

Abstract
Benzodiazepines exert their effects by binding to multiple subtypes of the GABAA receptor, the predominant subtypes in the brain being those that contain α1-, α2-, α3-, and α5-subunits. To understand the potentially different roles of these subtypes in the therapeutic and side effects of benzodiazepines, we evaluated GABAA receptor subtype-preferring compounds in nonhuman primate models predictive of anxiolytic, sedative, motor, subjective, and reinforcing effects of benzodiazepine-type drugs. These compounds included zolpidem, which shows preferential binding to GABAA receptors containing α1-subunits (α1GABAA receptors); L-838,417, which shows functional selectivity for α2GABAA, α3GABAA, and α5GABAA receptors; and nonselective conventional benzodiazepines. The results provide evidence in nonhuman primates that α1GABAA receptors do not play a key role in the anxiolytic and muscle-relaxant properties of benzodiazepine-type drugs; instead, these effects involve α2GABAA, α3GABAA, and/or α5GABAA subtypes. Our results also suggest that the α1GABAA receptor subtype might be critically involved in the subjective, sedative, and motor effects of benzodiazepine-type drugs. In contrast, stimulation of α1GABAA receptors is sufficient, but not necessary, for mediation of the abuse potential of these drugs.
Keywords: addiction, anxiety
Receptors for the neurotransmitter GABA, in particular the type A subtype (GABAA receptor), have received considerable attention as the site of action for drugs acting as anxiolytics, sedatives, anticonvulsants, and muscle relaxants. These clinically beneficial effects are exhibited by the benzodiazepines, which act by allosterically binding to GABAA receptors and enhancing the ability of GABA to increase chloride conductance. The therapeutic use of benzodiazepines is constrained, however, by other characteristic effects of these drugs, such as daytime drowsiness and impairment of motor coordination. Benzodiazepines additionally have subjective and reinforcing effects that might contribute to their widespread abuse (1). Recent studies have revealed the existence of multiple subtypes of the GABAA receptor (2, 3), and research with transgenic mice has postulated that the diverse behavioral effects of benzodiazepine-like drugs may reflect action at different subtypes of GABAA receptors (3-5). Although provocative, the extent to which these findings in transgenic mice are applicable to other species, especially primates, is not known. Moreover, virtually no information is available regarding the role of specific GABAA receptor subtypes in the addictive properties of benzodiazepines in any species.
The GABAA receptors in the central nervous system are pentamers constituted from structurally distinct proteins, with each protein family consisting of different subunits (for review, see ref. 3). The majority of GABAA receptors consist of α-, β-, and γ-subunit families, and benzodiazepine action appears to be determined by the presence of particular α-subunits. Benzodiazepine-like drugs bind predominantly to a site on the native GABAA receptor that occurs at the interface of the γ2-subunit with either α1-, α2-, α3-, or α5-subunits, whereas these drugs are inactive at α4-subunit- and α6-subunit-containing receptors. More than 90% of the GABAA receptors in the brain contain α1-, α2-, and α3-subunits (6), and GABAA receptors containing the α1-subunit (α1GABAA receptors) recently have been implicated in the sedative effects of benzodiazepines, whereas GABAA receptors containing α2- and α3-subunits (α2GABAA and α3GABAA receptors) have been implicated in the anxiolytic effects of benzodiazepines (4, 5). Receptors containing α5-subunits, in contrast, are a relatively minor population that might play a role in memory processes, but not anxiolysis or motor effects (7, 8).
Efforts to delineate the contribution of GABAA receptor subtypes in mediating the multiple effects of benzodiazepines have been hampered by the absence of compounds with substantial selectivity for the individual receptor subtypes. Recently, McKernan and colleagues (4) described the compound L-838,417 that, unlike previous benzodiazepine compounds, exhibits “functional selectivity” rather than binding selectivity for the α2GABAA, α3GABAA, and α5GABAA subtypes. That is, L-838,417 does not bind differentially to GABAA receptor subtypes; instead it is an agonist at GABAA receptors containing α2-, α3-, and α5-subunits, but an antagonist at α1GABAA receptors. Because L-838,417 exhibits no appreciable efficacy at α1GABAA receptors, the extent to which this compound lacks an effect characteristic of conventional benzodiazepines can be used to determine the role for the α1GABAA subtype in benzodiazepine agonist action. Using this approach, in the present study, we compared the ability of L-838,417 with those of the α1GABAA-preferring agonist zolpidem, as well as nonselective benzodiazepines to engender characteristic anxiolytic, motor, and sedative effects in monkeys. We also compared the effects of L-838,417 with reference drugs in primate models of the subjective and reinforcing effects of benzodiazepines to determine whether the unique subjective properties and the abuse potential associated with benzodiazepine-type drugs involve different GABAA receptor subtypes.
