

Abstract
ISOLATED ALTERATIONS OF BIOCHEMICAL MARKERS OF LIVER DAMAGE in a seemingly healthy patient can present a challenge for the clinician. In this review we provide a guide to interpreting alterations to liver enzyme levels. The functional anatomy of the liver and pathophysiology of liver enzyme alteration are briefly reviewed. Using a schematic approach that classifies enzyme alterations as predominantly hepatocellular or predominantly cholestatic, we review abnormal enzymatic activity within the 2 subgroups, the most common causes of enzyme alteration and suggested initial investigations.
Abnormal liver enzyme levels may signal liver damage or alteration in bile flow. Liver enzyme alteration may be either the accompanying biochemical picture in a patient with symptoms or signs suggestive of liver disease or an isolated, unexpected finding in a patient who has undergone a wide range of laboratory tests for a nonhepatic disease or for minor, vague complaints. The latter situation is a common clinical scenario today because of the routine incorporation of hepatic tests in automated blood chemistry panels. Isolated alterations of biochemical markers of liver damage in a seemingly healthy patient often represent a challenge even for the experienced clinician and usually set off a battery of further, costly tests1 and consultations that may ultimately prove unnecessary. The aim of this review is to provide physicians in general practice with a guide to interpreting liver enzyme alterations.
Background
The liver is a large, complex organ that is well designed for its central role in carbohydrate, protein and fat metabolism. It is the site where waste products of metabolism are detoxified through processes such as amino acid deamination, which produces urea. In conjunction with the spleen it is involved in the destruction of spent red blood cells and the reclamation of their constituents. It is responsible for synthesizing and secreting bile and synthesizing lipoproteins and plasma proteins, including clotting factors.2 It maintains a stable blood glucose level by taking up and storing glucose as glycogen (glycogenesis), breaking this down to glucose when needed (glycogenolysis) and forming glucose from noncarbohydrate sources such as amino acids (gluconeogenesis).
Many of these biosynthetic functions use the products of digestion. With the exception of most lipids, absorbed food products pass directly from the gut to the liver through the hepatic portal vein. At the microscopic level, the primary functional unit of the liver is the liver acinus, which is defined by the territory supplied by each terminal branch of the hepatic artery and hepatic portal vein. The portal tract forms the central axis of the acinus; hepatocytes are arranged in plates that radiate out from the portal triad. The acinus is divided into 3 zones on the basis of the distance from the supplying vessels: zone 3, for example, experiences the least oxygen perfusion and houses the most mitochondria. Bile is secreted into a network of minute bile caniculi situated between adjacent hepatocytes.3
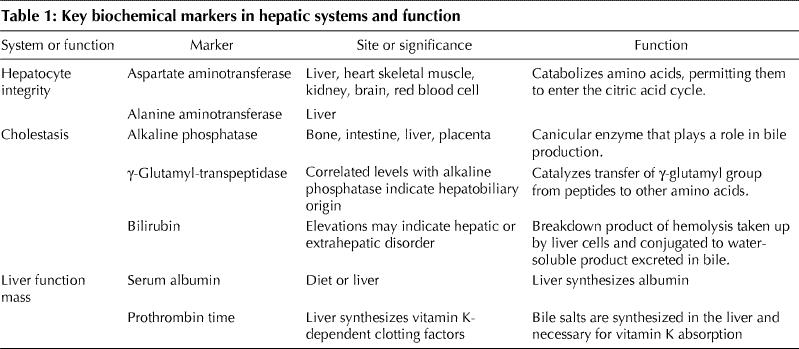
Liver disease is often reflected by biochemical abnormalities of 1 of 2 different hepatic systems or of liver function (Table 1). Although tests that measure the level of serum liver enzymes are commonly referred to as liver function tests, they usually reflect hepatocyte integrity or cholestasis rather than liver function. A change in serum albumin level or prothrombin time may be associated with a decrease in liver functioning mass, although neither is specific for liver disease.
