 s (Fig. 5, A–C). The fluorescence intensity at the plateau was comparable to the steady state measurements (Fig. 5D), indicating that the highest aggregation was achieved. Our LS experiments suggested a suppressive role of RNA in p53C aggregation (Fig. 3). In agreement with that, only a small increase in F340 was recorded when p53C was incubated in the presence of high concentration of the RNA (RNA:protein = 1:8). This result confirmed that RNA can effectively reduce the rate and extent of p53C aggregation. In contrast to LS data, low RNA concentrations did not alter the fluorescence emission significantly. In the presence of 0.1 μm RNA, the F340 gain was only marginally faster than when p53C was incubated alone. This obvious discrepancy between the two data sets stems from the fundamental difference between the two experimental approaches. Unlike light scattering, fluorescence intensity does not depend on particle size but only on the concentration of the fluorophore. Therefore, a lower RNA concentration promotes the formation of larger aggregates but does not affect the overall amount of aggregated protein. Our F340 kinetic data also indicate that p53C oligomerization commences within seconds after the protein is exposed to physiological temperature.
Characterization of p53C aggregation by cryo-TEM, thioflavin T fluorescence, and anti-amyloid dot-blot assay
Cryo-TEM is a well established microscopic approach allowing for size characterization and identification of protein aggregates of different morphologies (38). To study the effect of RNA on the morphology of p53C aggregates, we performed cryo-TEM on p53C (5 μm) samples incubated at 37 °C in the absence and presence of low (0.1 μm) and high (0.6 μm) concentrations of dom V rRNA or DHFR mRNA. Scattered, amorphous aggregates, varying in shape and size but larger than 500 nm on average, were clearly visible in the p53C no-RNA sample (Fig. 6A, 0 μm dom V rRNA). Similar in morphology, large aggregates were produced in the presence of RNA in low concentration (Fig. 6A, 0.1 μm dom V rRNA). By contrast, even after extended incubation periods, we could not detect any aggregates when a high concentration of RNA was used (Fig. 6, A and B, 0.6 μm dom V rRNA). Notably, no fibrillar aggregates were observed under our experimental conditions. These data served as a visual demonstration of the dual effect of RNA on p53C aggregation, which was facilitated and suppressed at low and high RNA concentrations, respectively.
Figure 6.

Cryo-TEM images of p53C aggregates. The p53C samples (4.5 μm) alone or with different concentrations of dom V rRNA were incubated for 120 min at 37 °C and then subjected to cryo-TEM. Blue arrows indicate ice crystals, whereas red arrows indicate p53C aggregates. Row A includes the global view of large aggregates, whereas row B is composed of images of smaller-sized aggregates. In the case of 0.6 μm dom V rRNA, no aggregates were detected. The scale bar corresponds to 100 nm.
In addition, we examined the ability of p53C aggregates to bind thioflavin T (ThT), a dye commonly used in protein aggregation studies that is known to interact with amyloid oligomers and fibers but not with amorphous aggregates (39). When in contact with amyloid aggregates, ThT emits a distinct fluorescence signal (at ∼486 nm) upon excitation at 446 nm. Similar to LS, ThT fluorescence increased with higher protein concentrations, suggesting that p53C aggregation leads to the formation of amyloid species (Fig. 7A). Once again, a shortening of the lag phase was observed with a gradual increase in protein concentration. Unlike LS, however, the saturation of ThT emission was reached at a much earlier stage. The affinity of ThT to purine oligoribonucleotides (40) unfortunately hindered the application of this probe in the presence of RNA. To circumvent nucleic acid interference, we resorted to the application of conformation-specific antibodies in the form of a dot-blot immunoassay.
Figure 7.

Characterization of p53C aggregation with ThT binding, dot-blot assay with A11 antibody and seeding experiment. A, time-resolved ThT fluorescence of increasing concentrations of p53C at 37 °C. The final ThT concentration was 115 μm. B, amyloid oligomer specific (A11) dot-blot analysis of p53C aggregates formed in the absence of RNA (top) or at the 1:50 (middle) and at 1:8 RNA to protein (bottom) ratios of DHFR mRNA (left) and dom V rRNA (right). C, time course of aggregation of p53C at 37 °C after seeding the reaction with 5% (v/v) supernatant from the end products of p53C aggregation without or with GFP+ mRNA in low and high RNA:protein ratios (see “Experimental Procedures” for details). AU, arbitrary units.
A11 is an antibody reported to selectively recognize soluble amyloid oligomers and prefibrillar aggregates independent of amino acid sequences (41, 42). The antibody does not recognize native protein or mature fibrils and is only specific to a surface epitope displayed by amyloid-like oligomeric species. To verify the nature of p53C aggregates produced in the absence or presence of both high and low RNA concentrations, we performed immunoblots with A11 antibody on samples generated after several hours of incubation at 37 °C. p53C alone produced very little signal. In comparison, all RNA-containing samples showed strong positive signals (Fig. 7B), indicating the formation of oligomeric species of amyloid nature. Samples incubated with a higher RNA concentration produced the highest signal. This result implies that higher RNA:protein ratio promotes the formation of amyloid oligomers of p53C but prevents the accumulation of large aggregates as suggested by LS and TEM. Although LS and TEM revealed extensive, amorphous aggregation in the presence of lower RNA concentrations, a positive A11 signal was observed for those samples as well. When we quantified the dot blot data, the most pronounced A11 reading (37.7% of the transthyretin standard) was produced by the 1:8 RNA to protein ratio. The low RNA and no-RNA samples were quantified at 16.7% and 5.1%, respectively. Collectively, our data indicate heterogeneous aggregation of p53C in the presence of low concentrations of RNA.
Seeding of p53C aggregation by amyloid oligomers formed in the presence of RNA
Our observations on the formation of amyloid oligomers, but not mature fibrils, of p53C prompted us to probe whether these can act as seeds for further aggregation. To that end, we designed a modified seeding experiment, where we incubated p53C with no RNA and with low and high concentrations of RNA at 37 °C. The samples were then RNase-treated and subsequently centrifuged to remove any remaining large aggregates. A fraction of the supernatant (5% of total sample volume) was then added to a fresh p53C dilution, and the aggregation kinetics was followed with LS.
