

Abstract
In Φ-value analysis, the effects of mutations on the folding kinetics are compared with the corresponding effects on thermodynamic stability to investigate the structure of the protein-folding transition state (TS). Here, molecular dynamics (MD) simulations (totaling 0.65 ms) have been performed for a large set of single-point mutants of a 20-residue three-stranded antiparallel β-sheet peptide. Between 57 and 120 folding events were sampled at near equilibrium for each mutant, allowing for accurate estimates of folding/unfolding rates and stability changes. The Φ values calculated from folding and unfolding rates extracted from the MD trajectories are reliable if the stability loss upon mutation is larger than ≈0.6 kcal/mol, which is observed for 8 of the 32 single-point mutants. The same heterogeneity of the TS of the wild type was found in the mutated peptides, showing two possible pathways for folding. Single-point mutations can induce significant TS shifts not always detected by Φ-value analysis. Specific nonnative interactions at the TS were observed in most of the peptides studied here. The interpretation of Φ values based on the ratio of atomic contacts at the TS over the native state, which has been used in the past in MD and Monte Carlo simulations, is in agreement with the TS structures of wild-type peptide. However, Φ values tend to overestimate the nativeness of the TS ensemble, when interpreted neglecting the nonnative interactions.
Keywords: peptide folding, transition state
The Φ-value analysis is a protein engineering approach to investigate the transition state (TS) ensemble in protein folding (1, 2). The Φ value of residue i, that is the ratio ΔΔGTS-D/ΔΔGN-D between the free energy change in the TS and native state (N) because of a mutation of the residue i [taking the denatured state (D) as a reference], represents the degree of nativeness of the structure around residue i in the TS. Observations derived from Φ-value analysis of many proteins, carried out in several research groups, have revealed that the TS is an ensemble of structures with an overall topology similar to the folded state, but with looser interactions (ref. 3 and references therein).
Φ values are usually interpreted in terms of native contacts (4). This description has been successfully used to obtain sets of conformations from the TS ensemble of several proteins (5–9) and to bias molecular dynamics (MD) trajectories toward the TS (10). On the other hand, specific nonnative interactions may be formed at both the TS and denatured-state ensemble and lead to a wrong picture of TS if not taken into account (11). Furthermore, different experimental conditions or mutations may determine detectable changes in the TS structure, showing the presence of parallel pathways (12, 13) and, thus, a heterogeneous TS. In addition, the ensemble average associated with the use of certain folding observables, like the degree of tryptophan burial, may disguise the presence of multiple folding pathways and folding intermediates (14). Namely, a recent study (15) suggests that not all conformations obtained in MD simulations by using Φ values as restraints on a subset of the native contacts belong to the TS.
The TS structures can be identified by MD simulations through the calculation of their folding probability Pfold (16), i.e., the probability that a trajectory started from a given structure reaches the folded state before unfolding. The concept of Pfold calculation was first introduced in a method for determining transmission coefficients, starting from a known TS (17), and used to identify TSs of simple conformational changes (e.g., tyrosine ring flips) (18). The approach has recently been used to study the otherwise very elusive folding TS by atomistic Monte Carlo off-lattice simulations of small proteins with a Go potential (6, 15) and a 21-residue polyalanine helix without Go potential (19) as well as by implicit solvent MD simulations with a physicochemical potential (8, 20). MD simulations are particularly useful to investigate structured peptides at atomic level of detail. Structured peptides usually form stable secondary structure elements, i.e., the building blocks of most of the larger proteins. Hence, they represent the simplest protein conformations. Understanding their process of folding will help to characterize the folding mechanism of larger proteins.
Here, we use MD simulations with an implicit model of the solvent to describe the TS ensemble and evaluate Φ values for several single-point mutants of Beta3s, a designed three-stranded antiparallel β-sheet peptide of 20 residues (21). Beta3s has been successfully characterized by MD simulations of reversible folding in which the native long-range nuclear Overhauser effect distance restraints are mostly satisfied (22). The length of the simulations in the present work has been chosen to achieve near-equilibrium sampling of the phase space of the peptides at the melting temperature of the wild type.
This work was inspired by the following questions: Is it possible to extract Φ values from trajectories near equilibrium? Are Φ values a measure of the extent of formation of contacts in the TS ensemble? How heterogeneous is the TS ensemble of a small structured peptide? Does the Φ-value analysis allow for the observation of any TS movement? What is the importance of nonnative contacts in the TS conformations? Analysis of the trajectories of Beta3s and its mutants allows for an atomic detailed picture of its phase space that is useful in answering these questions. In addition, the simulation results indicate that for the accuracy of a Φ value the threshold in the change of stability (0.6 kcal/mol) is smaller than postulated by Sanchez and Kiefhaber (1.7 kcal/mol) (23) and the same as suggested recently by Fersht and Sato (24).
Methods
Mutants of Beta3s. Thirty-two single-point mutations of the hydrophobic and aromatic side chains W2, I3, W10, Y11, I18, and Y19 were investigated (Fig. 1). The six sites of mutation are distributed along the sequence of the peptide, two for each strand. Between four and eight mutations have been studied for each site. Six of the 32 mutations are nondisruptive (I3A, I3V, Y11F, I18A, I18V, and Y19F), six mutations are conservative but change the steric properties of the side chain (I3M, Y11L, Y11M, I18M, Y19L, and Y19M), and the remaining 20 mutations are radical but acceptable because, in most of the cases, they do not change significantly the TS of the peptide, as showed in Results and Discussion. This result is probably due to the fact that the side chains of Beta3s are not fully buried in a densely packed hydrophobic core, as is the case in larger proteins (24).
Fig. 1.

Schematic representation of the Beta3s peptide, where the wild-type (WT) sequence and the mutants are indicated. The backbone HBs (dotted lines) and side-chain contacts (SC, dashed lines) common to most of the peptides are reported.
