

Abstract
In malaria, CD36 plays several roles, including mediating parasite sequestration to host organs, phagocytic clearance of parasites, and regulation of immunity. Although the functions of CD36 in parasite sequestration and phagocytosis have been clearly defined, less is known about its role in malaria immunity. Here, to understand the function of CD36 in malaria immunity, we studied parasite growth, innate and adaptive immune responses, and host survival in WT and Cd36−/− mice infected with a non-lethal strain of Plasmodium yoelii. Compared with Cd36−/− mice, WT mice had lower parasitemias and were resistant to death. At early but not at later stages of infection, WT mice had higher circulatory proinflammatory cytokines and lower anti-inflammatory cytokines than Cd36−/− mice. WT mice showed higher frequencies of proinflammatory cytokine-producing and lower frequencies of anti-inflammatory cytokine-producing dendritic cells (DCs) and natural killer cells than Cd36−/− mice. Cytokines produced by co-cultures of DCs from infected mice and ovalbumin-specific, MHC class II-restricted α/β (OT-II) T cells reflected CD36-dependent DC function. WT mice also showed increased Th1 and reduced Th2 responses compared with Cd36−/− mice, mainly at early stages of infection. Furthermore, in infected WT mice, macrophages and neutrophils expressed higher levels of phagocytic receptors and showed enhanced phagocytosis of parasite-infected erythrocytes than those in Cd36−/− mice in an IFN-γ-dependent manner. However, there were no differences in malaria-induced humoral responses between WT and Cd36−/− mice. Overall, the results show that CD36 plays a significant role in controlling parasite burden by contributing to proinflammatory cytokine responses by DCs and natural killer cells, Th1 development, phagocytic receptor expression, and phagocytic activity.
Keywords: cellular immune response, cytokine induction, humoral response, malaria, phagocytosis, CD36, phagocytic receptors, Plasmodium yoelii, immune regulation, resistance to disease
Introduction
Malaria is a major public health problem in many countries around the world, causing about ∼0.5 million deaths annually (1, 2). The disease is caused primarily by Plasmodium falciparum and Plasmodium vivax, which are the most prevalent malaria parasites in endemic regions (3, 4). Thus far, antimalarial drugs have been the only mode of treatment for malaria infections; however, in recent years, parasites have developed resistance, including to the more recently introduced artemisinins (5–7). Given that parasites are likely to develop resistance to newly introduced drugs, alternative strategies are required to treat malaria infections. Mass vaccination would be a better strategy, but an effective vaccine is not available, and its development remains challenging (8, 9). Gaining insight into the molecular and cellular processes involved in protective immunity and pathogenesis may help in the design of an efficacious vaccine and/or immunomodulation agents to treat malaria (10, 11).
Innate immunity plays crucial roles in the outcome of malaria (10, 12, 13). It functions as the first line of defense in controlling infection through phagocytic clearance of parasites and regulates the development of adaptive immunity. Dendritic cells (DCs)5 and macrophages (Mφs) are important early responders of the innate immune system. In malaria, Mφs are primarily involved in parasitemia control through phagocytic uptake of parasites (14–16), whereas DCs efficiently produce proinflammatory cytokines, such as TNF-α and IL-12, and present antigens. IL-12, a Th1-polariazing cytokine, activates NK cells to induce the secretion of IFN-γ, another Th1-promoting cytokine. IFN-γ, TNF-α, and IL-1 prime phagocytic cells to enhance phagocytic activity (17). The initial cytokines produced by DCs modulate T and B cell functions, contributing to the development of parasite-specific cellular and humoral immunity.
CD36 is a multifunctional class B scavenger receptor that also functions as a pattern recognition receptor in innate immune cells (18, 19). CD36 not only interacts with diverse ligands, including thrombospondin-1, long-chain fatty acids, certain oxidized lipids, types I and IV collagen, and β-amyloid, but also binds and mediates uptake of pathogens and apoptotic cells (18, 19). Many cell types, including platelets, Mφs, DCs, adipocytes, and endothelial, epithelial, and muscle cells, express CD36 (18, 19). The CD36-dependent internalization of pathogens and endogenous pathogenic molecules, such as oxidized low-density lipoprotein and β-amyloid protein, activate Src/Syk family non-receptor tyrosine kinases, ERK and Jun members of the MAPK signaling pathways, and NF-κB transcription factors, resulting in the production of proinflammatory mediators (16, 19, 20). Thus, CD36 is involved in several physiological and pathological processes, including immunity, lipid absorption, storage and metabolism, inflammation, and cardiovascular and Alzheimer's diseases (18, 19).
CD36 plays several important roles in malaria, including parasite sequestration in microvascular capillaries of various organs; in phagocytic clearance of parasites through uptake of infected erythrocytes; and in immune responses (20–27). Of these, it is best known for functioning as a receptor for several members of the P. falciparum erythrocyte membrane protein-1 (PfEMP1) family of variant proteins, which are expressed on the infected red blood cell (IRBC) surface (28–30). This interaction mediates the adherence of IRBCs to microvascular endothelia, enabling parasites to be sequestered in host organs and avoid clearance from the circulation. When immunity against an adherent PfEMP1 is present in the host, parasites expressing other PfEMP1 variants having different adherent specificity are sequestered in the host organs. The parasites multiply and accumulate in large numbers in organs, contributing to severe malaria pathogenesis (31–34).
Murine CD36 has ∼85% identity to human CD36 at the amino acid level. Although murine parasites do not express PfEMP1 orthologs, CD36 mediates the sequestration of Plasmodium berghei Antwerpen-Kasapa (ANKA), a cytoadherent murine malaria parasite strain, to lungs and adipose tissue of infected mice, presumably by interacting with unidentified structure(s) on the IRBC surface (35). Recent studies have shown that CD36-mediated sequestration of P. berghei ANKA strain causes acute liver injury (36, 37). Furthermore, both in human and mouse malaria, CD36 controls parasitemia through phagocytic uptake of IRBCs by Mφs and polymorphonuclear neutrophils (PMNs) (14, 16). Additionally, CD36 has been shown to regulate MAPK signaling and modulate TNF-α responses in mouse malaria infection and to modulate parasite glycosylphosphatidylinositol-induced cytokine responses by mouse Mφs and human blood DCs (16, 27, 38). In P. falciparum-endemic regions, single-nucleotide polymorphisms in the Cd36 gene have been linked to protection against cerebral and other severe malaria (39, 40), although another study has implicated mutations in Cd36 to susceptibility to malaria (41). Thus, CD36 plays an important role in immune responses to malaria, but insight into CD36-mediated regulation of immune responses to malaria remains less understood. Therefore, the main objective of this study was to determine the contribution of CD36 in malaria-induced immune responses and dissect the associated cellular mechanisms. To this end, we analyzed immune responses and assessed parasite growth kinetics and host survival in WT and Cd36−/− mice infected with a non-lethal, non-sequestering Plasmodium yoelii strain, which allows assessment of immune responses during the course of innate and adaptive immunity development. The results showed that CD36 significantly contributes to the production of proinflammatory cytokines by the cells of the innate immune system and Th1 development. CD36 also contributes to the suppression of anti-inflammatory cytokine production and Th2 development. Additionally, the CD36-dependent immune responses contribute to the expression of other phagocytic receptors and phagocytic activity, thereby significantly reducing parasite burden and malarial mortality. Overall, our results define the role of CD36 in malaria immunity, revealing the associated cellular mechanisms.
Results
CD36 contributes to malaria parasitemia control and resistance against mortality
In this study, we investigated the role of CD36 in immunity to blood stage malaria and in clinical outcomes by infecting WT and Cd36−/− mice with a non-sequestering P. yoelii 17XNL strain and analyzing various responses during the course of innate and adaptive immunity development. In mice infected with P. yoelii 17XNL strain, parasitemias reach a peak of 30–40% by about 20 days, and mice generally survive by clearing infection (42–44). In contrast, mice infected with the sequestering, lethal P. yoelii 17XL strain die at 6–10 days postinfection (pi), exhibiting peak parasitemias of 60–80% (pi) (45–47).
Survival analysis showed that 90% of WT mice infected with P. yoelii 17XNL survived, whereas the majority of infected Cd36−/− mice died between 17 and 24 days pi (Fig. 1A). Analysis of parasite growth kinetics showed, in both WT and Cd36−/− mice, slow parasite growth during the 1st week of infection and then a rapid increase, reaching peak parasitemias at 20–22 days pi (Fig. 1B). After this period, parasitemias were markedly decreased to basal levels in surviving mice. These observations agree with the previously reported growth pattern of P. yoelii 17XNL (42–44). Throughout the infection period, parasitemias were significantly higher in Cd36−/− mice than in WT mice. Although parasitemias were relatively low in both WT and Cd36−/− mice up to 10–12 days pi, the parasitemias in Cd36−/− mice were 57–125% higher than those in WT mice (Fig. 1C). The CD36-depedent control of parasitemia is consistent with the reported results that CD36 is efficiently expressed by Mφs and contributes to parasitemia control in mice infected with Plasmodium chabaudi (25). Because CD36 functions both as a scavenger and pattern recognition receptor, it is not surprising that P. yoelii IRBCs are recognized by CD36 on Mφs likely through relatively weak interactions and phagocytosed. Thus, the above data demonstrated that CD36 contributes to parasitemia control and resistance against malarial fatality.