Table 1
In order to accurately interpret biochemical abnormalities, it is necessary to understand how normal ranges are established and how to apply reference ranges. The performance characteristics (e.g., reproducibility, bias, total error) and reference limits for the most common liver tests have been thoroughly reviewed and guidelines established.4,5 An abnormal level is usually defined as a value exceeding the upper reference limit, since there is no clinical significance to the occurrence of low levels of biochemical markers, except for serum albumin. Because the reference limits for each test often vary among laboratories, specific ranges will not be provided here, in order to avoid generating confusion.
It is common practice when establishing laboratory parameters to define the “normal” range as the mean value within ± 2 standard deviations observed in the reference, “normal” population. Accordingly, as many as 2.5% of normal patients have “abnormal” aminotransferase levels. Moreover, at least 16% of patients with chronic hepatitis C infection and 13% of patients with nonalcoholic fatty liver disease have varying degrees of histological damage despite showing persistently normal aminotransferase levels.6,7 This apparent discrepancy may be explained by the fact that the healthy population that was recruited to establish the normal range could have included a small subset of patients with subclinical liver disease.
As well, aminotransferase levels vary according to age and sex, so care must be taken to use age- and sex-appropriate reference limits.4,5 The clinical context of patient presentation is also important when interpreting reference limits. Levels of both aspartate aminotransferase (AST) and alanine aminotransferase (ALT) may increase with strenuous exercise,4,8 and hospital admission has been observed to induce a 5% increase in AST levels (0.4%–9.6%, 95% confidence interval [CI]) and a 17.5% increase in ALT levels (9.1%–21.6%, 95% CI) in healthy subjects;9 restricted physical activity and hospital diet have been suggested as possible explanations for these increases.
Where, when and how: a schematic approach to liver enzyme alteration
The comprehensive evaluation of adult patients with abnormal liver enzyme levels is a multistep process. In moving through this process, the clinician can ask a series of questions categorized under where, when and how (Fig. 1).
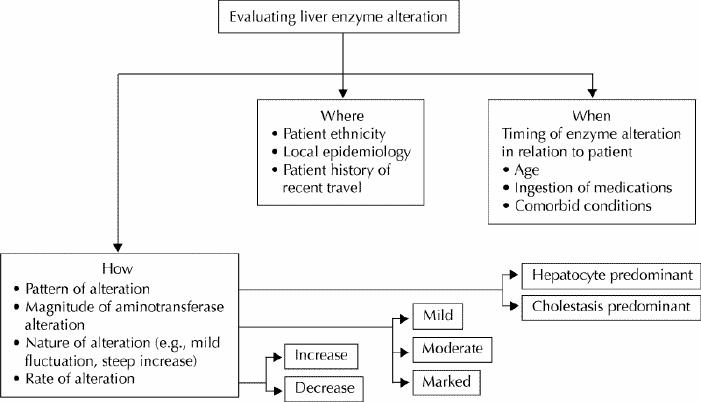
Fig. 1: Schematic representation of an approach to liver enzyme alteration. Specific modalities of enzyme alteration (how) and their relation with peculiar characteristics of the patient and locality (where and when) should be thoroughly assessed before the definitive diagnostic work-up is begun.
Where
Alterations in liver enzyme levels that are encountered in hospital centres may vary by the geographical location of the hospital and the ethnicity of the patients. For example, about 60% of cases of elevated hepatic AST levels in Wales can be attributed to ischemic or toxic liver injury,10 whereas orofecal transmittable hepatitis viruses are the major cause (> 60%) of sporadic acute and fulminant hepatitis in the Far East.11 The incidence of primary biliary cirrhosis ranges from 1.9–2.2 per 100 000 in Australia to 0.34–0.42 per 100 000 in Asia,12 and the prevalence of homozygosity for the C282Y mutation in the HFE gene can be found in about 5 per 1000 people of northern European descent13 but 0.0001 per 100 people of African American ethnicity.14 Physicians should be aware of the local epidemiological features of liver disease in the region where they practise in order to identify likely causes and reduce the number of unnecessary tests and the amount of time needed to make a diagnosis. Inquiry about the patient's recent travel history is also essential.