MD Simulations. All simulations and part of the analysis of the trajectories were performed with the program charmm (25). Beta3s was modeled by explicitly considering all heavy atoms and the hydrogen atoms bound to nitrogen or oxygen atoms [param19 force field (25)]. A mean field approximation based on the solvent-accessible surface was used to describe the main effects of the aqueous solvent on the solute (26). Ten MD runs of 2 μs each (total of 20 μs for each mutant) with different initial velocities were performed with the Berendsen thermostat at 330 K, which is close to the melting temperature of wild-type Beta3s (27). To improve sampling, the solute-solvent friction has been neglected that has no effect on the thermodynamic properties of the system (27). Despite the absence of collisions with water molecules, in the simulations with implicit solvent, relative rates are comparable with the values observed experimentally. Helices fold in ≈1 ns (28), β-hairpins in ≈10 ns (28) and triple-stranded β-sheets in ≈100 ns (27), whereas the experimental values are ≈0.1 (29), ≈1 (29), and ≈10 μs (21), respectively. Moreover, the effects of the viscosity on the folding and unfolding rates are essentially the same because the solvent-accessible surface and radius of gyration of Beta3s are only marginally larger in the 330 K denatured-state ensemble with respect to the native state (30). A time step of 2 fs was used and the coordinates were saved every 20 ps for a total of 106 conformations for each mutant. During the 20-μs simulation time, between 57 and 120 folding events were observed for every mutant (Table 1), thus providing sufficient statistical sampling for the kinetic analysis (see below for definition of folding event). This result is supported by the small difference in the native population measured for each individual mutant on two disjoint equal-size subsets of the trajectories (5% on average, the largest being 13%).
Table 1. Stability, folding/unfolding rates, and Φ values of the mutants.
| Mutation* | Whighq,† % | Nat. Cont.‡ | Wlowq,§ % | τf,¶ ns | Nf∥ | τu,** ns | Nu†† |
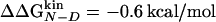 , kcal/mol‡‡ , kcal/mol‡‡
|
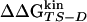 , kcal/mol‡‡ , kcal/mol‡‡
|
Φ‡‡§§ |
|---|---|---|---|---|---|---|---|---|---|---|
| WT | 21.4 | 19.3 ± 1.7 | 2.9 | 70 ± 10 | 92 | 67 ± 6 | 94 | |||
| W2A | 26.5 | 18.1 ± 2.3 | 3.5 | 107 ± 14 | 108 | 63 ± 6 | 114 | –0.32 ± 0.15 | –0.28 ± 0.13 | 0.87 ± 0.57 |
| W2F | 33.5 | 18.8 ± 2.2 | 3.4 | 106 ± 14 | 97 | 82 ± 8 | 103 | –0.14 ± 0.16 | –0.27 ± 0.13 | — |
| W2L | 24.9 | 18.2 ± 2.2 | 6.3 | 109 ± 16 | 101 | 63 ± 5 | 111 | –0.34 ± 0.16 | –0.30 ± 0.14 | 0.87 ± 0.57 |
| W2V | 23.6 | 18.3 ± 2.3 | 4.4 | 124 ± 17 | 95 | 62 ± 6 | 102 | –0.43 ± 0.16 | –0.38 ± 0.13 | 0.89 ± 0.45 |
| W2Y | 21.9 | 18.5 ± 2.4 | 6.4 | 129 ± 21 | 93 | 65 ± 6 | 98 | –0.43 ± 0.16 | –0.41 ± 0.14 | 0.95 ± 0.49 |
| 13A | 19.9 | 18.7 ± 2.2 | 3.9 | 137 ± 18 | 92 | 64 ± 5 | 101 | –0.48 ± 0.15 | –0.44 ± 0.13 | 0.93 ± 0.40 |
| 13F | 33.0 | 18.8 ± 2.1 | 3.3 | 121 ± 22 | 83 | 93 ± 8 | 91 | –0.15 ± 0.17 | –0.36 ± 0.15 | — |
| 13L | 28.5 | 18.5 ± 2.4 | 3.9 | 119 ± 19 | 94 | 72 ± 7 | 101 | –0.31 ± 0.17 | –0.35 ± 0.14 | 1.1 ± 0.77 |
| 13M | 30.2 | 18.9 ± 2.2 | 5.4 | 108 ± 19 | 94 | 81 ± 9 | 102 | –0.16 ± 0.17 | –0.29 ± 0.15 | — |
| 13V | 37.2 | 18.6 ± 2.1 | 5.2 | 124 ± 18 | 75 | 109 ± 10 | 83 | –0.06 ± 0.16 | –0.38 ± 0.14 | — |
| W10A | 31.8 | 19.5 ± 2.1 | 5.0 | 161 ± 21 | 74 | 95 ± 10 | 79 | –0.32 ± 0.16 | –0.55 ± 0.13 | 1.7 ± 0.93 |
| W10F | 41.3 | 18.7 ± 2.2 | 3.8 | 77 ± 9 | 120 | 78 ± 6 | 127 | 0.04 ± 0.14 | –0.06 ± 0.12 | — |
| W10G | 12.7 | 19.3 ± 2.2 | 3.1 | 212 ± 32 | 60 | 68 ± 9 | 69 | –0.72 ± 0.17 | –0.73 ± 0.14 | 1.0 ± 0.31 |
| W10I | 30.8 | 18.3 ± 2.1 | 6.0 | 129 ± 17 | 77 | 88 ± 9 | 83 | –0.23 ± 0.16 | –0.40 ± 0.13 | — |
| W10L | 20.8 | 18.8 ± 2.2 | 4.2 | 166 ± 22 | 81 | 58 ± 5 | 87 | –0.67 ± 0.16 | –0.57 ± 0.13 | 0.86 ± 0.28 |
| W10M | 18.4 | 19.0 ± 2.2 | 6.6 | 155 ± 21 | 82 | 52 ± 5 | 91 | –0.68 ± 0.16 | –0.52 ± 0.13 | 0.76 ± 0.26 |
| W10V | 17.2 | 17.8 ± 2.5 | 6.7 | 259 ± 40 | 57 | 65 ± 11 | 64 | –0.88 ± 0.19 | –0.86 ± 0.14 | 0.98 ± 0.26 |