Figure 1.

CD36 contributes to malaria parasitemia control and confers resistance to malaria mortality. In these and all other in vivo experiments, WT and Cd36−/− mice were infected by i.p. injection of P. yoelii IRBCs (1 × 106/mouse). Mice were monitored daily for mortality (A), and parasitemias (B) were assessed on alternative days by counting IRBCs in Giemsa-stained blood smears and mean parasitemia values of surviving mice were plotted. A, shown are the combined results of two separate experiments, one performed using seven WT and six Cd36−/− mice and the other using three WT and eight Cd36−/− mice. B, the results are representative of three independent experiments. Error bars indicate S.D. of surviving mice in each group. *, p < 0.05; **, p < 0.01; ***, p < 0.001. C, percent parasitemia in WT and Cd36−/− mice and percentage of difference in parasitemia between WT and Cd36−/− mice at the indicated time points.
CD36 contributes to malaria-induced cytokine production primarily at early stages of infection
Based on the results presented in Fig. 1, we predicted that CD36 contributes to malaria-induced immune responses that are involved in parasitemia control and resistance to disease severity. Proinflammatory cytokine responses induced at early stages of infection play important roles in either pathogenesis or resistance to malaria depending on whether or not parasites are sequestered in host organs (10, 12, 23). High levels of proinflammatory cytokines contribute to cerebral and other severe malaria in the context of parasite sequestration. However, in non-sequestering infections, cytokine responses are likely to be beneficial because IFN-γ, TNF-α, and IL-1 contribute to parasitemia control by priming phagocytic cells for enhanced phagocytic activity (17). Cytokines released early in infections also influence responses by other cells of innate immunity, such as NK cells, and contribute to Th1 and humoral responses (48–51), leading to infection-specific immune development. To determine the function of CD36 in malaria-induced cytokine responses and understand how these responses are involved in providing resistance to malaria, we analyzed a panel of cytokines produced during the course of P. yoelii infection. At 3 and 5 days pi, serum TNF-α, IL-1β, and IL-18 as well as Th1-promoting cytokines IFN-γ and IL-12 were significantly higher in infected WT mice than in infected Cd36−/− mice (Fig. 2, A–E). However, at 10 and 17 days pi, there were no differences in these cytokine levels between WT and Cd36−/− mice. In contrast to inflammatory cytokines, the level of Th2-promoting IL-4 was higher in infected Cd36−/− mice than that in infected WT mice throughout the infection period (Fig. 2F). At 5 and 10 days pi, the level of IL-10 in Cd36−/− mice was higher than in WT mice, but there was no difference between WT and Cd36−/− mice at 17 days pi (Fig. 2G). These results indicate that CD36-mediated enhanced proinflammatory and Th1-promoting cytokine production and decreased anti-inflammatory and Th2-promoting cytokine production mainly occur at relatively early stages of infection (Fig. 2).
Figure 2.

CD36 regulates cytokine responses to malaria infection. A–G, cytokines in sera prepared from the blood collected from uninfected (UI) mice (n = 3) and P. yoelii-infected WT and Cd36−/− mice (n = 4–5 mice/group) at the indicated time points were analyzed by ELISA. Sera of uninfected mice were analyzed as controls. In A–G, error bars indicate S.D. of mice in each group. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not statistically significant.
CD36 promotes immune responses to malaria by DCs
Based on the observed kinetics of serum cytokine responses in P. yoelii-infected WT and Cd36−/− mice, we predicted that the CD36-mediated responses primarily regulate innate immunity. Among the cells of the innate immune system, DCs and spleen red pulp Mφs, which have DC-like characteristics, are the earliest and major producers of cytokines in response to blood stage malaria infection (52–55). DCs regulate responses of other innate immune cells, such as NK cells, and bridge innate immunity to adaptive immunity (48–51). To determine the role of CD36 in the activation of DCs and their cytokine responses, first we analyzed the surface expression of DC costimulatory molecules CD80 and CD86 and cytokine responses by spleen DCs of infected mice by flow cytometry. At 4 days pi, DCs from spleen of infected Cd36−/− mice were less mature than those from WT mice as indicated by a lower surface expression of CD80 and CD86 (Fig. 3, A–C). Cytokine analysis showed that at an early time point (4 days pi) Cd36−/− mice had lower frequencies of TNF-α-producing DCs and higher frequencies of IL-10-producing DCs than WT mice (Fig. 3, D and E). At a later time point (7 days pi), however, there were no differences in the frequencies of either pro- or anti-inflammatory cytokine-producing DCs from infected Cd36−/− and WT mice. We also assessed cytokines secreted by DCs isolated from infected mice. Consistent with the above results, DCs isolated from Cd36−/− mice at 5 days pi secreted significantly lower levels of TNF-α and IL-12 and a higher level of IL-10 than DCs from infected WT mice (Fig. 3F). However, 10 days pi, there were no significant differences in the levels of TNF-α, IL-12, or IL-10 produced by DCs from WT and Cd36−/− mice. Together, the above results demonstrated that, at early stages of infection, CD36 promotes malaria-induced proinflammatory cytokines and down-regulates anti-inflammatory cytokines by DCs.
Figure 3.

CD36 regulates malaria infection-induced DC maturation and cytokine production. Mice were infected with P. yoelii as outlined in the legend to Fig. 1. A–E, total splenocytes from uninfected and P. yoelii-infected mice at 4 days pi were stained with antibodies against mouse CD80, CD86, DC surface markers, and cytokines. A–C, gating of DCs (A) and surface expression of CD80 and CD86 by gated DCs (B and C) are shown. B and C, left panels, show histograms of co-stimulatory molecule-expressing DCs from a representative mouse in each group (n = 3–6 mice/group). Shaded area, red line, and blue line, respectively, represent uninfected (UI), infected WT, and infected Cd36−/− mice. Right panels show the expression of CD80 and CD86 by DCs in each mouse group. D and E, the spleen cells from uninfected mice and infected WT and Cd36−/− mice were cultured in 24-well plates for 6 h in the presence of GolgiPlug, stained with antibodies against DC marker proteins, fixed, stained with antibodies against mouse cytokines, and analyzed by flow cytometry. Contour diagrams of cytokine-expressing spleen DCs from a representative mouse at 4 days pi (left panels) and plots showing the frequencies of cytokine-expressing DCs at 4 and 7 days pi in a representative mouse in each group (right panels) (n = 3–4 mice/group) are shown. DCs at 7 days pi were similarly gated for cytokine expression. The data are a representative of three independent experiments. F, pooled DCs isolated from the spleens of uninfected mice and P. yoelii-infected WT and Cd36−/− mice at 5 and 10 days pi in 96-well plates were incubated in complete DMEM at 37 °C. After 24 h, cytokines released into the culture medium were analyzed by ELISA. In B–F, the results are a representative of two independent experiments. Error bars indicate S.D. of mice in each group. *, p < 0.05; **, p < 0.01; ns, not statistically significant. d, days; FSC, forward scatter; SSC, side scatter; GeoMFI, geometric mean fluorescence intensity.
CD36-mediated immune responses confer DCs an ability to regulate Th responses
Given that DCs regulate adaptive immunity (50, 51), we tested DC-induced T cell responses in co-cultures of DCs isolated from mice at 5 days pi pulsed with ovalbumin peptide (OVA(323–339)) and T cells from uninfected OT-II transgenic mice expressing TCR for OVA(323–339) peptide on CD4 T cells. The levels of IFN-γ and TNF-α produced by the co-cultures of spleen DCs from Cd36−/− mice and OT-II T cells were significantly lower than those produced by the co-cultures of spleen DCs from WT mice and OT-II T cells (Fig. 4). On the other hand, the levels of IL-10 and IL-4 produced by the co-cultures of spleen DCs from Cd36−/− mice and OT-II T cells were significantly higher than those produced by the co-cultures of WT DCs and OT-II T cells (Fig. 4). Because DCs either secrete very low levels of IFN-γ and IL-4 or do not secrete these cytokines, the observed cytokines were most likely produced by OT-II T cells, reflecting the function of DCs. Thus, these results suggested that the CD36-regulated function of DCs contributes to immune responses of T cells.
Figure 4.
