


Abstract
We examined whether the human nucleotide excision repair complex, which is specialized on the removal of bulky DNA adducts, also displays a correcting activity on base mismatches. The cytosine/cytosine (C/C) lesion was used as a model substrate to monitor the correction of base mismatches in human cells. Fibroblasts with different repair capabilities were transfected with shuttle vectors that contain a site-directed C/C mismatch in the replication origin, accompanied by an additional C/C mismatch in one of the flanking sequences that are not essential for replication. Analysis of the vector progeny obtained from these doubly modified substrates revealed that C/C mismatches were eliminated before DNA synthesis not only in the repair-proficient background, but also when the target cells carried a genetic defect in long-patch mismatch repair, in nucleotide excision repair, or when both pathways were deleted. Furthermore, cells deficient for long-patch mismatch repair as well as a cell line that combines mismatch and nucleotide excision repair defects were able to correct multiple C/C mispairs, placed at distances of 21–44 nt, in an independent manner, such that the removal of each lesion led to individual repair patches. These results support the existence of a concurrent short-patch mechanism that rectifies C/C mismatches.
INTRODUCTION
Spontaneous DNA replication errors arising in both prokaryotic and eukaryotic cells are mainly processed by the long-patch mismatch repair (MMR) system (1–4). This highly conserved excision repair pathway for the correction of base mismatches, nucleotide insertions or deletions is able to distinguish between the parental template DNA and the newly synthesized daughter strands through its coupling to the replication machinery (5). Excision repair is directed to the newly synthesized strand in order to prevent the conversion of replication errors to irreversible mutations. The human long-patch MMR complex recognizes base mismatches and insertion/deletion loops using the dimeric complexes of hMSH2-hMSH6 (hMutSα) or hMSH2-hMSH3 (hMutSβ), which are homologs of the bacterial MutS protein (6,7). At least two homologs of bacterial MutL, acting as a hMLH1-hPMS2 dimer (hMutLα), are required for progression of the long-patch MMR pathway towards its exonucleolytic step (8). Eventually, a long stretch (up to 1000 bases) of the newly synthesized strand including the mispaired or unpaired site is degraded and DNA is re-synthesized by replicative DNA polymerases (9,10). Heterozygous germ line mutations in one of these human MutS or MutL homologs are associated with the hereditary non-polyposis colorectal cancer (HNPCC) syndrome. HNPCC is characterized by the early development of tumors in the colon, as well as in the endometrium, stomach, small intestine and ovaries. Patients affected by HNPCC have usually inherited one defective allele and one wild-type allele of a MutS or MutL homolog. Functional inactivation of the latter allele results in long-patch MMR deficiency, which generates tumor cells with a strong mutator phenotype and elevated microsatellite instability (11–14).
Alternative MMR activities, which are independent of DNA replication, have been described in several instances. A prominent case is provided by thymine in G/T mismatches, resulting from spontaneous deamination of 5′-methylcytosine. This faulty base is excised by thymine DNA glycosylase (TDG), thereby initiating a base excision repair mechanism that restores the correct G/C pair (15,16). Another short-patch MMR pathway has been found to process base mismatches generated in recombination intermediates (17). Subsequent studies showed that several nucleotide excision repair (NER) factors, including the homologs of the human genes XPA, XPC, XPF, ERCC 1 and XPG, are components of this short-patch MMR pathway in meiotic Schizosaccharomyces pombe cells (18–21). More recently, a short-patch MMR activity has been described in both meiotic and mitotic stages of Saccharomyces cerevisiae (22). Finally, a short-patch MMR activity that removes A/C mispairs has been detected in extracts of mouse fibroblasts (23). However, these alternative MMR activities of S.cerevisiae and mouse cell extracts seem to operate in the absence of NER factors (22,23).
The question of whether there is a DNA replication-independent MMR mechanism in humans is of primary interest. In fact, such a process could interfere with the maintenance of genome stability by eliminating small heterologies between recombining DNA sequences. When hybrid DNA is formed as part of the recombinational process, mispaired bases arise at sites of sequence divergence between two homologous chromosomes. The resolution of mismatched heteroduplex DNA in meiotic recombination intermediates is thought to account for gene conversion effects (17,22). Furthermore, the removal of sequence dissimilarities during mitotic recombination processes constitutes an important determinant for the loss of heterozygosity during tumor development. Previous reports indicate that the C/C mismatch represents a structurally unique type of mispair because it induces more helix destabilization than other sequence heterologies (24), but without being a substrate for long-patch MMR activity in Escherichia coli and yeast (25,26). These findings prompted us to use the C/C mismatch as a model lesion to search for alternative MMR reactions, which are not coupled to DNA replication in human cells.
MATERIALS AND METHODS
In vitro NER assay
Internally labeled 32P-DNA fragments of 147 bp containing site-directed mismatches, AAF or pivaloyl adducts, were constructed by ligation of six oligonucleotides with partially overlapping sequences as described previously (27–29). Excision assays were performed by incubating 50 μg of HeLa whole cell extract (30) with 147mer DNA substrates (0.5 ng; 75 000 d.p.m.) at 30°C for 40 min. The resulting excision products were resolved on 10% polyacrylamide denaturing gels and visualized by autoradiography. Radiolabeled 19mer and 30mer oligonucleotides were used as size markers.
In vitro MMR assay
The MMR efficiency of cell extracts (50 μg) was tested by incubation with 5 ng of bacteriophage 13mp2 DNA heteroduplex containing a mismatch in the lacZ α-complementation gene (31,32). The repaired DNA was purified and subsequently introduced by electroporation into E.coli NR9162 (a mutS strain), which was plated on to a minimal medium in a soft agar layer containing CSH50 (the α-complementation strain), 0.5 mg of isopropyl-β-d-thiogalactopyranoside and 2 mg of 5-bromo-4-chloro-3-indoyl-β-d-galactopyranoside. After 20 h at 37°C, the plaques were counted and classified as blue, colorless or mixed. Repair efficiency (%) was calculated as follows: 100 × [1 − (% mixed plaques in extract-treated sample)/(% mixed plaques in mock sample without extract)]. Recombinant hMutLα dimer was expressed in Spodoptera frugiperda cells (33).