When
The timing of liver enzyme abnormalities in relation to the age of the patient, comorbid conditions and ingestion of medications provides valuable information. For example, the likelihood that the alteration is due to a disease that usually manifests early in life, such as Wilson's disease, is higher in younger patients than in elderly ones.15 The course of all comorbid conditions must be fully explored, along with a detailed list of drugs being taken by the patient and the date they were started in relation to the onset of enzyme alterations or of signs or symptoms of disease. Almost any medication can alter liver enzyme levels.16 Use of herbal remedies and over-the-counter preparations is often overlooked by physicians but should be carefully recorded.17
How
The pattern of liver enzyme alterations is often the first piece of evidence that catches the physician's eye. This is because common causes of liver disease have typical patterns (Fig. 2, Table 2); however, sometimes the details of the pattern are not fully examined. Full assessment of enzyme abnormalities involves careful evaluation of (1) the predominant pattern of enzyme alteration (hepatocellular v. cholestatic); (2) the magnitude of enzyme alteration in the case of aminotransferases (< 5 times, 5–10 times or > 10 times the upper reference limit, or mild, moderate or marked); (3) the rate of change (increase or decrease over time); and (4) the nature of the course of alteration (e.g., mild fluctuation v. progressive increase).
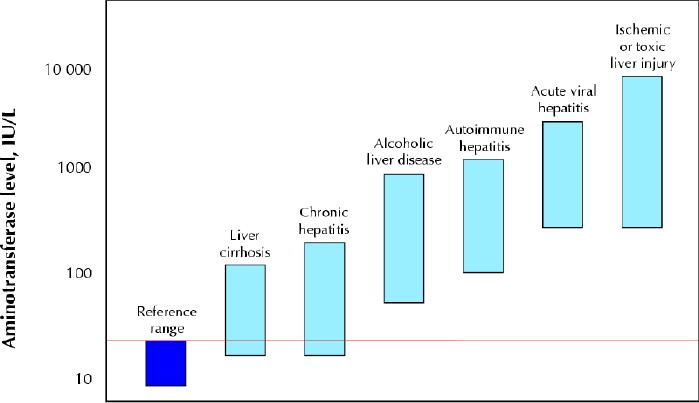
Fig. 2: Serum aminotransferase levels in various liver diseases. Patients with acute viral or ischemic or toxic liver injury reach the highest aminotransferase levels, but there is a broad overlap in aminotransferase values between patients with acute alcoholic hepatitis and autoimmune hepatitis as well as between patients with chronic hepatitis and liver cirrhosis. Both chronic hepatitis and cirrhotic patients may have aminotransferase levels within the reference range. The red line indicates the upper limit of the reference range.
Table 2
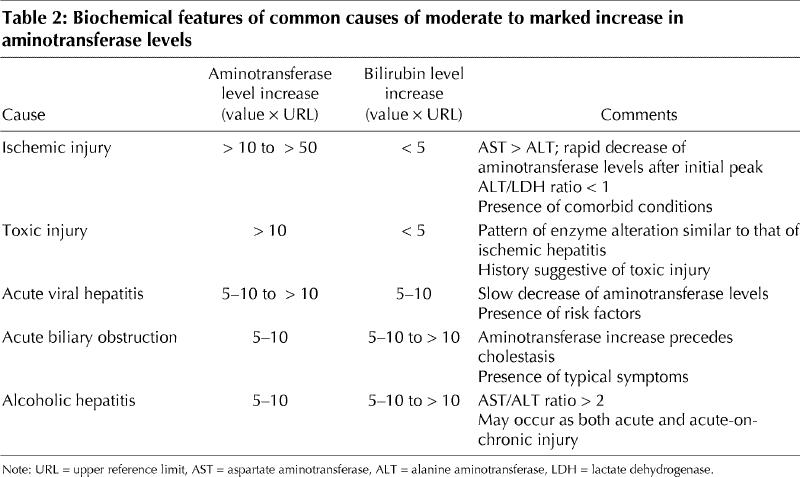
The most common alterations in enzyme levels encountered in clinical practice can be divided into 2 major subgroups: hepatocellular predominant and cholestatic predominant. Although certain liver diseases may display a mixed biochemical picture — usually elevated AST and ALT levels with mild abnormalities of alkaline phosphatase (ALP) and γ-glutamyl transpeptidase (GGT) levels — the ability to distinguish between the 2 subgroups is fundamental to narrowing down the differential diagnosis.
