

Abstract
Mutation is the ultimate source of genetic variation, and knowledge of mutation rates is fundamental for our understanding of all evolutionary processes. High throughput sequencing of mutation accumulation lines has provided genome wide spontaneous mutation rates in a dozen model species, but estimates from nonmodel organisms from much of the diversity of life are very limited. Here, we report mutation rates in four haploid marine bacterial-sized photosynthetic eukaryotic algae; Bathycoccus prasinos, Ostreococcus tauri, Ostreococcus mediterraneus, and Micromonas pusilla. The spontaneous mutation rate between species varies from μ = 4.4 × 10−10 to 9.8 × 10−10 mutations per nucleotide per generation. Within genomes, there is a two-fold increase of the mutation rate in intergenic regions, consistent with an optimization of mismatch and transcription-coupled DNA repair in coding sequences. Additionally, we show that deviation from the equilibrium GC content increases the mutation rate by ∼2% to ∼12% because of a GC bias in coding sequences. More generally, the difference between the observed and equilibrium GC content of genomes explains some of the inter-specific variation in mutation rates.
Keywords: spontaneous mutation rate, mutation accumulation, transcription-coupled DNA repair, deletion bias, GC content, GC bias, phytoplankton, Mamiellophyceae
Introduction
Mutations are responsible for genetic variation between organisms, which permits adaptation by natural selection. Thus, estimation of mutation rates (μ) is paramount for a better understanding of all evolutionary change. Because mutations are rare events, estimating their rate was difficult until recently. However, new high throughput sequencing technologies have allowed the investigation of mutations from either offspring-parent trios, in humans (Abecasis et al. 2010; Conrad et al. 2011) and mice (Adewoye et al. 2015; Uchimura et al. 2015), or mutation accumulation (MA) experiments (Halligan and Keightley 2009; Lynch et al. 2016) in organisms such as Drosophila melanogaster (Haag-Liautard et al. 2007; Keightley et al. 2009,2014a), Arabidopsis thaliana (Ossowski et al. 2010), Caenorhabditis elegans (Denver et al. 2004, 2009, 2012), unicellular eukaryotes such as Saccharomyces cerevisiae (Wloch et al. 2001; Lang and Murray 2008; Lynch et al. 2008; Zhu et al. 2014) and bacteria such as Escherichia coli (Lee et al. 2012) and Salmonella typhimurium (Lind and Andersson 2008). These studies have revealed a large variation of spontaneous mutation rates across the tree of life from 7.6 × 10−12 in Tetrahymena thermophila (Long et al. 2016) to 1.1 × 10−8 in humans (Abecasis et al. 2010; Conrad et al. 2011).
The variation of mutation rate between species appears to be correlated to two factors—genome size (Drake 1991; Drake et al. 1998), and in particular the size of the protein coding component of the genome (Lynch 2010; Lynch et al. 2016), and effective population size (Lynch 2010; Sung et al. 2012a). Both of these correlations may arise because of the limitations that genetic drift imposes on selection to minimize the mutation rate (Lynch 2010; Sung et al. 2012a). In sexual species, selection always acts to minimize the mutation rate because a modifier of the mutation rate only stays linked with the mutations it causes for a short period of time and deleterious mutations are more prevalent than advantageous mutations, increasing the genetic load (Leigh 1973). However, genetic drift ultimately limits the degree to which the mutation rate can be reduced (Lynch 2010), because the strength of selection acting on a modifier is equal to γ*U*s, where, γ is the proportional decrease in the mutation rate, U is the genomic rate of mutation and s is the average strength of selection against deleterious mutations. If γ*U*s < 1/Ne then selection will be ineffective against the modifier and the mutation rate cannot be reduced further. Hence we expect the per site rate of mutation to depend upon the effective population size (Lynch 2010)—species with larger Ne should have lower mutation rates—and genome size—the more selected sites there are, the lower the mutation rate should be. These predictions appear to be largely upheld Lynch et al. 2016). In asexual species, selection will favor an intermediate mutation rate, which generates sufficient advantageous, while not generating too many deleterious mutations (Paland and Lynch 2006; Henry et al. 2012). However, relatively few species appear to be truly asexual.
It has also been observed that there is variation in the mutation rate within a genome at a number of different scales, from differences between chromosomes, to variation between regions on a chromosome and variation between adjacent sites (Hodgkinson and Eyre-Walker 2011; Schrider et al. 2011; Ségurel et al. 2014). As an example, the Y-chromosome in humans and chimps mutates faster than the other chromosomes (Ebersberger et al. 2002). It is also known that mitochondria has a higher mutation rate than the nuclear genome in Caenorhabditis elegans (Denver et al. 2000, 2009), Homo sapiens (Rebolledo-Jaramillo et al. 2014) and Drosophila melanogaster (Haag-Liautard et al. 2008; Keightley et al. 2009). Within chromosomes, it has been shown that nucleotide context affects the mutability of a site in Chlamydomonas reinhardtii (Ness et al. 2015b), Bacillus subtilis (Sung et al. 2015), and humans (Aggarwala and Voight 2016). In mammals, the most conspicuous effect is the high mutability of CpG dinucleotides resulting from cytosine deamination (Coulondre et al. 1978; Fryxell and Zuckerkandl 2000), which leads to an 80% reduction in the frequency of CpG dinucleotides in the human genome (Lander et al. 2001).
Gene expression also affects the rate of mutation and its effect is controversial. Martincorena et al.’s (2012) analysis of polymorphisms suggested that the mutation rate in Escherichia coli is lower in highly expressed genes. However, analysis of MA lines in Escherichia coli suggests that the mutation rate actually increases with gene expression (Chen and Zhang 2013). Such a pattern has been observed in two other species, Saccharomyces cerevisiae and humans (Polak and Arndt 2008; Park et al. 2012). This phenomenon is known as transcription-associated mutagenesis (TAM) (Kim and Jinks-Robertson 2012).
In this study, we provide the first estimates of the spontaneous mutation rate in 4 species of haploid green algae [Chlorophyta, Mamiellophyceae (Marin and Melkonian 2010)]: Ostreococcus tauri RCC4221 (Blanc-Mathieu et al. 2014), O. mediterraneus RCC2590 (Subirana et al. 2013), Micromonas pusilla RCC299 (Worden et al. 2009), and Bathycoccus prasinos RCC1105 (Moreau et al. 2012), with compact genomes containing 83–84% coding sequences. Green algae constitute one of the most important photosynthetic groups on Earth, with an ubiquitous distribution in world’s oceans (de Vargas et al. 2015; Vannier et al. 2016), and play a fundamental role in foodweb and biogeochimical cycles (Collins et al. 2014). These green algae span a large evolutionary divergence, as revealed by a high proportion of species-specific genes, and high amino-acid divergence between orthologous genes (Šlapeta et al. 2006; Jancek et al. 2008). Their genome size ranges from 13 to 21 Mb and their average GC content ranges from 48% to 63%.