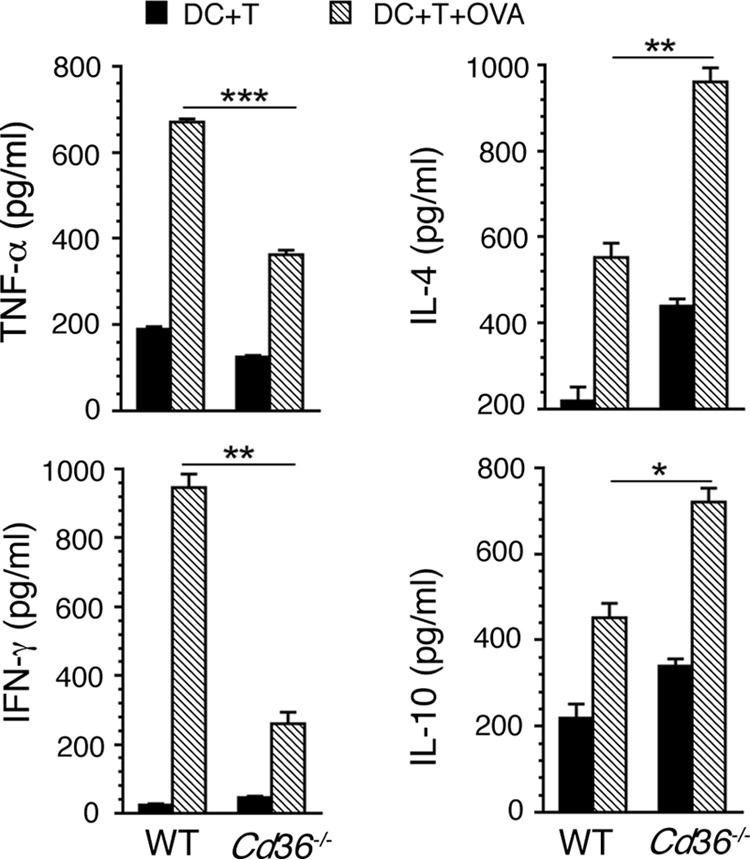
CD36-modulates cytokine responses by T cells. DCs (1 × 105 cells/well) isolated from the spleens of P. yoelii-infected WT and Cd36−/− mice at 5 days pi were co-cultured with OT-II T cells (0.5 × 105/well) from uninfected mice in 96-well plates in the presence of 2 μg/ml OVA(323–339) peptide for 72 h. Co-cultures of DCs and OT-II T cells without OVA(323–339) peptide treatment were used as controls. The cytokines in the culture medium were analyzed by ELISA. Data shown are a representative of two independent experiments. Error bars indicate S.D. of triplicate co-cultures. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
CD36 promotes proinflammatory responses by NK cells and Th1 development in malaria
To examine the role of CD36 in malaria-induced immune responses by NK and T cells, we analyzed cytokine and Th1- and Th2-promoting transcription factor responses in NK and T cells from the spleens of infected mice. At 4 days pi, Cd36−/− mice showed lower frequencies of TNF-α- and IFN-γ-producing NK cells than WT mice (Fig. 5, A–C). Cd36−/− mice also showed significantly lower frequencies of Tbet-expressing NK cells than WT mice (Fig. 5D). In contrast, the frequency of IL-10-producing NK cells was higher in Cd36−/− mice than in WT mice (Fig. 5E). At 7 days pi, there were no differences in either proinflammatory cytokine responses or Tbet expression by NK cells from WT and Cd36−/− mice. However, the frequency of IL-10-producing NK cells was higher in Cd36−/− mice than in WT mice (Fig. 5E). Together, the above data demonstrated that, at early stages of infection, CD36 contributes to inflammatory responses by NK cells.
Figure 5.

CD36 contributes to the malaria-induced immune responses by NK cells. Splenocytes from uninfected (UI) and P. yoelii-infected WT and Cd36−/− mice (n = 3 or 4 mice/group) at 4 and 7 days pi were cultured in complete DMEM in the presence of GolgiPlug for 6 h, stained with antibodies against NK cell markers, fixed, then stained with anti-cytokine and anti-Tbet antibodies, and analyzed by flow cytometry. A shows the gating of NK cells. B–E, analysis of gated cytokine- and Tbet-expressing NK cells. Left panels show contour diagrams of NK cells expressing TNF-α (B), IFN-γ (C), Tbet (D), and IL-10 (E) from a representative mouse in each group at 4 days pi; cells from infected mice at 7 days pi were similarly analyzed. Right panels represent frequencies of cytokine- and Tbet-expressing NK cells. Data are a representative of three independent experiments. Error bars indicate S.D. of mice in each group. *, p < 0.05; **, p < 0.01; ns, statistically not significant. d, days; FSC, forward scatter; SSC, side scatter.
CD36 contributes to malaria-induced T cell responses as well. At 4 days pi, analysis of gated T cells (Fig. 6A) showed significantly decreased numbers of IFN-γ-expressing and Th1-promoting master transcription factor Tbet-expressing CD4+ and CD8+ T cells in Cd36−/− mice compared with those in WT mice (Fig. 6, B and C), indicating that CD36 up-regulates Th1 responses. In contrast, Cd36−/− mice had higher numbers of IL-10- and IL-4-producing CD4+ and CD8+ T cells than WT mice (Fig. 6, D and E). Consistent with these results, at 4 days pi, the numbers of Th2-promoting transcription factor GATA3-expressing CD4+ and CD8+ T cells were significantly higher in Cd36−/− mice than in WT mice (Fig. 6F). Together, these results demonstrated that CD36 contributes significantly to Th1 development and that CD36 deficiency leads to early switching from Th1 to Th2 responses during malaria infection.
Figure 6.

CD36 contributes to malaria-induced T cell responses. Splenocytes from uninfected and P. yoelii-infected mice (n = 4–5 mice/group) at 4, 7, and 14 days pi were cultured in complete DMEM in the presence of GolgiPlug for 6 h and stained with antibodies against mouse T cell surface markers. The cells were fixed, stained with anti-cytokine and anti-transcription factor antibodies, and analyzed by flow cytometry. A shows the gating of T cells. B–F, the gated T cells from splenocytes of uninfected mice (UI) and infected WT and Cd36−/− mice at the indicated time points of infection were analyzed as outlined in Fig. 5 for NK cells, and the numbers of IFN-γ-, Tbet-, IL-10-, IL-4-, and GATA3-expressing cells were plotted. The data are a representative of three independent experiments. Error bars indicate S.D. of mice in each group. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, statistically not significant. d, days; FSC, forward scatter; SSC, side scatter.
At 7 and 14 days pi, however, the numbers of IFN-γ- and Tbet-expressing cells were comparable between WT and Cd36−/− mice (Fig. 6, B and C). This was despite the increased Th2 responses observed in Cd36−/− mice as indicated by the increased numbers of IL-4- and GATA3-expressing cells at 7 days pi (Fig. 6, E and F). The numbers of IL-4-expressing CD4+ and CD8+ T cells were also moderately increased in Cd36−/− mice compared with WT mice at 14 days pi, although the differences were not statistically significant (Fig. 6E). Moreover, at 7 days pi, the numbers of IL-10-producing CD4+ and CD8+ T cells were higher in Cd36−/− mice (see Fig. 6D), but at 14 days pi, the numbers IL-10-producing CD4+ and CD8+ T cells were actually lower in Cd36−/− mice than in WT mice. This was likely due to increased parasitemia in Cd36−/− mice, driving Th1 response and suppressing IL-10 production. The observed similar levels of proinflammatory cytokine production by CD4+ and CD8+ T cells in WT and Cd36−/− mice at 7 and 14 days pi are also probably a result of continued increased parasitemia in Cd36−/− mice, rendering enhanced uptake of parasite components by DCs. This results in DCs driving sustained high levels of Th1 responses in Cd36−/− mice at later stages of infections even in the face of increasing Th2 responses.
CD36-mediated immune responses up-regulate phagocytic activity and phagocytic receptor expression
Phagocytic clearance of IRBCs and merozoites is thought be the main mechanism that controls malaria parasitemia (14–16). Thus, we postulated that CD36, which is a scavenger receptor, is involved in phagocytosis of parasites. Accordingly, we assessed IRBC uptake in vivo by injecting IRBCs stained with CFSE, a fluorescent cell-staining dye, into infected mice to measure phagocytosed IRBCs and analyzing CFSE-positive spleen Mφs and PMNs. At 5 and 8 days pi, the uptake of IRBCs by spleen Mφs and PMNs (as gated in Fig. 7A) in WT mice was substantially higher than that by Mφs and PMNs in Cd36−/− mice (Fig. 7B), demonstrating that CD36 plays a significant role in parasite clearance. In addition to CD36, receptors such as complement receptors 1 and 2 (CR1/CR2) and Fc receptors (FcRs) also mediate phagocytic clearance of parasites. At 5 and 8 days pi, the expression of CR1/CR2, FcγII/IIIR, and Fcα/μR by spleen Mφs and PMNs in WT mice was substantially higher than that by spleen Mφs and PMNs in Cd36−/− mice (Fig. 7C). In contrast to 5 and 8 days pi, at 14 days pi, the uptake of IRBCs by Mφs and PNNs of WT mice was comparable with that of the respective cells of Cd36−/− mice (Fig. 7B). Notably, at 14 days pi, the capacity of Mφs and PMNs of both WT and Cd36−/− mice to take up IRBCs was substantially decreased compared with the phagocytic capacity of cells at 5 and 8 days pi. Consistent with these results, at 14 days pi, the expression levels of CR1/CR2, FcγII/IIIR, and Fcα/μR by spleen Mφs and PMNs in WT and Cd36−/− mice were not only comparable but also substantially decreased compared with levels at 5 and 8 days pi (Fig. 7C). The observed reduced phagocytic capacity of Mφs and PMNs at 14 days pi explains the exponential increase in parasitemia in both WT and Cd36−/− mice after 12–14 days infection (see Fig. 1B). It appears that the CD36-dependent increased control of parasitemia by WT mice during the early stages of infection allowed a sustained higher rate of parasite clearance in WT mice compared to Cd36−/− mice, although other mechanisms may also be involved. The reason for the observed reduced phagocytosis activity and markedly decreased phagocytic receptor expression at later stages of infection in this study is unclear. Substantial further studies are needed to understand the phenomena.
