

Abstract
Alcoholic liver disease is a major health problem in the United States and worldwide without successful treatments. Chronic alcohol consumption can lead to alcoholic liver disease (ALD), which is characterized by steatosis, inflammation, fibrosis, cirrhosis and even liver cancer. Recent studies suggest that alcohol induces both cell death and adaptive cell survival pathways in the liver, and the balance of cell death and cell survival ultimately decides the pathogenesis of ALD. This review summarizes the recent progress on the role and mechanisms of apoptosis, necroptosis and autophagy in the pathogenesis of ALD. Understanding the complex regulation of apoptosis, necrosis and autophagy may help to develop novel therapeutic strategies by targeting all three pathways simultaneously.
Keywords: alcohol, apoptosis, necroptosis, autophagy, liver injury
Alcoholic liver disease (ALD) is a major health problem and a significant source of liver injury in United States. The pathogenesis of ALD includes steatosis, fibrosis, acute alcoholic hepatitis, cirrhosis and eventual hepatocellular carcinoma. Careful research work from many labs in the past decades indicates that cell death including apoptotic and necrotic cell death contributes to the pathogenesis of ALD. Liver injury and disease can be triggered by a diverse set of metabolic, toxic, and inflammatory insults. These insults often lead to the activation of apoptosis, necrosis/necroptosis and autophagy in liver cells. It should be noted that liver contains multiple different cell types including parenchymal and non-parenchymal cells. Cell death of different cell types can all contribute to various liver diseases. In this review, we summarize the progress on the mechanisms by which alcohol induces cell death in hepatocytes. The death of non-parenchymal cells is beyond the topic of this review.
Apoptosis in ALD
Apoptosis is characterized by nuclear fragmentation, chromatin condensation, and cellular shrinkage, which is generally dependent on activation of caspases although caspase-independent apoptosis can also occur. Apoptotic cells can break apart into apoptotic bodies, which are membrane-enclosed particles containing intact organelles that are later phagocytosed by immune cells such as macrophages/Kupffer cells in the liver without inducing an inflammatory response. In general, apoptosis can either be triggered via intrinsic (mitochondria) or extrinsic (death receptor) pathway.
Activation of mitochondrial (intrinsic) apoptotic pathway by alcohol
The mitochondrial apoptotic pathway or the intrinsic pathway, is activated by various apoptotic stimuli, such as DNA damage, oxidative stress, or deprivation of hormone or growth factor (Yin and Ding, 2003). In this scenario, the death signals are transmitted to the mitochondria, which release a number of apoptotic factors, including cytochrome c, Smac/DIABLO, HtrA2/Omi, apoptosis inducing factor (AIF), and endonuclease G. Once released into the cytosol, cytochrome c binds to Apaf-1 and caspase-9 to form the “Apoptosome” to trigger caspase-9 activation in the presence of dATP (Li et al., 1997). Activated caspase-9 then activates downstream executioner caspases, such as caspase-3, -6 and -7. Smac/DIABLO (second mitochondria-derived activator of caspase), when released into the cytosol, binds to the inhibitor of apoptosis proteins (IAPs) such as XIAP to relieve their inhibitory effects on caspases. Activated caspases then cleave various cellular substrates, leading to the characteristic morphological features of apoptosis including DNA fragmentation, chromatin condensation, externalization of phosphatidylserine, and formation of apoptotic bodies. It is known that the metabolism of alcohol plays a critical role in alcohol-induced activation of mitochondrial apoptotic pathway and subsequent apoptosis. Ethanol is metabolized through two major oxidative and two minor non-oxidative pathways. Firstly, alcohol is metabolized by alcohol dehydrogenase (ADH) into the highly reactive acetaldehyde, which is further metabolized by cytosolic aldehyde dehydrogenase 1 (ALDH1) and mitochondrial ALDH2 into more harmless acetate (Crabb et al., 2004). The metabolism of ethanol through this process increases the conversion of nicotinamide adenine dinucleotide (NAD+) into its reduced form, NADH, resulting in alterations of cellular redox status and decreased NAD+-dependent enzyme activities. Increased ratio of NADH/NAD+, which promotes the excess flow of electrons in the mitochondrial respiratory chain resulting in accumulation and leakage of electrons at the mitochondria respiratory chain complex I and III to produce reactive oxygen species (ROS) (Bailey and Cunningham, 2002). Alcohol exposure also damages mitochondrial DNA and ribosomes leading to reduced mitochondrial protein and ATP synthesis (Coleman and Cunningham, 1991). Moreover, chronic alcohol exposure decreases hepatic mitochondrial respiration (state III) and increases sensitivity to Ca2+-mediated mitochondrial permeability transition induction resulting in mitochondrial-mediated apoptosis (King et al., 2014). Furthermore, chronic alcohol exposure decreases mitochondrial maximal oxygen consumption rate and in turn increases the susceptibility of hepatocytes to alcohol-induced hypoxia and liver injury (Zelickson et al., 2011).
In the second oxidative metabolism pathway, ethanol is metabolized by cytochrome P450 family 2, subfamily E, polypeptide 1 (Cyp2E1) and catalase. Acute or chronic ethanol exposure induces Cyp2E1, which can generate ROS (Lu and Cederbaum, 2008). Chronic alcohol exposure to rat also leads to the reduced antioxidant enzymes including catalase, superoxide dismutase, and glutathione peroxidase in liver (Bourogaa et al., 2013). Increased ROS production can directly damage mitochondrial proteins and mitochondria DNA, induce mitochondrial depolarization and onset of mitochondrial permeability transition (MPT) (Hoek et al., 2002). Induction of MPT then leads to more mitochondrial depolarization and ROS production to form a vicious feed forward loop (Zorov et al., 2000).
In addition to the oxidative metabolism, a small percentage of ethanol can also be metabolized via two non-oxidative pathways. In the first pathway, ethanol interacts with fatty acid and generates fatty acid ethyl ester (FAEE) through FAEE synthase (Zelner et al., 2013). Increasing evidence shows that FAEE exacerbates alcohol-induced injury in various tissues including liver (Wu et al., 2006), pancreas (Wu et al., 2008, Werner et al., 2002), and heart (Beckemeier and Bora, 1998, Wu et al., 2008, Wu et al., 2006). In the second pathway, ethanol reacts with phospholipase D (PLD) to generate phosphatidyl ethanol. Following chronic consumption of large amounts of alcohol, phosphatidyl ethanol accumulates to detectable levels due to its poor metabolism. While the effects of phosphatidyl ethanol on cellular functions are currently not clear (Zakhari, 2006), FAEE induces mitochondria damage by binding to mitochondria membrane and uncoupling oxidative phosphorylation (Lange and Sobel, 1983).