Methods
Animals. Subjects were adult rhesus monkeys (Macaca mulatta) for the conflict and self-administration procedures and adult squirrel monkeys (Saimiri sciureus) for the observation and drug discrimination studies. Monkeys in the conflict and discrimination studies were maintained at 85-95% of their free-feeding weights, whereas the other monkeys were not food-restricted. Monkeys were individually housed and maintained on a 12-h lights on/12-h lights off cycle, with water available continuously. Catheters were implanted into a major vein (jugular, brachial, or femoral) according to the procedures described by Carey and Spealman (9). Animals in this study were maintained in accordance with the guidelines of the Committee on Animals of the Harvard Medical School and the Guide for Care and Use of Laboratory Animals (National Research Council, Department of Health, Education and Welfare Publication no. NIH 85-23, revised 1996).
Conflict Procedure. Four rhesus monkeys (two males and two females) were trained under a multiple schedule of food reinforcement consisting of two components: (i) a schedule of food delivery and (ii) a schedule of food delivery plus a schedule of foot shock delivery. At the beginning of a session, monkeys were seated in restraint chairs (Crist Instruments, Hagerstown, MD) and placed in an experimental chamber (Med Associates, Georgia, VT). Four components were available in a session, separated by 10-min timeout periods in which responding had no programmed consequences. Responding was maintained in each component under a 18-response, fixed-ratio schedule of food pellet delivery (1 g, Bioserve, Frenchtown, NJ). Each component consisted of the schedule of food delivery signaled by red stimulus lights, followed immediately by the same schedule of food delivery combined with a 20-response, fixed-ratio schedule of foot shock delivery (1.5-3.0 mA, 0.25-s duration), signaled by green stimulus lights. Each response requirement was followed by a 10-s timeout. On training days, monkeys received i.v. injections of saline (2 ml) in the fifth minute of each 10-min timeout. On test days (Tuesdays and Fridays), i.v. injections of vehicle or drug were administered in the fifth minute. Data were expressed as the mean responses per s (±SEM) for each dose of test compound.
Observation Procedure. Four male squirrel monkeys were initially habituated to an observation arena (described in ref. 10) for ≈1 month. After habituation, 30-min observational sessions were conducted daily, during which the animal's behavior was videotaped continuously. Drug test sessions were conducted once or twice per week, with saline control sessions on intervening days. All drugs, as well as saline controls, were administered i.m. in a calf or thigh muscle. During the 6th, 18th, and 30th min of each 30-min session, the monkeys were removed briefly from the observation arena by a trained handler and evaluated for ataxia, defined as the inability to balance on a stainless-steel transport pole (length, 56.0 cm; diameter, 1.0 cm) held in the horizontal plane. During each ataxia assessment, a score of 0 indicated that the monkey was able to balance normally on the transport pole, a score of 1 indicated inability to balance (e.g., hang suspended by limbs below pole), and a score of 2 indicated that the monkey could neither balance on nor support its weight on the pole.
A different technique was included during the hands-on measurement of ataxia to evaluate the degree of muscle relaxation induced by the benzodiazepine-type drugs. After rating the ability of the monkey to balance on the pole, the experimenter then grasped one leg and gently extended it to assess the degree of flexion. A score of 0 indicated that the monkey retracted its leg normally, a score of 1 indicated delayed and/or reduced leg flexion, and a score of 2 indicated no flexion of the leg when held by the experimenter. For both ataxia and muscle relaxation, scores were cumulated for a maximum of 6, and data were expressed as the mean cumulative score (±SEM).
Scoring of videotapes was conducted by observers trained to use the behavioral scoring system described in ref. 10. The observer was not informed about the drugs under investigation. Four observers performed the videotape scoring for the duration of this study. To assure reliability across observers, all individuals underwent at least 20 h of training until they reached an interobserver reliability criterion of ≥90% based on percent agreement scores. The monkeys were scored for locomotor activity, defined as any two or more directed steps in the horizontal and/or vertical plane, and the appearance of sedation, defined as procumbent posture (i.e., loose-limbed, sprawled, unable to maintain an upright position). These behaviors were scored by recording their presence or absence in 15-s intervals during three 5-min observation periods across the session (0-5, 12-17, and 24-29 min). Frequency scores were calculated from these data as the proportion of 15-s intervals in which a particular behavior occurred, and the maximum possible score was 20.