Figure 7.

CD36 up-regulates phagocytic receptor expression and phagocytosis of malaria parasites. P. yoelii IRBCs isolated from the blood of infected WT mice were labeled with CFSE and injected into P. yoelii-infected WT and Cd36−/−mice (n = 3–4/group) at 4, 7, or 13 days pi. After 18 h at each time point, splenocytes from uninfected (UI) mice and infected WT and Cd36−/− mice were prepared and stained with antibodies against surface markers for Mφs and PMNs and the indicated phagocytic receptors. The cells were analyzed by flow cytometry. A, shown is the gating for CD11b+F4/80+ Mφs and CD11bhiLy6Ghi PMNs. B and C, shown are the numbers of CFSE+ IRBCs internalized by spleen Mφs and PMNs (B) and the numbers of the gated receptor-expressing spleen Mφs and PMNs (C). Error bars indicate S.D. of individual mice in each group. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, statistically not significant. d, days; FSC, forward scatter; SSC, side scatter.
Proinflammatory cytokines, especially IFN-γ, directly activate phagocytic cells and increase phagocytic activity (56, 57). Therefore, we tested the phagocytic uptake of P. yoelii IRBCs by untreated and IFN-γ-primed BMDMs derived from WT and Cd36−/− mice. In agreement with the in vivo data that CD36 contributes to IRBC uptake by Mφs and PMNs (see Fig. 7B), the phagocytosis of IRBCs by WT BMDMs was significantly higher than that by Cd36−/− BMDMs (Fig. 8A). The uptake of IRBCs by WT BMDMs treated with IFN-γ was also significantly higher than that by similarly treated Cd36−/− BMDMs and that by untreated WT BMDMs. Treatment with IFN-γ significantly increased the phagocytosis of IRBCs by Cd36−/− BMDMs compared with untreated Cd36−/− BMDMs (Fig. 8A). These results indicated that IFN-γ increases the phagocytic activity of both WT and Cd36−/− Mφs.
Figure 8.

IFN-γ up-regulates phagocytosis of IRBCs and phagocytic receptor expression. BMDMs (5 × 105 cells/well) in 24-well plates were treated with 20 units/ml IFN-γ at 37 °C for 24 h and incubated with CFSE-stained IRBCs (1 × 104 cell/well) for 1 h. The cells were stained with dye-conjugated antibodies against mouse CD11b, CR1/CR2, and Fcα/μR and analyzed by flow cytometry. A--C, shown are percent CFSE+ (A), CR1/CR2+ (B), and Fcα/μR+ (C) BMDMs. Error bars indicate S.D. of quadruplicate cultures. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To determine whether the observed IFN-γ-dependent enhanced phagocytosis is due to increased phagocytic receptor expression, we analyzed the expression of CR1/CR2 and Fcα/μR by IFN-γ-treated BMDMs. IFN-γ treatment significantly increased the expression of CR1/CR2 and Fcα/μR in both WT and Cd36−/− BMDMs (Fig. 8, B and C). However, in both untreated and IFN-γ-treated BMDMs, the expression of phagocytic receptors by WT cells was significantly higher than that by Cd36−/− cells. Together, these results demonstrated that, in addition to directly contributing to parasite clearance, CD36 indirectly contributes through the up-regulation of other phagocytic receptor expression as well as by the up-regulation of cytokines, such as IFN-γ, that promote phagocytic receptor expression and thus the phagocytosis of parasites.
CD36 has no role in humoral responses to malaria
Based on the observation that CD36 modulates immune responses to malaria mainly at early stages of infection, we envisaged that there would be a differential IgM response by Cd36−/− mice compared with WT mice. However, the serum IgM levels at 5 and 8 days pi were similar in Cd36−/− and WT mice (Fig. 9A). At early stages of infection, IgMs are produced mainly by B1 cells, which are considered to be innate immune cells, and are rapidly activated upon sensing pathogen (58). There are two B1 subpopulations, B1a and B1b, which respectively produce natural IgM and parasite antigen-specific IgM (59, 60). B1 cells (gated as shown in Fig. 9B) were significantly expanded in response to malaria infection (Fig. 9C). Furthermore, consistent with the observed similar IgM levels (see Fig. 9A), there were no differences in the numbers of either B1a (CD5+CD19hiCD23−) cells or B1b (CD5−CD19hiCD23−) cells between infected Cd36−/− mice and WT mice (Fig. 9C).
Figure 9.

CD36 has no role in humoral immunity. A and B, mice were infected with P. yoelii and sacrificed at 5 or 8 days pi, and sera from the total blood were prepared. A, the parasite-specific serum IgM levels of mice (n = 3 or 4 mice/group) were analyzed by ELISA. Sera from uninfected WT mice were used as a control. Data are a representative of three independent experiments. ns, statistically not significant. B and C, flow cytometry analysis of splenic B1a and B1b B cells of mice at 5 days pi. B1 cells from spleens of uninfected WT mice were analyzed as a control. Shown are the gating strategy for gating B1a and B1b cells (B) and splenic B1a and B1b cell numbers in infected mice and uninfected control mice (C). D and E, antibody titer (D) and Ig isotypes (E) in sera prepared from P. yoelii-infected WT and Cd36−/− mice (n = 4 or 5/group) at 26 days pi were analyzed by ELISA. Sera of uninfected mice were analyzed as controls. C–E, the data are a representative of two independent experiments. In A and C–E, error bars indicate S.D. of mice in each group. d, days; FSC, forward scatter; SSC, side scatter; FMO, fluorescence minus one.
Early immune responses to pathogenic infections, especially activation of DCs and cytokine responses by DCs and other innate immune cells, influence Th responses, which in turn contribute to B cell function (61). To determine the contribution of CD36 to malaria-induced antibody responses at later stages of infection, we analyzed serum antibody levels and Ig isotypes at 26 days pi. The IgG titer and Ig isotypes as well as IgM levels in the sera of Cd36−/− mice were similar to those in WT mice (Fig. 9, D and E). Thus, the above results suggested that CD36 has either a negligible or no role in humoral responses to malaria infection.
Discussion
Although CD36 plays a role in organ-related severe malaria through endothelial adherence-mediated sequestration of IRBCs (31–34), CD36 also substantially contributes to uncomplicated malaria and resistance to mortality (25, 62, 63). In P. falciparum infection, the majority of IRBCs exhibit CD36-binding specificity (64). Because high parasite burden is a risk factor for severe malaria, CD36-dependent phagocytic clearance of parasites and thus control of parasitemia are significantly protective. In addition to direct involvement in parasite clearance, CD36 also contributes to phagocytosis by other receptors by up-regulating their expression (see Fig. 7). The present study demonstrates that CD36 regulates immune responses to blood stage malaria and explains how CD36-regulated immune responses contribute to parasitemia control and protection against susceptibility to malaria under parasite-non-sequestering conditions.
The results presented here show that CD36 contributes to malaria-induced immune responses primarily at early stages of infection and minimally, at best, at later stages of infection. This conclusion is evident from the results that, at early stages of infection, serum levels of proinflammatory cytokines were substantially higher in infected WT mice than in infected Cd36−/− mice, whereas at later stages cytokine levels in WT mice were comparable with those in Cd36−/− mice. Also at early stages of infection, the serum levels of the anti-inflammatory cytokine IL-10 and the Th2 cytokine IL-4 were significantly lower in WT mice than in Cd36−/− mice. However, at later stages of infection, these cytokines were present at similar levels in WT and Cd36−/− mice. The contribution of CD36 to malaria-induced cytokine responses was also evident at the cellular level. At early stages of infection, DCs and NK and T cells in infected WT mice produced higher levels of proinflammatory and lower levels of anti-inflammatory cytokine responses than those in infected Cd36−/− mice. At later stages of infection, there were no differences in cytokine responses by DCs and NK and T cells in infected WT and Cd36−/− mice. The observed CD36-dependent immune responses mainly at early stages of infection are due to the fact that CD36 is expressed by the early responding innate phagocytic cells but not by the later responding adaptive immune cells (18, 19). Thus, the CD36-dependent immune responses to malaria are mainly associated with the innate immune system.