| W10Y | 26.2 | 19.0 ± 2.1 | 3.5 | 118 ± 15 | 94 | 77 ± 7 | 98 | –0.26 ± 0.15 | –0.35 ± 0.13 | — |
| Y11A | 5.7 | 18.1 ± 2.0 | 2.3 | 249 ± 38 | 64 | 30 ± 3 | 71 | –1.37 ± 0.17 | –0.84 ± 0.14 | 0.61 ± 0.13 |
| Y11F | 33.1 | 19.1 ± 2.2 | 4.4 | 138 ± 20 | 73 | 112 ± 12 | 79 | –0.11 ± 0.16 | –0.45 ± 0.14 | — |
| Y11L | 14.8 | 18.6 ± 2.1 | 4.8 | 169 ± 23 | 76 | 54 ± 6 | 83 | –0.72 ± 0.16 | –0.58 ± 0.13 | 0.81 ± 0.26 |
| Y11M | 11.3 | 18.0 ± 2.2 | 3.5 | 152 ± 24 | 95 | 35 ± 3 | 105 | –0.94 ± 0.16 | –0.51 ± 0.14 | 0.54 ± 0.18 |
| Y11V | 5.7 | 17.0 ± 2.7 | 7.4 | |||||||
| I18A | 12.3 | 18.5 ± 2.3 | 2.4 | 168 ± 22 | 80 | 53 ± 6 | 88 | –0.73 ± 0.16 | –0.58 ± 0.13 | 0.79 ± 0.25 |
| I18F | 21.3 | 19.0 ± 2.0 | 3.2 | 159 ± 23 | 74 | 72 ± 8 | 83 | –0.50 ± 0.17 | –0.54 ± 0.14 | 1.1 ± 0.46 |
| I18L | 22.2 | 19.0 ± 2.2 | 4.4 | 145 ± 19 | 73 | 94 ± 9 | 81 | –0.26 ± 0.16 | –0.48 ± 0.13 | — |
| I18M | 28.9 | 18.8 ± 2.2 | 4.8 | 97 ± 15 | 99 | 77 ± 6 | 106 | –0.13 ± 0.16 | –0.22 ± 0.14 | — |
| I18V | 29.6 | 18.8 ± 2.3 | 3.2 | 124 ± 20 | 87 | 86 ± 9 | 93 | –0.22 ± 0.17 | –0.38 ± 0.14 | — |
| Y19A | 20.7 | 18.6 ± 2.4 | 7.4 | 123 ± 18 | 90 | 84 ± 8 | 95 | –0.23 ± 0.16 | –0.37 ± 0.14 | — |
| Y19F | 29.2 | 18.4 ± 2.2 | 3.8 | 130 ± 18 | 92 | 71 ± 7 | 98 | –0.37 ± 0.16 | –0.41 ± 0.13 | 1.1 ± 0.59 |
| Y19L | 30.0 | 18.3 ± 2.2 | 3.2 | 117 ± 17 | 83 | 88 ± 8 | 89 | –0.17 ± 0.16 | –0.34 ± 0.13 | — |
| Y19M | 17.5 | 18.5 ± 2.3 | 6.2 | 155 ± 26 | 68 | 97 ± 10 | 76 | –0.28 ± 0.17 | –0.52 ± 0.15 | — |
Mutants in italics are radical but acceptable and mutations in Roman are conservative (see Methods and ref. 24)
Statistical weight of the three most populated clusters with Q ≥ 16/24
Average number of contacts in the three most populated clusters with Q ≥ 16/24
Statistical weight of the three most populated clusters with Q < 16/24
Average folding time
Number of folding events
Average unfolding time
Number of unfolding events
The SD have been obtained by propagation of the error on τf and τu
Dashes indicate unreliable Φ values because of 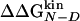 kcal/mol. The reliable Φ values and the corresponding large stability changes (24) are bold. The multipoint Φ values are 0.77, 0.60, 0.79, 0.46, 0.72, and 1.23 for W2, I3, W10, Y11, I18, and Y19, respectively
kcal/mol. The reliable Φ values and the corresponding large stability changes (24) are bold. The multipoint Φ values are 0.77, 0.60, 0.79, 0.46, 0.72, and 1.23 for W2, I3, W10, Y11, I18, and Y19, respectively
Clustering. The conformations of each peptide were clustered by the leader algorithm (31, 32) based on the distance rms (drms) deviation considering the Cα and Cβ atoms. The drms and rms deviations were recently shown to be highly correlated (15). This algorithm is very fast, even when analyzing sets of 106 structures like in the present work. The drms cutoff of 1.2 Å has been chosen on the basis of the distribution of the pairwise drms values in a subsample of the wild-type trajectories. The distribution shows two main peaks that originate from intra- and intercluster distances, respectively (data not shown). The cutoff is located at the minimum between the two peaks.
Native Contacts. As in our previous work (22), a hydrogen bond (HB) is defined as native if the distance between the hydrogen and oxygen atoms is <2.5 Å for more than two-thirds of the conformations belonging to the most populated cluster. A side-chain contact is defined as native if the distance between the center of mass of the two residues averaged over the most populated cluster is <6.5 Å. Seventeen native contacts are common to the wild type and all mutants (but Y11V, see Results and Discussion) and 24 are common to the wild type and more than half of the mutants (Fig. 1). The latter set of contacts has been chosen as the reference for assessing the degree of nativeness of the structures, measured by the fraction of native contacts (Q). The high number of common native contacts shows that the most populated cluster of each mutant (except Y11V) is structurally the same as the one of the wild type.