Drug Discrimination. Monkeys were previously trained to discriminate 0.03 mg/kg triazolam i.v. from saline (11). Briefly, five male squirrel monkeys were placed in restraint chairs (Med Associates), and each monkey was trained to respond on both levers under a fixed-ratio 10-response schedule of food reinforcement. Training sessions consisted of one to four 15-min cycles. A cycle consisted of a 10-min pretreatment period and a 5-min response period. During the pretreatment period, the chamber was dark and responses had no programmed consequence. An injection of either saline or drug (triazolam, 0.03 mg/kg, i.v.) was administered during the fifth minute of the pretreatment period. During the response period, stimulus lights were illuminated, and 10 consecutive responses on the lever designated correct by the injection administered during the pretreatment period of the cycle resulted in food delivery. A 10-s timeout occurred after food delivery or if the response requirement was not met within 60 s. Responses on the incorrect lever did not result in food delivery and reset the response requirement on the correct lever. Response periods ended after 5 min or the delivery of 10 pellets, whichever occurred first. The number of cycles per session and order of saline and drug cycles varied randomly with the constraint that if a drug cycle was scheduled it was always the last cycle of the session.
Drug test sessions were conducted once or twice per week with training sessions scheduled on intervening days. Test sessions were conducted if 80% or more of total responses occurred on the correct lever for at least four of five training sessions. Test sessions consisted of four cycles and were identical to the training sessions, except that 10 consecutive responses on either lever resulted in food delivery. Dose-response functions were determined by using a cumulative dosing procedure in which drug injections were administered during the fifth minute of each cycle. Testing with a particular drug continued up to the doses that engendered 80% or more of responses on the drug-appropriate lever or that decreased response rates to 20% or less of control response rates. Percent drug-lever responding was computed for individual subjects in each cycle of a test session by dividing the number of responses on the drug lever by the total number of responses on both levers and multiplying by 100. Discrimination data for an individual subject were excluded from the analyses if response rates were 20% or less of response rates during vehicle tests. Full substitution was defined as 80% or more drug-lever responding, partial substitution as 20-80% drug-lever responding, and no substitution as 20% or less drug-lever responding.
Self-Administration Procedure. Five rhesus monkeys (three male, two female) were trained to self-administer the short-acting barbiturate methohexital under a progressive-ratio schedule of i.v. drug injection as described (12). Monkeys were housed individually in stainless-steel primate cages (Harford Metal Products, Aberdeen, MD) that served as the experimental chambers. A removable panel was placed on the front of each cage and contained four stimulus lights (two red and two white; 3 cm, 1.1 W; Med Associates) and a response lever (Med Associates). Each monkey was fitted with a nylon-mesh jacket (Lomir Biomedical, Malone, NY) that was connected to a 1-m stainless-steel flexible tether (Lomir Biomedical). The monkey's catheter was routed through the tether and attached to a fluid swivel (Lomir Biomedical) on top of the cage. The swivel was attached to an injection pump (Med Associates) located on top of the cage, which could infuse drug solutions at a rate of 0.2 ml/s. The stimulus lights, response levers, and infusion pump were connected to interfaces (Med Associates) and PC-compatible computers located in an adjacent room.
At the beginning of a daily session, the white stimulus lights above the lever were illuminated to signal the start of a trial. Upon completion of the response requirement, the white lights were extinguished and the red stimulus lights were illuminated for 1 s, coinciding with a 1-s infusion. Each trial ended with either an injection or the expiration of a 30-min limited hold. Trials were separated by a 30-min timeout period, during which all lights were extinguished and responding had no programmed consequences.
Experimental sessions consisted of five components made up of four trials each, for a maximum possible of 20 trials per session. The response requirement remained constant for each of the four trials within a component and doubled during each successive component. The session ended when a monkey self-administered a maximum of 20 injections or when the response requirement was not completed for two consecutive trials. The number of trials per response requirement was chosen so that the maximum number of injections could be completed within a day (maximum session time was ≈10 h).