Fig. 7C shows that the inocula derived from the no-RNA and low-RNA reactions had almost no effect on the aggregation kinetics of p53C. By contrast, the inoculum from the p53C reaction with a high concentration of RNA accelerated the aggregation of the protein. This result suggests that in the presence of higher RNA concentrations p53C forms seed-like, oligomeric species that can promote the aggregation of p53C alone by shortening the lag phase and effectively eliminating the nucleation event.
Aggregation of FLp53 with RNA
To understand the biological significance of our results with p53C, we have tested whether RNA can influence the aggregation of FLp53. A time course of the LS measurements shows that FLp53 alone follows rather slow aggregation kinetics (Fig. 8). Similar to p53C, FLp53 aggregation also has three phases, but the lag phase is very short, and the late exponential phase takes much longer than p53C. When dom V rRNA (or DHFR mRNA; data not shown) was added in the reaction, the rate of aggregation accelerated dramatically (Fig. 8). The LS values reached a plateau within a minute after the manual mixing time and then decreased gradually, probably due to precipitation of the large FLp53 aggregates. The hydrodynamic radii (Rh) measured at the end points of the reactions with DLS supported the observation (Table 1). The FLp53 aggregates formed in the presence of the RNAs were largely polydiverse with Rh values more than two times larger than that of FLp53 alone. Thus, our results demonstrate that RNA can modulate the aggregation of FLp53. However, further experiments will be required to check whether, similar to p53C, RNA can also inhibit aggregation of FLp53 under specific conditions.
Figure 8.
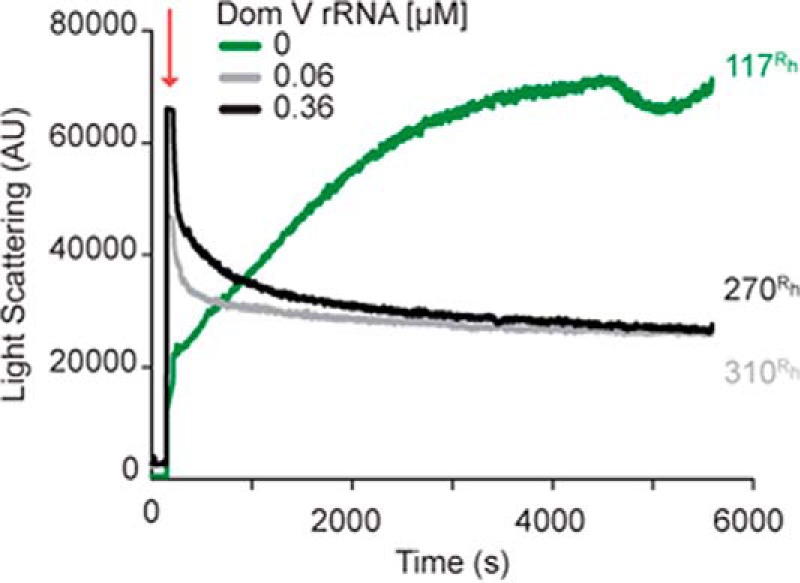
Aggregation of FLp53 followed by LS at 400 nm. Time course of FLp53 (3 μm) aggregation at 37 °C with or without dom V rRNA. The numbers adhering to the end points of the plots represent the Rh (nm) as measured by DLS. The red arrow indicates protein addition to the temperature-equilibrated sample mixture. AU, arbitrary units.
Table 1.
DLS analysis of FLp53 aggregation
Mean Rh and polydispersity values corresponding to the mean hydrodynamic radius of all components present in the full-length p53 (Flp53) samples acquired at the end point of each aggregation (kinetics) reaction presented in Fig 8.
| Sample | Hydrodynamic radius, Rh | Polydispersity |
|---|---|---|
| nm | % | |
| Aggregated Flp53 Sample | 116.94 ± 9.14 | 18.8 ± 8.7 |
| Flp53 + DHFR RNA (50:1) | 251.68 ± 18.94 | Multimodal |
| Flp53 + DHFR RNA (3:1) | 270.41 ± 130.03 | Multimodal |
| Flp53 + DomV RNA (50:1) | 310.27 ± 19.42 | Multimodal |
| Flp53 + DomV RNA (8:1) | 270.16 ± 4.02 | Multimodal |
Discussion
Over the last decade evidence has been accumulating in support of the hypothesis that various non-protein macromolecules are involved in prion formation and propagation (43, 44). Of the numerous possible adjuvants, nucleic acids have successfully been identified as the interaction partners of many disease-related proteins (24, 45–47). We previously showed that the RNA-mediated protein folding activity of the ribosome (48, 49) is a specific target for the anti-prion compounds 6-aminophenanthridine and guanabenz acetate (50–53). At the same time, it has been demonstrated that certain oligonucleotide sequences counteract prion infectivity ex vivo and in vivo (54). It is then obvious that an in-depth study of the role of DNA/RNA in protein folding-misfolding-aggregation is essential to uncover the underlying principles of degenerative proteinopathies and, in this way, the therapeutic potential of nucleic acids.
In line with earlier reports (5, 36), we found that p53C aggregation in vitro is highly dependent on protein concentration and temperature. A concentration of at least 3 μm and a temperature above 30 °C are required to promote p53C aggregation without agitation within our experimental timeframe (Fig. 2, A and B). Our cryo-TEM images revealed that p53C spontaneously forms large, amorphous aggregates at 37 °C and physiological pH (Fig. 6, 0 μm dom V rRNA). On the other hand, the increase in the ThT fluorescence signal under identical experimental conditions suggests that p53C can adopt alternative amyloid-like aggregation (Fig. 7A). While fibrillar structures could not be seen (Fig. 6), the generation of amyloids of a size below our detection limit cannot be excluded. These results are in good agreement with earlier indications of p53C producing a complex mixture of amorphous, oligomeric, and fibrillar aggregates (20). Interestingly, our dot-blot result indicated that p53C alone does not form much oligomeric amyloid species (Fig. 7B). This observation was consolidated by our seeding experiment (Fig. 7C), which demonstrated that supernatants from a p53C (no RNA) aggregation reaction did not facilitate the de novo aggregation of the protein. We, therefore, conclude that p53C, at physiological temperature and pH, in at least μm concentration forms a heterogeneous mixture of predominantly amorphous aggregates.