Results
Mutation Rates in Mamiellophyceae
To estimate spontaneous mutation rates, we sequenced 150 mutation accumulation lines in four species of green algae (supplementary table S1, Supplementary Material online, 36–40 lines per species). All together, we found 238 single nucleotide mutations and 48 indels, summarized in table 1. The numbers of synonymous and nonsynonymous mutations are as expected if mutations are randomly distributed across sites for all species (table 2), consistent with an absence of selection against nonsynonymous mutations. We thus assume that selection played a minimal role in the pattern of mutations in these MA experiments.
Table 1.
Summary of Spontaneous Mutation Rates in Four Mamiellophyceae Species.
| Species | TotGen | G (Mb) | BS | Ins | Del | μbs−10 | μID−10 | μtot−10 |
|---|---|---|---|---|---|---|---|---|
| Ostreococcus tauri | 17,250 | 12.46 | 91 | 5 | 8 | 4.19 | 0.60 | 4.79 (3.91–5.80) |
| Ostreococcus mediterraneus | 8,380 | 13.34 | 54 | 3 | 8 | 4.92 | 1.00 | 5.92 (4.57–7.55) |
| Bathycoccus prasinos | 4,145 | 14.96 | 22 | 5 | 5 | 3.02 | 1.37 | 4.39 (3.00–6.20) |
| Micromonas pusilla | 4,994 | 20.99 | 71 | 2 | 12 | 8.15 | 1.61 | 9.76 (7.80–12.07) |
Note.—BS is the number of base-substitution mutations, Ins the number of insertions and Del the number of deletions. G is the genome size in Mb and μ the mutation rate per nucleotide per genome per generation. TotGen is the total number of generations accumulated per species. Confidence interval for μtot is given under the assumption of a Poisson distribution of the mutations.
Table 2.
Mutation Rate Variation between Coding and Intergenic Sequences.
| Species | % coding genome | μ × 10−10 coding regions | μ × 10−10 intergenic regions | Numbers of mutations syn:nonsyn |
|---|---|---|---|---|
| O. tauri | 81.6 | 3.9 | 8.9 | 19:42 |
| O. mediterraneus | 84.4 | 5.0 | 11.7 | 9:34 |
| B. prasinos | 83.1 | 3.4 | 14.7 | 5:10 |
| M. pusilla | 81.9 | 8.2 | 16.1 | 15:41 |
Note.—The bias of mutation toward intergenic sequences is significant (Chi-squared test, P-value < 0.01). Syn and nonsyn are the synonymous and nonsynonymous point mutations.
The base substitution mutation rate (μbs) and the insertion-deletion mutation rate (μID) per nucleotide per generation were estimated on callable sites (G*), which represented 97–99% of the complete genome sequence (supplementary table S2, Supplementary Material online). There is no difference in G* between coding and intergenic regions. The total mutation rate, μtot, is the sum of μbs and μID. Mutation rates varied over 2-fold from 4.4 × 10−10 mutations per site per generation in B. prasinos to 9.8 × 10−10 in M. pusilla. The complete list of mutations for each of the four MA experiments is provided in supplementary tables S3–S8, Supplementary Material online. There is no significant difference in mutation rates between chromosomes (Chi-Squared test, ns).
The number of plastic genomes per cell is known to depend on tissue type in plants (Ma and Li 2015) and on growth and redox status in the freshwater green alga C. reinhardtii (Lau et al. 2000). The coverage of chloroplastic (cpDNA) and mitochondrial DNA (mtDNA) relative to the coverage of the nuclear genome can be taken as a proxy for cpDNA, mtDNA, and nuclear genome copy number stoichiometry in Mamiellophyceae. O. tauri and B. prasinos have 2:3:1 mtDNA, cpDNA, nuclear genome coverage on average, whereas these relative ratios are 3:4:1 in O. mediterraneus and 4:7:1 in M. pusilla RCC299. No mutation candidate was found in organellar genomes, even when the mutation detection threshold was lowered to take the genome copy number into account. This implies that the spontaneous mutation rates of organelles are less than 9.6 × 10−10 for the mitochondria and 4.8 × 10−10 for the chloroplast, so that we cannot estimate the nuclear versus organellar mutation rates in Mamiellophyceae. The organelle mutation rates have been estimated to be lower than the nuclear mutation rates in higher plants (Wolfe et al. 1987; Smith 2015), while similar mutation rates in the chloroplast and the nuclear genome have been recently estimated in the green algae Chlamydomonas reinhardtii (Ness et al. 2015a).
Nonrandom Mutation Events in the Genome
The analysis of the distribution of mutations across these four species reveals significant deviations from a uniform distribution of mutations along the genome.
First, mutation events tend to cluster within adjacent nucleotides: of our 238 base substitution mutations across all species, 32 occurred within 30 bp of another mutation. These clustered mutations probably represent single mutational events since all adjacent mutations are found within the same strain. These multinucleotide mutations (MNM) were not part of homopolymeric nucleotides.
Second, there is an excess of mutations in intergenic regions, with a two-fold increase as compared to coding regions (Chi-Square, P-value < 0.01) (table 2). In addition, transcription levels seems to impact the mutability: in intergenic regions, mutated sites have on average 2.6–5.7 fold less RNAseq read coverage (for B. prasinos and O. tauri, respectively) than nonmutated sites (supplementary table S9, Supplementary Material online, Wilcoxon test, P-value < 0.001). However, there is no significant RNAseq coverage variation between mutated and nonmutated sites in coding sequences in B. prasinos and O. tauri.
Third, there are significantly more deletions than insertions over the four experiments (Binomial test, P-value < 0.05). The deletion over insertion ratio is 2.2, and the average length of insertions versus deletions is 1.9 and 9.8 bp, respectively. This corresponds to a net loss of 0.004 bp per genome per generation in O. tauri to 0.03 bp per generation in M. pusilla. This deletion bias has been previously noted in species such as Chlamydomonas reinhardtii (Ness et al. 2015b), with a net loss per genome per generation of 0.022 bp (for 72.5% of the genome).