Folding/Unfolding Events and Rates. The fraction of native contacts Q has been computed along the trajectories of all peptides. A folding (unfolding) event occurs when, along the trajectory, Q first reaches values >0.85 (<0.15) immediately after a previous unfolding (folding) event (22). All of the trajectories are started from the folded state, thus, the first event is always an unfolding. The average time separation between a folding (unfolding) event and the previous unfolding (folding) event is the folding (unfolding) time τf (τu). The folding and unfolding rates are kf = 1/τf and ku = 1/τu, respectively.
Φ Values Calculated from Folding/Unfolding Rates. As in the kinetic experiments used to measure Φexp values, free energy changes with respect to wild type are computed from the folding and unfolding rates with the free energy of the denatured state as reference.
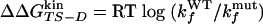 |
[1] |
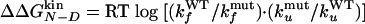 |
[2] |
The Φ value is 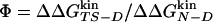 . Values of
. Values of 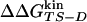 and
and 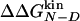 from multiple mutations at the same site can be displayed on a single plot. The slope of the corresponding regression line is called the multipoint Φ value (23, 24).
from multiple mutations at the same site can be displayed on a single plot. The slope of the corresponding regression line is called the multipoint Φ value (23, 24).
Folding Probability and Definition of Native, TS, and Denatured-State Ensemble. The native state of the peptides consists of rapidly interconverting clusters, and the same holds for the denatured state. The following approach is used to group them together. The segment of MD trajectory after each snapshot is analyzed until it first reaches a Q value of >0.85 (i.e., the snapshot leads to folding) or <0.15 (unfolding). For each cluster, the ratio between the snapshots that lead to folding and the total number of snapshots in the cluster is defined as the cluster Pfold. This value is assumed as an approximation of the Pfold of any single structure of the cluster. We have recently shown that cluster Pfold values evaluated with this procedure correlate well with the Pfold values estimated by starting several MD simulations from different structures of a given cluster and counting the fraction of those that fold (F.R., G.S., and A.C., unpublished work and Supporting Text, which is published as supporting information on the PNAS web site).
The native state, the TS, and the denatured-state ensemble consist of the snapshots in the clusters with Pfold ≥ 0.51, 0.49 ≤ Pfold <0.51, and Pfold <0.49, respectively (see Figs. 7 and 8, which are published as supporting information on the PNAS web site). Their statistical weights are WN, WTS, and WD, respectively; these values can be used to evaluate relative free energies by a different equation with respect to the kinetically evaluated ΔΔGkin. In the canonical ensemble, 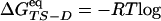 (WTS/WD) and
(WTS/WD) and 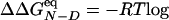 (WN/WD). An excellent match is observed between the
(WN/WD). An excellent match is observed between the 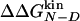 and
and 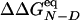 values (correlation coefficient of 0.99) and a good correlation between
values (correlation coefficient of 0.99) and a good correlation between 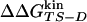 and
and 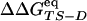 (correlation coefficient of 0.73) (See Fig. 8). The agreement represents a consistency check for the parameters used to define folding and unfolding events. That activation free energy differences computed with the two sets of data show larger discrepancies than do changes in stability is because of the difficulty in sampling the TS ensemble. Note that the
(correlation coefficient of 0.73) (See Fig. 8). The agreement represents a consistency check for the parameters used to define folding and unfolding events. That activation free energy differences computed with the two sets of data show larger discrepancies than do changes in stability is because of the difficulty in sampling the TS ensemble. Note that the 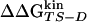 vs.
vs. 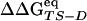 correlation increases by decreasing until 0.02 the interval width of cluster Pfold values defining the TS ensemble (data not shown). The
correlation increases by decreasing until 0.02 the interval width of cluster Pfold values defining the TS ensemble (data not shown). The 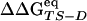 is only very slightly affected by the width of this interval because of the much larger number of structures in the denatured and native states than in the TS.
is only very slightly affected by the width of this interval because of the much larger number of structures in the denatured and native states than in the TS.
Structural Φ Values Based on Atomic Contacts. In each snapshot, a van der Waals contact is defined when the distance between two heavy atoms is <6 Å. pN(i) and pTS(i) measure the fraction of native and TS structures, respectively, in which the contact i is formed. If pN(i) >0.66, the contact i belongs to the set of the native contacts (NC). The structural Φ value
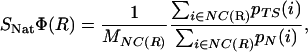 |
[3] |
where MNC(R) is the number of native contacts of residue R, represents an estimate of the degree of nativeness of residue R at the TS ensemble. This measure has been used in the past to give a structural interpretation to experimental Φ values (4, 5, 10). An estimate of the relevance of nonnative interactions at the TS is obtained by extending the computation to all possible contacts (AC), including contacts not present in the NC set
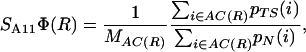 |
[4] |
Results and Discussion
MD Simulations of Reversible Folding. The native structure of the wild type, i.e., the three-stranded antiparallel β-sheet with turns at G6-S7 and G14-S15, is also the most populated in all of the mutants, as shown by the cluster analysis of the trajectories (Table 1). The only exception is Y11V, which has a more distorted native state and has not been considered for further analysis. Moreover, there is no predominant structure in the denatured state for any of the mutants. The number of folding and unfolding events observed along the trajectories ranges from 57 to 120 and from 64 to 127, respectively (Table 1). Interestingly, the values of the stability change upon mutation, calculated with Eq. 2, show that all mutants are less stable than wild-type Beta3s, except for W10F and I3V, which are essentially as stable as Beta3s. This result is not unexpected because Beta3s is a designed peptide whose sequence was carefully optimized for its fold (21).