The presence of RNA appears to largely influence the aggregation of p53C. While a low RNA:protein ratio (RNA:p53C ≤ 1:50) facilitated the formation of large aggregates (Fig. 6, 0.1 μm dom V rRNA), an increase in the RNA concentration led to a gradual and substantial decrease in p53C aggregation (Figs. 3A and 6). At a molar ratio of 1:5 or 1:8 between RNA and p53C (referred to as high RNA concentration), protein aggregation was minimal, and almost no particles could be detected by TEM (Fig. 6, 0.6 μm dom V rRNA). To understand whether our observations with p53C are of biological significance, we studied FLp53 aggregation with or without two different RNAs. We found that RNA accelerated FLp53 aggregation substantially (Fig. 8). To our knowledge, this is the first report on RNA influence on p53C and, more importantly, FLp53 aggregation.
The effect of RNA on p53C aggregation could be reproduced with various RNAs of different origin, sequence, and cellular function, indicating that the p53C-RNA interaction is not dependent on specific nucleotide sequences. However, the facts that this effect could be abrogated by RNase treatment and that rNTPs did not influence p53C aggregation suggest that the suppression of p53C aggregation requires intact RNAs with folded structures. In addition, our results imply that there could be a size cut-off for RNA to be an effective modulator of p53C aggregation. For shorter RNAs, such as RNA1 and tRNAs, a higher concentration was necessary to achieve the same level of aggregation suppression compared to the larger RNAs, such as dom V rRNA and DHFR mRNA (Fig. 3A). Moreover, equimolar concentrations of double-stranded DNA (GFP+ and p53C), lacking the consensus sequence required for specific p53 binding, did not interfere with p53C aggregation. On the contrary, increasing concentrations of DNA appeared to stimulate aggregation (as measured by LS; Fig. 3B). Single-stranded DNA initially increased the LS signal produced by p53C aggregates (Fig. 3B). However, a ssDNA:protein ratio of 2:1, equal to the highest tRNA concentration tested, only reverted p53C aggregation to its native level, thereby emphasizing the unique, suppressive effect of RNA on the aggregation of p53C. At this stage, our data on the role of metal ions and different salts in this process remain inconclusive and in need of elaboration.
When comparing the LS kinetics of p53C aggregation with those obtained by fluorescence, a discrepancy between the two sets of data was detected. While LS was increased rapidly in the presence of the lower RNA concentration (Fig. 2, C and D) in relation to the no-RNA sample, almost no change in the kinetics of the two samples was recorded with F340 (Fig. 5, A–C). Furthermore, our TEM images displayed large amorphous aggregates of p53C with a low concentration of RNA, similar to the no-RNA sample (Fig. 6, 0 and 0.1 μm dom V rRNA), whereas the dot-blot assay indicated the presence of amyloid oligomers (Fig. 7B). As discussed previously, the discrepancy between the two kinetic datasets is most likely an attribute of the mechanistic differences in signal acquisition between the two approaches. While LS is a function of both amount and size of dispersed particles, intrinsic fluorescence depends entirely on the concentration of signal-inducing fluorophores. Consequently, nanomolar concentrations of RNA do not necessarily induce a higher extent of p53C aggregation but likely lead to the formation of larger-in-size aggregates (as seen by TEM; Fig. 6). Altogether, our data suggest that in the presence of a low RNA concentration, p53C forms both large amorphous aggregates and amyloid oligomers, the former being the predominant species and likely incorporating the latter (Fig. 9). It therefore seems plausible that RNA at a low RNA:protein ratio aids the process of amorphous aggregation of p53C.
Figure 9.

Aggregation scheme of p53C with and without RNA. On its own, p53C spontaneously forms large, heterogeneous aggregates under physiological conditions in vitro (no-RNA aggregation scenario). RNA serves as a scaffold for the aggregation-prone p53C molecules, increasing the chance of amyloid-like aggregation by providing the spatial and energetic requirements for specific protein-protein interactions (both low- and high-RNA:protein scenario). With increasing RNA concentrations, the equilibrium shifts toward amyloid oligomerization (high RNA:protein scenario). Such oligomers could function as seeds, nucleating the de novo aggregation of the protein (dotted line).
Inversely, high (micromolar) concentrations of RNA showed a strong, preventive effect on the amorphous aggregation of p53C, as demonstrated by LS (Fig. 3A), intrinsic fluorescence (Fig. 5, A–C) and TEM (Fig. 6). At a high concentration, RNA appears to predominantly stimulate the formation of small, oligomeric species of amyloid nature (Fig. 7B) that are capable of seeding the de novo aggregation of p53C (Figs. 7C and 9). This newly discovered role of RNA is highly intriguing, although the exact interpretation will require many more experiments. One possible explanation is that the binding of p53C to RNA likely occludes the protein-protein interaction interface, thereby limiting the available aggregation pathways and/or inducing structural perturbations that affect the kinetic stability of the bound species (55). With their large surface area and charge, structured RNAs might act as globular nanoparticles, which have been shown to enforce significant changes in the structure of bound proteins, in some cases suppressing aggregation, and in other cases, inducing fibrillation (56, 57). This premise can explain why longer RNAs are more effective modulators of p53C aggregation than shorter ones (Fig. 3A). Perhaps, below a certain size, RNA cannot form an adequate globule and fails to prevent aggregation, regardless of the concentration. Similar to polyphosphate chaperones (58), the polyanion RNA chain could also act as a chemical scaffold for the intrinsically unstable and aggregation-prone p53C, favoring local, protein-protein interactions and preventing long-distance ones. In this context, it is worth mentioning that rRNA has been shown to facilitate protein folding (48, 59, 60) as well as to prevent aggregation of partially folded protein intermediates (61). The parallels between the preventive role of RNA in p53C aggregation and protein folding activity of rRNA (53, 62) remain to be elucidated.