Finally, mutations are over-represented in subtelomeric regions—within 1 kb of the start of the nucleotides AAACCCT telomere repeats (22 mutations identified, considering indels and substitutions). These mutations do not appear in homopolymeric nucleotides, and were identified in the four species. The overrepresentation of mutations in subtelomeric regions is highly significant in O. tauri (10 vs. 94 mutations in subtelomeric versus nonsubtelomeric regions, Fisher exact test, P-value <10−12), B. prasinos (8 vs. 24 mutations, Fisher exact test, P-value <10−12) and M. pusilla (4 vs. 81 mutations, Fisher exact test, P-value <10−4). It is nonsignificant in O. mediterraneus (1 vs. 64 mutations in subtelomeric versus nonsubtelomeric regions).
The Direction of Base-Substitution Mutations
The mutation rate between the four nucleotides is biased from GC to AT mutations (fig. 1). The equilibrium GC content (GCeq), that is the GC content where the number of mutations from GC to AT is equal to AT to GC, is substantially lower than the observed GC content in all Mamiellophyceae (GCeq = 36.8% vs. GCobs = 59.0% for O. tauri, 43.5% vs. 56.0% for O. mediterraneus, 46.2% vs. 63.8 for M. pusilla, and 36.8% vs. 48.0% for B. prasinos). This discrepancy implies either that the mutational spectrum has recently changed in all four species, or that nonmutational processes are acting to maintain the GC content above its mutational equilibrium value. Interestingly, one chromosome has a GC content that is 10% points lower than the others in Mamiellophyceae: 51.3% in M. pusilla, 49.9% in O. mediterraneus, 54.3% in O. tauri, and 41.9% in B. prasinos (Piganeau et al. 2011). Despite this variation, no significant difference in the distribution of GC to AT and AT to GC mutations between high and low GC content chromosomes was observed. Surprisingly, this GC bias seemed to be stronger in coding sequences: the mutation rate from GC to AT was higher than the mutation rate from AT to GC in coding region, whereas it is not significantly biased in intergenic regions (supplementary table S10, Supplementary Material online).
Fig. 1.
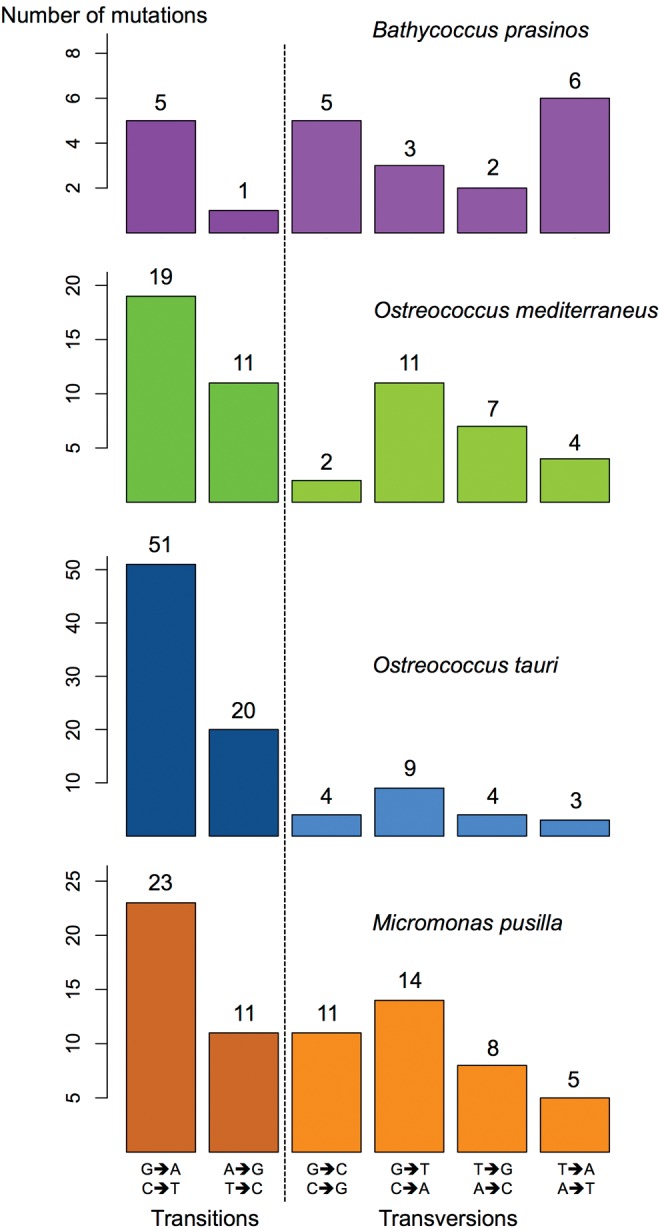
Mutation patterns in the four species. GC to AT bias is significant in O. tauri and M. pusilla (Binomial test, P = 0.0001 and 0.02, respectively).
Inter-genomic Variation in the Mutation Rate
Several ecological and biological factors have been proposed to explain the variation in the mutation rate between species, such as genome size and effective population size (Lynch et al. 2016 for a review). We compiled the available estimates of the spontaneous mutation rate from the literature (supplementary table S11, Supplementary Material online), and combined them with the mutation rate estimates from this study. There is a significant decrease of the mutation rate with the effective population size (n = 18, Pearson correlation, ρ = −0.78, P < 0.0001) (fig. 2, supplementary table S11, Supplementary Material online), and the one Mamiellophyceae for which we have diversity data (O. tauri), and hence an estimate of Ne fits in with this pattern. This negative correlation supports the drift barrier hypothesis (Lynch 2010; Sung et al. 2012; Lynch et al. 2016). There is a negative correlation between genome size (G) and mutation rate (μ) in bacteria (n = 10, Pearson correlation, ρ = −0.95, P = 0.001) and all micro-organisms (n = 19, Pearson correlation, ρ = −0.57, P = 0.01). In contrast, the correlation between G and μ in multicellular eukaryotes is positive (supplementary fig. S1 and table S11, Supplementary Material online) (n = 10, Pearson correlation, ρ = 0.67, P < 0.03).
Fig. 2.

Mutation rates versus effective population size (n = 18, Pearson correlation, ρ = −0.78, P < 0.0001, data from supplementary table S11, Supplementary Material online).
A biased mutation pattern can potentially cause differences between the observed genomic GC content and the equilibrium GC content (supplementary table S12, Supplementary Material online). This in turn can alter the mutation rate. For example, if the mutation pattern is biased towards AT then elevating the GC content above its mutational equilibrium increases the mutation rate above the mutation rate you would get at equilibrium, μeq. In Mamiellophyceae, the deviation from the equilibrium GC content leads to a modest increase in the mutation rate from 2% (O. mediterraneus) to 12% (O. tauri). However, using data from MA experiments in 20 other species (supplementary table S12, Supplementary Material online) the increase of μobs relative to μeq can be much larger; up to 64% in the eukaryote A. thaliana, and 160% in the prokaryote Mesoplasma florum. More generally we find that the mutation rate is positively correlated to the ratio of the observed GC-content to its equilibrium value (fig. 3, Pearson correlation, n = 23, ρ = 0.55, P = 0.007).