Accuracy of Two-Point and Multipoint Φ Values. Fig. 2 shows the Φ values extracted from the simulations as a function of the change in free energy of folding upon mutation (see also Table 1). Because of the difficulties in the interpretation of Φ values, as many mutants as possible have been considered and the resulting Φ values divided into classes of reliable, tolerable, and unreliable, according to the size of the induced stability change 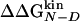 . The deviations from the 0–1 range are large for unreliable Φ values, i.e., for mutations with
. The deviations from the 0–1 range are large for unreliable Φ values, i.e., for mutations with 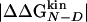 <0.3 kcal/mol, in agreement with previous observations (23). Indeed, in the unreliable class, the deviation can be observed for both radical mutations (e.g., I3F, W10A, and Y19A) and for nondisruptive mutations (e.g., I3V, Y11F, and I18V). For tolerable Φ values, i.e., 0.3 kcal/mol ≤
<0.3 kcal/mol, in agreement with previous observations (23). Indeed, in the unreliable class, the deviation can be observed for both radical mutations (e.g., I3F, W10A, and Y19A) and for nondisruptive mutations (e.g., I3V, Y11F, and I18V). For tolerable Φ values, i.e., 0.3 kcal/mol ≤ 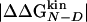 <0.6 kcal/mol, the deviation from the 0–1 interval is less frequent but the relative error is large. The eight reliable Φ values (
<0.6 kcal/mol, the deviation from the 0–1 interval is less frequent but the relative error is large. The eight reliable Φ values (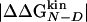 ≥ 0.6 kcal/mol) are all in the range of 0–1 and have a small SD. In a small structured peptide like Beta3s, most residues have a relatively large exposed surface area in the folded state so that conservative mutations generally induce small free-energy changes. Indeed, among the six conservative mutations, only I18A falls into the reliable class. For this reason, more radical mutations have been also investigated.
≥ 0.6 kcal/mol) are all in the range of 0–1 and have a small SD. In a small structured peptide like Beta3s, most residues have a relatively large exposed surface area in the folded state so that conservative mutations generally induce small free-energy changes. Indeed, among the six conservative mutations, only I18A falls into the reliable class. For this reason, more radical mutations have been also investigated.
Fig. 2.

Φ values as a function of change in the native state stability upon mutation. The shadowed horizontal region indicates 1 SD around the multipoint Φ value. The Φ values span a wide range and become anomalous for 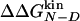 smaller than ≈0.3 kcal/mol. The Φ values corresponding to mutations with
smaller than ≈0.3 kcal/mol. The Φ values corresponding to mutations with 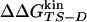 are mainly in the normal range, i.e., between 0 and 1, and are in agreement with the multipoint Φ value. Vertical dashed lines are drawn at
are mainly in the normal range, i.e., between 0 and 1, and are in agreement with the multipoint Φ value. Vertical dashed lines are drawn at 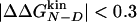 and
and 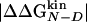 . The Φ value of mutations I3V, W10F, and Y11F are located outside of the plot boundaries. The graphs are ordered according to the antiparallel β-sheet topology of Beta3s with vertical orientation of the three strands, and the N (Left Upper) and C (Right Lower) termini, respectively.
. The Φ value of mutations I3V, W10F, and Y11F are located outside of the plot boundaries. The graphs are ordered according to the antiparallel β-sheet topology of Beta3s with vertical orientation of the three strands, and the N (Left Upper) and C (Right Lower) termini, respectively.
The multipoint Φ of Beta3s as extracted from the simulations are reported in Fig. 3. The good linear relationship between 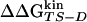 and
and 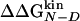 , observed in mutants of W2, W10, Y11, and Y19, supports the validity of the multipoint analysis for these residues and indicates a substantial similarity among the folding TS ensembles of those peptides. In mutants of I3, the linear correlation is weaker than the others, and in I18, there is a change in the slope for
, observed in mutants of W2, W10, Y11, and Y19, supports the validity of the multipoint analysis for these residues and indicates a substantial similarity among the folding TS ensembles of those peptides. In mutants of I3, the linear correlation is weaker than the others, and in I18, there is a change in the slope for 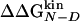 < -0.3 kcal/mol. A possible explanation for the presence of a linear relationship in the multipoint plots is the partial flexibility of the native state of Beta3s (20). Its partially exposed nonpolar side chains that have been mutated in this work are involved in less-specific interactions with the rest of the peptide than buried side chains in the hydrophobic core of larger proteins. Because of the partial flexibility, the mutations do not affect only specific interactions but produce an effect that is spread over the large available set of contacts and thus averaged over them. This averaging of the effects of mutations in the native state may translate into a simple linear dependence of the effects in the TS. In this context, deviations from linearity may indicate TS shifts (see Heterogeneity of the TS Ensemble).
< -0.3 kcal/mol. A possible explanation for the presence of a linear relationship in the multipoint plots is the partial flexibility of the native state of Beta3s (20). Its partially exposed nonpolar side chains that have been mutated in this work are involved in less-specific interactions with the rest of the peptide than buried side chains in the hydrophobic core of larger proteins. Because of the partial flexibility, the mutations do not affect only specific interactions but produce an effect that is spread over the large available set of contacts and thus averaged over them. This averaging of the effects of mutations in the native state may translate into a simple linear dependence of the effects in the TS. In this context, deviations from linearity may indicate TS shifts (see Heterogeneity of the TS Ensemble).
Fig. 3.

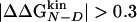 plotted vs.
plotted vs. 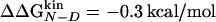 for all of the mutants grouped according to the mutation site along the structure of Beta3s. The optimal regression line (including the wild-type data point) is plotted, and its slope, i.e., the multipoint Φ value, is reported in the lower right corner of each graph with the SD derived from the fit in parentheses. The correlation coefficient is 0.91, 0.67, 0.93, 0.86, 0.87, and 0.88 for W2, I3, W10, Y11, I18, and Y19 mutants, respectively.
for all of the mutants grouped according to the mutation site along the structure of Beta3s. The optimal regression line (including the wild-type data point) is plotted, and its slope, i.e., the multipoint Φ value, is reported in the lower right corner of each graph with the SD derived from the fit in parentheses. The correlation coefficient is 0.91, 0.67, 0.93, 0.86, 0.87, and 0.88 for W2, I3, W10, Y11, I18, and Y19 mutants, respectively.