The distinct aggregation propensity of p53 is likely one of the many facets of its regulatory network. It has been proposed that the prion-like effect of aggregated p53 can be associated with tumor formation and malignancy (63). FLp53 is known to interact with different p53 isoforms (8, 64). Some of these isoforms, Δ133p53β, Δ133p53γ, Δ160p53β, and Δ160p53γ, are often altered in cancer cells (65, 66), implying their significance for cancer development. Although p53C is not exactly similar to the disease-related p53 isoforms described to date, isoforms Δ133p53β and Δ133p53γ retain almost the entire p53-DNA binding domain present in p53C; compared with p53C, they lack 39 N-terminal amino acid residues but possess an extra 29 and 34 C-terminal residues, respectively (7). Additionally, they show comparable overall charges (pI 8.83 for p53C, 8.04 for Δ133p53β, and 8.33 for Δ133p53γ). Therefore, we expect that the interaction of RNA with these p53 isoforms would not be substantially altered in comparison with p53C. Thus, our observations with p53C and FLp53 as model systems can add valuable insights concerning the aggregation of p53 and its isoforms in the cell.
In conclusion, with its general impact on p53C aggregation, particularly on amyloid-like oligomerization, RNA appears to be a key modulator of this process. Studies on the aggregation of p53 isoforms and co-aggregation with FLp53 (mutant and WT forms) as well as p53 paralogs (p63 and p73) in the presence of RNAs could provide fundamental information on cancer pathogenesis and progression. Because amyloid oligomers have been identified as the likely precursors and prevalent constituents of p53 aggregates in vivo (30), the potential role of non-coding RNAs as modulators for p53 aggregation in the cell should not be overlooked.
Experimental procedures
Protein expression and purification
Wild-type human FLp53 and p53C (corresponding to CCD, residues 94–312) were overexpressed in E. coli BL21(DE3) from plasmid clones using the vectors pET15b and pET11a (Novagen), respectively. p53C purification was performed as described by Bullock et al. (36). FLp53 overexpression was induced with 0.75 mm isopropyl 1-thio-β-d-galactopyranoside at an A600 of 0.6 after supplementing the medium with 0.02 mm ZnSO4. The cells were grown overnight at 15 °C before harvesting by centrifugation. The cell pellets were resuspended in lysis buffer (50 mm Tris-HCl, 50 mm NaCl, 5 mm dithioerythrol and 5% glycerol (v/v), pH 7.2) supplemented with 1 mm PMSF and a protease inhibitor mixture from Roche Applied Science. The suspension was sonicated on ice for cell lysis. FLp53 was purified from the cleared lysate using ion exchange (SP-Sepharose) followed by a Superdex 75 gel filtration column. Protein samples were stored in 50 mm Tris-HCl, pH 7.2, 150 mm NaCl, 5 mm dithioerythrol, and 5% glycerol (v/v) at −80 °C.
RNA and DNA sequences
All RNAs used here were produced by T7 transcription in vitro (67) from the corresponding linear DNA templates. The templates for domains V, II, and IV of the 23S rRNA (E. coli) were produced by PCR amplification of the respective gene sequence from a linearized DNA cassette generated from the plasmid pGEM4Z containing the full-length, E. coli 23S rRNA sequence (60). Similarly, the template for RNA1, the 337-nucleotide long, central-loop fragment of dom V of 23S rRNA (B. subtilis) was PCR-amplified from a plasmid pDK106 (59). The templates for HCA, DHFR. and GFP+ mRNAs were generated from their respective plasmid clones (pET24b). Bulk-tRNA was isolated from E. coli as described in Holm et al. (68). GFP+ dsDNA was generated through PCR amplification as described above. The same procedure was adopted for the production of p53C dsDNA from the plasmid clone encoding the WT p53C sequence. The 82-nt-long ssDNA oligonucleotide sequence (TACCGTTGGCGCGGGCGTTGTAGCAAAAGTTCTGAGCTAAAAACAGTAATACAAGGGGTGTTATGAGCCATATTCAACGGGA) was purchased from Sigma. None of the DNA sequences contained typical p53 consensus binding sites (PuPuPuC(A/T)).
p53C aggregation and denaturation
For aggregation reactions, p53C samples (final concentration: 4.5 μm, unless otherwise mentioned) were incubated in 50 mm Tris-HCl, pH 7.5, 10 mm NaCl, 5 mm dithioerythrol, and 5% glycerol (v/v) at 37 °C without agitation. FLp53 aggregation was performed under identical conditions at 3 μm protein concentration. For cryo-TEM, the sample buffer did not contain glycerol. The protein stocks were cleared by spinning at 14,000 rpm for at least 30 min before the experiments. For equilibrium aggregation, p53C was incubated at 37 °C for 1.5 h. To acquire the denatured state, fresh p53C was incubated in 6 m urea overnight at 37 °C.
LS and fluorescence measurements
Spectroscopic measurements were conducted using a Hitachi F-7000 steady state fluorescence spectrophotometer fitted with a Julabo F25-ME temperature-controlled circulating water bath. For LS measurements, the samples were excited at 400 nm, and the emission was recorded at the same wavelength. Tyrosine/tryptophan-based fluorescence experiments were performed with an excitation at 280 nm and emission at 340 nm. The emission spectra were recorded between 300 and 420 nm. For ThT measurements, the excitation and emission wavelengths were 446 nm and 486 nm, respectively. The final ThT concentration was 115 μm. p53C aggregation kinetics were performed at 37 °C. In all cases the background scattering/fluorescence contribution by nucleic acids was measured in the absence of p53C and was then accounted for in the final data. Using a 10-mm path-length cuvette, the fluorescence emission and absorption of 0.6 μm RNA samples were collected and fitted in the equation for the inner filter effect correction (35),
| (Eq. 1) |
where Fobserved and Fcorrected are the observed and corrected fluorescence intensity values, respectively, and Absex and Absem are the absorbance values at the excitation and emission wavelengths, respectively.