Monkeys were trained to self-administer 0.3 mg/kg per injection of methohexital under a progressive-ratio schedule beginning with a response requirement of 40 responses per injection. Thus, the sequence of possible response requirements was 40, 80, 160, 320, and 640 responses per injection. Once performance was stable under these conditions (no increasing or decreasing trend in the number of injections per session for three consecutive sessions), saline was substituted for methohexital until responding declined to low levels and was stable. Methohexital was again made available for at least three sessions, and doses of benzodiazepine agonists were available in each monkey for the same number of sessions required for responding to decline under conditions of saline availability. The number of injections per session and the break points (BPs) were determined for individual monkeys under each test condition. BP, defined as the highest response requirement completed during a test session, was calculated under each test condition, and used to calculate the maximum BP (BPmax), which was the highest BP maintained by individual subjects for a drug, irrespective of dose. The reinforcing effects of zolpidem and L-838,417 were compared with those of diazepam and midazolam, the latter chosen because of its similar duration of action to both zolpidem and L-838,417 (unpublished observations).
Data Analysis and Drug Preparation. Effects of doses of drugs in the behavioral procedures were evaluated by conducting a priori Dunnett's or Bonferroni tests. For Dunnett's tests, individual doses were compared with control conditions (saline or vehicle injection). For all comparisons, the α level was set at P ≤ 0.05.
Zolpidem and L-838,417 were provided by Merck, Sharp, and Dohme Research Laboratories; all other compounds were obtained from Tocris-Cookson (Ellisville, MO). All compounds were dissolved in propylene glycol and diluted in sterile saline (final concentration of propylene glycol of 50%) and sterilized by filtration. Injection volumes ranged from 0.25 to 1.0 ml/kg.
Results
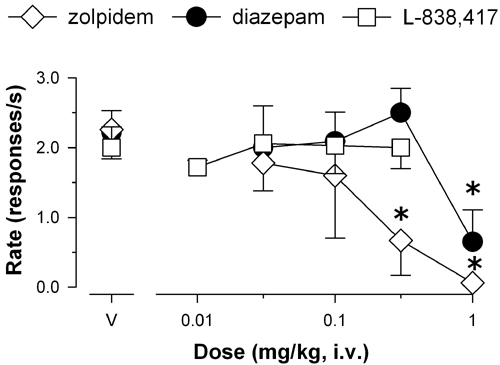
Anxiolytic-Like Effects. The reference benzodiazepine diazepam induced anticonflict effects, characterized as a reliable increase in food-maintained behavior that was suppressed by electric shock presentations (Fig. 1, •; Dunnett's tests, P < 0.05). Similarly, the functionally selective α2,3,5GABAA agonist L-838,417 induced anticonflict behavior to the same degree as diazepam (Fig. 2, □). In contrast, the preferential α1GABAA agonist zolpidem lacked anticonflict effects (Fig. 2, Dunnett's tests, P > 0.05), implying that preferential action at α2GABAA, α3GABAA, and/or α5GABAA receptors mediates the anxiolytic-like effects of benzodiazepine-type drugs.
Fig. 1.

Effects of zolpidem (⋄, α1GABAA-preferring agonist), L-838,417 (□, agonist with functional selectivity for α2GABAA, α3GABAA, and α5GABAA receptors), and diazepam (•, nonselective agonist) on behavior maintained by food presentation that was suppressed by presentation of mild electric shock (n = 4 rhesus monkeys). V, vehicle (50% propylene glycol, 50% saline). *, P <0.05 vs. vehicle, Dunnett's tests.
Fig. 2.

Ability of zolpidem (⋄), L-838,417 (□), and diazepam (•) to engender muscle relaxation (a) and ataxia (b) in squirrel monkeys (n = 4). Data are the mean cumulative score (±SEM). *, P < 0.05 vs. vehicle, Dunnett's tests.
Conventional benzodiazepines characteristically have anticonflict effects at doses lower than those that disrupt nonconflict behavior, a pattern of effects that was observed in the present study with diazepam (Dunnett's tests, P < 0.05; data not shown, see Fig. 7, which is published as supporting information on the PNAS web site). Zolpidem also disrupted nonconflict behavior at the two highest doses tested (Dunnett's tests, P < 0.05; data not shown; see Fig. 7). However, over the dose range tested L-838,417 did not reliably disrupt nonconflict behavior.