As discussed above, it is clear that both oxidative and non-oxidative metabolism of ethanol can lead to mitochondria dysfunction and induction of MPT resulting in the release of mitochondria apoptotic factors such as cytochrome c and SMAC/DIABLO (Figure 1). In cultured rat hepatocytes, it is shown that ethanol treatment induces MPT and triggers release of mitochondrial cytochrome c, which can be inhibited by cyclosporin A, a MPT inhibitor, and by several antioxidants (Higuchi et al., 2001). Using an intravital confocal/multiphoton microscopy approach, Zhong et al found that acute alcohol caused reversible hepatic mitochondrial depolarization and onset of MPT that was dependent on ethanol metabolism in vivo in mouse livers (Zhong et al., 2014). The finding that depolarized mitochondria in mouse liver are reversible is very intriguing, suggesting that mitochondria may adapt to ethanol-induced damage. Indeed, it has been well documented that acute or chronic alcohol exposure can alter liver mitochondria structures (e.g., enlarged mitochondria) and functions in both animal models and human alcoholics (Garcia-Ruiz et al., 2013). Several adaptive mechanisms in the liver can be activated in response to acute or chronic alcohol-induced mitochondrial damage and metabolic stress. For example, alcohol-induced damaged mitochondria can be removed via Parkin-mediated mitophagy (Williams et al., 2015, Williams and Ding, 2015). Chronic alcohol exposure also enhances PGC-1α-mediated mitochondrial biogenesis, increases mitochondrial fusion and mitochondrial respiration in mice (Han et al., 2012). These adaptive mechanisms and mitochondrial plasticity suggest that there is a balance between alcohol-induced mitochondrial damage and repair/biogenesis. Disruption of the balance may result in accumulated damaged mitochondria and subsequent apoptosis and liver injury.
Figure 1. Possible pathways leading to alcohol-induced apoptosis and necrosis.
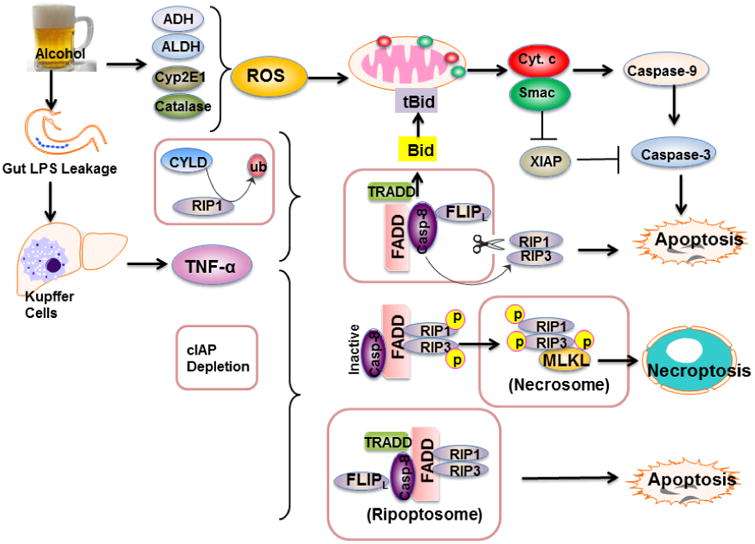
Alcohol metabolism via ADH, ALDH, Cyp2E1 and catalase increases ROS production that triggers mitochondrial damage and onset of MPT resulting in the release of mitochondrial Cyto c and Smac. Released Cyto c promotes the activation of caspase-9 and caspase-3, whereas Smac inhibits XIAP to remove its inhibition on caspases. Activated caspase-3 then leads to hepatocyte apoptosis. Alcohol consumption also increases gut permeability resulting in elevated influx of LPS into the liver. LPS activates Kupffer cells to produce TNF-α. TNF-α binds to its receptor (TNFR1), which further recruits TRADD, FADD, caspase-8 and FLIPl resulting in caspase-8 activation when RIP1 is de-ubiquitinated by CYLD. Activated caspase-8 cleaves Bid to activate the mitochondrial apoptotic pathway and trigger apoptosis. Activated caspase-8 also cleaves RIP1 and RIP3 to inactivate RIP1-RIP3-mediated necroptosis. When cIAPs are depleted and caspase-8 is inhibited, RIP1 and RIP3 interact with each other via RHIM domains to from the amyloid-like necrosome. Auto- and transphosphorylated RIP1 and RIP3 then further recruit and phosphorylate downstream MLKL to initiate necroptosis. In the absence of cIAPs, RIP1, RIP3, TRADD, caspase-8 and FLIPl form a complex called the, which induces caspase-8 activation and apoptosis and depends on RIP1 kinase activity.
Other organelle stress-mediated apoptosis in ALD
Accumulating evidence indicates that alcohol and its metabolites directly or indirectly affect proteostasis resulting in induction of endoplasmic reticulum (ER) stress that leads to liver injury (Ji and Kaplowitz, 2003, Ji et al., 2005). Alcohol and its metabolites such as acetaldehyde are highly reactive and can modify protein structures or form protein adducts resulting in misfolded protein accumulation and triggering ER stress. Moreover, increased ROS or peroxynitrite production can also disturb the redox status of ER and impair the proper protein folding within the ER lumen to cause ER stress. Indeed, ER stress markers are associated with severe steatosis, apoptosis and necroinflammatory foci in the mouse livers fed with chronic alcohol (Ji and Kaplowitz, 2003). Cells can adapt to ER stress by activating the unfolded protein response (UPR), which is mediated by three arms of specialized transcriptional programs including inositol requiring enzyme 1 α (IRE1α), activating transcription factor 6 (ATF6), and PKR-like ER localized eIF2α kinase (PERK). Activation of UPR can attenuate ER stress and restore ER homeostasis by decreasing general protein translation, increasing protein folding capacity by promoting expression of chaperone proteins, and enhancing degradation of misfolded proteins by ER-associated protein degradation (ERAD) via proteasome or by ER stress-mediated compensatory autophagy (Walter and Ron, 2011, Ding and Yin, 2008). However, when ER stress is too severe, UPR may also trigger cell death by activating caspase-12 (murine) or-4 (human), inducing IRE1α-mediated JNK activation or CHOP-mediated transcription of pro-apoptotic cell death genes. Interestingly, CHOP knockout mice protect against chronic alcohol-induced liver apoptosis but have no effect on alcohol-induced steatosis and ER stress (Ji et al., 2005). These data suggest that targeting one arm of UPR may not be sufficient to protect against alcohol-induced liver pathogenesis. Furthermore, recent evidence also suggests that ethanol-induced endoplasmic reticulum (ER) stress activates interferon regulator factor 3 (IRF3) in order to initialize ethanol-induced hepatocyte apoptosis in mice (Petrasek et al., 2013).