The CD36-dependent cytokine responses observed at early stages of malaria infection are collectively a result of CD36 directly regulating the function of DCs and indirectly regulating the function of NK and T cells through DC-NK cell and DC-T cell cross-talk. Several lines of evidence support this conclusion. First, CD36 is expressed by DCs but not by NK cells (65). Although effector and regulatory CD8 T cells have been reported to express CD36 (66, 67), it seems unlikely that T cells interact with IRBCs through CD36 and that CD36 directly alters responses of T cells to reciprocally regulate DC responses. Second, co-cultures of OT-II T cells with DCs from WT mice at early stages of infection produced higher levels of TNF-α and IFN-γ and lower levels of IL-10 than co-cultures of OT-II cells with DCs from infected Cd36−/− mice, reflecting the CD36-regulated functions of DCs. Third, a previous in vitro study by us indicated that NK cells from uninfected mice co-cultured with WT DCs stimulated with P. falciparum IRBCs produced about 2-fold higher levels of IFN-γ than NK cells co-cultured with CD36-deficient DCs stimulated with IRBCs (38). Because DCs produce little or no IFN-γ, the increased production of IFN-γ by the co-cultures was from NK and T cells. Given that NK cells do not express CD36, and it is unlikely that CD36 is involved in parasite-NK cell and parasite-T cell interaction, it is evident that CD36 in DCs indirectly regulates the functions of NK and T cells. In the present study, the increased expression of TNF-α, IFN-γ, and Tbet and reduced expression of IL-10 by NK cells from infected WT mice compared with Cd36−/− mice reflected the CD36-dependent function of WT DCs, specifically CD36-dependent up-regulation of proinflammatory and down-regulation of anti-inflammatory cytokine responses by DCs. The lower proinflammatory cytokine responses and higher anti-inflammatory cytokine responses by NK cells and lower Th1 and higher Th2 responses in Cd36−/− mice are also consistent with the CD36-dependent function of DCs. Also consistent with these results were the increased expression of TNF-α, IFN-γ, and Tbet and reduced expression of IL-10, IL-4, and GATA3 in T cells from infected WT mice compared with Cd36−/− mice. Considering that DCs shape NK and T cell immune responses to infections, including malaria (48, 49, 68), collectively our data showed that CD36 directly regulates malaria parasite-induced immune responses by DCs and that CD36-dependent modulation of DC functions confers to NK and T cells the ability to produce CD36-dependent immune responses.
An important question that arises from the present study is why are immune responses to malaria independent of CD36 at later stages of infection as shown by similar proinflammatory cytokine responses by NK and T cells and comparable serum inflammatory cytokine levels in WT and CD36-deficient mice by 10 days pi? We have previously reported that CD36-dependent immune responses are parasite dose-dependent and that the role of CD36 becomes minimal at the threshold parasite load required for maximal responses (38). This is because phagocytic cells express several phagocytic receptors besides CD36, such as other scavenger receptors and Fc and complement receptors. As infection progresses and parasitemia increases, phagocytosis of parasites by DCs through receptors other than CD36 also becomes prominent, generating saturated levels of immune responses. Under such conditions, it is likely that DCs continue to produce high levels of proinflammatory responses despite the absence of CD36. Thus, at later stages of infection, when parasitemia is relatively high, the uptake of parasites by CD36-deficient DCs reaches minimum threshold levels required for maximum responses, and thus the cells produce sustained high levels of proinflammatory cytokine responses despite producing increased levels of anti-inflammatory cytokines. Eventually, the proinflammatory responses by DCs and those by activated NK and T cells due to DC function-specific immune responses become similar in WT and Cd36−/− mice. Alternatively, it is possible that myeloid cells newly released into blood from bone marrow in response to increased parasitemia produce proinflammatory cytokines and induce type 1 responses, whereas those that have been already exposed to parasites produce anti-inflammatory cytokines and induce type 2 responses. This may result in overall comparable levels of pro-/anti-inflammatory and Th1/Th2 responses at later stages.
The results of this study also indicate that the CD36-dependent up-regulated production of IFN-γ by NK cells and Th1 development contribute to resistance against malaria mortality by controlling parasitemia through phagocytic clearance of parasites. IFN-γ is an important cytokine that is known to activate Mφs and PMNs, enhancing phagocytic activity (56, 57). IFN-γ also promotes Th1 development, which leads to further IFN-γ production by Th1 cells. Additionally, IL-12 and IL-18 promote IFN-γ production by NK and T cells. The IFN-γ produced by NK and T cells in turn enhances DC maturation, which leads to increased IL-12 production. The enhanced production of IFN-γ through this cellular response loop, imprinted through CD36-regulated DC function, contributes to an efficient priming of Mφs, leading to control of parasitemia and resistance to malaria. These conclusions are consistent with the results of previous studies that showed that IL-12 and IFN-γ play protective roles in malaria as indicated by the fact that mice deficient in these cytokines had increased susceptibility to malaria. It has been shown that cross-talk between DCs and NK cells and that between DCs and T cells contribute to IL-12- and IFN-γ-induced adaptive immunity (49, 68). At early stages of malaria infection, IL-12 is produced mainly by DCs and red pulp Mφs (52, 53). IL-12-induced IFN-γ production by NK cells results in enhanced DC maturation and enhanced production of IL-12, which promotes IFN-γ secretion by T cells. Together, these responses contribute to protection against malaria severity. Consistent with these results, in the present study, IFN-γ-treated Mφs showed increased phagocytic receptor expression and enhanced phagocytic activity (see Fig. 8). These results and our observation that CD36-deficiency results in reduced IL-12 production by DCs, decreased IFN-γ responses by NK and T cells at early stages of infection, and early switching from a Th1 to a Th2 are consistent with the notion that CD36 contributes to parasitemia control and resistance to death. Furthermore, it has been shown that IL-10 suppresses the phagocytic activity of Mφs and PMNs (69, 70). In agreement with these observations, we found that CD36 deficiency causes a substantial increase in IL-10 production. Thus, overall our results demonstrated that CD36 plays an important role in protective immunity to malaria.
Our findings have important implications for P. falciparum infection. Because the majority of parasites in infected people express CD36-binding PfEMP1, it is likely that CD36 play a significant role in parasitemia control, although sequestration may tend to favor parasite growth to a certain degree (62). The dynamics between parasitemia control and parasite growth promotion may depend on host factors, such as immune status and receptor polymorphisms. When parasites are sequestered in organs, increased proinflammatory responses locally due to increased parasite biomass may contribute to severe pathology and organ dysfunction. In contrast, the CD36-dependent enhancement of immune responses and its contribution to increased parasite clearance are likely to lower the risk of severe disease. Thus, the clinical outcomes to malaria infection depend on a balance between these dual roles of CD36. It appears that, in sequestering parasite infection, CD36 more often promotes pathology (36, 37, 39, 40, 71), although in some situations it can be protective (41). In parasite-non-sequestering situations, however, as we observed here in non-lethal mouse malaria, CD36-dependent interaction is likely to be beneficial. Further detailed studies on CD36-mediated signaling events in immune responses to malaria will likely enhance our knowledge of the role of CD36 in malaria immunity and pathogenesis.
Experimental procedures
Reagents
Non-essential amino acids for culture media, and Percoll® were from Sigma-Aldrich. DMEM, RPMI 1640 medium, and penicillin/streptomycin solution were from Gibco Life Technologies. Fetal bovine serum was from Atlanta Biologicals (Lawrenceville, GA). Cell TraceTM CFSE cell staining kit was from Molecular Probes, Inc. (Eugene, OR). Collagenase D was from Roche Applied Science. Chicken ovalbumin peptide, OVA(323–339), was from InvivoGen (San Diego, CA). GolgiPlug was from BD Biosciences. Mouse IFN-γ was from PeproTech (Rocky Hill, NJ). ELISA kits for analyzing mouse TNF-α, IL-12p40, IL-18, IL-1β, IL-10, IL-4, and IFN-γ were from R&D Systems (Minneapolis, MN). Mouse Ig isotyping kit was from BD Biosciences. Mouse CD11c-conjugated microbeads for DC isolation and CD90.2-conjugated microbeads for T cell isolation and magnetic columns for cell separation were from Miltenyi Biotec Inc. (Auburn, CA).
Antibodies for flow cytometry analysis
The following anti-mouse antibody conjugates were used for flow cytometry: PE- and APC-anti-CD11c (418N), Brilliant Violet (BV)421-anti-I-A/I-E (M5/114.15.2), BV421-anti-CD49b (DX5), PE-Cy5-anti-CD80 (16-10A1), PE-Cy7-anti-IL-12 (C17.8), APC-eFluor 780-anti-CD5 (clone 53-7.3), APC-eFluor 780-anti-F4/80 (BM8), PE- and PerCP-Cy5.5-anti-CD11b (M1/70), PerCP-Cy5.5-anti-CD16/32 (clone 93), and PerCP-Cy5.5-anti-Tbet (eBio4B10) were from e-Biosciences (San Diego, CA). Anti-CD16/32 (2.4G2), APC-Cy7-anti-CD3ϵ (145-2C11), Alexa Fluor 700-anti-CD4 (RM4–5), BV605-anti-CD8α (53–6.7), BV510-anti-CD86 (GL1), PE-anti-NK1.1 (PK136), APC- and PerCP-Cy5.5-anti-TCRβ (H57-597), PE-Cy7- and PerCP-Cy5.5-anti-IL-4 (11B11), PE-anti-IL-10 (JES5-16E3), APC-anti-IFN-γ (XMG1.2), PE- and PE-CF594-anti-CD19 (1D3), BV510-anti-CD21/35 (7G6), Alexa Fluor 700-anti-Ly6G (1A8), FITC-anti-CD23 (B3B4), and FITC-anti-TNF-α (MP6-XT22) antibodies were from BD Biosciences. PE-anti-Fcα/μR (TX61) and Alexa Fluor 488-conjugated anti-GATA3 antibody (16E10A23) were from BioLegend (San Diego, CA).
Mice and parasite infection
WT mice and OT-II transgenic mice expressing TCR for OVA(323–339) peptide on CD4 T cells were from The Jackson Laboratory (Bar Harbor, ME). Cd36−/− mice were generated in the laboratory of Dr. Maria Febbraio. All mice were in a C57BL/6J background. Mice were housed in a pathogen-free environment and bred in house, and 10–12-week-old mice were used for the study. To measure DC-induced T cell responses in co-cultures, spleen DCs from infected mice and T cells from uninfected OT-II transgenic mice were used. Non-lethal P. yoelii 17XNL strain (Bei Research Resources-ATCC, Manassas, VA) was passaged through donor mice by i.p. administration of parasites. For experimental infections, blood from donor mice containing 1 × 106 IRBCs suspended in 100 μl of saline was administered i.p., and survival was monitored daily. Parasitemia was assessed by light microscopic examination of Giemsa-stained thin smears of blood.