Fig. 3.

Correlation between mutation rate and the ratio of the observed versus the equilibrium GC-content (n = 23, Pearson correlation, ρ = 0.55, P = 0.007, data from supplementary table S12, Supplementary Material online).
Discussion
We have performed mutation accumulation experiments in four species of pico-phytoplankton followed by whole genome sequencing of 150 lines. In total, we have observed 238 point mutations and 48 indels, which we have used to analyse various aspects of the mutation rate. The genome coverage of each mutation accumulation lines is higher than 97% and mutation rates vary from μ = 4.4 × 10−10 to 9.8 × 10−10 mutations per nucleotide per generation.
Within Genome Variation of Mutation Rate
Coding Sequences Bias
We observed a 2- to 3-fold difference in the mutation rate of coding and intergenic regions. There are three possible explanations for this observation.
First, this could simply reflect selection against spontaneous mutations in coding regions. The MA experiment was designed such that all but the most strongly deleterious mutations would accumulate. However, strongly deleterious mutations will lead to line loss, which is something we observed (Krasovec et al. 2016). In coding regions, approximately one third of nucleotide positions are synonymous and are thus expected to have little consequence on fitness. If selection occurred during the MA experiments, mutations in coding regions should be biased towards synonymous mutations, but there is no excess of synonymous mutations in any of the MA experiments (Chi-squared test, ns) (table 1), consistent with a lack of selection against nonsynonymous mutations during the experiment.
Second, the lower mutation rate in coding regions could reflect a difference in the efficiency of mismatch repair (MMR) between coding and intergenic regions (Kunkel and Erie 2015), provided the MMR efficiency is optimized in coding region of the genome (Lee et al. 2012; Foster et al. 2015). Indeed, an experiment comparing wild type and MMR deficient lines in E. coli show that the difference in mutation rates between coding and intergenic sequence vanishes in MMR deficient lines (Foster et al. 2015). Seven genes of the MutS repair machinery have been predicted in the four Mamiellophyceae species of this study [MutS gene family from the picoPLAZA website (Vandepoele et al. 2013)].
Third, the lower mutation rate in coding regions could be due to transcription-coupled DNA repair (TCR) (Hanawalt and Spivak 2008). This system allows the repair of lesions in the DNA that are encountered during transcription and hence is more likely in the regions of the genome that are expressed. Consistent with TCR, mutated intergenic sites have ∼3- to 6-fold lower transcription rates than nonmutated intergenic sites in B. prasinos and O. tauri (supplementary table S9, Supplementary Material online). Genes coding for TCRs have been identified in these four species [rad26 gene family from the picoPLAZA website (Vandepoele et al. 2013)]. However, this bias is not observed in coding sequences, suggesting a threshold effect. When a minimum level of transcription is reached, the probability of repair by TCR becomes too high to detect significant variation between two transcription rates above the threshold. Interestingly, the transcription effect on the mutation rate varies between species, depending of the difference between TCR and TAM (Lynch et al. 2016). Our data suggest that TCR in Mamiellophyceae species compensates and significantly reduces the TAM effect.
In conclusion, both MMR and TCR are likely mechanisms involved in lower mutation rates in coding regions in Mamiellophyceae. It is worth noting that the difference in the mutation rate between coding and intergenic regions may affect genome-wide mutation rate estimates in other species, if coding and intergenic sites are unequally represented in re-sequenced MA lines. De novo mutations were estimated from only 78% of the genome in A. thaliana (Ossowski et al. 2010), 75% in C. reinhardtii (Ness et al. 2012, 2015b) and 46% in Heliconius melpomene (Keightley et al. 2014b). These estimates might thus be biased if there is a tendency for de novo mutations to be easier to identify in the coding fraction of the genome.
Multinucleotide Mutations
Multinucleotide mutations (MNM) are known to be responsible of ∼2% to ∼16% of the total number of mutations in humans, S. cerevisiae, D. melanogaster, A. thaliana, and C. elegans (Schrider et al. 2011). Multinucleotide mutations might have implications for the molecular clock, and for the evolution of amino acid sequences by allowing jumps between amino acids that are separated by more than one mutation (Schrider et al. 2011; Besenbacher et al. 2016). In the case of Mamiellophycae, ∼5% of mutation events are MNM. Several hypotheses were proposed to explain the origin of MNM, such as a defect in transcription or translation of a polymerase (Ninio 1991), replication timing (Stamatoyannopoulos et al. 2009) or simply that single mutation may induce other mutations at adjacent sites (Tian et al. 2008). Our data do not permit us to explore these hypotheses, but the MNM rate in Mamiellophyceae has to be taken into account to re-assess estimation of speciation times in this genera (Šlapeta et al. 2006).
Inter-Specific Mutation Rate Variation
The genomic mutation rate varies by about ∼10,000-fold amongst the tree of life. As previously reported, the effective population size appears to be a key correlate of the mutation rate variations, probably because of the drift barrier, which imposes a limit to the lower mutation rate that can be possibly reached by selection (Lynch 2010; Sung et al. 2012a,b; Lynch et al. 2016). The increase of the mutation rate with genome size observed in multicellular eukaryotes might thus simply result from the negative correlation between genome size and the effective population size (Lynch and Conery 2003). Microbial eukaryotes have large effective population sizes (supplementary table S11, Supplementary Material online), and it is thus expected that mutation rates decrease with genome sizes, as reported for bacteria. The available data does not support this, as there is no significant relationship between genome size and mutation rates in microbial eukaryotes (n = 10, supplementary fig. S1, Supplementary Material online, Pearson correlation ρ = −0.47, ns), in sharp contrast to the strong negative relationship in bacteria (n = 10, supplementary fig. S1, Supplementary Material online, Pearson correlation ρ = −0.95, P = 0.001). Differences in the life cycle, such as the frequency of clonal versus sexual reproduction, or in genome biology, such as the nontranscription of the germline genome in ciliates (as opposed to the somatic genome) (Sung et al. 2012a,b; Long et al. 2016), may blur the expected relationship between μ and genome size in microbial eukaryotes.