In addition to ER, alcohol consumption can alter the activities of lysosomes by elevating lysosomal pH (Kharbanda et al., 1997), which leads to the reduction in protein degradation resulting in excessive protein accumulation. Accumulation of abnormal protein aggregates within hepatocytes can be harmful to the hepatocytes through the proteotoxicity. Accumulation of cytokeratin 8 and 18 (CK8/18) positive cytoplasmic inclusion bodies in hepatocytes, termed Mallory bodies (MBs), has been well documented in ALD patients and animal models (Stumptner et al., 2000, Zatloukal et al., 2007). Rats fed with liquid ethanol diet for one to five weeks causes decreased lysosomal acid phophatase and β-galactosidase activities, redistribution of lysosome enzymes cathepsin B and cathepsin L to lower density cellular compartments and impairment of processing procathepsin L to its mature enzyme (Kharbanda et al., 1996). Mannose 6-phosphate receptor (M6P) is a protein that targets the acid hydrolases to the lysosome. Chronic ethanol feeding lowers the M6P activity and content in hepatocytes isolated from ethanol-fed rats (Haorah et al., 2002, Haorah et al., 2003). This reduction may account for the impaired processing and delivery of acid hydrolases to lysosomes. In addition to lysosome, it has also been reported that chronic ethanol administration impairs receptor-mediated endocytosis, which is mainly due to a decreased number of cell surface receptors in ethanol-fed animals and to a lesser extent, due to defective receptor-ligand internalization leading to decreased ligand degradation (Casey et al., 1987, Dalke et al., 1990). While direct effects of ethanol-impaired lysosomal function and endocytosis on ethanol-induced apoptosis have not been examined, it is likely that dysfunctional lysosome and endocytosis may impair autophagy, which relies on lysosomal functions. Impaired autophagy may indirectly exacerbate ethanol-induced apoptosis and liver injury (see below discussion).
Activation of death receptor (extrinsic) apoptotic pathway by alcohol
The death receptor pathway, or the extrinsic pathway, is mainly initiated by the binding of the death receptor ligands to the death receptors. Death receptors are cell surface cytokine receptors mainly belong to the TNF receptor super-family protein that has almost 30 different members sharing sequence homology in the cysteine-rich extracellular domains. These receptors also share additional sequence homology at the intracellular death domain, which include TNF-receptor 1 (TNFR1), Fas, TRAIL-receptor 1 (DR-4), and TRAIL receptor 2 (DR5), the ligands of which are TNFα, FasL and TRAIL, respectively (Yin and Ding, 2003).
The intracellular events could be quite different upon different ligand/receptor interactions. Some signaling events are quite conserved leading to executioner caspase activation, as exemplified in the Fas-mediated cell death. When FasL or the agonistic antibody binds with Fas, it triggers the homotrimeric association of the receptors. The clustering of the death domain in the intracellular portion of the receptors recruits FADD (Fas-associated protein with death domain) and caspase-8 to form the so called death-inducing signaling complex (DISC), which induces the activation of caspase-8. It should be noted that the FLICE (FADD-like IL-1β-converting enzyme)-like inhibitory protein long (FLIPl), a caspase-8 inhibitory protein, is also recruited to the DISC by binding to the death effector domain (DED) of FADD. Activated caspase-8 then cleaves and activates downstream effector caspases such as caspase-3 to trigger apoptosis in Type I cells such as lymphocytes (Yin and Ding, 2003). However, in some cell types (Type II cells), such as hepatocytes, the direct activation of caspase-3 by caspase-8 is weak and not sufficient to induce apoptosis (Yin and Ding, 2003, Ding and Yin, 2004). Activated caspase-8 also cleaves Bid, a BH3-only proapoptotic Bcl-2 family protein. The cleaved truncated Bid (tBid) then translocates to mitochondria and activates the mitochondrial apoptotic pathway by inducing release of mitochondria apoptotic factors, such as cytochrome c and Smac/DIABLO (Yin et al., 1999b). Once in the cytosol, cytochrome c then promotes caspase-9 activation that further induce caspase-3 activation whereas Smac/DIABLO release the inhibition of XIAP on caspase activation (Li et al., 1997, Du et al., 2000, Verhagen et al., 2000).
Compared with FasL-induced apoptotic signaling pathways, TNFα-induced caspase activation and apoptosis follows a little bit more complicated different course. After TNF-α binds to TNF-α receptor 1 (TNFR1), it forms the TNFR complex I by recruiting downstream factors such as TNFR-associated death domain (TRADD), receptor interacting protein kinase 1 (RIP1), TNFR-associated factor 2 (TRAF2), and cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1/2) (Vucic et al., 2011, Zhou et al., 2012). RIP1 is ubiquitinated by the E3 ligases cIAP1/2 and a secondary E3 ligase complex of LUBAC (linear ubiquitin chain assembly complex). Ubiquitinated RIP1 then recruits transforming growth factor β-activated kinase 1 (TAK1) and the ubiquitin binding partners TAB2 (TAK1 binding protein 2) and TAB3 that serves as a platform for the downstream IκB kinase (IKK) complex to activate the NF-κB pathway, which upregulates the expression of genes for cell survival and inflammation (Zhou et al., 2012, Vucic et al., 2011). In contrast, cylindromatosis (CLYD) de-ubiquitinates RIP1, which then recruits TRADD, the Fas-associated protein with a death domain (FADD), FLIPl and caspase-8 to form the pro-death complex II to initiate caspase-8 activation. FLIPl is transcriptionally regulated by NF-κB and acts as a negatively regulator on caspase-8 activation. Similar to FasL-induced apoptosis, activated caspase-8 then cleaves Bid and tBid translocates to mitochondria resulting in the activation of the mitochondrial-apoptotic pathway (Ding and Yin, 2004).
It is reported that acute alcohol exposure to mice increased Fas ligand-mediated apoptosis, which could be inhibited by the treatment with zinc (Lambert et al., 2003). Recently it was shown that chronic alcohol increased expression of microRNA 21 (miR-21) that negatively regulates the expression of Fas ligand (TNF superfamily, member 6) (FASLG) and death receptor 5 (DR5) in the mouse livers. Inhibition of miR-21 by specific Vivo-Morpholino in ethanol-treated mice increased the expression of DR5 and FASLG and exacerbated ethanol-induced liver injury. These findings suggest that miR-21 may act as an adaptive protective mechanism against alcohol-induced apoptosis (Francis et al., 2014). In addition to Fas-mediated apoptosis, compelling evidence supports that TNFα-mediated cell death is critical in the pathogenesis of ALD (Yin et al., 1999a, Nagy et al., 2016). Accumulating evidence indicates that acute or chronic alcohol exposure increases intestinal permeability and elevated systemic levels of gut-derived endotoxins and other microbial products (Hartmann et al., 2015). Endotoxin (LPS) activates Kupffer cells (the resident macrophage in the liver) to induce the production of TNFα and subsequent TNFα-mediated apoptosis in the liver. In addition to inducing apoptosis in hepatocytes, alcohol exposure also induces apoptosis and inflammation in adipose tissue. Intriguingly, while Bid-deficient mice are found to be resistant to alcohol-induced apoptosis in both the adipose and liver tissue, Bid-deficient mice are not protected from alcohol-induced liver injury and steatosis (Sebastian et al., 2011, Roychowdhury et al., 2012). Moreover, administration of VX166 (a pan-caspase inhibitor) also fails to attenuate alcohol-induced liver injury and steatosis (Roychowdhury et al., 2012). Taken together, while it is clear that hepatoccyte apoptosis contributes to alcohol-induced liver injury through both mitochondrial and death receptor-mediated apoptotic pathways, other forms of cell death are also involved in the pathogenesis of ALD in addition to apoptosis.