Motor Effects. Using a behavioral scoring system to quantify drug effects analogous to muscle relaxation and ataxia in monkeys, we found that both diazepam and the subtype selective benzodiazepine agonists induced reliable and comparable levels of muscle relaxation (Fig. 2a, Dunnett's tests, P < 0.05). In contrast, maximum scores for ataxia were observed with diazepam and zolpidem only, whereas L-838,417 had no reliable ataxic effects (Fig. 2b). In addition, diazepam and zolpidem reliably suppressed locomotor activity (Fig. 3a, Dunnett's tests, P < 0.05); whereas L-838,417 was ineffective up to 10 mg/kg (Fig. 3a). We recently have reported an observable measure of sedation in monkeys, characterized as an immobile and flaccid posture (10). As shown in Fig. 3b, diazepam and zolpidem induced reliable levels of sedation by using this measure, whereas L-838,417 did not induce sedation up to a relatively large dose of 10 mg/kg. Overall, these results suggest that in primates, stimulation of GABAA receptors containing α2-, α3-, and α5-subunits results in muscle relaxation but relatively few of the other motor side effects typical of benzodiazepine-type drugs.
Fig. 3.

Effects of zolpidem (⋄), L-838,417 (□), and diazepam (•) on observable measures related to sedation, including locomotor activity (a) and procumbent posture (sedation, b), in squirrel monkeys (n = 4). Data are expressed as the mean frequency score (±SEM). *, P < 0.05 vs. vehicle, Dunnett's tests.
Discriminative Stimulus Effects. Benzodiazepines produce a unique profile of subjective effects that are distinguishable from other drugs, and the subjective effects of benzodiazepines likely contribute both to their clinically beneficial properties and their side effects. The subjective effects of benzodiazepines are studied in the laboratory by using drug discrimination procedures, and in our previous drug discrimination studies, we have postulated that the subjective effects of benzodiazepines involve stimulation of the α1GABAA receptor (11, 13). Consistent with this idea, the α1GABAA-preferring agonist zolpidem, like diazepam, substituted in monkeys trained to discriminate triazolam (Fig. 4), a prototypical benzodiazepine that is an agonist at all GABAA receptor subtypes. In contrast, L-838,417 did not substitute for triazolam up to a dose of 10 mg/kg (Fig. 4).
Fig. 4.

Effects of selective and nonselective benzodiazepine agonists in squirrel monkeys (n = 5) trained to discriminate triazolam (0.03 mg/kg) from vehicle. Data are mean (±SEM) percentage of responding on the triazolam-associated lever. ▾, Triazolam; •, diazepam; ⋄, zolpidem; □, L-838,417.
Because L-838,417 has very low efficacy at α1GABAA receptors, we next examined the extent to which this compound could antagonize the discriminative stimulus effects of triazolam. We found that pretreatments with L-838,417 shifted the dose-response function for triazolam to the right (Fig. 5). Therefore, over a dose range that did not substitute for triazolam, L-838,417 was a pharmacological antagonist of the subjective effects of a reference agonist.
Fig. 5.

Antagonism of the effects of triazolam by L-838,417 in squirrel monkeys (n = 4) trained to discriminate triazolam (0.03 mg/kg) from vehicle. Data are mean (±SEM) percentage of responding on the triazolam-associated lever. ▾, Triazolam alone; □, +0.1 L-838,417;▵, +0.3 L-838,417; ⋄, +1.0 L-838,417.
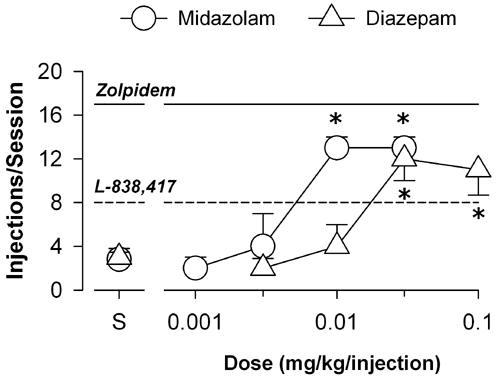
Self-Administration. To date, very little information exists regarding the role of different GABAA receptor subtypes in mediating the addictive potential of benzodiazepine-type drugs. We evaluated the ability of zolpidem and the functionally selective α2,3,5GABAA agonist L-838,417 to maintain responding in a progressive-ratio schedule of i.v. methohexital delivery. Zolpidem maintained near maximum performance, with a mean number of injections per session of 17 (of 20) at a dose of 0.03 mg/kg per injection (Fig. 6a). L-838,417 also maintained self-administration reliably above vehicle levels, with a maximum mean number of injections per session of 8.0 (Dunnett's tests, P < 0.05; Fig. 6a). For comparison, diazepam and the short duration of action benzodiazepine midazolam increased the mean number of injections per session to maximum averages (±SEM) of 12 (±1.5) and 13 (±1.0) injections per session, respectively (data not shown, see Fig. 8, which is published as supporting information on the PNAS web site). These findings indicate that similar to nonselective benzodiazepine agonists both zolpidem and L-838,417 had reinforcing effects. Thus, our results suggest that stimulation of α1GABAA receptors is sufficient, but not necessary, to maintain self-administration, whereas activity at α2GABAA, α3GABAA, and/or α5GABAA receptor subtypes might result in abuse potential.