In addition to the effect of effective population size, the departure from equilibrium GC base composition is responsible for a substantial part of mutation rate variation between species (Smith et al. 2002), as a consequence of the positive relationship between the mutation rate and the departure of the GC content from equilibrium. Indeed, most species have a higher GC content than expected from the GC->AT versus AT->GC mutation rates, and this increases the mutation rate. The mechanisms responsible for increasing the genomic GC content thus contribute to increasing the spontaneous mutation rate in most species. Two mechanisms could move the GC content of a genome away from its equilibrium value; selection or biased gene conversion.
Selection can act on protein coding sequences, synonymous codon use (Ikemura 1981) and gene regulatory sequences, in a manner which is expected to lead to less biased base composition than the mutational spectrum would cause. The GC bias is stronger in coding sequences, constituting indirect evidence supporting selection on coding sequence composition.
Biased gene conversion, a by-product of recombination, has been identified in many organisms from bacteria (Lassalle et al. 2015) and yeast (Harrison and Charlesworth 2011; Lesecque et al. 2013) to humans (Duret and Arndt 2008; Duret and Galtier 2009). There is indirect evidence of recombination and biased gene conversion in O. tauri (Jancek et al. 2008; Grimsley et al. 2010), so that GC biased gene conversion is likely involved in increasing the GC content of Mamiellophyceae genomes.
Conclusion
Our study provides four novel spontaneous mutation rate estimates in unicellular eukaryotes. Spontaneous mutations rates were assessed over 97–99.5% of the nuclear genomes, they are higher in intergenic than in coding sequences and 5% of mutation events affect multiple nucleotides. Combined with previous spontaneous mutation rate estimates from 20 species, our data also provides evidence that the spontaneous mutation rate increases with the deviation of the genome GC-content from its equilibrium GC-content.
Materials and Methods
Mutation Accumulation Experiments
Mutation accumulation experiments were performed on four haploid marine green algae (Chlorophyta): Ostreococcus tauri RCC4221, O. mediterraneus RCC2590, Micromonas pusilla RCC299, and Bathycoccus prasinos RCC1105. All strains are maintained in the Roscoff Culture Collection (RCC), in France (http://roscoff-culture-collection.org/). Classically, in MA experiments of unicellular organisms, a colony of cells is transferred to a fresh agar plate at each bottleneck to allow the separation of the cells and the random sampling of a new cell. However, this is not possible in these pico-algae as they do not grow on the surface of gelled media, in contrast to Saccharomyces cerevisiae, Dictyostelium discoideum or Chlamydomonas reinhardtii (Wloch et al. 2001; Hall et al. 2013; Morgan et al. 2014). Nevertheless, they are easily cultured in liquid medium in the laboratory. We therefore developed an experimental protocol combining flow cytometry, which has the advantages of counting individual cells while verifying cell size and fluorescence, and transfer of single cells in liquid media (Krasovec et al. 2016). Briefly, MA lines were started from one single cell from a clonal population and maintained in L1 liquid medium in 24 wells of a microtiter plate with a one-cell bottleneck every 14 days. Serial bottlenecks allowed to largely remove the influence of natural selection, the average effective population size, estimated with the harmonic mean of cell number, varied between 6 and 9 across the four species (supplementary table S1, Supplementary Material online).
Cell concentrations of MA lines were measured by flow cytometry using a FACSCanto II flow cytometer (Becton Dickinson, Franklin Lakes, NJ), relative to their natural chlorophyll fluorescence (FL3 acquisition at 670 nm) and size scatter (SSC) acquisitions. Depending of cell concentration, the volume corresponding to one cell was inoculated into a new well plate with new media. The number of generations per day, D, was estimated each 14 days at bottleneck time as follows:
| (1) |
Nt is the number of cells in the well at bottleneck time, and t = 14 days. MA experiments were performed over a period of 224–378 days depending on the species. MA lines accumulated between 80 and 500 independent generations (supplementary table S1, Supplementary Material online).
Sequencing
DNA of ancestral types and MA lines were extracted as described previously (Winnepenninckx et al. 1993) and sequenced with Illumina technology by GATC biotech® (Konstanz, Germany). Two different sequencing technologies were used: MiSeq for O.tauri and O. mediterraneus, and HiSeq for B. prasinos and M. pusilla. Reads from ancestral types and MA lines were aligned to the reference genomes using BWA (Li and Durbin 2010) (M. pusilla: GCA_000151265.1; O. tauri: GCF_000214015.2); B. prasinos (ORCAE database; Sterck et al. 2012); O. mediterraneus (S Yau et al. in preparation), and SAMtools (Li et al. 2009) were used to obtained bam and mpileup files. The four ancestral types and 150 MA lines were sequenced: 40 for O. tauri, 37 for O. mediterraneus, 37 for M. pusilla, and 36 for B. prasinos.
Mutation Calling
Mutations were called from mpileup files (Li et al. 2009) using GATK (DePristo et al. 2011). The final mutation candidates were filtered to remove low mapping quality regions (MQ < 50), low coverage regions (<5 reads), and shared mutations between all MA lines. The number of callable sites per genome above these thresholds was computed to estimate the per base pair mutation rate [97–99% of the genomes are callable (supplementary table S2)). All alignments around mutations were checked manually. All mutation candidates were compared with the ancestral type to discard spurious candidates that result from substitutions between the reference genome sequence and the genome of the strain at the start of the MA experiment. Additionally, 22 mutation candidates were chosen randomly for Sanger re-sequencing. All re-sequencing confirmed the predicted mutations (true positive rate = 100%, false negative rate < 1 × 10−4). The type of mutation (nonsynonymous, synonymous, intronic, or intergenic) was extracted with snpEff (Cingolani et al. 2012) using the annotation files of each genome [available via ORCAE (Sterck et al. 2012)]. The same method was used for base substitutions and indels mutations.
Mutation Rate at Equilibrium GC Content
Let R1 be equal to the rate of mutation from GC to AT, R2 from AT to GC, R3 the rate of transversions between A and T, and R4 be the rate of transversions between G and C:
| (2) |
NNn is the number of GC or AT sites in the genome. Then it is straightforward to show that the GC-content at mutational equilibrium (Sueoka 1962) is:
| (3) |
Assuming that R1, R2, R3, and R4 are constant, the expected mutation rate at equilibrium is
| (4) |
with
| (5) |
it may be written
| (6) |
where
| (7) |
Mutation Spectrum Analysis
To investigate the effect of context we extracted the 10 bp at either side of each mutated site and used binomial tests to investigate whether a particular trinucleotide, either NXN or NNX, where X is the mutated site, has a significantly higher or lower mutation rate.