Hepatocellular predominance
Injury to the liver, whether acute or chronic, eventually results in an increase in serum concentrations of aminotransferases. AST and ALT are enzymes that catalyze the transfer of α-amino groups from aspartate and alanine to the α-keto group of ketoglutaric acid to generate oxalacetic and pyruvic acids respectively, which are important contributors to the citric acid cycle. Both enzymes require pyridoxal-5'-phosphate (vitamin B6) in order to carry out this reaction, although the effect of pyridoxal-5'-phosphate deficiency is greater on ALT activity than on that of AST.4,18 This has clinical relevance in patients with alcoholic liver disease, in whom pyridoxal-5'-phosphate deficiency may decrease ALT serum activity and contribute to the increase in the AST/ALT ratio that is observed in these patients.19,20
Both aminotransferases are highly concentrated in the liver. AST is also diffusely represented in the heart, skeletal muscle, kidneys, brain and red blood cells, and ALT has low concentrations in skeletal muscle and kidney;21 an increase in ALT serum levels is, therefore, more specific for liver damage. In the liver, ALT is localized solely in the cellular cytoplasm, whereas AST is both cytosolic (20% of total activity) and mitochondrial (80% of total activity).22 Zone 3 of the hepatic acinus has a higher concentration of AST, and damage to this zone, whether ischemic or toxic, may result in greater alteration to AST levels. Aminotransferase clearance is carried out within the liver by sinusoidal cells.23 The half-life in the circulation is about 47 hours for ALT, about 17 hours for total AST and, on average, 87 hours for mitochondrial AST.4
The magnitude of aminotransferase alteration can be classified as “mild” (< 5 times the upper reference limit), “moderate” (5–10 times the upper reference limit) or “marked” (> 10 times the upper reference limit). This classification is somewhat arbitrary, since no uniform definition exists and various reviews of the subject use different cut-off points.1,24,25 Marked and moderate increases are discussed together because the clinical distinction between them is especially grey.
Marked and moderate aminotransferase increase
Patients with a marked increase in aminotransferase levels (> 10 times the upper reference limit) typically have acute hepatic injury. However, data from a series of patients with acute hepatic injury due to viral hepatitis suggest that the most sensitive and specific aminotransferase threshold level to identify acute injury lies within the moderate range of increase (5–10 times the upper reference limit, at 200 IU/L for AST [sensitivity 91%, specificity 95%] and 300 IU/L for ALT [sensitivity 96%, specificity 94%]).26 Thus, the academic attribution of cause and “severity” of acute damage on the basis of the magnitude of enzyme elevation may sometimes be misleading, since there can be grey areas in which a range of causes overlap (Fig. 2). Moreover, the degree of elevation varies during the course of injury and depends on when the enzyme levels were tested (Fig. 3).
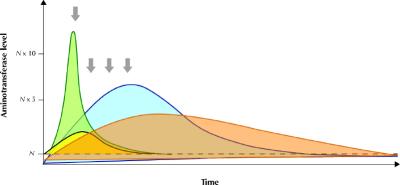
Fig. 3: Schematic representation of the rate of change of aminotransferase and bilirubin levels in a patient with acute ischemic hepatitis (green area, yellow area respectively) and acute viral hepatitis (blue area, orange area respectively). It is important to underscore that the pattern of enzyme alteration may vary and occasionally appear similar if a single observation point is taken into consideration (arrows).