Construction of pS189 derivatives
The single-stranded form of pS189 was prepared using the helper phage R408. Site-specific modifications were introduced into covalently closed pS189 DNA by annealing and extending mutagenic primers (34). The sequence of each phosphorylated oligonucleotide is shown in Table 1. Oligonucleotides P1 and P2 were used to synthesize the unmodified control substrate. P3 and P4 were used for the construction of substrate 1, P3 and P5 for substrate 2, P6 and P7 for substrate 3, P8 and P9 for substrate 4, P9 and P10 for substrate 5, and P5 and P11 for substrate 6. The primers were annealed in a 10-fold molar excess to 40 μg (0.1 mg/ml) single-stranded pS189 DNA (in 50 mM NaCl, 10 mm Tris–HCl, 10 mM MgCl2 and 1 mM dithiothreitol, pH 7.9) by heating to 65°C for 8 min, then cooling slowly to 4°C. The primer extension and ligation reactions were performed with 35 U of T4 DNA polymerase (Roche) and 70 U of T4 DNA ligase (New England Biolabs), following the manufacturer's instructions. The substrates were methylated by incubation with dam methylase and S-adenosylmethionine (New England Biolabs). Closed circular DNA was then isolated by CsCl density gradient centrifugation, purified by gel filtration (MicroSpin S-400 HR columns, Amersham Biosciences) and concentrated in a centrifugal filter device (Microcon YM-30, Millipore).
Table 1. Oligonucleotide primers used to synthesize the pS189 vector derivatives.
| Primer | Sequencea |
|---|---|
| P1 | 5′-CCTCACTACTTCTGGAATAGCTCAGAGGCCGAGGCGGCCTCGGCCTCTGC-3′ |
| P2 | 5′-GGTTCTTTCCGCCTCAGAAGGTACCTAACCAAGTTCCTC-3′ |
| P3 | 5′-CCTCACTACTTCTGGAATAGCTCAGAGCCCGAGGCGGCCTCGGCCTCTGC-3′ |
| P4 | 5′-GGTTCTTTCCGCCTCAGAAGCTACCTAACCAAGTTCCTC-3′ |
| P5 | 5′-GCAAAAGCCTACGCCTCCAAAAAAGCCTCC-3′ |
| P6 | 5′-CCTCACTACTTCTGGAATAGCTCAGAGACCGAGGCGGCCTCGGCCTCTGC-3′ |
| P7 | 5′-GGTTCTTTCCGCCTCAGAAGATACCTAACCAAGTTCCTC-3′ |
| P8 | 5′-CCTCACTACTTCTGGAATAGCTCAGAGCCGAGGCGGCCTCGGCCTCTGC-3′ |
| P9 | 5′-GGTTCTTTCCGCCTCAGAAGTACCTAACCAAGTTCCTC-3′ |
| P10 | 5′-CCTCACTACTTCTGGAATAGCTCAGAGCTTGCCGAGGCGGCCTCGGCCTCTGC-3′ |
| P11 | 5′-CCTCACTACTTCTGGAATAGCTCAGAGCCCGAGGCGGCCTCGGCCTCTCCTAAATAAAAAAAATTAGTCAGCC-3′ |
aNucleotides that substitute for G and thus result in mismatches are underlined. P8 introduces a 1-nucleotide deletion, P10 a 3-nucleotide insertion in the SfiI site.
Host cell reactivation assay
The SV40-transformed human fibroblasts (GM00637, GM04312C, GM08437 and GM14931) were purchased from the Coriell Institute for Medical Research (Camden, NJ). The 293T human embryonic kidney cell line was a gift from Dr K. Ballmer, and the XP12RO and XP12ROB4 fibroblast cell lines were a gift from Dr P. Karran. The cell lines were grown in Dulbecco's modified Eagle's medium supplemented with 10% FCS and, prior to each experiment, seeded into 35-mm wells until the cultures reached 80% confluence. The pS189 vector (0.25 μg DNA per well) was transfected using the Lipofectamine Plus reagent from Invitrogen. Cells were harvested 48 h later, subjected to alkaline lysis and progeny plasmids were isolated using QIAprep spin columns (Qiagen). The DNA was treated with DpnI (New England Biolabs) to digest the parental strands carrying the bacterial dam methylation pattern. An aliquot of the DpnI digest was used to transform competent E.coli DH5α. Transformants were plated onto Luria–Bertani (LB) agar plates containing ampicillin (100 μg/ml) and replication efficiency was determined by counting the number of ampicillin-resistant colonies obtained from each 35-mm well. A fraction of colonies were picked, grown in LB medium containing ampicillin, and the respective plasmids were isolated by QIAprep spin columns. The samples were then subjected to restriction analysis using the indicated enzymes (New England Biolabs). Sequencing of the clones was performed by Microsynth (Switzerland).
RESULTS
In vitro assay for human NER activity
An oligonucleotide excision assay performed in HeLa whole cell extracts was first used to test whether base mismatches are susceptible to correction by the human NER complex. Site-specific lesions (mismatches or DNA adducts) were placed in the BstBI recognition sequence (5′-T1T2C3G4A5A6-3′) in the center of 147 bp long substrates. These linear DNA fragments were constructed to contain a 32P-labeled residue near the site of modification (27). Repair reactions performed with the site-directed substrate containing an acetylaminofluorene (AAF) adduct at position G4 yielded characteristic excision products that migrated in polyacrylamide gels as an oligomeric ladder with lengths ranging from 25 to ∼30 nt (Figure 1, lane 8). Similarly, a pivaloyl adduct in combination with a T/T mismatch at the position T2 (28) induced substantial oligonucleotide excision in the cell extract (Figure 1, lane 9). In contrast, G/A or G/G mismatches (at the position G4), a C/C mismatch at the position C3, as well as T/T or T/C mismatches at the position T2 of the BstBI sequence, were unable to elicit any excision products that could be distinguished from the background nuclease ladder generated from the homoduplex control (Figure 1, lanes 1–7). These results suggest that the human NER system fails to exert a substantial activity towards base mismatches, including the C/C lesion.
Figure 1.