Dynamic light scattering
The size distribution and Rh of Flp53 samples after each aggregation kinetics were evaluated using the DynaPro Nanostar system (Wyatt) equipped with a 630-nm laser at 25 °C. The size distribution graph expressed the individual populations identified with their specific polydispersity values and Rh values presented in Table 1, corresponding to the mean of 10 cumulants for each measurement (average from all the populations identified in each sample).
Cryo-transmission electron microscopy
Cryo-TEM analysis was performed using a Zeiss TEM Libra 120 instrument (Carl Zeiss NTS, Oberkochen, Germany) operated at 80 kV in the zero-loss bright-field mode. Digital images were recorded under low-dose conditions using a TRS slow scan CCD camera system (TRS GmbH, Moorenweis, Germany) and iTEM software (Olympus Soft Imaging Solutions GmbH, Münster, Germany). An underfocus of 1–3 μm was used to enhance the contrast. Samples were prepared in a climate chamber with the temperature fixed at 25 °C and relative humidity close to saturation as described in Almgren et al. (69). p53C protein (4.5 μm) was incubated at 37 °C without or with various RNAs for 2 h to achieve equilibrium aggregation. A small droplet removed from each aggregation reaction was added on a copper grid-supported, perforated, polymer film covered with thin carbon layers on both sides. The droplet was then blotted with filter paper to allow thin film formation (10–500 nm) over the perforations in the polymer film. Immediately after blotting, the samples were vitrified by plunging them into liquid ethane kept just above its freezing point. Samples were kept below −165 °C and protected against atmospheric conditions during transfer to the TEM and examination.
Dot-blot assay
p53C samples with a final concentration of 15 μm were incubated at 37 °C for 2 h either alone or in the presence of DHFR mRNA or dom V rRNA in a 1:50 and 1:8 RNA-to-protein ratio. The whole-sample volume was loaded onto an activated and semi-dry PVDF membrane. The membrane was blocked with TBS-T (10 mm Tris-HCl, 150 mm NaCl, pH 8.0, 0.1% Tween 20) containing 5% nonfat milk for 1 h at room temperature, after which it was incubated for 18 h at 4 °C with the primary antibody A11 (rabbit, polyclonal antibody, producer: Invitrogen, catalogue number AHB0052) at a 1:1000 dilution. The membrane was then washed with TBS-T and incubated with a HRP-conjugated secondary antibody at a dilution of 1:3000 in TBS-T with 5% nonfat milk for 1 h at room temperature. After several TBS washes, the membrane was developed using an ECL kit (Bio-Rad). We used RNA, bovine serum albumin, and soluble p53C as negative controls and the amyloidogenic protein transthyretin as a positive control (ProSpec-TechnoGene Ltd.). Dot-blot quantification was performed using Image Lab Software (version 5.2.1, Bio-Rad) following the manufacturer's protocol.
Seeding experiments
For seeding experiments, p53C samples (4.5 μm) aggregated at equilibrium (without and with RNA) were treated with 5 μm RNase A (Thermo Fisher Scientific) for 2 h at 37 °C. The samples were then centrifuged at 14,000 rpm for 30 min to remove all large aggregates. A 20-fold dilution of the resulting supernatant was then added to the new p53C samples (4.5 μm), and the aggregation kinetics was followed in the fluorimeter by measuring LS against time at 37 °C.
Nucleic acid secondary structure prediction
The secondary structure maps for all rRNAs used in this study were procured from the rRNA secondary Structures (RibosomeGallery) database (see Ref. 70). The secondary structures for all other single-stranded polynucleotide sequences (DHFR mRNA, GFP+ mRNA, HCA mRNA, and ssDNA) were generated using the Vienna RNA websuite (71).
Author contributions
S. S. conceived the idea for this work and led the project together with J. L. S. and Y. C. The authors S. S. and P. S. K. designed the experiments, and P. S. K. conducted most of the experiments. D. B. and L. P. R. conducted some of the experiments on the kinetics of p53C aggregation. J. E. and K. E. conducted the TEM experiments. M. M. P. and M. M. D. C. M.-D. assisted P. S. K. to purify FLp53. P. S. K. and S. S. analyzed the results and wrote the manuscript. All authors contributed to the data analysis and manuscript preparation.
Acknowledgment
We thank Professor Chanchal DasGupta for providing us with the plasmid pGEM4Z. We also thank Raymond Fowler for editing the text of this manuscript.
This work was supported by research grants from the Swedish Research Council (2016-06264, 2013-8778, 2010-2619), the Swedish Foundation for International Cooperation in Research and Higher Education (STINT- Joint Brazilian-Swedish Research Collaboration; BR2013-5223 (to S. S.)), the Carlos Chagas Filho Foundation for Research Support in the State of Rio de Janeiro (FAPERJ; 170.027/2008), Coordination for the Improvement of Higher Education Personnel (CAPES; 1698/2012), and the National Council of Technological and Scientific Development (CNPql 573767/2008-4, 467500/2014-2, 470994/2012-6; to J. S.). The authors declare that they have no conflicts of interest with the contents of this article.