In multipoint plots, different local probes of the same residue are forced in a single fit that can yield wrong estimates (33). As an example, in the I → V → A → G mutation series. the I → V measures interactions originating from tertiary structure contacts, the V → A measures a mixture of tertiary and secondary structure interactions, whereas the A → G reports almost exclusively on secondary structure formation (33).
In a framework (34) or diffusion-collision (35) mechanism of folding, the tertiary Φ values will most probably be lower than secondary Φ values, even for the same residue. In the case of Beta3s, where the formation of β-sheet backbone HBs and long-range contacts between side chains are concomitant events (see figure 4 in ref. 22), different mutations probe the formation of the same level of structure (i.e., the β-sheet) with no distinction between secondary and tertiary components. This result supports the validity of the multipoint analysis for Beta3s that we do not want to generalize to proteins with more complex folds.
Given the peculiarities of Beta3s, i.e., concomitant formation of secondary and tertiary structure and partial flexibility of its folded state, multipoint Φ values may add information on the accuracy of the two-point Φ values. Indeed, reliable and tolerable Φ values fall mostly within an SD from the corresponding multipoint Φ value (Fig. 2), whereas unreliable Φ values show large deviations. Five of the six multipoint Φ values of Beta3s are >0.5. For diffuse TS ensembles of proteins of ≈100 residues, Φ values of ≈0.2–0.3 have been measured experimentally (36, 37). The high Φ values of Beta3s are probably because of the small size of the peptide. Because of its small size, a large part of the native interactions of the hydrophobic residues is already present in the rate-limiting step (see below).
Heterogeneity of the TS Ensemble. In the wild-type Beta3s, two parallel folding pathways were identified (22, 38). They correspond approximately to conformations having either of the two native β-hairpins formed and the remaining strand unstructured as revealed by the fraction of contacts formed in the two hairpins Q12 and Q23. The TS conformations of the mutants have been analyzed and a similar scenario has been found in all of them. However, the relative abundance of the two pathways is different for different mutants. In most of the mutants, the most populated (thus, rate-limiting) pathway corresponds to the formation of the β-hairpin 2–3, followed by the formation of the β-hairpin 1–2, as in the wild type. In some of the mutants of I3, W10, and I18, the relative weight of the two pathways is inverted. In the multipoint plot of I18 (Fig. 3), the wild-type and the less destabilized mutants (i.e., I18V and I18M) lie on a much steeper line (slope = 1.8) than the more unstable I18F and I18A (slope = 0.2). I18L lies on the crossing of the two lines. The presence of a kink in the linear relationship in the multipoint plot indicates a shift in the folding pathway (24, 39), as confirmed by structural analysis of the TS ensemble of wild type and mutants of I18 (Fig. 4) Wild type, I18V, and I18M have a TS ensemble with β-hairpin 2–3 that is more structured than β-hairpin 1–2 (i.e., Q23 > Q12). On the other hand, for the remaining mutants, the population of the pathways is either similar (I18A), or β-hairpin 1–2 is more structured than β-hairpin 2–3 (I18F and I18L), revealing a shift in the folding pathway determined by the destabilization of β-hairpin 2–3. This destabilization could be a consequence of different steric requirements of γ-branched side chains (Leu and Phe) with respect to β-branched (Val) or unbranched (Ala and Met). A similar shift is observed for the mutants of I3, where a destabilization >0.3 kcal/mol leads to a structural change of the TS (data not shown). Whereas for mutants of I3 and I18, the TS shift can be inferred from the multipoint plot, this is not the case for the W10L, W10Y, and W10V mutants, whose distribution of Q12 and Q23 at the TS (data not shown) indicates a more frequent folding pathway through early formation of β-hairpin 1–2.
Fig. 4.

Distribution of the fraction of native contacts in the N-terminal β-hairpin (Q12) and C-terminal β-hairpin (Q23) for the TS ensemble in the wild-type and I18 mutants. The color indicates the density of conformations and it changes from blue to red as the density increases. The two separated maxima correspond to the two possible folding pathways. (Upper) The more stable species. (Lower) The less stable species. A destabilization of >0.3 kcal/mol for I18 mutants results in a shift of the TS from β-hairpin 2–3 to β-hairpin 1–2.
The structural Φ values, i.e., the amount of contacts formed at the TS ensemble relative to the native state, provide a precise indication of the distribution of structure at TS with respect to the native state. The SxΦ profiles can be divided in two major classes (Fig. 5). The first class (I) contains all of the mutants with a TS that is more structured around the C-terminal G14-S15 turn, according to the SNatΦ values, whereas the structure around the N-terminal turn features many nonnative interactions, according to the large SAllΦ. This class contains the wild type, the mutants of W2, Y11 and Y19, and the mutants I3V, I3M, W10G, I18V, and I18M. The second class (II) contains the mutants that have a TS more structured around the N-terminal turn, as reported by the SNatΦ values, whereas the C-terminal turn is involved in many nonnative interactions, as shown by the SAllΦ profile. This class contains the mutants I3A, W10L, W10Y, W10V, I18F, and I18L. The remaining mutants show SxΦ profiles that lie between the two major classes (data not shown).
Fig. 5.

Heterogeneity and nonnative structure of TS. The labels on the right indicate mutants with the C-terminal β-hairpin more structured at the TS (I) and mutants with the N-terminal β-hairpin more structured at the TS (II). Each solid curve corresponds to a single-point mutant and the lines are drawn to help the eye. Vertical lines indicate the position of the G6-S7 and G14-S15 turns. In the first four rows, the structural Φ values (SNat Φ and SAll Φ) are the ratio between the number of contacts formed in the TS and native state. SNat Φ takes into account only native contacts, whereas SAll Φ also includes nonnative contacts at the TS and can assume values >1. In the last two rows, ΔCX(R) =Σi ∈ C(X,R) [pTS(i) - pN(i)] is the difference between the contacts formed in the TS and in the native state between residue X and R. Positive values indicate that in the TS, there are more contacts than in the native state. Both S7 and S15, if the corresponding hairpin is not native (i.e., in the class of mutants I and II, respectively), have a larger number of contacts in the TS than in the native state with K9 and the residue in position 10, and K17 and the residue in position 18, respectively. A smaller number of contacts in the TS than in the native state is observed with Q4 and Q12, respectively.