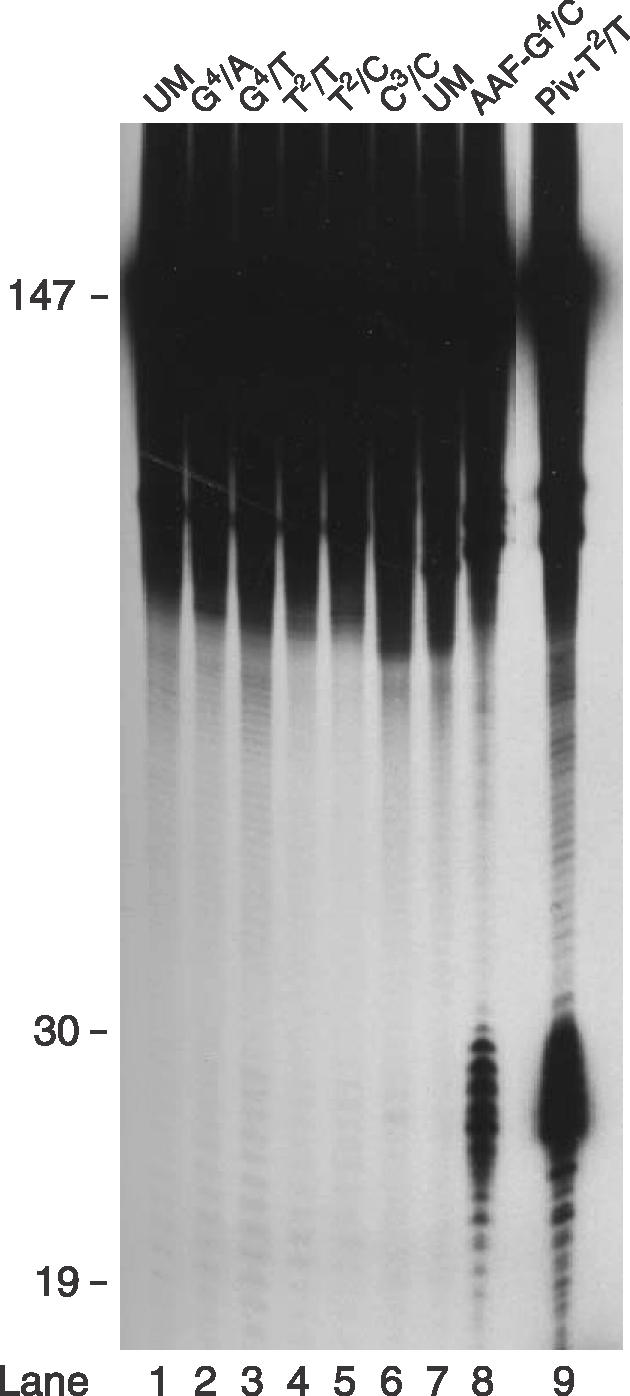
Assay for in vitro human NER activity. Internally 32P-labeled linear substrates of 147 bp were incubated with HeLa whole cell extract. The substrate contained various mismatches as indicated (lanes 2–6), a site-specific AAF-dG adduct (lane 8) or a site-specific pivaloyl backbone adduct (lanes 9) in the central BstBI sequence. UM, unmodified homoduplex substrate. The gel has been overexposed to demonstrate the absence of detectable activity in response to the mismatches.
In vitro assay for human long-patch MMR activity
The repair susceptibility of C/C mismatches was further tested in vitro using extracts from human cells that are either proficient (HeLa) or deficient (293T, LoVo and HCT15) for long-patch MMR activity. The 293T human embryonic kidney cell line lacks expression of the hMLH1 gene due to promoter hypermethylation and, as a consequence, its interaction partner hPMS2 is degraded (35). LoVo and HCT15 cells are deficient for MSH2 and MSH6, respectively. The different cell extracts were incubated with bacteriophage heteroduplex DNA containing a single G/T or C/C mispair and a DNA nick on the 3′ side relative to the mismatch. MMR activity was evaluated by bacteriophage plaque analysis following transfection of an E.coli mutS strain with the resulting DNA products (32). As expected, cytoplasmic extracts obtained from HeLa cells were able to rectify a G/T mismatch located in the coding sequence of the bacteriophage lacZ α-complementation gene (Figure 2). This activity was also observed in HeLa whole cell and nuclear extracts, although less efficiently then in the corresponding cytoplasmic fraction. Presumably, the reduced efficiency of these extracts is due to nuclear DNA-binding factors that might compete with MMR proteins in vitro (31). Importantly, the loss of G/T correcting activity in the 293T cytoplasmic extract could be complemented by the addition of recombinant hMutLα protein. However, neither the long-patch MMR-proficient HeLa cell extracts nor the MMR-deficient LoVo and HCT15 cell extracts were unable to process a C/C mispair above the background level detected in 293T cytoplasmic extract devoid of hMutLα (Figure 2). Also, biochemical complementation of the 293T cell extract with hMutLα failed to restore the correction of C/C mispairs. Identical results were obtained in experiments performed with bacteriophage DNA substrate containing a 5′ nick (data not shown).
Figure 2.

Excision of mismatches in an in vitro assay for human MMR activity. Heteroduplex bacteriophage substrates, containing a G/T or C/C mismatch, were incubated with cytoplamic extracts (CE), whole cell extracts (WCE) or nuclear extracts (NE) prepared from different cell lines. MMR efficiency was quantified after transformation of an E.coli mutS strain with the in vitro repair products. The mock-treated sample resulted from an incubation without cell extract. Complementation was performed by the addition of hMutLα to 293T cytoplasmic extracts. Lovo and HCT15 are human cell lines deficient for hMSH2 and hMSH6, respectively.
Characterization of doubly mismatched pS189 substrates
The findings obtained from the in vitro assays for NER and long-patch MMR activity prompted us to monitor the fate of C/C mismatches in living human cells. For that purpose, we constructed derivatives of shuttle vector pS189, containing site-specific C/C mismatches, that were tagged by treatment with dam methylase and S-adenosylmethionine before transfection. A direct approach, based on the selective amplification of repair products, was used to discriminate between the progeny of repaired templates from the contaminating DNA arising from unrepaired templates. In fact, the modified vectors carried a site-directed C/C lesion, termed C/CSfiI, in the single SfiI restriction sequence of the simian virus 40 (SV40) origin. The position of C/CSfiI (nucleotide 3578 in the pS189 vector) corresponds to nucleotide 5235 of the SV40 genome, which is part of the large T antigen binding site in the core origin (36) and, hence, essential for the initiation of DNA replication (Figure 3A). Because a mismatch in this location disrupts the origin function, the SV40-based vector can be replicated only after removal of the lesion and full restoration of the wild-type origin sequence.
Figure 3.

Vector pS189 derivatives containing two C/C mismatches. (A) Scheme illustrating the location of C/CSfiI (position 3578) in the SV40 origin of pS189. The pentanucleotide recognition sequences for T antigen binding are indicated by the arrows. (B) Composition of substrates 1 and 2. (C) Restriction analysis of substrate 1 (lanes 4–6) and substrate 2 (lanes 9–11). The homoduplex vector was prepared by extension and ligation of unmodified primers. Control, no restriction endonuclease added.