- p53
- tumor suppressor protein
- MDM2
- human double minute 2 homolog
- CCD/p53C
- central core domain of tumor suppressor protein
- FLp53
- full-length p53
- TEM
- transmission electron microscopy
- ThT
- thioflavin T
- F340
- fluorescence emission at 340 nm
- LS
- light scattering at 400 nm
- dom V rRNA
- domain V of E. coli 23S rRNA
- dom IV rRNA
- domain IV of E. coli 23S rRNA
- dom II rRNA
- domain II of E. coli 23S rRNA
- dom II rRNA
- domain II of E. coli 23S rRNA
- RNA1
- central-loop region of dom V of B. subtilis 23S rRNA
- HCA
- human carbonic anhydrase
- GFP+
- green fluorescent protein +
- DHFR
- dihydrofolate reductase
- rNTPs
- ribonucleoside tri-phosphates
- PrP
- major prion protein
- ssDNA
- single-stranded DNA
- DLS
- dynamic light scattering
- Rh
- hydrodynamic radii.
References
- 1. Vousden K. H., and Lane D. P. (2007) P53 in health and disease. Nat. Rev. Mol. Cell Biol. 8, 275–283 [DOI] [PubMed] [Google Scholar]
- 2. Prives C., and Hall P. (1999) The p53 pathway. J. Pathol. 187, 112–126 [DOI] [PubMed] [Google Scholar]
- 3. Vogelstein B., Lane D., and Levine A. J. (2000) Surfing the p53 network. Nature 408, 307–310 [DOI] [PubMed] [Google Scholar]
- 4. Muller P. A., and Vousden K. H. (2013) p53 mutations in cancer. Nat. Cell Biol. 15, 2–8 [DOI] [PubMed] [Google Scholar]
- 5. Ishimaru D., Andrade L. R., Teixeira L. S., Quesado P. A., Maiolino L. M., Lopez P. M., Cordeiro Y., Costa L. T., Heckl W. M., Weissmüller G., Foguel D., and Silva J. L. (2003) Fibrillar aggregates of the tumor suppressor p53 core domain. Biochemistry 42, 9022–9027 [DOI] [PubMed] [Google Scholar]
- 6. Joerger A. C., and Fersht A. R. (2008) Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 77, 557–582 [DOI] [PubMed] [Google Scholar]
- 7. Khoury M. P., and Bourdon J.-C. (2011) p53 Isoforms: an intracellular microprocessor? Genes Cancer 2, 453–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Moore H. C., Jordan L. B., Bray S. E., Baker L., Quinlan P. R., Purdie C. A., Thompson A. M., Bourdon J.-C., and Fuller-Pace F. V. (2010) The RNA helicase p68 modulates expression and function of the Δ133 isoform(s) of p53 and is inversely associated with Δ133p53 expression in breast cancer. Oncogene 29, 6475–6484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gong L., and Chen J. (2016) Δ113p53/Δ133p53 converts P53 from a repressor to a promoter of DNA double-stand break repair. Mol. Cell Oncol. 3, e1033587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davidson W. R., Kari C., Ren Q., Daroczi B., Dicker A. P., and Rodeck U. (2010) Differential regulation of p53 function by the N-terminal ΔNp53 and Δ113p53 isoforms in zebrafish embryos. BMC Dev. Biol. 10, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Olivares-Illana V., and Fåhraeus R. (2010) p53 isoforms gain functions. Oncogene 29, 5113–5119 [DOI] [PubMed] [Google Scholar]
- 12. Giaccia A. J., and Kastan M. B. (1998) The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev. 12, 2973–2983 [DOI] [PubMed] [Google Scholar]
- 13. Vousden K. H., and Vande Woude G. F. (2000) The ins and outs of p53. Nat. Cell Biol. 2, E178–E180 [DOI] [PubMed] [Google Scholar]
- 14. Lane D. P. (1992) p53, guardian of the genome. Nature 358, 15–16 [DOI] [PubMed] [Google Scholar]
- 15. Bell S., Klein C., Müller L., Hansen S., and Buchner J. (2002) p53 contains large unstructured regions in its native state. J. Mol. Biol. 322, 917–927 [DOI] [PubMed] [Google Scholar]
- 16. Friedler A., Veprintsev D. B., Hansson L. O., and Fersht A. R. (2003) Kinetic instability of p53 core domain mutants: implications for rescue by small molecules. J. Biol. Chem. 278, 24108–24112 [DOI] [PubMed] [Google Scholar]
- 17. Butler J. S., and Loh S. N. (2003) Structure, function, and aggregation of the zinc-free form of the p53 DNA binding domain. Biochemistry 42, 2396–2403 [DOI] [PubMed] [Google Scholar]
- 18. Ishimaru D., Maia L. F., Maiolino L. M., Quesado P. A., Lopez P. C., Almeida F. C., Valente A. P., and Silva J. L. (2003) Conversion of wild-type p53 core domain into a conformation that mimics a hot-spot mutant. J. Mol. Biol. 333, 443–451 [DOI] [PubMed] [Google Scholar]
- 19. Rangel L. P., Costa D. C., Vieira T. C., and Silva J. L. (2014) The aggregation of mutant p53 produces prion-like properties in cancer. Prion 8, 75–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ano Bom A. P., Rangel L. P., Costa D. C., de Oliveira G. A., Sanches D., Braga C. A., Gava L. M., Ramos C. H., Cepeda A. O., Stumbo A. C., De Moura Gallo C. V., Cordeiro Y., and Silva J. L. (2012) Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: implications for cancer. J. Biol. Chem. 287, 28152–28162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Forget K. J., Tremblay G., and Roucou X. (2013) p53 Aggregates penetrate cells and induce the co-aggregation of intracellular p53. PLoS ONE 8, e69242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Silva J. L., De Moura Gallo C. V., Costa D. C., and Rangel L. P. (2014) Prion-like aggregation of mutant p53 in cancer. Trends Biochem. Sci. 39, 260–267 [DOI] [PubMed] [Google Scholar]
- 23. Macedo B., Millen T. A., Braga C. A., Gomes M. P., Ferreira P. S., Kraineva J., Winter R., Silva J. L., and Cordeiro Y. (2012) Nonspecific prion protein-nucleic acid interactions lead to different aggregates and cytotoxic species. Biochemistry 51, 5402–5413 [DOI] [PubMed] [Google Scholar]