Necrosis and necroptosis in ALD
Necrosis is characterized by cell swelling, membrane rupture, and release of cell contents that leads to a subsequent inflammatory response (Malhi et al., 2010, Malhi et al., 2006). However, recent evidence suggests that necrosis can also be highly regulated, which involves the RIP1-RIP3-MLKL (mixed lineage kinase domain-like protein)-mediated necrotic cascade, a process also referred to as necroptosis or programmed necrosis (Degterev et al., 2005, Zhang et al., 2009, Cho et al., 2009, He et al., 2009). Necroptosis is similar in nature to necrosis, but is a caspase-independent programmed form of cell death that requires initiation by death receptors, similar to the extrinsic apoptotic pathway. As we discussed above, upon TNFα binding to its receptor TNFR1, it forms several complexes by recruiting different components to either trigger NFκB pathway or induce apoptosis depending on the ubiquitination levels of RIP1. Highly ubiquitinated RIP1 activates NFκB-mediated cell survival and inflammatory pathways whereas de-ubiquitinated RIP1 form the pro-death complex II resulting the activation of caspase-8. RIP3 can also be recruited to complex II to form complex IIb (also called the necrosome), which includes RIP1, RIP3, and MLKL.
Interestingly, activated caspase-8 also cleaves RIP3 and RIP1 to inactivate them, suggesting that induction of apoptosis can suppress necroptosis (Vandenabeele et al., 2010). When caspase-8 activity is inhibited either by a genetic defect in FADD-caspase-8 signaling or by pharmacological inhibition of caspase-8, RIP1 interacts with RIP3 via their RIP homotypic interaction motif (RHIM) and forms an amyloid-like structure termed the necrosome, which is stabilized by phosphorylated RIP1 and RIP3. Activated RIP3 then recruits and phosphorylates MLKL protein to promote its oligomerization and translocation to plasma membranes resulting in eventual membrane rupture and necrosis (Weinlich and Green, 2014, Vanden Berghe et al., 2014). Intriguingly, in the absence of cIAPs, RIP1, RIP3, FADD, caspase-8 and FLIPL form a large complex known as the ripoptosome (also called complex IIb), which activates caspase-8 to trigger apoptosis (Dillon et al., 2014, Mandal et al., 2014, Tenev et al., 2011). Notably, RIP1 knockout mice die perinatally whereas RIP3 knockout mice are viable without obvious phenotypes, suggesting that RIP1 may have a paradoxical cell survival role (Weinlich and Green, 2014). The kinase-dead RIP3D161N mice die at E10.5 whereas kinase-dead RIP1D138N mice are viable and healthy. The kinase-dead RIP3D161N mice are protected by germline deletion of caspase-8 but not by the kinase-dead RIP1D138N, suggesting that formation of the ripoptosome seems to be independent of RIP1 and RIP3 kinase activity but dependent on their scaffolding functions (Newton et al., 2014). Therefore, it appears that RIP1 and RIP3 may have multiple roles in regulating apoptosis, necroptosis, cell survival and inflammation (Figure 1).
RIP3-mediated necroptosis has recently been shown to play a role in ALD (Roychowdhury et al., 2013). Nagy and colleagues showed that RIP3 was induced by ethanol feeding in mouse livers. ALD patients had increased hepatic expression of RIP3 compared to control patients. Furthermore, ethanol-induced liver injury, steatosis, and inflammation were decreased in RIP3 KO mice compared to control mice, verifying the importance of RIP3 in mediating ethanol-induced liver injury and progression of ALD (Roychowdhury et al., 2013). We recently confirmed these findings using the chronic ethanol feeding plus acute binge model (Gao-binge model). We found that Gao-binge alcohol treatment decreased protein levels of proteasome subunit alpha type-2 (PSMA2) and proteasome 26S subunit ATPase 1 (PSMC1) resulting in reduced hepatic proteasome function. Genetic depletion of hepatic PSMC1 or pharmacological inhibition of proteasome by the proteasome inhibitor bortezomib also increased protein level of RIP3. These data suggest that RIP3 is degraded through ubiquitin proteasome system and impaired proteasomal function induced by Gao-binge alcohol treatment may account for the hepatic accumulation of RIP3. Notably, human patients with ALD also showed increased RIP3 expression and decreased PSMC1 expression in the liver (Wang et al., 2016). Intriguingly, inhibition of RIP1 kinase activity by 7-Cl-O-Nec-1 (a potent specific RIP1 inhibitor) blunted Gao-binge alcohol-induced hepatic inflammation but did not protect against chronic ethanol feeding-induced steatosis and liver injury, suggesting that alcohol-induced RIP3-mediated necroptosis is independent of RIP1. Therefore it seems that RIP3 plays a more critical role in alcohol-induced steatosis and cell death whereas RIP1 is more important in regulating inflammation.
It should be noted that RIP1 and RIP3 are predominantly expressed in the thymus and spleen, and their expression levels in the liver are relatively low (He et al., 2009, Dara et al., 2015, Wang et al., 2016). In addition to alcohol, acetaminophen treatment also increased hepatic RIP3 proteins and RIP3 contributes to the early phase of acetaminophen-induced liver injury (Ramachandran et al., 2013). Another recent study also reported that mice with knockdown of hepatic RIP1 by using a RIP1 antisense are resistant to acetaminophen-induced liver injury (Dara et al., 2015). These data thus support that RIP1-RIP3-mediated necroptosis is pathologically and physiologically relevant in liver diseases. However, the liver often has increased infiltration of inflammatory cells after either alcohol or acetaminophen exposure, which makes it difficult to conclude that the increased levels RIP3 are from hepatocytes or from the inflammatory cells. Future work to use liver-specific RIP1 KO or RIP3 KO mice that fed with alcohol may be able to help to further clarify the role of RIP1 and RIP3 in alcohol-induced necrosis and liver pathogenesis.
Since apoptosis normally suppresses necrosis by caspase-mediated cleavage of RIP3, it is intriguing that both apoptosis and necrosis occur during the pathogenesis of ALD. It is well known that liver has the unique zones that have different levels of oxygen, nutrient and metabolic enzymes. We previously demonstrated that autophagy induction differs in different zones of the mouse liver after acetaminophen administration (Ni et al., 2013), it is possible that apoptosis and necrosis may also occur in different zones of the liver. Immunohistochemistry co-staining for apoptotic (such as activated caspase-3) and necrotic markers (such as RIP3 and HMGB1) may help to confirm this hypothesis. Moreover, it is also likely that different cell death modes may occur during different stages of liver pathogenesis of ALD. For instance, it is possible that apoptosis occurs in early ALD, such as in steatosis, and necrosis occurs in later stages of ALD, such as in alcoholic hepatitis. Future work is needed to further elucidate these possibilities.