Specific Nonnative Structure in the TS. The large number of nonnative interactions made by S7 and S15 in peptides of class I and II, respectively, at the TS (Fig. 5) is mainly constituted by contacts with the lysine residue in position i + 2 (K9 and K17) and with the residue in position i + 3. On the other hand, the contacts of S7 and S15 with Q4 and Q12, respectively, are significantly less in the TS than in the native state. The secondary structure analysis of the G6-S7/G14-S15 residues in the disordered hairpin at TS indicates them as forming a turn in most of the conformations. However, the HBs between residues N5 and T8 (N13 and T16), characterizing the native type II′ turn, are present only in 34% (40%) of the TS structures of the mutants of class I (II). Furthermore, no other specific backbone HBs are formed that define different types of turn. All these data indicate that the precursors of the type II′ turn, formed by the G-S pair of amino acids, are prevalently loose turns devoid of a specific backbone HB pattern that are shifted by one residue to the C terminus. Nonnative interactions, thus, are specifically involved in determining the commitment to fold of a conformation.
Structural Interpretation of Φ Values. Both SNatΦ and SAllΦ profiles of wild-type Beta3s provide a detailed picture of its TS. A comparison has been made with the reliable Φ values derived from mutations that do not change significantly the TS of the peptide (i.e., W10M, W10G, Y11A, Y11L, Y11M, and I18A), as indicated by the similarity of the SxΦ profiles (namely, none of these mutants belong to class II, see above). This analysis allows for the assessment of the common interpretation of the Φ as a ratio between contacts formed at the TS and native states (Fig. 6). The comparison reveals that, within their error, the two-point Φ values are in agreement with both SxΦs. However, the former tend to overestimate the degree of native structure present at the TS ensemble (i.e., reliable Φ > SNatΦ) because specific nonnative interactions are formed at the TS.
Fig. 6.

Comparison between reliable two-point Φ values (filled squares) of mutants with a TS similar to wild type, and the structure of wild-type TS as measured by SxΦ values (open symbols). The structural Φ values are the ratio between the number of contacts formed in the TS and native state. SNatΦ takes into account only native contacts, whereas SAllΦ includes native and nonnative contacts. The two-point Φ values tend to overestimate the degree of nativeness of the TS (measured by SNatΦ) because of the presence of specific nonnative interactions.
Conclusions
The near-equilibrium MD simulations of Beta3s and eight single-point mutants have provided an accurate estimate of Φ values for the mutations with stability changes of >0.6 kcal/mol. For such mutations, the SD on the value of Φ is relatively small, and the two-point Φ value is close to the corresponding multipoint Φ value, and to the structural Φ value that is a measure of the amount of contacts in the TS relative to the native state. In the other cases, the error is large and the estimate is less reliable. The value of the stability change threshold (0.6 kcal/mol) obtained from the simulation results of Beta3s and its mutants is smaller than the one proposed by Sanchez and Kiefhaber (1.7 kcal/mol) (23). Although it is not possible to extrapolate the simulation results to larger proteins with well defined hydrophobic cores, it is reassuring that the same validity threshold was suggested recently by Fersht and Sato (24) for Φ values of nondisruptive deletion mutations, and was used in a study of the CspB protein (40), whereas a very close threshold was used for the immunity proteins Im7 and Im9 (0.7 kcal/mol) (41).
The cluster Pfold progress variable has been used for the identification of TS structures. The TS ensemble of Beta3s and its single-point mutants is made up of two sets of conformations with either of the two β-hairpins folded. A TS shift from structured β-hairpin 2–3 to structured β-hairpin 1–2 has been observed for some of the mutants with different steric properties of the side chain, e.g., β-branched vs. γ-branched. Furthermore, the important role of specific nonnative interactions in the TS has been revealed. Indeed, when either of the two hairpins is formed in the TS, the residues corresponding to the native type II′ turn assume in the unstructured hairpin mainly the conformation of a loose turn shifted by one residue in the C-terminal direction. Specific nonnative contacts distinguish the TS conformations from other structures having the same native interactions but having different nonnative interactions. Hence, neglecting nonnative interactions may prevent a complete understanding of the factors that are responsible for protein folding.
Supplementary Material
Acknowledgments
We thank E. Guarnera and Dr. E. Paci for interesting discussions. The MD simulations were performed on the Matterhorn Beowulf cluster at the Informatikdienste of the University of Zurich. We also thank C. Bollinger, Dr. T. Steenbock, and Dr. A. Godknecht (University of Zürich, Zürich) for setting up and maintaining the cluster. This work was supported by the Swiss National Science Foundation.
Author contributions: G.S. and A.C. designed research; G.S. performed research; G.S. and F.R. contributed new reagents/analytic tools; G.S. and A.C. analyzed data; and G.S. and A.C. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: TS, transition state; MD, molecular dynamics; HB, hydrogen bond.
References
- 1.Fersht, A. R., Matouschek, A. & Serrano, L. (1992) J. Mol. Biol. 224, 771-782. [DOI] [PubMed] [Google Scholar]
- 2.Fersht, A. R. (1999) Structure and Mechanism in Protein Science: Guide to Enzyme Catalysis and Protein Folding (Freeman, New York).