To investigate whether gene expression affects the rate of mutation we used STAR (Dobin et al. 2013) to compute the coverage of the genome by RNAseq data, available for B. prasinos (Moreau et al. 2012) and O. tauri (Blanc-Mathieu et al. 2014). Statistical analyses were performed with R (version 3.1.1) (R Development Core Team, 2014).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We are grateful to Claire Hemon, Elodie Desgranges and Christophe Salmeron for technical assistance with the mutation accumulation experiments from 2012 to 2015 and to the Genomics of Phytoplankton lab for support and stimulating discussions. We acknowledge the O. mediterraneus genome consortium for access to the complete genome data and the GenoToul Bioinformatics platform from Toulouse, France, for bioinformatics analysis support and cluster availability. This work was funded by ANRJCJC-SVSE6-2013-0005 to GP and SSF.
Conflict of interest statement. None declared.
References
- Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM.. 2010. 1000 Genomes Project Consortium. Nature 467:1061–1073.20981092 [Google Scholar]
- Adewoye AB, Lindsay SJ, Dubrova YE, Hurles ME.. 2015. The genome-wide effects of ionizing radiation on mutation induction in the mammalian germline. Nat Commun. 6:6684.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwala V, Voight BF.. 2016. An expanded sequence context model broadly explains variability in polymorphism levels across the human genome. Nat Genet. 48:349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besenbacher S, Sulem P, Helgason A, Helgason H, Kristjansson H, Jonasdottir A, Jonasdottir A, Magnusson OT, Thorsteinsdottir U, Masson G, et al. 2016. Multi-nucleotide de novo mutations in humans. PLoS Genet. 12:e1006315.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc-Mathieu R, Verhelst B, Derelle E, Rombauts S, Bouget F-Y, Carré I, Château A, Eyre-Walker A, Grimsley N, Moreau H, et al. 2014. An improved genome of the model marine alga Ostreococcus tauri unfolds by assessing Illumina de novo assemblies. BMC Genomics 15:1103.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Zhang J.. 2013. No gene-specific optimization of mutation rate in Escherichia coli. Mol Biol Evol. 30:1559–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM.. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly (Austin) 6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins S, Rost B, Rynearson TA.. 2014. Evolutionary potential of marine phytoplankton under ocean acidification. Evol Appl. 7:140–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Keebler JEM, DePristo MA, Lindsay SJ, Zhang Y, Cassals F, Idaghdour Y, Hartl CL, Torroja C, Garimella KV, et al. 2011. Variation in genome-wide mutation rates within and between human families. Nat Genet. 43:712–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulondre C, Miller JH, Farabaugh PJ, Gilbert W.. 1978. Molecular basis of base substitution hotspots in Escherichia coli. Nature 274:775–780. [DOI] [PubMed] [Google Scholar]
- de Vargas C, Audic S, Henry N, Decelle J, Mahé F, Logares R, Lara E, Berney C, Le Bescot N, Probert I, et al. 2015. Ocean plankton. Eukaryotic plankton diversity in the sunlit ocean. Science 348:1261605. [DOI] [PubMed] [Google Scholar]
- Denver DR, Dolan PC, Wilhelm LJ, Sung W, Lucas-Lledó JI, Howe DK, Lewis SC, Okamoto K, Thomas WK, Lynch M, et al. 2009. A genome-wide view of Caenorhabditis elegans base-substitution mutation processes. Proc Natl Acad Sci U S A. 106:16310–16314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver DR, Morris K, Lynch M, Thomas WK.. 2004. High mutation rate and predominance of insertions in the Caenorhabditis elegans nuclear genome. Nature 430:679–682. [DOI] [PubMed] [Google Scholar]
- Denver DR, Morris K, Lynch M, Vassilieva LL, Thomas WK.. 2000. High direct estimate of the mutation rate in the mitochondrial genome of Caenorhabditis elegans. Science 289:2342–2344. [DOI] [PubMed] [Google Scholar]
- Denver DR, Wilhelm LJ, Howe DK, Gafner K, Dolan PC, Baer CF.. 2012. Variation in base-substitution mutation in experimental and natural lineages of Caenorhabditis nematodes. Genome Biol Evol. 4:513–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR.. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinform Oxf Engl. 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW. 1991. A constant rate of spontaneous mutation in DNA-based microbes. Proc Natl Acad Sci U S A. 88:7160–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Charlesworth B, Charlesworth D, Crow JF.. 1998. Rates of spontaneous mutation. Genetics 148:1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Arndt PF.. 2008. The impact of recombination on nucleotide substitutions in the human genome. PLOS Genet. 4:e1000071.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Galtier N.. 2009. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 10:285–311. [DOI] [PubMed] [Google Scholar]
- Ebersberger I, Metzler D, Schwarz C, Pääbo S.. 2002. Genomewide comparison of DNA sequences between humans and chimpanzees. Am J Hum Genet. 70:1490–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PL, Lee H, Popodi E, Townes JP, Tang H.. 2015. Determinants of spontaneous mutation in the bacterium Escherichia coli as revealed by whole-genome sequencing. Proc Natl Acad Sci U S A. 112:E5990–E5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryxell KJ, Zuckerkandl E.. 2000. Cytosine deamination plays a primary role in the evolution of mammalian isochores. Mol Biol Evol. 17:1371–1383. [DOI] [PubMed] [Google Scholar]
- Grimsley N, Péquin B, Bachy C, Moreau H, Piganeau G.. 2010. Cryptic sex in the smallest eukaryotic marine green alga. Mol Biol Evol. 27:47–54. [DOI] [PubMed] [Google Scholar]
- Haag-Liautard C, Coffey N, Houle D, Lynch M, Charlesworth B, Keightley PD.. 2008. Direct estimation of the mitochondrial DNA mutation rate in Drosophila melanogaster. PLoS Biol. 6:e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag-Liautard C, Dorris M, Maside X, Macaskill S, Halligan DL, Houle D, Charlesworth B, Keightley PD.. 2007. Direct estimation of per nucleotide and genomic deleterious mutation rates in Drosophila. Nature 445:82–85. [DOI] [PubMed] [Google Scholar]
- Hall DW, Fox S, Kuzdzal-Fick JJ, Strassmann JE, Queller DC.. 2013. The rate and effects of spontaneous mutation on fitness traits in the social amoeba, Dictyostelium discoideum. G3 GenesGenomesGenetics 3:1115–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halligan DL, Keightley PD.. 2009. Spontaneous mutation accumulation studies in evolutionary genetics. Annu Rev Ecol Evol Syst. 40:151–172. [Google Scholar]