Other forms of regulated programmed cell death in ALD
Pyroptosis
Pyroptosis is a type of programmed cell death that depends on the activation of caspase-1, a pro-inflammatory caspase which is not required for apoptosis to occur (Miao et al., 2010). Pyroptosis is associated with cell swelling and rapid plasma membrane lysis due to the formation of pores on the plasma membrane in a caspase-1-dependent manner. Similar to apoptosis, cells undergo pyroptosis also have extensive nuclear DNA fragmentation but the underlying mechanism of DNA fragmentation for pyroptosis does not depend on CAD, the DNase activated by apoptotic caspases. Caspase-1 is activated at complexes termed inflammasome. It has been reported that chronic alcohol exposure activated inflammasome in mouse livers. Thus it is likely that alcohol may also induce caspase-1 mediated pyropotsis in the liver (Petrasek et al., 2012).
Ferroptosis
Ferroptosis is a regulated form of necrosis that is morphologically, biochemically, and genetically distinct from apoptosis, necrosis, and autophagy (Yang and Stockwell, 2016). Ferroptosis depends upon intracellular iron, but not other metals (Dixon et al., 2012). Ferropotsis is associated with plasma membrane rupture due to lipid peroxidation and is driven by loss of activity of the lipid repair enzyme glutathione peroxidase 4 (GPX4) (Yang et al., 2014). Either lipophilic antioxidants or iron chelators can strongly suppress ferroptosis, revealing the important roles of ROS and cellular iron in ferroptosis (Yang and Stockwell, 2016). Iron overload and increased oxidative stress have been well documented in ALD (Williams et al., 2014, Nagy et al., 2016, Kohgo et al., 2007). Therefore it will be interesting to determine whether ferroptosis would play a role in the pathogenesis of ALD.
Autophagy and cell death in ALD
In addition to the mutual regulation of apoptosis and necrosis, accumulating evidence indicates that autophagy and cell death can also regulate each other (Kroemer G et al. 2010, Luo et al. 2010). In fact, many cell death stimuli induce both cell death (apoptosis and necrosis) and autophagy at the same time or even at the same cell. Autophagy involves the formation of the double-membrane autophagosomes that traffic and fuse with lysosomes to form autolysosomes where the autophagic cargos are degraded. Autophagy generally acts as a pro-survival mechanism and plays a critical role in normal liver physiology and liver diseases (Czaja et al., 2013, Yin et al., 2008). Autophagy may regulate alcohol-induced cell death and protect against the pathogenesis of ALD in the following three aspects. First, as discussed above, mitochondrion is a central executioner for regulating apoptosis by controlling the release of apoptotic factors, and mitochondrial damage plays a key role in alcohol-induced cell death. Therefore, it is not surprising that removing alcohol-induced damaged mitochondria by mitophagy can protect against alcohol-induced liver pathogenesis. Increasing evidence indicates that Pink1 (tensin homolog-induced putative kinase 1)-Parkin axis plays a critical role in selective mitophagy in mammalian cells (Ding and Yin, 2012). Parkin is an evolutionarily conserved E3 ligase, which is recruited to depolarized damaged mitochondria by Pink1 to initiate ubiquitination of mitochondrial outer membrane proteins and subsequent mitochondrial degradation by mitophagy (Ding and Yin, 2012). Gao-binge alcohol treatment increased mitochondrial translocation of Parkin in hepatocytes. More importantly, alcohol caused greater mitochondrial damage, steatosis and liver injury in Parkin knockout mouse livers compared to wild type mouse livers (Williams et al., 2015). Second, autophagy also helps to remove alcohol-induced excess lipid droplet (lipophagy) to ameliorate alcohol-induced steatosis. We previously demonstrated that pharmacological induction of autophagy markedly reduced alcohol-induced steatosis and liver injury, and the autophagsomes are often found to enwrap lipid droplets in acute alcohol-treated mouse livers (Ding et al., 2010). Similar findings were also reported in chronic ethanol fed rats for 10 weeks (Eid et al., 2013). Third, autophagy removes protein aggregates and relieves ER stress in cultured cells (Ding et al., 2007). Since alcohol induces ER stress, it is likely that autophagy can also help to attenuate alcohol-induced ER stress and subsequent cell death. However, no studies have been conducted to test this hypothesis.
In addition to hepatocytes, autophagy in other cell types such as hepatic stellate cells (HSC) and macrophages in the liver also plays a critical role in the pathogenesis of ALD. Autophagy in HSC promotes liver fibrosis by increasing lipid droplet degradation via lipophagy resulting in HSC activation. Stellate cell-specific Atg7 knockout mice are resistant to CCl4-induced fibrosis in vivo (Hernandez-Gea et al., 2012). In rats that were fed with chronic ethanol diet for 8 weeks, it is found that alcohol-induced ER stress activates autophagy, which may promote chronic ethanol-induced fibrosis (Hernandez-Gea et al., 2013). In contrast to HSC, macrophage-specific autophagy-deficient mice are more susceptible to CCl4-induced fibrosis and endotoxin-induced liver injury (Lodder et al., 2015, Ilyas et al., 2015). Whether impaired macrophage autophagy would also exacerbate alcohol-induced liver injury remains to be determined.
While autophagy can protect against cell death, apoptosis can also suppress autophagy by inducing caspase-mediated cleavage of essential autophagy proteins such as Beclin 1 (Li et al., 2011). In addition, RIP1 also activates ERK and represses basal autophagy by inhibiting TFEB-mediated expression of autophagy-related and lysosomal genes (Yonekawa et al., 2015). Therefore, there is a complicated mutual regulatory network among autophagy, apoptosis and necroptosis. After alcohol exposure, a cell's fate is decided by the balance of autophagy versus apoptosis/necroptosis. Disruption of the balance from cell survival autophagy towards cell injury (apoptosis/necroptosis) will eventually lead to liver injury after alcohol exposure.
Concluding remarks and future perspective
ALD is a major health problem in the United States and worldwide with no successful treatments. Alcohol consumption can activate cell death and cell adaptive survival pathways such as autophagy in the liver. Thus the balance between cell death and autophagy may decide the pathogenesis of ALD. While apoptosis in ALD has been well documented, emerging evidence supports that RIP1-RIP3-mediated necroptosis also contributes to alcohol-induced steatosis, inflammation and liver injury. Because apoptosis, necrosis and autophagy are interlinked and all of them are involved in the pathogenesis of ALD, an ideal development to treat ALD should consider target all of them. Future works are needed to determine whether simultaneously inhibition of apoptosis and necroptosis with concomitant induction of autophagy would offer the maximized beneficial effects against alcohol-induced liver injury.