- 24. Silva J. L., and Cordeiro Y. (2016) The “Jekyll and Hyde” actions of nucleic acids on the prion-like aggregation of proteins. J. Biol. Chem. 291, 15482–15490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fontoura B. M., Atienza C. A., Sorokina E. A., Morimoto T., and Carroll R. B. (1997) Cytoplasmic p53 polypeptide is associated with ribosomes. Mol. Cell Biol. 17, 3146–3154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mosner J., Mummenbrauer T., Bauer C., Sczakiel G., Grosse F., and Deppert W. (1995) Negative feedback regulation of wild-type p53 biosynthesis. EMBO J. 14, 4442–4449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yoshida Y., Izumi H., Torigoe T., Ishiguchi H., Yoshida T., Itoh H., and Kohno K. (2004) Binding of RNA to p53 regulates its oligomerization and DNA-binding activity. Oncogene 23, 4371–4379 [DOI] [PubMed] [Google Scholar]
- 28. Riley K. J., and Maher L. J. 3rd (2007) p53 RNA interactions: new clues in an old mystery. RNA 13, 1825–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Riley K. J., Ramirez-Alvarado M., and Maher L. J. 3rd (2007) RNA-p53 interactions in vitro. Biochemistry 46, 2480–2487 [DOI] [PubMed] [Google Scholar]
- 30. Levy C. B., Stumbo A. C., Ano Bom A. P., Portari E. A., Cordeiro Y., Carneiro Y., Silva J. L., and De Moura-Gallo C. V. (2011) Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int. J. Biochem. Cell Biol. 43, 60–64 [DOI] [PubMed] [Google Scholar]
- 31. Pennisi E. (1996) Filling in the blanks in the p53 protein structure. Science 274, 921–922 [DOI] [PubMed] [Google Scholar]
- 32. Ishimaru D., Ano Bom A. P., Lima L. M., Quesado P. A., Oyama M. F., de Moura Gallo C. V., Cordeiro Y., and Silva J. L. (2009) Cognate DNA stabilizes the tumor suppressor p53 and prevents misfolding and aggregation. Biochemistry 48, 6126–6135 [DOI] [PubMed] [Google Scholar]
- 33. Wang G., and Fersht A. R. (2015) Mechanism of initiation of aggregation of p53 revealed by Φ-value analysis. Proc. Natl. Acad. Sci. U.S.A. 112, 2437–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ishimaru D., Lima L. M., Maia L. F., Lopez P. M., Ano Bom A. P., Valente A. P., and Silva J. L. (2004) Reversible aggregation plays a crucial role on the folding landscape of p53 core domain. Biophys. J. 87, 2691–2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy (Lakowicz J. R., ed), Springer US, Boston, MA: 10.1007/978-0-387-46312-4 [DOI] [Google Scholar]
- 36. Bullock A. N., Henckel J., DeDecker B. S., Johnson C. M., Nikolova P. V., Proctor M. R., Lane D. P., and Fersht A. R. (1997) Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. U.S.A. 94, 14338–14342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Munishkina L. A., and Fink A. L. (2007) Fluorescence as a method to reveal structures and membrane-interactions of amyloidogenic proteins. Biochim. Biophys. Acta 1768, 1862–1885 [DOI] [PubMed] [Google Scholar]
- 38. Bruggink K. A., Müller M., Kuiperij H. B., and Verbeek M. M. (2012) Methods for analysis of amyloid-β aggregates. J. Alzheimers Dis. 28, 735–758 [DOI] [PubMed] [Google Scholar]
- 39. Biancalana M., and Koide S. (2010) Molecular mechanism of thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 1804, 1405–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sugimoto S., Arita-Morioka K.-I., Mizunoe Y., Yamanaka K., and Ogura T. (2015) Thioflavin T as a fluorescence probe for monitoring RNA metabolism at molecular and cellular levels. Nucleic Acids Res. 43, e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., and Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 42. Glabe C. G. (2004) Conformation-dependent antibodies target diseases of protein misfolding. Trends Biochem. Sci. 29, 542–547 [DOI] [PubMed] [Google Scholar]
- 43. Silva J. L., Lima L. M., Foguel D., and Cordeiro Y. (2008) Intriguing nucleic-acid-binding features of mammalian prion protein. Trends Biochem. Sci. 33, 132–140 [DOI] [PubMed] [Google Scholar]
- 44. Silva J. L., Gomes M. P., Vieira T. C., and Cordeiro Y. (2010) PrP interactions with nucleic acids and glycosaminoglycans in function and disease. Front. Biosci. (Landmark Ed) 15, 132–150 [DOI] [PubMed] [Google Scholar]
- 45. Lima L. M., Cordeiro Y., Tinoco L. W., Marques A. F., Oliveira C. L., Sampath S., Kodali R., Choi G., Foguel D., Torriani I., Caughey B., and Silva J. L. (2006) Structural insights into the interaction between prion protein and nucleic acid. Biochemistry 45, 9180–9187 [DOI] [PubMed] [Google Scholar]
- 46. Liu C., and Zhang Y. (2011) Nucleic acid-mediated protein aggregation and assembly. Adv. Protein Chem. Struct. Biol. 84, 1–40 [DOI] [PubMed] [Google Scholar]
- 47. Cordeiro Y., Macedo B., Silva J. L., and Gomes M. P. B. (2014) Pathological implications of nucleic acid interactions with proteins associated with neurodegenerative diseases. Biophys. Rev. 6, 97–110 10.1007/s12551-013-0132-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sanyal S. C., Pal S., Chowdhury S., and DasGupta C., and Chaudhuri S. (2002) 23S rRNA assisted folding of cytoplasmic malate dehydrogenase is distinctly different from its self-folding. Nucleic Acids Res. 30, 2390–2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chattopadhyay S., Das B., Bera A. K., Dasgupta D., and Dasgupta C. (1994) Refolding of denatured lactate dehydrogenase by Escherichia coli ribosomes. Biochem. J. 300, 717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pang Y., Kurella S., Voisset C., Samanta D., Banerjee D., Schabe A., Das Gupta C., Galons H., Blondel M., and Sanyal S. (2013) The antiprion compound 6-aminophenanthridine inhibits the protein folding activity of the ribosome by direct competition. J. Biol. Chem. 288, 19081–19089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reis S. D., Pang Y., Vishnu N., Voisset C., Galons H., Blondel M., and Sanyal S. (2011) Mode of action of the antiprion drugs 6AP and GA on ribosome assisted protein folding. Biochimie 93, 1047–1054 [DOI] [PubMed] [Google Scholar]