- Hanawalt PC, Spivak G.. 2008. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 9:958–970. [DOI] [PubMed] [Google Scholar]
- Harrison RJ, Charlesworth B.. 2011. Biased gene conversion affects patterns of codon usage and amino acid usage in the Saccharomyces sensu stricto group of yeasts. Mol Biol Evol. 28:117–129. [DOI] [PubMed] [Google Scholar]
- Henry L, Schwander T, Crespi BJ.. 2012. Deleterious mutation accumulation in asexual timema stick insects. Mol Biol Evol. 29:401–408. [DOI] [PubMed] [Google Scholar]
- Hodgkinson A, Eyre-Walker A.. 2011. Variation in the mutation rate across mammalian genomes. Nat Rev Genet. 12:756–766. [DOI] [PubMed] [Google Scholar]
- Ikemura T. 1981. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes: a proposal for a synonymous codon choice that is optimal for the E. coli translational system. J Mol Biol. 151:389–409. [DOI] [PubMed] [Google Scholar]
- Jancek S, Gourbière S, Moreau H, Piganeau G.. 2008. Clues about the genetic basis of adaptation emerge from comparing the proteomes of two Ostreococcus ecotypes (Chlorophyta, Prasinophyceae). Mol Biol Evol. 25:2293–2300. [DOI] [PubMed] [Google Scholar]
- Keightley PD, Ness RW, Halligan DL, Haddrill PR.. 2014a. Estimation of the spontaneous mutation rate per nucleotide site in a Drosophila melanogaster full-sib family. Genetics 196:313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, Pinharanda A, Ness RW, Simpson F, Dasmahapatra KK, Mallet J, Davey JW, Jiggins CD.. 2014b. Estimation of the spontaneous mutation rate in Heliconius melpomene. Mol Biol Evol. 32:239–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keightley PD, Trivedi U, Thomson M, Oliver F, Kumar S, Blaxter ML.. 2009. Analysis of the genome sequences of three Drosophila melanogaster spontaneous mutation accumulation lines. Genome Res. 19:1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Jinks-Robertson S.. 2012. Transcription as a source of genome instability. Nat Rev Genet. 13:204–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasovec M, Eyre-Walker A, Grimsley N, Salmeron C, Pecqueur D, Piganeau G, Sanchez-Ferandin S.. 2016. Fitness effects of spontaneous mutations in picoeukaryotic marine green Algae. G3 6:2063–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel TA, Erie DA.. 2015. Eukaryotic mismatch repair in relation to DNA replication. Annu Rev Genet. 49:291–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409:860–921. [DOI] [PubMed] [Google Scholar]
- Lang GI, Murray AW.. 2008. Estimating the per-base-pair mutation rate in the yeast Saccharomyces cerevisiae. Genetics 178:67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassalle F, Périan S, Bataillon T, Nesme X, Duret L, Daubin V.. 2015. GC-content evolution in bacterial genomes: the biased gene conversion hypothesis expands. PLoS Genet. 11:e1004941.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau KWK, Ren J, Wu M.. 2000. Redox modulation of chloroplast DNA replication in Chlamydomonas reinhardtii. Antioxid Redox Signal. 2:529–535. [DOI] [PubMed] [Google Scholar]
- Lee H, Popodi E, Tang H, Foster PL.. 2012. Rate and molecular spectrum of spontaneous mutations in the bacterium Escherichia coli as determined by whole-genome sequencing. Proc Natl Acad Sci U S A. 109:E2774–E2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh EG. 1973. The evolution of mutation rates. Genetics 73:1–18. [PubMed] [Google Scholar]
- Lesecque Y, Mouchiroud D, Duret L.. 2013. GC-biased gene conversion in yeast is specifically associated with crossovers: molecular mechanisms and evolutionary significance. Mol Biol Evol. 30:1409–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2010. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinform Oxf Engl. 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind PA, Andersson DI.. 2008. Whole-genome mutational biases in bacteria. Proc Natl Acad Sci U S A. 105:17878–17883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long H, Winter DJ, Chang AY-C, Sung W, Wu SH, Balboa M, Azevedo RBR, Cartwright RA, Lynch M, Zufall RA.. 2016. Low base-substitution mutation rate in the germline genome of the ciliate Tetrahymena thermophila. Genome Biol Evol. evw223. doi: 10.1093/gbe/evw223. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M. 2010. Evolution of the mutation rate. Trends Genet. 26:345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Ackerman MS, Gout J-F, Long H, Sung W, Thomas WK, Foster PL.. 2016. Genetic drift, selection and the evolution of the mutation rate. Nat Rev Genet. 17:704–714. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS.. 2003. The origins of genome complexity. Science 302:1401–1404. [DOI] [PubMed] [Google Scholar]
- Lynch M, Sung W, Morris K, Coffey N, Landry CR, Dopman EB, Dickinson WJ, Okamoto K, Kulkarni S, Hartl DL, et al. 2008. A genome-wide view of the spectrum of spontaneous mutations in yeast. Proc Natl Acad Sci U S A. 105:9272–9277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Li X-Q.. 2015. Organellar genome copy number variation and integrity during moderate maturation of roots and leaves of maize seedlings. Curr Genet. 61:591–600. [DOI] [PubMed] [Google Scholar]
- Marin B, Melkonian M.. 2010. Molecular phylogeny and classification of the Mamiellophyceae class. nov. (Chlorophyta) based on sequence comparisons of the nuclear- and plastid-encoded rRNA operons. Protist 161:304–336. [DOI] [PubMed] [Google Scholar]
- Martincorena I, Seshasayee ASN, Luscombe NM.. 2012. Evidence of non-random mutation rates suggests an evolutionary risk management strategy. Nature 485:95–98. [DOI] [PubMed] [Google Scholar]
- Moreau H, Verhelst B, Couloux A, Derelle E, Rombauts S, Grimsley N, Van Bel M, Poulain J, Katinka M, Hohmann-Marriott MF, et al. 2012. Gene functionalities and genome structure in Bathycoccus prasinos reflect cellular specializations at the base of the green lineage. Genome Biol. 13:R74.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AD, Ness RW, Keightley PD, Colegrave N.. 2014. Spontaneous mutation accumulation in multiple strains of the green alga, Chlamydomonas reinhardtii. Evolution 68:2589–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Kraemer SA, Colegrave N, Keightley PD.. 2015a. Direct estimate of the spontaneous mutation rate uncovers the effects of drift and recombination in the Chlamydomonas reinhardtii plastid genome. Mol Biol Evol. 33:800–808. [DOI] [PubMed] [Google Scholar]