Additionally, the substrates were constructed to contain another C/C mismatch outside the core origin sequence at positions that are not essential for SV40 replication. This second C/C mismatch was used as an internal reference to confirm that the modified vectors were indeed repaired prior to replication. In substrate 1, the second C/C mismatch (C/CAcc65I) was incorporated at a distance of 303 nt on the 3′ side to C/CSfiI, i.e. in the single Acc65I restriction site (Figure 3B). Substrate 2 contained the second C/C mismatch at a different position located 44 nt on the 5′ side to C/CSfiI, within a single AvrII site (Figure 3B). Accordingly, the second mismatch in substrate 2 is referred to as C/CAvrII. To verify the presence of these mismatches at the expected positions, each substrate was subjected to digestion by incubation with the corresponding restriction endonucleases. The homoduplex control vector, which was constructed by annealing fully complementary primers to the SfiI and Acc65I regions, was linearized by SfiI and Acc65I (Figure 3C, lanes 2 and 3). In contrast, substrate 1 was resistant to SfiI and Acc65I digestion due to the presence of a C/C mismatch in both restriction sites (lanes 5 and 6). Similarly, substrate 2 was resistant to SfiI and AvrII (lanes 10 and 11), whereas the unmodified substrate was linearized by these restriction enzymes (lanes 7 and 8).
Repair of C/C mismatches in normal human fibroblasts
The pS189 substrates containing C/CSfiI and a second C/C mismatch (either C/CAcc65I or C/CAvrII) were transfected into SV40-transformed human fibroblasts. Parallel control experiments were performed by transfection of unmodified control vectors. Dose dependence experiments with unmodified vectors demonstrated that the number of progeny colonies reached a plateau when fibroblast cultures with 80% confluence (in 35-mm wells) were transfected with DNA, amounts of 0.5 μg or higher (data not shown). Therefore, subsaturating quantities of substrate (0.25 μg DNA per 35-mm well) were used to ensure that even a partial loss of origin activity would result in a measurable reduction of the final colony counts. After an incubation period of 48 h, the newly synthesized progeny molecules were extracted and separated from unreplicated DNA by treatment with DpnI, which eliminates parental strands carrying the bacterial dam methylation pattern. After transfection of E.coli with the DpnI-resistant fraction, the number of progeny was determined by quantification of bacterial colonies. A direct comparison showed that the mismatched substrates 1 and 2 were replicated in human fibroblasts as efficiently as the unmodified vector, indicating that the C/CSfiI lesion in the origin sequence is rapidly repaired.
Four randomly selected colonies resulting from substrate 1 were amplified for restriction analysis. All four clones were cleaved by SfiI, as expected if replication occurs only after exact restoration of the origin sequence (Figure 4A). Three of these four clones (numbers 2, 3 and 4) were sensitive to both enzymes, SfiI and Acc65I, suggesting that both C/C mismatches in substrate 1 had been converted into wild-type C/G bp, as illustrated in the upper panel of Figure 4B. In principle, these SfiI- and Acc65I-sensitive products could also be generated by replication of the parental strand carrying the wild-type sequence without any previous repair action. However, we found that clone number 1 was sensitive to SfiI but resistant to Acc65I (Figure 4A), indicating that the SfiI site was corrected to yield the wild-type sequence, while the second mismatch resulted in a G to C transversion (lower panel of Figure 4B). This particular product can be formed if C/CSfiI is processed but C/CAcc65I is left unrepaired or, alternatively, if C/CSfiI and C/CAcc65I are both repaired but with opposite strand polarity. Thus, the appearance of such SfiI-sensitive and Acc65I-resistant clones indicates that the C/C mismatches must have been corrected prior to replication.
Figure 4.

Independent processing of two neighboring C/C mismatches in human fibroblasts. (A) SfiI and Acc65I restriction analysis of clones 1–4 isolated after transfection of substrate 1 in wild-type fibroblasts. Control, no enzyme added. (B) Schematic view of the two different repair products isolated after transfection of substrate 1. (C) AvrII restriction analysis of clones number 1–14 isolated after transfection of substrate 2 in wild-type fibroblasts. (D) Scheme of the two different repair products of substrate 2.
As in the experiments with substrate 1, all products generated by in vivo replication of substrate 2 in normal fibroblasts were linearized by SfiI (data not shown). Out of 14 clones investigated, two were resistant to AvrII (Figure 4C, clones 6 and 12), indicating independent processing of the two mismatches, which in substrate 2 were separated by only 44 nt (Figure 4D). The nucleotide sequencing of these two representative clones confirmed that the SfiI site in the origin was restored and also proved the predicted G to C transversion in the AvrII restriction site. In summary, our results show that C/C mismatches are efficiently repaired in normal human fibroblasts. Also, the observation that two neighboring sites (C/CSfiI and C/CAvrII), with an intervening sequence of only 44 nt, can be processed in an independent manner suggests that the removal of C/C mismatches may involve the synthesis of rather short repair patches.
Repair of C/C mismatches in the absence of long-patch MMR
Apart from normal human fibroblasts, the pS189 vector constructs were tested in cell lines with different repair defects. To determine the potential contribution of the long-patch MMR system to C/C removal, we transfected substrate 1 into MMR-deficient 293T cells. These cells were still able to replicate the vectors containing C/CSfiI, but with slightly reduced efficiency (78% colony yield compared to the unmodified control substrate containing no C/C mismatches). Other pS189 derivatives were constructed to carry, instead of C/C mismatches, either A/C mismatches (substrate 3) or single nucleotide deletions (substrate 4) in the SfiI and Acc65I restriction sites (Figure 5A). Substrate 5 contained a 3-nt insertion in the SfiI sequence. Substrates 3, 4 and 5 were replicated as efficiently as unmodified vectors in normal fibroblasts, but the yield of progeny (3–26% of the unmodified control) was significantly reduced in 293T cells (Figure 5B). These quantitative differences are in agreement with A/C mismatches and small deletions/insertions being processed primarily by the long-patch MMR system, and show that inactivation of long-patch MMR has more severe consequences for the removal of A/C mismatches and small deletions/insertions than for the correction of C/C lesions.
Figure 5.

Processing of different lesions in long-patch MMR-deficient 293T cells. (A) Scheme of substrate 3 (pS189 containing two neighboring A/C mismatches) and substrate 4 (pS189 containing two neighboring single nucleotide deletions). (B) Relative yield of progeny after transfection of pS189 derivatives, carrying the indicated lesions, in 293T cells. Colony numbers (mean values of two independent experiments) are expressed as the percentages of bacterial colonies obtained after transfection of the same cells with unmodified control vector.