Acknowledgments
The research was supported in part by the NIAAA funds R01 AA020518, R01 DK102142, National Center for Research Resources P20RR021940, the National Institute of General Medical Sciences P20 GM103549, and the Cystic Fibrosis Foundation (RCD).
List of Abbreviations
- ACC
acetyl-CoA carboxylase
- ADH
alcohol dehydrogenase
- AIF
apoptosis inducing factor
- ALD
Alcoholic liver disease
- ALDH1
aldehyde dehydrogenase 1
- AMPK
AMP-activated protein kinase
- ATF6
activating transcription factor 6
- cIAP1
cellular inhibitor of apoptosis proteins 1
- Cyp2E1
cytochrome P450 family 2, subfamily E, polypeptide 1
- CPTI
carnitine palmitoyltransferase I
- CYLD
cylindromatosis
- DIABLO
second mitochondria-derived activator of caspase
- ER
endoplasmic reticulum
- ERAD
ER-associated protein degradation
- FAEE
fatty acid ethyl ester
- FLIPL
FLICE (FADD-like IL-1β-converting enzyme)-like inhibitory protein large
- FADD
Fas-associated protein with a death domain
- GSH
glutathione
- HSC
hepatic stellate cells
- IRE1 α
inositol requiring enzyme 1 α
- IAPs
inhibitor of apoptosis proteins
- IRF3
interferon regulator factor 3
- KO
knockout
- LPS
lipopolysaccharide
- MLKL
mixed lineage kinase domain-like protein
- NAD
nicotinamide adenine dinucleotide
- PERK
PKR-like ER localized eIF2α kinase
- PGC-1α
PPARγ co-activator-1α
- PPARα
peroxisome proliferator-activated receptor α
- ROS
reactive oxygen species
- SREBP-1
sterol regulatory element-binding protein-1
- STING
stimulator of interferon genes
- RIP
receptor-interacting protein kinase
- TNFR1
TNF-α receptor 1
- TRADD
TNFR-associated death domain, TRAF2, TNFR-associated factor 2
- TAK1
transforming growth factor β-activated kinase 1
- TAB2
TAK1 binding protein 2
- TNF-α
tumor necrosis factor-α
- UPR
unfolded protein response
Footnotes
All the authors have no conflict of interest to claim.
References
- Bailey SM, Cunningham CC. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med. 2002;32:11–16. doi: 10.1016/s0891-5849(01)00769-9. [DOI] [PubMed] [Google Scholar]
- Beckemeier ME, Bora PS. Fatty acid ethyl esters: potentially toxic products of myocardial ethanol metabolism. Journal of molecular and cellular cardiology. 1998;30:2487–2494. doi: 10.1006/jmcc.1998.0812. [DOI] [PubMed] [Google Scholar]
- Bourogaa E, Nciri R, Mezghani-Jarraya R, Racaud-Sultan C, Damak M, El Feki A. Antioxidant activity and hepatoprotective potential of Hammada scoparia against ethanol-induced liver injury in rats. J Physiol Biochem. 2013;69:227–237. doi: 10.1007/s13105-012-0206-7. [DOI] [PubMed] [Google Scholar]
- Casey CA, Kragskow SL, Sorrell MF, Tuma DJ. Chronic ethanol administration impairs the binding and endocytosis of asialo-orosomucoid in isolated hepatocytes. J Biol Chem. 1987;262:2704–2710. [PubMed] [Google Scholar]
- Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman WB, Cunningham CC. Effect of chronic ethanol consumption on hepatic mitochondrial transcription and translation. Biochim Biophys Acta. 1991;1058:178–186. doi: 10.1016/s0005-2728(05)80235-x. [DOI] [PubMed] [Google Scholar]
- Crabb DW, Matsumoto M, Chang D, You M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc Nutr Soc. 2004;63:49–63. doi: 10.1079/pns2003327. [DOI] [PubMed] [Google Scholar]
- Czaja MJ, Ding WX, Donohue TM, Jr, Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, Perlmutter DH, Randall G, Ray RB, Tsung A, Yin XM. Functions of autophagy in normal and diseased liver. Autophagy. 2013;9:1131–1158. doi: 10.4161/auto.25063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalke DD, Sorrell MF, Casey CA, Tuma DJ. Chronic ethanol administration impairs receptor-mediated endocytosis of epidermal growth factor by rat hepatocytes. Hepatology. 1990;12:1085–1091. doi: 10.1002/hep.1840120502. [DOI] [PubMed] [Google Scholar]
- Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D, Kaplowitz N. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology. 2015 doi: 10.1002/hep.27939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Dillon CP, Weinlich R, Rodriguez DA, Cripps JG, Quarato G, Gurung P, Verbist KC, Brewer TL, Llambi F, Gong YN, Janke LJ, Kelliher MA, Kanneganti TD, Green DR. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157:1189–1202. doi: 10.1016/j.cell.2014.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Li M, Chen X, Ni HM, Lin CW, Gao W, Lu B, Stolz DB, Clemens DL, Yin XM. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139:1740–1752. doi: 10.1053/j.gastro.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Yin XM. Dissection of the multiple mechanisms of TNF-alpha-induced apoptosis in liver injury. J Cell Mol Med. 2004;8:445–454. doi: 10.1111/j.1582-4934.2004.tb00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008;4:141–150. doi: 10.4161/auto.5190. [DOI] [PubMed] [Google Scholar]
- Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393:547–564. doi: 10.1515/hsz-2012-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Eid N, Ito Y, Maemura K, Otsuki Y. Elevated autophagic sequestration of mitochondria and lipid droplets in steatotic hepatocytes of chronic ethanol-treated rats: an immunohistochemical and electron microscopic study. Journal of molecular histology. 2013;44:311–326. doi: 10.1007/s10735-013-9483-x. [DOI] [PubMed] [Google Scholar]
- Francis H, McDaniel K, Han Y, Liu X, Kennedy L, Yang F, McCarra J, Zhou T, Glaser S, Venter J, Huang L, Levine P, Lai JM, Liu CG, Alpini G, Meng F. Regulation of the extrinsic apoptotic pathway by microRNA-21 in alcoholic liver injury. J Biol Chem. 2014;289:27526–27539. doi: 10.1074/jbc.M114.602383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Ruiz C, Kaplowitz N, Fernandez-Checa JC. Role of Mitochondria in Alcoholic Liver Disease. Curr Pathobiol Rep. 2013;1:159–168. doi: 10.1007/s40139-013-0021-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D, Ybanez MD, Johnson HS, McDonald JN, Mesropyan L, Sancheti H, Martin G, Martin A, Lim AM, Dara L, Cadenas E, Tsukamoto H, Kaplowitz N. Dynamic adaptation of liver mitochondria to chronic alcohol feeding in mice: biogenesis, remodeling, and functional alterations. J Biol Chem. 2012;287:42165–42179. doi: 10.1074/jbc.M112.377374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haorah J, MacDonald RG, Stoner JA, Donohue TM., Jr Ethanol consumption decreases the synthesis of the mannose 6-phosphate/insulin-like growth factor II receptor but does not decrease its messenger RNA. Biochemical pharmacology. 2003;65:637–648. doi: 10.1016/s0006-2952(02)01605-2. [DOI] [PubMed] [Google Scholar]