Fig. 6.

Self-administration of benzodiazepine agonists by rhesus monkeys trained under a progressive-ratio schedule of i.v. drug delivery. (a) Dose-response functions for the mean number of injections per session (±SEM) maintained by zolpidem and L-838,417 (n = 5 monkeys). *, P < 0.05 compared to saline availability, Dunnett's tests. ⋄, Zolpidem; □, L-838,417. (b) Maximum BP irrespective of dose (BPmax). Data are mean + SEM for n = 5 monkeys. Lines represent reliable differences from L-838,417, Bonferroni tests.
To determine the extent to which zolpidem and L-838,417 differ in terms of their effectiveness as reinforcers, we compared the BPmax values (i.e., maximum BP irrespective of dose) among these compounds, as well as to the values obtained with nonselective agonists. Zolpidem maintained the highest BPmax values, followed by midazolam, diazepam, and L-838,417 (Fig. 6b). The mean BPmax value for L-838,417 was reliably lower than those obtained for zolpidem or the nonselective benzodiazepines (Bonferroni tests, P < 0.05; Fig. 6b). These findings suggest that although activation of α2GA BAA, α3GA BAA, and/or α5GABAA receptors is sufficient to engender self-administration, activation of the α1GABAA receptor might enhance the reinforcing effects of benzodiazepine ligands. Alternatively, the intermediate intrinsic efficacy of L-838,417 might account for this compound's reduced reinforcing effectiveness compared with other benzodiazepine agonists.
Discussion
The results of the present study provide important evidence for a differential role of GABAA receptor subtypes in the anxiolytic, motor, subjective, and reinforcing effects of benzodiazepines in nonhuman primates. Specifically, the anxiolytic effects of benzodiazepine-like drugs appear to involve α2,3,5GABAA receptors, whereas the subjective, ataxic, and sedative properties of benzodiazepine-like drugs likely are mediated by α1GABAA receptors. In contrast, the reinforcing effects of these drugs might involve all receptor subtypes. These findings demonstrate GABAA receptor subtype-specific behaviors of benzodiazepines in nonhuman primate species.
Although the most notable difference between L-838,417 and zolpidem is their actions at α1GABAA receptors, another difference between the two compounds is that L-838,417 is active at α5GABAA receptors, whereas zolpidem is not (3). Although differences in the anxiolytic effects of L-838,417 and zolpidem might be attributable to differences in action at α5GABAA receptors, this possibility seems unlikely because α5GABAA receptors exist almost exclusively in the hippocampus (6), and mice with a point mutation rendering this receptor subtype insensitive to benzodiazepines show normal anxiolytic-like responses to diazepam (7, 8). Moreover, experiments with transgenic mice point to the α2GABAA subtype in mediating anxiolytic effects. In this regard, transgenic mice with a point mutation rendering the α2GABAA receptor insensitive to benzodiazepines showed no anxiolytic response to diazepam (5). On the contrary, a compound with inverse agonist effects at the α3GABAA receptor but not the α2GABAA receptor was anxiogenic in rodents (14). These latter findings raise the possibility that the exclusive role for α2GABAA receptors in mediating anxiolytic effects in genetically modified mice might not generalize to nongenetically modified animals. Alternatively, the α2GABAA and α3GABAA receptor subtypes might interact in mediating benzodiazepine-induced anxiolysis. Regardless, our findings support a key role for α2,3GABAA receptors in mediating the anxiolytic effects of benzodiazepine-type drugs in primates.
In addition to reducing anxiety, benzodiazepines are effective clinically as muscle relaxants. A unique property of L-838,417 reported here is its ability to engender muscle relaxation in the absence of other motor effects, a finding that we have not observed in this procedure with any other type of compound to date (including opiates, barbiturates, and dopamine antagonists). This finding is consistent with the observation that diazepam is ineffective as a muscle relaxant in mutant mice in which the α3GABAA receptor is insensitive to diazepam because of a point mutation (15) and provides evidence in primates for a role of a specific GABAA receptor in the muscle relaxation induced by conventional benzodiazepines.