Restriction analysis of four randomly selected clones obtained from the replication of substrate 1 (containing C/C mismatches) in 293T cells revealed that all four tested progeny molecules were sensitive to SfiI, but three clones were resistant to Acc65I (Figure 6A). Similarly, all 20 tested clones resulting from replication of substrate 2 in 293T cells were SfiI-sensitive (data not shown), but a large proportion (60%) of this progeny was resistant to AvrII (Figure 6B). Nucleotide sequencing of representative clones confirmed the restoration of the SfiI site as well as the predicted G to C transversion in the Acc65I and AvrII targets. These results support the existence of an alternative short-patch pathway by which even closely spaced C/C mismatches can be processed in an independent manner.
Figure 6.

Repair of C/C mismatches in long-patch MMR-deficient 293T cells. (A) SfiI and Acc65I restriction analysis of clones 1–4 isolated after transfection of substrate 1 in 293T cells. (B) Restriction analysis by AvrII of clones 1–20 isolated after transfection of substrate 2 in 293T cells.
Repair of C/C mismatches in the absence of NER factors
We took advantage of xeroderma pigmentosum (XP) cell lines to determine whether the repair of C/C mismatches requires NER factors. When substrate 1 was transfected into XP-A (GM04312C and XP12RO), XP-F (GM08437) or XP-G (GM14931) human fibroblasts, all cell lines were able to replicate the modified vector with normal efficiency. Four individual clones obtained from each transfection were amplified and subjected to restriction analysis. The sensitivity to SfiI was reestablished in all cases but the progeny of each cell line contained 1–2 clones (in a total of four clones analyzed) that were resistant to Acc65I (Table 2).
Table 2. Restriction analysis of random clones obtained after transfection of substrate 1.
| Cell line | Defect | Clones tested | Sensitive to SfiI | Resistant to Acc65I |
|---|---|---|---|---|
| GM00637 | Wild-type | 4 | 4 | 1 |
| 293T | hMLH1-hPMS2 | 4 | 4 | 3 |
| GM04312C | XPA | 4 | 4 | 2 |
| XP12RO | XPA | 4 | 4 | 2 |
| GM08437 | XPF | 4 | 4 | 1 |
| GM14931 | XPG | 4 | 4 | 1 |
| XP12ROB4 | XPA/hMSH2 | 4 | 4 | 2 |
It appears from the results obtained so far that C/C mismatches are efficiently corrected even in the absence of a functional long-patch MMR system or in the absence of NER activity in XP cells. It remained possible, however, that both pathways are able to process C/C mismaches and that they can substitute for each other if only one of the two mechanisms is eliminated. To rule out this scenario, we tested a cell line (XP12ROB4) that combines a long-patch MMR deficiency with a mutation that eliminates NER activity. This particular cell line carries the same XPA defect as XP12RO but, additionally, fails to express hMSH2 (37). Interestingly, substrate 1 was replicated with normal efficiency regardless of whether the host fibroblasts carried only the XPA mutation (XP12RO) or the combined NER and MMR defect (XP12ROB4). Four individual colonies obtained from the XP12ROB4 cells were randomly selected for restriction analysis. All four clones were sensitive to SfiI, but two clones were resistant to Acc65I, indicating independent repair of the mismatches located in different sites (Table 2). Similarly, all 20 tested clones resulting from replication of substrate 2 in XP12ROB4 fibroblasts were SfiI-sensitive, but a considerable fraction (6 clones) was refractory to digestion by AvrII (Table 3). Sequencing of this AvrII-resistant progeny confirmed full restoration of the SfiI sequence and the presence of a G to C transversion at the expected position in the AvrII site. Taken together, these results are consistent with the correction of C/C mismatches by an alternative MMR system for which both long-patch MMR and NER factors are dispensable.
Table 3. Restriction analysis of random clones obtained from transfection of substrate 2.
| Cell line | Defect | Clones tested | Sensitive to SfiI | Resistant to AvrII |
|---|---|---|---|---|
| GM00637 | Wild-type | 14 | 14 | 2 |
| 293T | hMLH1-hPMS2 | 20 | 20 | 12 |
| XP12ROB4 | XPA/hMSH2 | 20 | 20 | 6 |
Molecular analysis of C/C repair in long-patch MMR- and NER-deficient cells
In the cell lines that carry a defect in the long-patch MMR system, we noted that the two closely spaced C/CSfiI and C/CAvrII lesions were processed independently with a high frequency. This observation led us to test whether the alternative human system for C/C repair may involve the formation of short repair patches, as it has been described previously in yeast (18,22). For that purpose, a new vector derivative (substrate 6) was designed where C/CSfiI in the SV40 origin is flanked by two additional C/C mismatches just outside the large T antigen binding site (Figure 7A). C/CAvrII was located at a distance of 44 nt on the 5′ side, as in substrate 2. In addition, a third lesion (C/C3599) was positioned on the 3′ side, 21 nt away from the central C/CSfiI lesion.
Figure 7.

Short-patch repair of C/C mismatches. (A) Schematic view of substrate 6 containing three closely spaced C/C mismatches. (B) Sequence of a representative clone isolated after transfection of substrate 6 in MMR- and NER-defective XP12ROB4 fibroblasts. Red arrows indicate the original position of C/C mismatches in the substrate.
Substrate 6 (carrying the three closely spaced C/C mismatches) was used to transfect XP12ROB4 cells, in which long-patch MMR and NER are inactive. All 24 tested clones resulting from replication of this composite substrate were SfiI-sensitive, indicating restoration of the correct origin sequence. Approximately 50% (13 out of 24) of these clones were not cleaved by AvrII, indicating a G to C transversion in the most 5′ site (data not shown). These SfiI-sensitive but AvrII-resistant clones were sequenced to identify molecules with a second G to C transversion in the 3′ site. Interestingly, 6 out of a total of 13 AvrII-resistant clones analyzed contained such a G to C conversion at the position 3599, flanking the SfiI site (Figure 7B). These results indicate that only the central C/CSfiI was repaired while the flanking C/C mismatches were left unrepaired or, alternatively, that all three sites were corrected but with alternating strand polarity. Such an independent processing of closely spaced C/C mismatches was also detected after transfection of substrate 6 in the MutLα-deficient 293T cells, thus confirming the presence of a short-patch C/C repair activity.
DISCUSSION
The SV40 derivative of this study was designed to contain a C/C lesion in the SfiI site of its origin sequence, such that the repaired DNA molecules were directly amplified by in vivo replication. Accordingly, in all daughter clones examined the C/CSfiI lesion was corrected to the wild-type G/C base pair at position 3578 of the core origin sequence. The repair products with opposite strand polarity were not detected in our assay, because a G to C conversion at position 3578, corresponding to nucleotide 5235 of the SV40 genome, disrupts the origin activity (36) and, hence, is not compatible with amplification.