- Ness RW, Morgan AD, Colegrave N, Keightley PD, Ness RW, Morgan AD, Colegrave N, Keightley PD.. 2012. Estimate of the spontaneous mutation rate in Chlamydomonas reinhardtii. Genetics 192:1447–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ness RW, Morgan AD, Vasanthakrishnan RB, Colegrave N, Keightley PD.. 2015b. Extensive de novo mutation rate variation between individuals and across the genome of Chlamydomonas reinhardtii. Genome Res. 25:1739–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninio J. 1991. Transient mutators: a semiquantitative analysis of the influence of translation and transcription errors on mutation rates. Genetics 129:957–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski S, Schneeberger K, Lucas-Lledó JI, Warthmann N, Clark RM, Shaw RG, Weigel D, Lynch M.. 2010. The rate and molecular spectrum of spontaneous mutations in Arabidopsis thaliana. Science 327:92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paland S, Lynch M.. 2006. Transitions to asexuality result in excess amino acid substitutions. Science 311:990–992. [DOI] [PubMed] [Google Scholar]
- Park C, Qian W, Zhang J.. 2012. Genomic evidence for elevated mutation rates in highly expressed genes. EMBO Rep. 13:1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piganeau G, Grimsley N, Moreau H.. 2011. Genome diversity in the smallest marine photosynthetic eukaryotes. Res Microbiol. 162:570–577. [DOI] [PubMed] [Google Scholar]
- Polak P, Arndt PF.. 2008. Transcription induces strand-specific mutations at the 5’ end of human genes. Genome Res. 18:1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2014. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
- Rebolledo-Jaramillo B, Su MS-W, Stoler N, McElhoe JA, Dickins B, Blankenberg D, Korneliussen TS, Chiaromonte F, Nielsen R, Holland MM, et al. 2014. Maternal age effect and severe germ-line bottleneck in the inheritance of human mitochondrial DNA. Proc Natl Acad Sci U S A. 111:15474–15479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrider DR, Hourmozdi JN, Hahn MW.. 2011. Pervasive multinucleotide mutational events in eukaryotes. Curr Biol. 21:1051–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ségurel L, Wyman MJ, Przeworski M.. 2014. Determinants of mutation rate variation in the human germline. Annu Rev Genomics Hum Genet. 15:47–70. [DOI] [PubMed] [Google Scholar]
- Šlapeta J, López-García P, Moreira D.. 2006. Global dispersal and ancient cryptic species in the smallest marine eukaryotes. Mol Biol Evol. 23:23–29. [DOI] [PubMed] [Google Scholar]
- Smith DR. 2015. Mutation rates in plastid genomes: they are lower than you might think. Genome Biol Evol. 7:1227–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith NGC, Webster MT, Ellegren H.. 2002. Deterministic mutation rate variation in the human genome. Genome Res. 12:1350–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatoyannopoulos JA, Adzhubei I, Thurman RE, Kryukov GV, Mirkin SM, Sunyaev SR.. 2009. Human mutation rate associated with DNA replication timing. Nat Genet. 41:393–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterck L, Billiau K, Abeel T, Rouzé P, Van de Peer Y.. 2012. ORCAE: online resource for community annotation of eukaryotes. Nat Methods 9:1041.. [DOI] [PubMed] [Google Scholar]
- Subirana L, Péquin B, Michely S, Escande M-L, Meilland J, Derelle E, Marin B, Piganeau G, Desdevises Y, Moreau H, et al. 2013. Morphology, genome plasticity, and phylogeny in the genus ostreococcus reveal a cryptic species, O. mediterraneus sp. nov. (Mamiellales, Mamiellophyceae). Protist 164:643–659. [DOI] [PubMed] [Google Scholar]
- Sueoka N. 1962. On the genetic basis of variation and heterogeneity of DNA base composition. Proc Natl Acad Sci U S A. 48:582–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, Ackerman MS, Gout J-F, Miller SF, Williams E, Foster PL, Lynch M.. 2015. Asymmetric context-dependent mutation patterns revealed through mutation–accumulation experiments. Mol Biol Evol. 32:1672–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, Ackerman MS, Miller SF, Doak TG, Lynch M.. 2012a. Drift-barrier hypothesis and mutation-rate evolution. Proc Natl Acad Sci U S A. 109:18488–18492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung W, Tucker AE, Doak TG, Choi E, Thomas WK, Lynch M.. 2012b. Extraordinary genome stability in the ciliate Paramecium tetraurelia. Proc Natl Acad Sci U S A. 109:19339–19344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian D, Wang Q, Zhang P, Araki H, Yang S, Kreitman M, Nagylaki T, Hudson R, Bergelson J, Chen J-Q.. 2008. Single-nucleotide mutation rate increases close to insertions/deletions in eukaryotes. Nature 455:105–108. [DOI] [PubMed] [Google Scholar]
- Uchimura A, Higuchi M, Minakuchi Y, Ohno M, Toyoda A, Fujiyama A, Miura I, Wakana S, Nishino J, Yagi T.. 2015. Germline mutation rates and the long-term phenotypic effects of mutation accumulation in wild-type laboratory mice and mutator mice. Genome Res. 25:1125–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandepoele K, Van Bel M, Richard G, Van Landeghem S, Verhelst B, Moreau H, Van de Peer Y, Grimsley N, Piganeau G.. 2013. pico-PLAZA, a genome database of microbial photosynthetic eukaryotes. Environ Microbiol. 15:2147–2153. [DOI] [PubMed] [Google Scholar]
- Vannier T, Leconte J, Seeleuthner Y, Mondy S, Pelletier E, Aury J-M, Vargas C de, Sieracki M, Iudicone D, Vaulot D, et al. 2016. Survey of the green picoalga Bathycoccus genomes in the global ocean. Sci. Rep. 6:37900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnepenninckx B, Backeljau T, De Wachter R.. 1993. Extraction of high molecular weight DNA from molluscs. Trends Genet. 9:407.. [DOI] [PubMed] [Google Scholar]
- Wloch DM, Szafraniec K, Borts RH, Korona R.. 2001. Direct estimate of the mutation rate and the distribution of fitness effects in the yeast Saccharomyces cerevisiae. Genetics 159:441–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, Li WH, Sharp PM.. 1987. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Natl Acad Sci U S A. 84:9054–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worden AZ, Lee J-H, Mock T, Rouzé P, Simmons MP, Aerts AL, Allen AE, Cuvelier ML, Derelle E, Everett MV, et al. 2009. Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes micromonas. Science 324:268–272. [DOI] [PubMed] [Google Scholar]
- Zhu YO, Siegal ML, Hall DW, Petrov DA.. 2014. Precise estimates of mutation rate and spectrum in yeast. Proc Natl Acad Sci U S A. 111:E2310–E2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.