It is well documented that the clinical use of benzodiazepines is restricted by their undesirable side effects. For example, treatment of anxiety disorders with benzodiazepines often is hampered by motor incoordination, which can prevent a patient from engaging in important activities such as driving an automobile. In the present study, both the α1GABAA-preferring agonist zolpidem and the nonselective benzodiazepine diazepam induced a marked impairment in the ability of monkeys to balance on a pole, indicative of ataxia. In contrast, L-838,417, which lacks activity at α1GABAA receptors, did not engender ataxia over the dose range tested. These results agree with our previous finding that benzodiazepine-induced ataxia in monkeys was blocked by a α1GABAA receptor-selective antagonist (10). Thus, our findings suggest that this receptor subtype plays a key role in motor impairments often observed with clinical use of benzodiazepines. It should be noted, however, that L-838,417 possesses intermediate efficacy at α2GABAA, α3GABAA, and α5GABAA receptors, raising the possibility that a lack of ataxia may reflect this compound's relatively lower efficacy at GABAA receptors compared with conventional benzodiazepines.
Another commonly observed effect that limits the use of benzodiazepines as anxiolytics and muscle relaxants is the occurrence of daytime drowsiness. Sedative effects are a documented property of α1GABAA-preferring drugs, and our finding that zolpidem suppressed locomotor activity and induced procumbent posture in monkeys is consistent with the reported sedative properties of this drug. In contrast, L-838,417 lacked sedative effects over the doses tested. Thus, as with studies using mutant mice, our findings support the idea that the sedative properties of benzodiazepine-type drugs involve the α1GABAA subtype primarily, whereas stimulating α2- and α3-subunit-containing GABAA receptors results in little or no sedation (3, 4).
In addition to motor side effects, concerns of abuse, dependence, and illicit diversion have limited the use of benzodiazepine-type drugs in psychiatric medicine. In fact, the addictive potential of these drugs has led to scheduling of all prescription benzodiazepines by the Drug Enforcement Agency of the United States and similar agencies worldwide. Thus, the development of benzodiazepine-type drugs lacking some or all abuse-related effects would represent a significant improvement in treating anxiety disorders. We report here that L-838,417 lacked triazolam-like discriminative stimulus effects and proved to be an antagonist of the effects of triazolam over dose ranges similar to those producing anxiolytic-like effects. These results are consistent with our previous findings that suggest a critical role for α1GABAA receptors in the discriminative stimulus effects of benzodiazepines (11, 13), although, as with the ataxia measure, the lack of effects of L-838,417 might reflect this compound's intermediate efficacy at α2GABAA, α3GABAA, and/or α5GABAA subtypes. To the extent that discriminative stimulus effects reflect subjective effects associated with the abuse of benzodiazepines, these findings suggest that unique subjective effects induced by selective stimulation of GABAA receptor subtypes might result in compounds with reduced abuse potential compared with conventional benzodiazepines.
To evaluate the abuse potential of subtype-selective benzodiazepine-type drugs directly, we conducted experiments in which the compounds were available for self-administration under a progressive-ratio schedule of drug delivery (12). L-838,417 maintained reliable self-administration, indicating that this compound can function as a positive reinforcer. Because this compound exhibits virtually no efficacy at α1GABAA receptors in vitro, activity at GABAA receptors containing α2-, α3-, and/or α5-subunits might be sufficient to engender reinforcing effects. Moreover, based on our analysis of BPs, the reinforcing effectiveness of L-838,417 was less than that of zolpidem and benzodiazepines such as diazepam and midazolam. This finding may be attributable to L-838,417 having no efficacy at α1GABAA receptors and/or to the compound having intermediate efficacy at α2GABAA, α3GABAA, and/or α5GABAA receptors. Interestingly, zolpidem maintained the highest levels of self-administration of any benzodiazepine-type drug tested (see ref. 1). Thus, selective stimulation of the α1GABAA receptor, although not necessary for self-administration, might result in reinforcing effects that are greater than those induced by stimulation of α2GABAA, α3GABAA, and/or α5GABAA receptor subtypes.