- Haorah J, McVicker DL, Byrd JC, MacDonald RG, Donohue TM., Jr Chronic ethanol administration decreases the ligand binding properties and the cellular content of the mannose 6-phosphate/insulin-like growth factor II receptor in rat hepatocytes. Biochemical pharmacology. 2002;63:1229–1239. doi: 10.1016/s0006-2952(02)00877-8. [DOI] [PubMed] [Google Scholar]
- Hartmann P, Seebauer CT, Schnabl B. Alcoholic liver disease: the gut microbiome and liver cross talk. Alcohol Clin Exp Res. 2015;39:763–775. doi: 10.1111/acer.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Hernandez-Gea V, Ghiassi-Nejad Z, Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ, Friedman SL. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142:938–946. doi: 10.1053/j.gastro.2011.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Gea V, Hilscher M, Rozenfeld R, Lim MP, Nieto N, Werner S, Devi LA, Friedman SL. Endoplasmic reticulum stress induces fibrogenic activity in hepatic stellate cells through autophagy. J Hepatol. 2013;59:98–104. doi: 10.1016/j.jhep.2013.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi H, Adachi M, Miura S, Gores GJ, Ishii H. The mitochondrial permeability transition contributes to acute ethanol-induced apoptosis in rat hepatocytes. Hepatology. 2001;34:320–328. doi: 10.1053/jhep.2001.26380. [DOI] [PubMed] [Google Scholar]
- Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: a dysfunctional relationship. Gastroenterology. 2002;122:2049–2063. doi: 10.1053/gast.2002.33613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilyas G, Zhao E, Liu K, Lin Y, Tesfa L, Tanaka KE, Czaja MJ. Macrophage autophagy limits acute toxic liver injury in mice through down regulation of interleukin-1beta. J Hepatol. 2015 doi: 10.1016/j.jhep.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C, Kaplowitz N. Betaine decreases hyperhomocysteinemia, endoplasmic reticulum stress, and liver injury in alcohol-fed mice. Gastroenterology. 2003;124:1488–1499. doi: 10.1016/s0016-5085(03)00276-2. [DOI] [PubMed] [Google Scholar]
- Ji C, Mehrian-Shai R, Chan C, Hsu YH, Kaplowitz N. Role of CHOP in hepatic apoptosis in the murine model of intragastric ethanol feeding. Alcohol Clin Exp Res. 2005;29:1496–1503. doi: 10.1097/01.alc.0000174691.03751.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharbanda KK, McVicker DL, Zetterman RK, Donohue TM., Jr Ethanol consumption alters trafficking of lysosomal enzymes and affects the processing of procathepsin L in rat liver. Biochim Biophys Acta. 1996;1291:45–52. doi: 10.1016/0304-4165(96)00043-8. [DOI] [PubMed] [Google Scholar]
- Kharbanda KK, McVicker DL, Zetterman RK, MacDonald RG, Donohue TM., Jr Flow cytometric analysis of vesicular pH in rat hepatocytes after ethanol administration. Hepatology. 1997;26:929–934. doi: 10.1002/hep.510260419. [DOI] [PubMed] [Google Scholar]
- King AL, Swain TM, Mao Z, Udoh US, Oliva CR, Betancourt AM, Griguer CE, Crowe DR, Lesort M, Bailey SM. Involvement of the mitochondrial permeability transition pore in chronic ethanol-mediated liver injury in mice. Am J Physiol Gastrointest Liver Physiol. 2014;306:G265–277. doi: 10.1152/ajpgi.00278.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohgo Y, Ikuta K, Ohtake T, Torimoto Y, Kato J. Iron overload and cofactors with special reference to alcohol, hepatitis C virus infection and steatosis/insulin resistance. World J Gastroenterol. 2007;13:4699–4706. doi: 10.3748/wjg.v13.i35.4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Zhou Z, Kang YJ. Suppression of Fas-mediated signaling pathway is involved in zinc inhibition of ethanol-induced liver apoptosis. Exp Biol Med (Maywood) 2003;228:406–412. doi: 10.1177/153537020322800411. [DOI] [PubMed] [Google Scholar]
- Lange LG, Sobel BE. Mitochondrial dysfunction induced by fatty acid ethyl esters, myocardial metabolites of ethanol. J Clin Invest. 1983;72:724–731. doi: 10.1172/JCI111022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang P, Sun Q, Ding WX, Yin XM, Sobol RW, Stolz DB, Yu J, Zhang L. Following cytochrome c release, autophagy is inhibited during chemotherapy-induced apoptosis by caspase 8-mediated cleavage of Beclin 1. Cancer Res. 2011;71:3625–3634. doi: 10.1158/0008-5472.CAN-10-4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Lodder J, Denaes T, Chobert MN, Wan J, El-Benna J, Pawlotsky JM, Lotersztajn S, Teixeira-Clerc F. Macrophage autophagy protects against liver fibrosis in mice. Autophagy. 2015;11:1280–1292. doi: 10.1080/15548627.2015.1058473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med. 2008;44:723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhi H, Gores GJ, Lemasters JJ. Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology. 2006;43:S31–44. doi: 10.1002/hep.21062. [DOI] [PubMed] [Google Scholar]
- Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev. 2010;90:1165–1194. doi: 10.1152/physrev.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, Lich JD, Finger J, Kasparcova V, Votta B, Ouellette M, King BW, Wisnoski D, Lakdawala AS, DeMartino MP, Casillas LN, Haile PA, Sehon CA, Marquis RW, Upton J, Daley-Bauer LP, Roback L, Ramia N, Dovey CM, Carette JE, Chan FK, Bertin J, Gough PJ, Mocarski ES, Kaiser WJ. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell. 2014;56:481–495. doi: 10.1016/j.molcel.2014.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy LE, Ding WX, Cresci G, Saikia P, Shah VH. Linking Pathogenic Mechanisms of Alcoholic Liver Disease With Clinical Phenotypes. Gastroenterology. 2016 doi: 10.1053/j.gastro.2016.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, Komuves L, Ferrando RE, French DM, Webster J, Roose-Girma M, Warming S, Dixit VM. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science. 2014;343:1357–1360. doi: 10.1126/science.1249361. [DOI] [PubMed] [Google Scholar]