Ethics statement
The Animal Care and Use Committee of the Pennsylvania State University College of Medicine, Hershey, PA approved the protocol (Number 46661) for animal experiments.
Analysis of spleen cell immune responses
Single-cell suspensions of spleens from infected and uninfected mice (control) were prepared as described previously (72). The cells were suspended in incomplete DMEM, stained with antibodies against cell surface marker proteins and the desired intracellular proteins, and analyzed by flow cytometry.
Isolation of spleen DCs and T cells
DCs from spleen cell suspensions were isolated by magnetic cell sorting using anti-mouse CD11c-conjugated microbeads as reported previously (72). OT-II T cells from the spleen cell suspensions were isolated using anti-mouse CD90.2 antibody-conjugated magnetic beads.
Isolation of P. yoelii IRBCs
Blood from the P. yoelii-infected mice was diluted with 2 volumes of PBS, pH 7.4, and centrifuged on Isolymph (CTL Scientific Supply, Deer Park, NY) at 1200 × g for 15 min. The cell pellets were suspended in 2 volumes of PBS and centrifuged on 70% Percoll cushions at 1200 × g for 15 min. The IRBCs on the top of Percoll cushions were collected and washed with PBS.
Preparation of bone marrow-derived macrophages (BMDMs)
Mouse BMDMs were generated by culturing bone marrow cells in DMEM containing 20% FBS, 1 mm sodium pyruvate, 50 μm 2-mercaptoethanol, 1% non-essential amino acids, 10 units/ml penicillin/streptomycin (complete DMEM), and 30% L929 cell culture-conditioned medium. After 7 days, BMDMs were harvested by scraping, suspended in complete DMEM, and used for experimental purposes (52, 72).
Analysis of phagocytic receptor expression and IRBC phagocytosis
P. yoelii IRBCs were prepared as described previously (62). The purified IRBCs (3 × 107 cells/ml) were suspended in PBS and stained with 2 μm CFSE (for in vivo phagocytosis analysis) or 0.5 μm CFSE (for in vitro phagocytosis assay) at room temperature. After 10 min, 200 μl of fetal bovine serum were added and incubated for 5 min, and cells were washed three times with PBS. The CFSE-labeled IRBCs (∼3 × 107) were injected intravenously into mice at 4, 7, or 13 days pi. After overnight, spleen cells were prepared and stained with antibodies against cell surface marker proteins of PMNs and Mφs with dye-conjugated antibodies against mouse CD11b, F4/80, Ly6G, and CR1/CR2, FcγII/IIIR, and Fcα/μR. The CFSE+ and phagocytic receptor-positive cells were analyzed by flow cytometry.
For the analysis of IRBC phagocytosis in vitro, BMDMs in ultralow-binding 24-well plates were incubated with CFSE-labeled P. yoelii IRBCs at Mφ to IRBC ratios of 50:1 at 37 °C for 1 h. The cells were stained with dye-conjugated anti-CD11b antibody, and CFSE+ cells were analyzed by flow cytometry.
Analysis of phagocytic receptor expression and IRBC phagocytosis in IFN-γ-primed Mφs
BMDMs (5 × 105 cells/well) in ultralow-binding 24-well plates were cultured overnight and treated with 20 units/ml IFN-γ at 37 °C for 24 h. To each well was added CFSE-labeled P. yoelii IRBCs (1 × 104 cells/well) at Mφ to IRBC ratios of 50:1. After 1 h, the plates were chilled on ice for 15 min, and the cells were stained with antibodies against CD11b, CR1/CR2, and Fcα/μR at 4 °C for 15 min. The cells were analyzed by flow cytometry.
DC and T cell co-culturing
In 96-well U-bottomed plates, spleen DCs (105 cells/well) from P. yoelii-infected WT or Cd36−/− mice at 5 and 10 days pi and OT-II T cells (0.5 × 105/well) isolated from uninfected mice were cultured in the presence or absence of 2 μg/ml OVA(323–339) peptide for 36 h. Cytokines in culture supernatants were analyzed by ELISA.
Flow cytometry
Total spleen cells from uninfected or infected mice were treated with Fc Block (anti-mouse CD16/32 antibody), except in the case of FcγII/IIIR analysis, for 10 min; stained with antibodies against cell surface marker proteins at 4 °C for 20 min; and analyzed by flow cytometry. For analysis of intracellular proteins, cells were first surface-stained for marker proteins, fixed with 2% paraformaldehyde for 30 min at room temperature, and then stained with antibodies against the specific proteins in PBS containing 0.5% saponin and 1% BSA at room temperature for 30 min. For cytokine analysis, spleen cells (1 × 107 cells/well) in 24-well plates were cultured for 4–6 h in the presence of GolgiPlug and then treated with Fc Block. The cells were stained with cell surface marker proteins, fixed with 2% paraformaldehyde, stained with antibodies against cytokines, and analyzed by flow cytometry using a BD LSRII flow cytometer. The data were analyzed using FlowJo software.
Analysis of serum antibodies by ELISA
The serum IgG titer was analyzed by ELISA as described previously (72). Ig isotypes and IgM were analyzed by using a mouse immunoglobulin isotyping ELISA kit according to the manufacturer's instructions as reported previously (72). Briefly, 96-well microtiter plates were coated with total P. yoelii-IRBC proteins (30 μg of protein/ml) in PBS, pH 7.2; incubated overnight at 4 °C; and blocked with 1% BSA in PBS. The plates were incubated with 1:5000-diluted mouse sera at room temperature for 1 h, washed, and treated with 1:5-diluted isotype-specific rat anti-mouse antibodies for 1 h at room temperature. After washing, the bound Igs were measured using 1:3000-diluted HRP-conjugated goat anti-rat antibodies.
Statistical analysis
Statistical analyses of data were performed using GraphPad Prism version 6.01. Mouse survival data were analyzed by log-rank (Mantel-Cox) test. The data from all other experiments were analyzed by one-way analysis of variance followed by Newman-Keuls test (Figs. 3, B–E, and 5–9) or by unpaired two-tailed t test (Figs. 1B, 2, 3F, and 4). The results are presented as the mean ± S.D. p values <0.05 were considered statistically significant.
Author contributions
D. C. G., X. W., and N. M. G. conceived the study and designed the experiments. D. C. G. coordinated the study. R. P. T., N. M. G., X. W., K. P., and S. E. N. performed the experiments. X. W., D. C. G., R. P. T., and N. M. G. analyzed the data. M. F. provided CD36 knock-out mice. D. C. G., X. W., R. P. T., N. M. G., and M. F. wrote the paper.
Acknowledgments
The following reagent was obtained through BEI Resources, NIAID, and National Institutes of Health as part of the Human Microbiome Project: P. yoelii 17XNL strain.
This work was supported in part by NIAID, National Institutes of Health Grant AI 41139. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- DC
- dendritic cell
- IRBC
- infected red blood cell
- PfEMP1
- P. falciparum erythrocyte membrane protein-1
- Mφ
- macrophage
- PMN
- polymorphonuclear neutrophil
- BMDM
- M-CSF-differentiated mouse bone marrow cell
- CR1/CR2
- complement receptors 1 and 2
- pi
- postinfection
- CFSE
- carboxyfluorescein succinimidyl ester
- OVA
- ovalbumin
- NK
- natural killer
- TCR
- T cell receptor
- OT-II T cell
- OVA-specific, MHC class II-restricted α/β T cell
- FcR
- Fc receptor
- Th
- T helper cell
- Tbet
- T-box transcription factor encoded by Tbx21
- PE
- phycoerythrin
- APC
- allophycocyanin
- BV
- Brilliant Violet
- PerCP
- peridinin-chlorophyll protein complex.