Anxiety disorders are some of the most frequently diagnosed disorders in psychiatric medicine worldwide. Although conventional benzodiazepines are effective anxiolytics, their use is restricted in part because of the occurrence of undesirable side effects. The identification of α2,3GABAA receptors as important for mediating the anxiolytic effects of benzodiazepines, in combination with the possibility that subjective and motor effects might involve action primarily at α1GABAA receptors, provides an important framework for developing improved medications for the treatment of anxiety disorders. As a note of caution, however, our results suggest that intrinsic efficacy at α2GABAA, α3GABAA, and/or α5GABAA receptor subtypes is sufficient for a benzodiazepine-type compound to possess some abuse potential.
Supplementary Material
Acknowledgments
We thank A. Martino and A. Duggan for technical assistance and Drs. R. D. Spealman and D. Reynolds for comments on the manuscript. This work was supported by U.S. Public Health Service Grants DA11792, DA13591, and RR00168.
Author contributions: J.K.R., D.M.P., J.R.A., and G.R.D. designed research; D.M.P. and S.L. performed research; J.K.R., D.M.P., and S.L. analyzed data; J.R.A. and G.R.D. contributed new reagents/analytic tools; and J.K.R. wrote the paper.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviation: BP, break point.
References
- 1.Griffiths, R. R. & Weerts, E. M. (1999) Psychopharmacology 134, 1-37. [DOI] [PubMed] [Google Scholar]
- 2.Pritchett, D. B., Lüddens, H. & Seeburg, P. H. (1989) Science 245, 1389-1392. [DOI] [PubMed] [Google Scholar]
- 3.Rudolph, U., Crestani, F. & Möhler, H. (2000) Trends Pharmacol. Sci. 22, 188-194. [DOI] [PubMed] [Google Scholar]
- 4.McKernan, R. M., Rosahl, T. W., Reynolds, D. S., Sur, C., Wafford, K. A., Atack, J. R., Farrar, S., Myers, J., Cook, G., Ferris, P., et al. (2000) Nat. Neurosci. 3, 587-592. [DOI] [PubMed] [Google Scholar]
- 5.Löw, K., Crestani, F., Keist, R., Benke, D., Brünig, I., Benson, J. A., Fritschy, J.-M., Rülicke, T., Bluethmann, H., Möhler, H. & Rudolph, W. (2000) Science 290, 131-134. [DOI] [PubMed] [Google Scholar]
- 6.McKernan, R. M. & Whiting, P. J. (1996) Trends Pharmacol. Sci. 19, 139-143. [DOI] [PubMed] [Google Scholar]
- 7.Collinson, N., Kuenzi, F. M., Jarolimek, W., Maubach, K. A., Cothliff, R., Sur, C., Smith, A., Otu, F. M., Howell, O., Atack, J. R., et al. (2002) J. Neurosci. 22, 5572-5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crestani, F., Keist, R., Fritschy, J.-M., Benke, D., Vogt, K., Prut, L., Blüthmann, H., Möhler, H. & Rudolph, U. (2002) Proc. Natl. Acad. Sci. USA 99, 8980-8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carey, G. & Spealman, R. D. (1998) in Current Protocols in Pharmacology, eds. Enna, S., Williams, M., Ferkany, J., Kenakin, T., Porsolt, R. & Sullivan, J. (Wiley, New York), pp. 10.5.1-10.5.15.
- 10.Platt, D. M., Rowlett, J. K., Spealman, R. D., Cook, J. M. & Ma, C. (2002) Psychopharmacology 164, 151-159. [DOI] [PubMed] [Google Scholar]
- 11.Lelas, S., Rowlett, J. K. & Spealman, R. D. (2001) Psychopharmacology 154, 96-104. [DOI] [PubMed] [Google Scholar]
- 12.Rowlett, J. K., Rodefer, J. S. & Spealman, R. D. (2002) Exp. Clin. Psychopharmacol. 10, 367-375. [DOI] [PubMed] [Google Scholar]
- 13.Lelas, S., Rowlett, J. K., Spealman, R. D., Cook, J. M., Ma, C. & Yin, W. (2002) Psychopharmacology 161, 180-188. [DOI] [PubMed] [Google Scholar]
- 14.Atack, J. R., Hutson, P. H., Collinson, N., Marshall, G., Bentley, G., Moyes, C., Cook, S. M., Collins, I., Wafford, K. A., McKernan, R. M. & Dawson, G. R., Br. J. Pharmacol., in press. [DOI] [PMC free article] [PubMed]
- 15.Crestani, F., Löw, K., Keist, R., Mandelli, M.-J., Möhler, H. & Rudolph, U. (2001) Mol. Pharmacol. 59, 442-445. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}