- Ni HM, Williams JA, Jaeschke H, Ding WX. Zonated induction of autophagy and mitochondrial spheroids limits acetaminophen-induced necrosis in the liver. Redox Biol. 2013;1:427–432. doi: 10.1016/j.redox.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, Barrieau M, Min SY, Kurt-Jones EA, Szabo G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitisin mice. J Clin Invest. 2012;122:3476–3489. doi: 10.1172/JCI60777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, Fitzgerald KA, Szabo G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis inearly alcoholic liver disease. Proc Natl Acad Sci U S A. 2013;110:16544–16549. doi: 10.1073/pnas.1308331110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, McGill MR, Xie Y, Ni HM, Ding WX, Jaeschke H. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology. 2013;58:2099–2108. doi: 10.1002/hep.26547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychowdhury S, Chiang DJ, Mandal P, McMullen MR, Liu X, Cohen JI, Pollard J, Feldstein AE, Nagy LE. Inhibition of apoptosis protects mice from ethanol-mediated acceleration of early markers of CCl4 -induced fibrosis but not steatosis or inflammation. Alcohol Clin Exp Res. 2012;36:1139–1147. doi: 10.1111/j.1530-0277.2011.01720.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57:1773–1783. doi: 10.1002/hep.26200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian BM, Roychowdhury S, Tang H, Hillian AD, Feldstein AE, Stahl GL, Takahashi K, Nagy LE. Identification of a cytochrome P4502E1/Bid/C1q-dependent axis mediating inflammation in adipose tissue after chronic ethanol feeding to mice. J Biol Chem. 2011;286:35989–35997. doi: 10.1074/jbc.M111.254201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumptner C, Omary MB, Fickert P, Denk H, Zatloukal K. Hepatocyte cytokeratins are hyperphosphorylated at multiple sites in human alcoholic hepatitis and in a mallory body mouse model. Am J Pathol. 2000;156:77–90. doi: 10.1016/S0002-9440(10)64708-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, MacFarlane M, Cain K, Meier P. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43:432–448. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- Vandenabeele P, Declercq W, Van Herreweghe F, Vanden Berghe T. The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci Signal. 2010;3:re4. doi: 10.1126/scisignal.3115re4. [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Vucic D, Dixit VM, Wertz IE. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat Rev Mol Cell Biol. 2011;12:439–452. doi: 10.1038/nrm3143. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang S, Ni HM, Dorko K, Kumer SC, Schmitt TM, Nawabi A, Komatsu M, Huang H, Ding WX. Increased hepatic receptor interacting protein kinase 3 expression due to impaired proteasomal functions contributes to alcohol-induced steatosis and liver injury. Oncotarget. 2016 doi: 10.18632/oncotarget.6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinlich R, Green DR. The two faces of receptor interacting protein kinase-1. Mol Cell. 2014;56:469–480. doi: 10.1016/j.molcel.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner J, Saghir M, Warshaw AL, Lewandrowski KB, Laposata M, Iozzo RV, Carter EA, Schatz RJ, Fernandez-Del Castillo C. Alcoholic pancreatitis in rats: injury from nonoxidative metabolites of ethanol. Am J Physiol Gastrointest Liver Physiol. 2002;283:G65–73. doi: 10.1152/ajpgi.00419.2001. [DOI] [PubMed] [Google Scholar]
- Williams JA, Ding WX. Targeting Pink1-Parkin-mediated mitophagy for treating liver injury. Pharmacol Res. 2015;102:264–269. doi: 10.1016/j.phrs.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Manley S, Ding WX. New advances in molecular mechanisms and emerging therapeutic targets in alcoholic liver diseases. World J Gastroenterol. 2014;20:12908–12933. doi: 10.3748/wjg.v20.i36.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Ni HM, Ding Y, Ding WX. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am J Physiol Gastrointest Liver Physiol. 2015;309:G324–340. doi: 10.1152/ajpgi.00108.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Bhopale KK, Ansari GA, Kaphalia BS. Ethanol-induced cytotoxicity in rat pancreatic acinar AR42J cells: role of fatty acid ethyl esters. Alcohol Alcohol. 2008;43:1–8. doi: 10.1093/alcalc/agm044. [DOI] [PubMed] [Google Scholar]
- Wu H, Cai P, Clemens DL, Jerrells TR, Ansari GA, Kaphalia BS. Metabolic basis of ethanol-induced cytotoxicity in recombinant HepG2 cells: role of nonoxidative metabolism. Toxicology and applied pharmacology. 2006;216:238–247. doi: 10.1016/j.taap.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, Brown LM, Girotti AW, Cornish VW, Schreiber SL, Stockwell BR. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016;26:165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, Thurman RG. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology. 1999a;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- Yin XM, Ding WX. Death receptor activation-induced hepatocyte apoptosis and liver injury. Curr Mol Med. 2003;3:491–508. doi: 10.2174/1566524033479555. [DOI] [PubMed] [Google Scholar]
- Yin XM, Ding WX, Gao W. Autophagy in the liver. Hepatology. 2008;47:1773–1785. doi: 10.1002/hep.22146. [DOI] [PubMed] [Google Scholar]
- Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA, Korsmeyer SJ. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999b;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- Yonekawa T, Gamez G, Kim J, Tan AC, Thorburn J, Gump J, Thorburn A, Morgan MJ. RIP1 negatively regulates basal autophagic flux through TFEB to control sensitivity to apoptosis. EMBO Rep. 2015;16:700–708. doi: 10.15252/embr.201439496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakhari S. Overview: how is alcohol metabolized by the body? Alcohol research & health : the journal of the National Institute on Alcohol Abuse and Alcoholism. 2006;29:245–254. [PMC free article] [PubMed] [Google Scholar]
- Zatloukal K, French SW, Stumptner C, Strnad P, Harada M, Toivola DM, Cadrin M, Omary MB. From Mallory to Mallory-Denk bodies: what, how and why? Exp Cell Res. 2007;313:2033–2049. doi: 10.1016/j.yexcr.2007.04.024. [DOI] [PubMed] [Google Scholar]
- Zelickson BR, Benavides GA, Johnson MS, Chacko BK, Venkatraman A, Landar A, Betancourt AM, Bailey SM, Darley-Usmar VM. Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes. Biochim Biophys Acta. 2011;1807:1573–1582. doi: 10.1016/j.bbabio.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelner I, Matlow JN, Natekar A, Koren G. Synthesis of fatty acid ethyl esters in mammalian tissues after ethanol exposure: a systematic review of the literature. Drug metabolism reviews. 2013;45:277–299. doi: 10.3109/03602532.2013.795584. [DOI] [PubMed] [Google Scholar]
- Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Ramshesh VK, Rehman H, Liu Q, Theruvath TP, Krishnasamy Y, Lemasters JJ. Acute ethanol causes hepatic mitochondrial depolarization in mice: role of ethanol metabolism. PLoS One. 2014;9:e91308. doi: 10.1371/journal.pone.0091308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Han V, Han J. New components of the necroptotic pathway. Protein Cell. 2012;3:811–817. doi: 10.1007/s13238-012-2083-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]