- 52. Tribouillard-Tanvier D., Dos Reis S., Gug F., Voisset C., Béringue V., Sabate R., Kikovska E., Talarek N., Bach S., Huang C., Desban N., Saupe S. J., Supattapone S., Thuret J.-Y., Chédin S., et al. (2008) Protein folding activity of ribosomal RNA is a selective target of two unrelated antiprion drugs. PLoS ONE 3, e2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Banerjee D., and Sanyal S. (2014) Protein folding activity of the ribosome (PFAR): a target for antiprion compounds. Viruses 6, 3907–3924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kocisko D. A., Vaillant A., Lee K. S., Arnold K. M., Bertholet N., Race R. E., Olsen E. A., Juteau J. M., and Caughey B. (2006) Potent antiscrapie activities of degenerate phosphorothioate oligonucleotides. Antimicrob. Agents Chemother. 50, 1034–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cordeiro Y., and Silva J. L. (2005) The hypothesis of the catalytic action of nucleic acid on the conversion of prion protein. Protein Pept. Lett. 12, 251–255 [DOI] [PubMed] [Google Scholar]
- 56. Colvin V. L., and Kulinowski K. M. (2007) Nanoparticles as catalysts for protein fibrillation. Proc. Natl. Acad. Sci. U.S.A. 104, 8679–8680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Brambilla D., Le Droumaguet B., Nicolas J., Hashemi S. H., Wu L. P., Moghimi S. M., Couvreur P., and Andrieux K. (2011) Nanotechnologies for Alzheimer's disease: diagnosis, therapy, and safety issues. Nanomedicine 7, 521–540 [DOI] [PubMed] [Google Scholar]
- 58. Gray M. J., Wholey W. Y., Wagner N. O., Cremers C. M., Mueller-Schickert A., Hock N. T., Krieger A. G., Smith E. M., Bender R. A., Bardwell J. C., Jakob U. (2014) Polyphosphate is a primordial chaperone. Mol. Cell 53, 689–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pal S., Chandra S., Chowdhury S., Sarkar D., Ghosh A. N., and Gupta C. D. (1999) Complementary role of two fragments of domain V of 23 S ribosomal RNA in protein folding. J. Biol. Chem. 274, 32771–32777 [DOI] [PubMed] [Google Scholar]
- 60. Samanta D., Mukhopadhyay D., Chowdhury S., Ghosh J., Pal S., Basu A., Bhattacharya A., Das A., Das D., and DasGupta C. (2008) Protein folding by domain V of Escherichia coli 23S rRNA: specificity of RNA-protein interactions. J. Bacteriol. 190, 3344–3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pathak B. K., Mondal S., Ghosh A. N., and Barat C. (2014) The ribosome can prevent aggregation of partially folded protein intermediates: studies using the Escherichia coli ribosome. PLoS ONE 9, e96425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Das D., Das A., Samanta D., Ghosh J., Dasgupta S., Bhattacharya A., Basu A., Sanyal S., and Das Gupta C. (2008) Role of the ribosome in protein folding. Biotechnol. J. 3, 999–1009 [DOI] [PubMed] [Google Scholar]
- 63. Costa D. C., de Oliveira G. A., Cino E. A., Soares I. N., Rangel L. P., and Silva J. L. (2016) Aggregation and prion-like properties of misfolded tumor suppressors: is cancer a prion disease? Cold Spring Harb. Perspect. Biol. 8, a023614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kim S., and An S. S. (2016) Role of p53 isoforms and aggregations in cancer. Medicine 95, e3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Avery-Kiejda K. A., Morten B., Wong-Brown M. W., Mathe A., and Scott R. J. (2014) The relative mRNA expression of p53 isoforms in breast cancer is associated with clinical features and outcome. Carcinogenesis 35, 586–596 [DOI] [PubMed] [Google Scholar]
- 66. Takahashi R., Giannini C., Sarkaria J. N., Schroeder M., Rogers J., Mastroeni D., and Scrable H. (2013) p53 isoform profiling in glioblastoma and injured brain. Oncogene 32, 3165–3174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Banerjee D., Vovusha H., Pang Y., Oumata N., Sanyal B., and Sanyal S. (2014) Spectroscopic and DFT studies on 6-Aminophenanthridine and its derivatives provide insights in their activity towards ribosomal RNA. Biochimie 97, 194–199 [DOI] [PubMed] [Google Scholar]
- 68. Holm M., Borg A., Ehrenberg M., and Sanyal S. (2016) Molecular mechanism of viomycin inhibition of peptide elongation in bacteria. Proc. Natl. Acad. Sci. U.S.A. 113, 978–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Almgren M., Edwards K., and Karlsson G. (2000) Cryo transmission electron microscopy of liposomes and related structures. Colloids and Surfaces 174, 3–21 [Google Scholar]
- 70. Petrov A. S., Bernier C. R., Gulen B., Waterbury C. C., Hershkovits E., Hsiao C., Harvey S. C., Hud N. V., Fox G. E., Wartell R. M., and Williams L. D. (2014) Secondary structures of rRNAs from all three domains of life. PLoS ONE 9, e88222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gruber A. R., Lorenz R., Bernhart S. H., Neuböck R., and Hofacker I. L. (2008) The Vienna RNA websuite. Nucleic Acids Res. 36, W70–W74 [DOI] [PMC free article] [PubMed] [Google Scholar]