To prove that repair of the C/C mismatch indeed occurs before the first round of DNA replication, an additional C/C lesion (C/CAcc65I) was introduced at a distance of 303 nt in a 3′ region of the pS189 substrate that is not essential for origin activity. Alternatively, a second control mismatch (C/CAvrII) was placed at a distance of 44 nt in a 5′ region of pS189, which is again not required for origin function. These additional C/C mismatches are converted to both the wild-type (G/C) and mutated (C/G) base pairs in the respective Acc65I or AvrII restriction sequences. In the case that replication could occur without prior repair of the C/CSfiI lesion, only the strand carrying the wild-type sequence would give rise to daughter DNA that can be further replicated. This unlikely event should result in the recovery of progeny vectors that contain an identical pattern of SfiI and Acc65I (or AvrII) sensitivity. Instead, a considerable proportion (on the average 50%) of the progeny was characterized by the restored SfiI sequence and a mutated Acc65I site, as expected if the C/CSfiI lesion has undergone a local repair event before replication. Similarly, AvrII-resistant clones were detected but at variable frequencies depending on the long-patch MMR proficiency of the host cells (see below).
The efficient correction of C/C mismatches in wild-type cells is in agreement with the report of Brown and Jiricny (38), who estimated that the majority of SV40 genomes containing a C/C mismatch were readily repaired in monkey kidney cells. Surprisingly, the correction of C/C mismatches was only slightly reduced in the hMutLα-deficient background of 293T cells, and no reduction of repair activity was observed in another long-patch MMR-deficient cell line (XP12ROB4), lacking MSH2 expression. However, these findings cannot be taken as an evidence that the long-patch MMR system exerts no activity on C/C substrates. In fact, the proportion of progeny vectors, in which the closely spaced C/CSfiI and C/CAvrII were processed independently, was higher in both long-patch MMR-deficient cells compared with the MMR-proficient fibroblasts (Tables 2 and 3). Apparently, some of these adjacent mismatches were processed by long-patch MMR and hence removed simultaneously with identical strand polarity. Thus, our results support a partial contribution of long-patch MMR to the removal of C/C mismatches in human cells. Such an involvement has already been proposed by Genschel et al. (39), who reported that the long-patch MMR system is able to correct C/C lesions in an in vitro assay performed with human cell extracts. We found that C/C substrates were not processed by long-patch MMR in a similar in vitro assay (Figure 2). Since different substrates were used in these studies, distinct flanking of sequence effects may provide an explanation for the dissenting results. In any case, the observation that C/C lesions can be corrected in human cells despite a long-patch MMR-deficient background prompted us to test whether, like in S.pombe (18), a mechanism involving NER factors may act as a backup system to remove such mismatches. However, the repair of C/C lesions remained unaffected in XP cells of complementation group A, F and G, indicating that the NER system does not play a prominent role in the removal of C/C lesions. This finding is in agreement with the results of an in vitro excision assay, indicating that the human NER complex exerts only poor activities against base mismatches (Figure 1). Using a similar excision assay, Huang et al. (29) found a weak, although detectable, NER activity mainly in response to mismatches involving purine bases. In view of the low-intrinsic susceptibility of pyrimidine mismatches to the removal by NER activity, we tested whether human cells, like S.cerevisiae (22), may display a short-patch MMR mechanism that is independent of NER factors. For this purpose, we constructed a pS189 derivative that contains three consecutive C/C mismatches separated by only 21 and 44 nt. Indeed, a substantial fraction (25%) of repair products obtained after transfection of MMR- and NER-deficient XP12ROB4 cells displayed restriction patterns that could be generated only by completely independent repair events, with limited patch size, at each of the three mismatched sites. In principle, the correction of C/C mismatches could be a result of random endonuclease cleavage, generating single strand nicks, and subsequent nick translation along the DNA substrate. However, the observation that C/C mismatches are corrected by a mechanism that involves very short repair patches argues against such a non-specific reaction and supports the conclusion that a specialized process is responsible for recognition and excision of these particular lesions.
The protein factors that are responsible for the short-patch correction of C/C mismatches in human cells have not yet been identified. However, other base mismatches are thought to be removed by DNA glycosylases (16,40). The human homolog of bacterial MutY, which excises adenine from A/G, A/8-oxoG and, less efficiently, from A/C (41), could be responsible for the correction of A/C heterologies. In fact, two separate studies provide evidence for an alternative pathway that repairs A/C mismatches in mammalian cells. Heywood and Burke (42) used a shuttle vector assay in SV40-transformed human cells to demonstrate that the removal of A/C mismatches involves repair patches shorter than 40 nt. Oda et al. (23) detected an A/C repair activity with bias towards the replacement of adenine in long-patch MMR-deficient mouse cell extracts. To date, a comparable activity of DNA glycosylases on cytosines has not been demonstrated, but several factors have been shown to bind preferentially to C/C and, with variable efficiencies, to other pyrimidine-containing mismatches. Examples of DNA glycosylases that recognize C/C mismatches include the E.coli MutM and its counterpart OGG1 in S.cerevisiae (43), as well as S.pombe Mag1 (44) and its mammalian functional homolog MPG (45). However, neither MutM nor MPG exert any detectable incision activity on C/C substrates in vitro (43,45). The observation that MPG and similar enzymes release intact bases (mainly guanines) from DNA, on the other hand, prompted Berdal et al. (46) to postulate that DNA glycosylases may cleave base-sugar bonds without absolute damage specificity. Possible candidates for the excision of cytosines include the pyrimidine DNA glycosylase hNTH1 (47) or one of the recently identified NEIL glycosylases (48,49).
In the absence of a molecular signal for strand discrimination, alternative short-patch MMR activities may act in a mutagenic manner by processing randomly one or the other partner of mismatched bases. This activity could play a role in HNPCC cells that are deficient for the main long-patch MMR system, thereby accelerating the conversion of replication errors to mutations. Short-patch MMR activities are also likely to influence the frequency of meiotic and mitotic recombination events. In fact, it has been shown that long-patch MMR components are involved in preventing the recombination between divergent sequences (50). If mismatches formed in recombination intermediates are eliminated by an MMR-independent system, the sequences of the pairing DNA molecules become more similar, which increases the probability for recombination to occur. In pre-carcinogenic cells, where one copy of a particular tumor suppressor gene is mutated, this short-patch MMR-mediated mechanism could enhance the rate by which the heterozygous state is lost.