References
- 1. World Health Organization (2015) World Malaria Report 2015, World Health Organization, Geneva [Google Scholar]
- 2. Murray C. J., Rosenfeld L. C., Lim S. S., Andrews K. G., Foreman K. J., Haring D., Fullman N., Naghavi M., Lozano R., and Lopez A. D. (2012) Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 379, 413–431 [DOI] [PubMed] [Google Scholar]
- 3. Battle K. E., Guerra C. A., Golding N., Duda K. A., Cameron E., Howes R. E., Elyazar I. R., Baird J. K., Reiner R. C. Jr., Gething P. W., Smith D. L., and Hay S. I. (2015) Global database of matched Plasmodium falciparum and P. vivax incidence and prevalence records from 1985–2013. Sci. Data 2, 150012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Howes R. E., Battle K. E., Mendis K. N., Smith D. L., Cibulskis R. E., Baird J. K., and Hay S. I. (2016) Global epidemiology of Plasmodium vivax. Am. J. Trop. Med. Hyg. 95, (suppl.) 15–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baird J. K. (2009) Resistance to therapies for infection by Plasmodium vivax. Clin. Microbiol. Rev. 22, 508–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ashley E. A., Dhorda M., Fairhurst R. M., Amaratunga C., Lim P., Suon S., Sreng S., Anderson J. M., Mao S., Sam B., Sopha C., Chuor C. M., Nguon C., Sovannaroth S., Pukrittayakamee S., et al. (2014) Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 371, 411–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Faway E., Musset L., Pelleau S., Volney B., Casteras J., Caro V., Menard D., Briolant S., and Legrand E. (2016) Plasmodium vivax multidrug resistance-1 gene polymorphism in French Guiana. Malar. J. 15, 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stanisic D. I., and Good M. F. (2015) Whole organism blood stage vaccines against malaria. Vaccine 33, 7469–7475 [DOI] [PubMed] [Google Scholar]
- 9. Neafsey D. E., Juraska M., Bedford T., Benkeser D., Valim C., Griggs A., Lievens M., Abdulla S., Adjei S., Agbenyega T., Agnandji S. T., Aide P., Anderson S., Ansong D., Aponte J. J., et al. (2015) Genetic diversity and protective efficacy of the RTS,S/AS01 malaria vaccine. N. Engl. J. Med. 373, 2025–2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stevenson M. M., and Riley E. M. (2004) Innate immunity to malaria. Nat. Rev. Immunol. 4, 169–180 [DOI] [PubMed] [Google Scholar]
- 11. Augustine A. D., Hall B. F., Leitner W. W., Mo A. X., Wali T. M., and Fauci A. S. (2009) NIAID workshop on immunity to malaria: addressing immunological challenges. Nat. Immunol. 10, 673–678 [DOI] [PubMed] [Google Scholar]
- 12. Schofield L., and Grau G. E. (2005) Immunological processes in malaria pathogenesis. Nat. Rev. Immunol. 5, 722–735 [DOI] [PubMed] [Google Scholar]
- 13. Gazzinelli R. T., Kalantari P., Fitzgerald K. A., and Golenbock D. T. (2014) Innate sensing of malaria parasites. Nat. Rev. Immunol. 14, 744–757 [DOI] [PubMed] [Google Scholar]
- 14. Smith T. G., Serghides L., Patel S. N., Febbraio M., Silverstein R. L., and Kain K. C. (2003) CD36-mediated nonopsonic phagocytosis of erythrocytes infected with stage I and IIA gametocytes of Plasmodium falciparum. Infect. Immun. 71, 393–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Patel S. N., Serghides L., Smith T. G., Febbraio M., Silverstein R. L., Kurtz T. W., Pravenec M., and Kain K. C. (2004) CD36 mediates the phagocytosis of Plasmodium falciparum-infected erythrocytes by rodent macrophages. J. Infect. Dis. 189, 204–213 [DOI] [PubMed] [Google Scholar]
- 16. Cabrera A., Neculai D., and Kain K. C. (2014) CD36 and malaria: friends or foes? A decade of data provides some answers. Trends Parasitol. 30, 436–444 [DOI] [PubMed] [Google Scholar]
- 17. Mandell G. L. (1995) Cytokines, phagocytes, and pentoxifylline. J. Cardiovasc. Pharmacol. 25, Suppl. 2, S20–S22 [DOI] [PubMed] [Google Scholar]
- 18. Febbraio M., Hajjar D. P., and Silverstein R. L. (2001) CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J. Clin. Investig. 108, 785–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Silverstein R. L., and Febbraio M. (2009) CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal. 2, re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stewart C. R., Stuart L. M., Wilkinson K., van Gils J. M., Deng J., Halle A., Rayner K. J., Boyer L., Zhong R., Frazier W. A., Lacy-Hulbert A., El Khoury J., Golenbock D. T., and Moore K. J. (2010) CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 11, 155–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pasloske B. L., and Howard R. J. (1994) Malaria, the red cell, and the endothelium. Annu. Rev. Med. 45, 283–295 [DOI] [PubMed] [Google Scholar]
- 22. Day K. P., Hayward R. E., Smith D., and Culvenor J. G. (1998) CD36-dependent adhesion and knob expression of the transmission stages of Plasmodium falciparum is stage specific. Mol. Biochem. Parasitol. 93, 167–177 [DOI] [PubMed] [Google Scholar]
- 23. Weatherall D. J., Miller L. H., Baruch D. I., Marsh K., Doumbo O. K., Casals-Pascual C., and Roberts D. J. (2002) Malaria and the red cell. Hematology Am. Soc. Hematol. Educ. Program 2002, 35–57 [DOI] [PubMed] [Google Scholar]
- 24. Hoebe K., Georgel P., Rutschmann S., Du X., Mudd S., Crozat K., Sovath S., Shamel L., Hartung T., Zähringer U., and Beutler B. (2005) CD36 is a sensor of diacylglycerides. Nature 433, 523–527 [DOI] [PubMed] [Google Scholar]
- 25. Patel S. N., Lu Z., Ayi K., Serghides L., Gowda D. C., and Kain K. C. (2007) Disruption of CD36 impairs cytokine response to Plasmodium falciparum glycosylphosphatidylinositol and confers susceptibility to severe and fatal malaria in vivo. J. Immunol. 178, 3954–3961 [DOI] [PubMed] [Google Scholar]
- 26. Erdman L. K., Cosio G., Helmers A. J., Gowda D. C., Grinstein S., and Kain K. C. (2009) CD36 and TLR interactions in inflammation and phagocytosis: implications for malaria. J. Immunol. 183, 6452–6459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Urban B. C., Willcox N., and Roberts D. J. (2001) A role for CD36 in the regulation of dendritic cell function. Proc. Natl. Acad. Sci. U.S.A. 98, 8750–8755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Baruch D. I., Gormely J. A., Ma C., Howard R. J., and Pasloske B. L. (1996) Plasmodium falciparum erythrocyte membrane protein 1 is a parasitized erythrocyte receptor for adherence to CD36, thrombospondin, and intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. U.S.A. 93, 3497–3502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hsieh F. L., Turner L., Bolla J. R., Robinson C. V., Lavstsen T., and Higgins M. K. (2016) The structural basis for CD36 binding by the malaria parasite. Nat. Commun. 7, 12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Smith J. D., Chitnis C. E., Craig A. G., Roberts D. J., Hudson-Taylor D. E., Peterson D. S., Pinches R., Newbold C. I., and Miller L. H. (1995) Switches in expression of Plasmodium falciparum var genes correlate with changes in antigenic and cytoadherent phenotypes of infected erythrocytes. Cell 82, 101–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ochola L. B., Siddondo B. R., Ocholla H., Nkya S., Kimani E. N., Williams T. N., Makale J. O., Liljander A., Urban B. C., Bull P. C., Szestak T., Marsh K., and Craig A. G. (2011) Specific receptor usage in Plasmodium falciparum cytoadherence is associated with disease outcome. PLoS One 6, e14741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bernabeu M., Danziger S. A., Avril M., Vaz M., Babar P. H., Brazier A. J., Herricks T., Maki J. N., Pereira L., Mascarenhas A., Gomes E., Chery L., Aitchison J. D., Rathod P. K., and Smith J. D. (2016) Severe adult malaria is associated with specific PfEMP1 adhesion types and high parasite biomass. Proc. Natl. Acad. Sci. U.S.A. 113, E3270–E3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jespersen J. S., Wang C. W., Mkumbaye S. I., Minja D. T., Petersen B., Turner L., Petersen J. E., Lusingu J. P., Theander T. G., and Lavstsen T. (2016) Plasmodium falciparum var genes expressed in children with severe malaria encode CIDRα1 domains. EMBO Mol. Med. 8, 839–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Magistrado P. A., Staalsoe T., Theander T. G., Hviid L., and Jensen A. T. (2008) CD36 selection of 3D7 Plasmodium falciparum associated with severe childhood malaria results in reduced VAR4 expression. Malar. J. 7, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Franke-Fayard B., Janse C. J., Cunha-Rodrigues M., Ramesar J., Büscher P., Que I., Löwik C., Voshol P. J., den Boer M. A., van Duinen S. G., Febbraio M., Mota M. M., and Waters A. P. (2005) Murine malaria parasite sequestration: CD36 is the major receptor, but cerebral pathology is unlinked to sequestration. Proc. Natl. Acad. Sci. U.S.A. 102, 11468–11473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lagassé H. A., Anidi I. U., Craig J. M., Limjunyawong N., Poupore A. K., Mitzner W., and Scott A. L. (2016) Recruited monocytes modulate malaria-induced lung injury through CD36-mediated clearance of sequestered infected erythrocytes. J. Leukoc. Biol. 99, 659–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Brugat T. (2016) Editorial: CD36: Russian roulette of host and parasites during malaria infection. J. Leukoc. Biol. 99, 643–645 [DOI] [PubMed] [Google Scholar]