- 3.Daggett, V. & Fersht, A. (2003) Nat. Rev. Mol. Cell Biol. 4, 497-502. [DOI] [PubMed] [Google Scholar]
- 4.Li, A. J. & Daggett, V. (1996) J. Mol. Biol. 257, 412-429. [DOI] [PubMed] [Google Scholar]
- 5.Vendruscolo, M., Paci, E., Dobson, C. M. & Karplus, M. (2001) Nature 409, 641-645. [DOI] [PubMed] [Google Scholar]
- 6.Li, L. & Shakhnovich, E. I. (2001) Proc. Natl. Acad. Sci. USA 98, 13014-13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paci, E., Vendruscolo, M., Dobson, C. M. & Karplus, M. (2002) J. Mol. Biol. 324, 151-163. [DOI] [PubMed] [Google Scholar]
- 8.Gsponer, J. & Caflisch, A. (2002) Proc. Natl. Acad. Sci. USA 99, 6719-6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lindorff-Larsen, K., Vendruscolo, M., Paci, E. & Dobson, C. M. (2004) Nat. Struct. Mol. Biol. 11, 443-449. [DOI] [PubMed] [Google Scholar]
- 10.Settanni, G., Gsponer, J. & Caflisch, A. (2004) Biophys. J. 86, 1691-1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho, J. H., Sato, S. & Raleigh, D. P. (2004) J. Mol. Biol. 338, 827-837. [DOI] [PubMed] [Google Scholar]
- 12.Nauli, S., Kuhlman, B. & Baker, D. (2001) Nat. Struct. Biol. 8, 602-605. [DOI] [PubMed] [Google Scholar]
- 13.Wright, C. F., Lindorff-Larsen, K., Randles, L. G. & Clarke, J. (2003) Nat. Struct. Biol. 10, 658-662. [DOI] [PubMed] [Google Scholar]
- 14.Shimada, J. & Shakhnovich, E. I. (2002) Proc. Natl. Acad. Sci. USA 99, 11175-11180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hubner, I. A., Shimada, J. & Shakhnovich, E. I. (2004) J. Mol. Biol. 336, 745-761. [DOI] [PubMed] [Google Scholar]
- 16.Du, R., Pande V. S., Grosberg, A. Y., Tanaka, T. & Shakhnovich, E. I. (1998) J. Chem. Phys. 108, 334-350. [Google Scholar]
- 17.Chandler, D. (1978) J. Chem. Phys. 68, 2959-2970. [Google Scholar]
- 18.Northrup, S. H., Pear, M. R., Lee, C. Y., McCammon, J. A. & Karplus, M. (1982) Proc. Natl. Acad. Sci. USA 79, 4035-4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lenz, P., Zagrovic, B., Shapiro, J. & Pande, V. S. (2004) J. Chem. Phys. 120, 6769-6778. [DOI] [PubMed] [Google Scholar]
- 20.Rao, F. & Caflisch, A. (2004) J. Mol. Biol. 342, 299-306. [DOI] [PubMed] [Google Scholar]
- 21.De Alba, E., Santoro, J., Rico, M. & Jiménez, M. A. (1999) Protein Sci. 8, 854-865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrara, P. & Caflisch, A. (2000) Proc. Natl. Acad. Sci. USA 97, 10780-10785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanchez, I. E. & Kiefhaber, T. (2003) J. Mol. Biol. 334, 1077-1085. [DOI] [PubMed] [Google Scholar]
- 24.Fersht, A. R. & Sato, S. (2004) Proc. Natl. Acad. Sci. USA 101, 7976-7981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S. & Karplus, M. (1983) J. Comput. Chem. 4, 187-217. [Google Scholar]
- 26.Ferrara, P., Apostolakis, J. & Caflisch, A. (2002) Proteins 46, 24-33. [DOI] [PubMed] [Google Scholar]
- 27.Cavalli, A., Ferrara, P. & Caflisch, A. (2002) Proteins 47, 305-314. [DOI] [PubMed] [Google Scholar]
- 28.Ferrara, P., Apostolakis, J. & Caflisch, A. (2000) J. Phys. Chem. B 104, 5000-5010. [Google Scholar]
- 29.Eaton, W. A., Munoz, V., Hagen, S, J., Jas, G. S., Lapidus, L. J., Henry, E. R. & Hofrichter, J. (2000) Annu. Rev. Biophys. Biomol. Struct. 29, 327-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferrara, P. & Caflisch, A. (2001) J. Mol. Biol. 306, 837-850. [DOI] [PubMed] [Google Scholar]
- 31.Hartigan, J. A. (1975) Clustering Algorithms (Wiley, New York).
- 32.Tou, J. T. & Gonzalez, R. C. (1974) Pattern Recognition Principles (Addison–Wesley, Reading, MA).
- 33.Fersht, A. R. (2004) Proc. Natl. Acad. Sci. USA 101, 14338-14342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baldwin, R. L. & Rose, G. D. (1999) Trends Biochem. Sci. 24, 77-83. [DOI] [PubMed] [Google Scholar]
- 35.Karplus, M. & Weaver, D. L. (1976) Nature 260, 404-406. [DOI] [PubMed] [Google Scholar]
- 36.Itzhaki, L. S., Otzen, D. E. & Fersht, A. R. (1995) J. Mol. Biol. 254, 260-288. [DOI] [PubMed] [Google Scholar]
- 37.Daggett, V., Li, A. J., Itzhaki, L. S., Otzen, D. E. & Fersht, A. R. (1996) J. Mol. Biol. 257, 430-440. [DOI] [PubMed] [Google Scholar]
- 38.Davis, R., Dobson, C. M. & Vendruscolo, M. (2002) J. Chem. Phys. 117, 9510-9517. [Google Scholar]
- 39.Warshel, A., Schweins, T. & Fothergill, M. (1994) J. Am. Chem. Soc. 116, 8437-8442. [Google Scholar]
- 40.Garcia-Mira, M. M., Boehringer, D. & Schmid, F. X. (2004) J. Mol. Biol. 339, 555-569. [DOI] [PubMed] [Google Scholar]
- 41.Friel, C. T., Capaldi, A. P. & Radford, S. E. (2003) J. Mol. Biol. 326, 293-305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.