Acknowledgments
ACKNOWLEDGEMENTS
We kindly thank Dr M. Seidman for plasmid pS189, Dr P. Karran for XP12RO and XP12ROB4 cells, Dr K. Ballmer for 293T cells, and Dr J. Enzlin for helper phage R408. We also thank Dr P. Schär for critical reading of the manuscript. This work was supported by the Swiss National Science Foundation grant 3100A0-101747.
REFERENCES
- 1.Kolodner R. (1996) Biochemistry and genetics of eukaryotic mismatch repair. Genes Dev., 10, 1433–1442. [DOI] [PubMed] [Google Scholar]
- 2.Schär P. and Jiricny,J. (1998) Eukaryotic mismatch repair. In Eckstein,F.A. and David,M.J. (eds), Nucleic Acids and Molecular Biology. Springer, NY, Vol. XII, pp. 199–246. [Google Scholar]
- 3.Marra G. and Schär,P. (1999) Recognition of DNA alterations by the mismatch repair system. Biochem. J., 338, 1–13. [PMC free article] [PubMed] [Google Scholar]
- 4.Marti T.M., Kunz,C. and Fleck,O. (2002) DNA mismatch repair and mutation avoidance pathways. J. Cell. Physiol., 191, 28–41. [DOI] [PubMed] [Google Scholar]
- 5.Modrich P. and Lahue,R. (1996) Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem., 65, 101–133. [DOI] [PubMed] [Google Scholar]
- 6.Acharya S., Wilson,T., Gradia,S., Kane,M.F., Guerrette,S., Marsischky,G.T., Kolodner,R. and Fishel,R. (1996) hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl Acad. Sci. USA, 93, 13629–13634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drummond J.T., Li,G.M., Longley,M.J. and Modrich,P. (1995) Isolation of an hmsh2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science, 268, 1909–1912. [DOI] [PubMed] [Google Scholar]
- 8.Li G.M. and Modrich,P. (1995) Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc. Natl Acad. Sci. USA, 92, 1950–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cooper D.L., Lahue,R.S. and Modrich,P. (1993) Methyl-directed mismatch repair is bidirectional. J. Biol. Chem., 268, 11823–11829. [PubMed] [Google Scholar]
- 10.Fang W.H. and Modrich,P. (1993) Human strand-specific mismatch repair cccurs by a bidirectional mechanism similar to that of the bacterial reaction. J. Biol. Chem., 268, 11838–11844. [PubMed] [Google Scholar]
- 11.Fishel R., Lescoe,M.K., Rao,M.R., Copeland,N.G., Jenkins,N.A., Garber,J., Kane,M. and Kolodner,R. (1993) The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell, 75, 1027–1038. [DOI] [PubMed] [Google Scholar]
- 12.Hemminki A., Peltomaki,P., Mecklin,J.P., Jarvinen,H., Salovaara,R., Nystrom-Lahti,M., de la Chapelle,A. and Aaltonen,L.A. (1994) Loss of the wild type MLH1 gene is a feature of hereditary nonpolyposis colorectal cancer. Nature Genet., 8, 405–410. [DOI] [PubMed] [Google Scholar]
- 13.Leach F.S., Nicolaides,N.C., Papadopoulos,N., Liu,B., Jen,J., Parsons,R., Peltomaki,P., Sistonen,P., Aaltonen,L.A., Nystrom-Lahti,M. et al. (1993) Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell, 75, 1215–1225. [DOI] [PubMed] [Google Scholar]
- 14.Parsons R., Li,G.M., Longley,M.J., Fang,W.H., Papadopoulos,N., Jen,J., de la Chapelle,A., Kinzler,K.W., Vogelstein,B. and Modrich,P. (1993) Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell, 75, 1227–1236. [DOI] [PubMed] [Google Scholar]
- 15.Hardeland U., Bentele,M., Lettieri,T., Steinacher,R., Jiricny,J. and Schär,P. (2001) Thymine DNA glycosylase. Prog. Nucleic Acid Res. Mol. Biol., 68, 235–253. [DOI] [PubMed] [Google Scholar]
- 16.Wiebauer K. and Jiricny,J. (1990) Mismatch-specific thymine DNA glycosylase and DNA polymerase beta mediate the correction of G/T mispairs in nuclear extracts from human cells. Proc. Natl Acad. Sci. USA, 87, 5842–5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schär P., Munz,P. and Kohli,J. (1993) Meiotic mismatch repair quantified on the basis of segregation patterns in Schizosaccharomyces pombe. Genetics, 133, 815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fleck O., Lehmann,E., Schär,P. and Kohli,J. (1999) Involvement of nucleotide-excision repair in msh2 pms1-independent mismatch repair. Nature Genet., 21, 314–317. [DOI] [PubMed] [Google Scholar]
- 19.Kunz C. and Fleck,O. (2001) Role of the DNA repair nucleases Rad13, Rad2 and Uve1 of Schizosaccharomyces pombe in mismatch correction. J. Mol. Biol., 313, 241–253. [DOI] [PubMed] [Google Scholar]
- 20.Hohl M., Christensen,O., Kunz,C., Naegeli,H. and Fleck,O. (2001) Binding and repair of mismatched DNA mediated by Rhp14, the fission yeast homologue of human XPA. J. Biol. Chem., 276, 30766–30772. [DOI] [PubMed] [Google Scholar]
- 21.Marti T.M., Kunz,C. and Fleck,O. (2003) Repair of damaged and mismatched DNA by the XPC homologues Rhp41 and Rhp42 of fission yeast. Genetics, 164, 457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coïc E., Gluck,L. and Fabre,F. (2000) Evidence for short-patch mismatch repair in Saccharomyces cerevisiae. EMBO J., 19, 3408–3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oda S., Humbert,O., Fiumicino,S., Bignami,M. and Karran,P. (2000) Efficient repair of A/C mismatches in mouse cells deficient in long-patch mismatch repair. EMBO J., 19, 1711–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boulard Y., Cognet,J.A. and Fazakerley,G.F. (1997) Solution structure as a function of pH of two central mismatches, C/T and C/C, in the 29 to 39 K-ras gene sequence, by nuclear magnetic resonance and molecular dynamics. J. Mol. Biol., 268, 331–347. [DOI] [PubMed] [Google Scholar]
- 25.Kramer B., Kramer,W. and Fritz,H.J. (1984) Different base/base mismatches are corrected with different efficiencies by the methyl-directed DNA mismatch-repair system of E. coli. Cell, 38, 879–887. [DOI] [PubMed] [Google Scholar]