- 38. Gowda N. M., Wu X., Kumar S., Febbraio M., and Gowda D. C. (2013) CD36 contributes to malaria parasite-induced pro-inflammatory cytokine production and NK and T cell activation by dendritic cells. PLoS One 8, e77604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pain A., Urban B. C., Kai O., Casals-Pascual C., Shafi J., Marsh K., and Roberts D. J. (2001) A non-sense mutation in Cd36 gene is associated with protection from severe malaria. Lancet 357, 1502–1503 [DOI] [PubMed] [Google Scholar]
- 40. Omi K., Ohashi J., Patarapotikul J., Hananantachai H., Naka I., Looareesuwan S., and Tokunaga K. (2003) CD36 polymorphism is associated with protection from cerebral malaria. Am. J. Hum. Genet. 72, 364–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aitman T. J., Cooper L. D., Norsworthy P. J., Wahid F. N., Gray J. K., Curtis B. R., McKeigue P. M., Kwiatkowski D., Greenwood B. M., Snow R. W., Hill A. V., and Scott J. (2000) Malaria susceptibility and CD36 mutation. Nature 405, 1015–1016 [DOI] [PubMed] [Google Scholar]
- 42. Butler N. S., Moebius J., Pewe L. L., Traore B., Doumbo O. K., Tygrett L. T., Waldschmidt T. J., Crompton P. D., and Harty J. T. (2011) Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat. Immunol. 13, 188–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stegmann K. A., De Souza J. B., and Riley E. M. (2015) IL-18-induced expression of high-affinity IL-2R on murine NK cells is essential for NK-cell IFN-γ production during murine Plasmodium yoelii infection. Eur. J. Immunol. 45, 3431–3440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sebina I., James K. R., Soon M. S., Fogg L. G., Best S. E., Labastida Rivera F., Montes de Oca M., Amante F. H., Thomas B. S., Beattie L., Souza-Fonseca-Guimaraes F., Smyth M. J., Hertzog P. J., Hill G. R., Hutloff A., et al. (2016) IFNαR1-signalling obstructs ICOS-mediated humoral immunity during non-lethal blood-stage Plasmodium infection. PLoS Pathog. 12, e1005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Imai T., Shen J., Chou B., Duan X., Tu L., Tetsutani K., Moriya C., Ishida H., Hamano S., Shimokawa C., Hisaeda H., and Himeno K. (2010) Involvement of CD8+ T cells in protective immunity against murine blood-stage infection with Plasmodium yoelii 17XL strain. Eur. J. Immunol. 40, 1053–1061 [DOI] [PubMed] [Google Scholar]
- 46. Couper K. N., Blount D. G., Hafalla J. C., van Rooijen N., de Souza J. B., and Riley E. M. (2007) Macrophage-mediated but γ interferon-independent innate immune responses control the primary wave of Plasmodium yoelii parasitemia. Infect. Immun. 75, 5806–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kaul D. K., Nagel R. L., Llena J. F., and Shear H. L. (1994) Cerebral malaria in mice: demonstration of cytoadherence of infected red blood cells and microrheologic correlates. Am. J. Trop. Med. Hyg. 50, 512–521 [DOI] [PubMed] [Google Scholar]
- 48. Ing R., and Stevenson M. M. (2009) Dendritic cell and NK cell reciprocal cross talk promotes γ interferon-dependent immunity to blood-stage Plasmodium chabaudi AS infection in mice. Infect Immun. 77, 770–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Su Z., and Stevenson M. M. (2002) IL-12 is required for antibody-mediated protective immunity against blood-stage Plasmodium chabaudi AS malaria infection in mice. J. Immunol. 168, 1348–1355 [DOI] [PubMed] [Google Scholar]
- 50. Walsh K. P., and Mills K. H. (2013) Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol. 34, 521–530 [DOI] [PubMed] [Google Scholar]
- 51. Granucci F., Zanoni I., and Ricciardi-Castagnoli P. (2006) Natural killer (NK) cell functions can be strongly boosted by activated dendritic cells (DC). Eur. J. Immunol. 36, 2819–2820 [DOI] [PubMed] [Google Scholar]
- 52. Wu X., Gowda N. M., and Gowda D. C. (2015) Phagosomal acidification prevents macrophage inflammatory cytokine production to malaria, and dendritic cells are the major source at the early stages of infection: implication for malaria protective immunity development. J. Biol. Chem. 290, 23135–23147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim C. C., Nelson C. S., Wilson E. B., Hou B., DeFranco A. L., and DeRisi J. L. (2012) Splenic red pulp macrophages produce type I interferons as early sentinels of malaria infection but are dispensable for control. PLoS One 7, e48126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yu X., Cai B., Wang M., Tan P., Ding X., Wu J., Li J., Li Q., Liu P., Xing C., Wang H. Y., Su X. Z., and Wang R. F. (2016) Cross-regulation of two type I interferon signaling pathways in plasmacytoid dendritic cells controls anti-malaria immunity and host mortality. Immunity 45, 1093–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Spaulding E., Fooksman D., Moore J. M., Saidi A., Feintuch C. M., Reizis B., Chorro L., Daily J., and Lauvau G. (2016) STING-licensed macrophages prime type I IFN production by plasmacytoid dendritic cells in the bone marrow during severe Plasmodium yoelii malaria. PLoS Pathog. 12, e1005975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fernandez-Boyanapalli R., McPhillips K. A., Frasch S. C., Janssen W. J., Dinauer M. C., Riches D. W., Henson P. M., Byrne A., and Bratton D. L. (2010) Impaired phagocytosis of apoptotic cells by macrophages in chronic granulomatous disease is reversed by IFN-γ in a nitric oxide-dependent manner. J. Immunol. 185, 4030–4041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Marchi L. F., Sesti-Costa R., Ignacchiti M. D., Chedraoui-Silva S., and Mantovani B. (2014) In vitro activation of mouse neutrophils by recombinant human interferon-gamma: increased phagocytosis and release of reactive oxygen species and pro-inflammatory cytokines. Int. Immunopharmacol. 18, 228–235 [DOI] [PubMed] [Google Scholar]
- 58. Tangye S. G. (2013) To B1 or not to B1: that really is still the question! Blood 121, 5109–5110 [DOI] [PubMed] [Google Scholar]
- 59. Racine R., and Winslow G. M. (2009) IgM in microbial infections: taken for granted? Immunol. Lett. 125, 79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Baumgarth N. (2011) The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat. Rev. Immunol. 11, 34–46 [DOI] [PubMed] [Google Scholar]
- 61. Reinhardt R. L., Liang H. E., and Locksley R. M. (2009) Cytokine-secreting follicular T cells shape the antibody repertoire. Nat. Immunol. 10, 385–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fonager J., Pasini E. M., Braks J. A., Klop O., Ramesar J., Remarque E. J., Vroegrijk I. O., van Duinen S. G., Thomas A. W., Khan S. M., Mann M., Kocken C. H., Janse C. J., and Franke-Fayard B. M. (2012) Reduced CD36-dependent tissue sequestration of Plasmodium-infected erythrocytes is detrimental to malaria parasite growth in vivo. J. Exp. Med. 209, 93–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Rogerson S. J., Tembenu R., Dobaño C., Plitt S., Taylor T. E., and Molyneux M. E. (1999) Cytoadherence characteristics of Plasmodium falciparum-infected erythrocytes from Malawian children with severe and uncomplicated malaria. Am. J. Trop. Med. Hyg. 61, 467–472 [DOI] [PubMed] [Google Scholar]
- 64. Robinson B. A., Welch T. L., and Smith J. D. (2003) Widespread functional specialization of Plasmodium falciparum erythrocyte membrane protein 1 family members to bind CD36 analysed across a parasite genome. Mol. Microbiol. 47, 1265–1278 [DOI] [PubMed] [Google Scholar]
- 65. Chen Q., Ye W., Jian Tan W., Mei Yong K. S., Liu M., Qi Tan S., Loh E., Te Chang K., Chye Tan T., Preiser P. R., and Chen J. (2015) Delineation of natural killer cell differentiation from myeloid progenitors in human. Sci. Rep. 5, 15118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. O'Sullivan D., van der Windt G. J., Huang S. C., Curtis J. D., Chang C. H., Buck M. D., Qiu J., Smith A. M., Lam W. Y., DiPlato L. M., Hsu F. F., Birnbaum M. J., Pearce E. J., and Pearce E. L. (2014) Memory CD8+ T cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 41, 75–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Cipolletta D., Feuerer M., Li A., Kamei N., Lee J., Shoelson S. E., Benoist C., and Mathis D. (2012) PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486, 549–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Su Z., and Stevenson M. M. (2000) Central role of endogenous gamma interferon in protective immunity against blood-stage Plasmodium chabaudi AS infection. Infect. Immun. 68, 4399–4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chung Y., Zhang N., and Wooten R. M. (2013) Borrelia burgdorferi elicited-IL-10 suppresses the production of inflammatory mediators, phagocytosis, and expression of co-stimulatory receptors by murine macrophages and/or dendritic cells. PLoS One 8, e84980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Laichalk L. L., Danforth J. M., and Standiford T. J. (1996) Interleukin-10 inhibits neutrophil phagocytic and bactericidal activity. FEMS Immunol. Med. Microbiol. 15, 181–187 [DOI] [PubMed] [Google Scholar]
- 71. Lovegrove F. E., Gharib S. A., Peña-Castillo L., Patel S. N., Ruzinski J. T., Hughes T. R., Liles W. C., and Kain K. C. (2008) Parasite burden and CD36-mediated sequestration are determinants of acute lung injury in an experimental malaria model. PLoS Pathog. 4, e1000068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gowda N. M., Wu X., and Gowda D. C. (2012) TLR9 and MyD88 are crucial for the development of protective immunity to malaria. J. Immunol. 188, 5073–5085 [DOI] [PMC free article] [PubMed] [Google Scholar]