- 26.Schär P. and Kohli,J. (1993) Marker effects of G to C transversions on intragenic recombination and mismatch repair in Schizosaccharomyces pombe. Genetics, 133, 825–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hess M.T., Schwitter,U., Petretta,M., Giese,B. and Naegeli,H. (1996) Site-specific DNA substrates for human excision repair: comparison between deoxyribose and base adducts. Chem. Biol., 3, 121–128. [DOI] [PubMed] [Google Scholar]
- 28.Hess M.T., Schwitter,U., Petretta,M., Giese,B. and Naegeli,H. (1997) Bipartite substrate discrimination by human nucleotide excision repair. Proc. Natl Acad. Sci. USA, 94, 6664–6669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang J.-C., Hsu,D.S., Kazantsev,A. and Sancar,A. (1994) Substrate spectrum of human excinuclease: repair of abasic sites, methylated bases, mismatches, and bulky adducts. Proc. Natl Acad. Sci. USA, 91, 12213–12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manley J.L., Fire,A., Cano,A., Sharp,P.A. and Gefter,M.L. (1980) DNA-dependent transcription of adenovirus genes in a soluble whole-cell extract. Proc. Natl Acad. Sci. USA, 77, 3855–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thomas D.C., Roberts,J.D. and Kunkel,T.A. (1991) Heteroduplex repair in extracts of human HeLa cells. J. Biol. Chem., 266, 3744–3751. [PubMed] [Google Scholar]
- 32.Marra G., Iaccarino,I., Lettieri,T., Roscilli,G., Delmastro,P. and Jiricny,J. (1998) Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proc. Natl Acad. Sci. USA, 95, 8568–8573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raeschle M., Marra,G., Nyström-Lahti,M., Schär,P. and Jiricny,J. (1999) Identification of hMutLbeta, a heterodimer of hMLH1 and hPMS1. J. Biol. Chem., 274, 32368–32375. [DOI] [PubMed] [Google Scholar]
- 34.Shivji M.K., Moggs,J.G., Kuraoka,I. and Wood,R.D. (1999) Dual-incision assays for nucleotide excision repair using DNA with a lesion at a specific site. Methods Mol. Biol., 113, 373–392. [DOI] [PubMed] [Google Scholar]
- 35.Trojan J., Zeuzem,S., Randolph,A., Hemmerle,C., Brieger,A., Raedle,J., Plotz,G., Jiricny,J. and Marra,G. (2002) Functional analysis of hMLH1 variants and HNPCC-related mutations using a human expression system. Gastroenterology, 122, 211–219. [DOI] [PubMed] [Google Scholar]
- 36.Dean F.B., Borowiec,J.A., Ishimi,Y., Deb,S., Tegtmeyer,P. and Hurwitz,J. (1987) Simian virus 40 large tumor antigen requires three core replication origin domains for DNA unwinding and replication in vitro. Proc. Natl Acad. Sci. USA, 84, 8267–8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Driscoll M., Martinelli,S., Ciotta,C. and Karran,P. (1999) Combined mismatch and nucleotide excision repair defects in a human cell line: mismatch repair processes methylation but not UV- or ionizing radiation-induced DNA damage. Carcinogenesis, 20, 799–804. [DOI] [PubMed] [Google Scholar]
- 38.Brown T.C. and Jiricny,J. (1988) Different base/base mispairs are corrected with different efficiencies and specificities in monkey kidney cells. Cell, 54, 705–711. [DOI] [PubMed] [Google Scholar]
- 39.Genschel J., Littman,S.J., Drummond,J.T. and Modrich,P. (1998) Isolation of MutSβ from human cells and comparison of the mismatch repair specificities of MutSβ and MutSα. J. Biol. Chem., 273, 19895–19901. [DOI] [PubMed] [Google Scholar]
- 40.Krokan H.E., Nilsen,H., Skorpen,F., Otterlei,M. and Slupphaug,G. (2000) Base excision repair of DNA in mammalian cells. FEBS Lett., 476, 73–77. [DOI] [PubMed] [Google Scholar]
- 41.McGoldrick J.P., Yeh,Y.C., Solomon,M., Essigmann,J.M. and Lu,A.L. (1995) Characterization of a mammalian homolog of the Escherichia coli MutY mismatch repair protein. Mol. Cell. Biol., 15, 989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heywood L.A. and Burke,J.F. (1990) Repair of single nucleotide DNA mismatches transfected into mammalian cells can occur by short-patch excision. Mutat. Res., 236, 59–66. [DOI] [PubMed] [Google Scholar]
- 43.Nakahara T., Zhang,Q.M., Hashiguchi,K. and Yonei,S. (2000) Identification of proteins of Escherichia coli and Saccharomyces cerevisiae that specifically bind to C/C mismatches in DNA. Nucleic Acids Res., 28, 2551–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fleck O., Kunz,C., Rudolph,C. and Kohli,J. (1998) The high mobility group domain protein Cmb1 of Schizosaccharomyces pombe binds to cytosines in base mismatches and opposite chemically altered guanines. J. Biol. Chem., 273, 30398–30405. [DOI] [PubMed] [Google Scholar]
- 45.Biswas T., Clos,L.J.,II, SantaLucia,J.,Jr, Mitra,S. and Roy,R. (2002) Binding of specific DNA base-pair mismatches by N-methylpurine-DNA glycosylase and its implication in initial damage recognition. J. Mol. Biol., 320, 503–513. [DOI] [PubMed] [Google Scholar]
- 46.Berdal K.G., Johansen,R.F. and Seeberg,E. (1998) Release of normal bases from intact DNA by a native DNA repair enzyme. EMBO J., 17, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilson D.M. III, Sofinowski,T.M. and McNeill,D.R. (2003) Repair mechanisms for oxidative DNA damage. Front. Biosci., 8, D963–D981. [DOI] [PubMed] [Google Scholar]
- 48.Hazra T.K., Izumi,T., Boldogh,I., Imhoff,B., Kow,Y.W., Jaruga,P., Dizdaroglu,M. and Mitra,S. (2002) Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl Acad. Sci. USA, 99, 3523–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morland I., Rolseth,V., Luna,L., Rognes,T., Bjoras,M. and Seeberg,E. (2002) Human DNA glycosylases of the bacterial Fpg/MutM superfamily: an alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res., 30, 4926–4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.de Wind N., Dekker,M., Berns,A., Radman,M. and te Riele,H. (1995) Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell, 82, 321–330. [DOI] [PubMed] [Google Scholar]