Despite these ambiguities, the magnitude and rate of change of aminotransferase alteration may provide initial insight into a differential diagnosis. Very high aminotransferase levels (> 75 times the upper reference limit) indicate ischemic or toxic liver injury in more than 90% of cases of acute hepatic injury, whereas they are less commonly observed with acute viral hepatitis.4 In ischemic or toxic liver injury, AST levels usually peak before those of ALT because of the enzyme's peculiar intralobular distribution.27,28,29 Zone 3 of the acinus is more vulnerable to both hypoxic (hepatocytes are exposed to an already oxygen-poor milieu) and toxic (hepatocytes are richer in microsomal enzymes) damage. Furthermore, in ischemic injury aminotransferase levels tend to decrease rapidly after peaking (Fig. 3). In about 80% of patients with ischemic injury, the serum bilirubin level is lower than 34 μmol/L, and lactate dehydrogenase (LDH), a marker of ischemic damage, may reach very high concentrations (ALT/LDH ratio < 1).28,29,30 It is important to stress that a decrease in aminotransferase levels alone after a marked increase does not have prognostic meaning, since both resolution and massive hepatic necrosis may draw a similar biochemical picture. In this setting, patients with high bilirubin serum levels and prolonged prothrombin time should be closely monitored for the risk of hepatic failure.
In cases of acute viral hepatitis, aminotransferase levels usually peak before jaundice appears and have a more gradual decrease thereafter, and there is a greater increase in serum bilirubin levels (Fig. 3).31 Jaundice occurs in about 70% of cases of acute hepatitis A infection, 33%–50% of cases of acute hepatitis B infection and 20%–33% of cases of acute hepatitis C infection.5 LDH concentration is altered in about 50% of the patients, with values typically only slightly above the reference limit.28,29 The entire alphabet of viral hepatitis (A, B, C, D and E) may be responsible for a marked increase in aminotransferase levels, although the increase associated with hepatitis C infection tends to be more modest than that associated with hepatitis A or B.32 Patients with acute viral hepatitis may lack a history of exposure to risk factors and may have nonspecific (fatigue, arthralgias, low-grade fever) or specific (jaundice) symptoms of liver disease; the diagnosis may be made following routine analysis with asymptomatic hypertransaminasemia. The presence of symptoms is more common in patients with acute hepatitis A (70%–80% of infected adults) or B (30%–50% of infected adults) infection than in patients with acute hepatitis C (20% of patients) infection.5,33 Obviously, the presence of risk factors such as travel to areas endemic for viral hepatitis or parenteral exposure may help identify the cause and should drive the subsequent investigation. Patients with suspected acute viral hepatitis should be tested for IgM antibodies to hepatitis A and hepatitus B core virus, hepatitis B surface antigens and hepatitis C virus (HCV) antibodies. Testing for hepatitis D infection should be limited to patients with evidence of hepatitis B surface antigens. If test results for HCV antibodies are negative and there is no evidence of acute hepatitis A or B infection, testing for HCV RNA may be a useful strategy since a recent study has demonstrated that, although these patients are rarely at risk of acute liver failure, they may benefit from early antiviral treatment.34
Ischemic and hypoxic acute liver damage is more likely in patients with concomitant clinical conditions such as sepsis or low-flow hemodynamic state.10,28,30 Careful investigation of the patient's pharmacologic history is essential in order to identify medications and herbal products associated with hepatotoxicity.16,17,35 Acetaminophen-induced hepatic damage causes 54% and 16% of the cases of acute liver failure in the United Kingdom and United States respectively.36Although ischemic damage can be suspected on a clinical basis alone, and the patient history may suggest drug- or toxin-induced liver damage, there are no specific serologic tests for either condition, except in the case of acetaminophen poisoning, in which plasma acetaminophen levels may also represent a helpful guide to therapy.37 Alcohol-induced acute hepatic damage represents a distinct situation. It can be encountered in clinical practice both as acute and as acute-on-chronic liver injury. The biochemical picture is characteristic (e.g., GGT/ALP ratio > 2.5; jaundice may occur in more than 60% of patients38,39). In a landmark study by Cohen and Kaplan, the increase in AST levels was less than 6–7 times the upper reference value in 98% of the patients with alcoholic liver disease, and the AST/ALT ratio was greater than 1 in 92% and greater than 2 in 70% of the patients.19 The presence of physical signs of chronic liver disease and the patient's history may help distinguish between acute and acute-on-chronic damage.
After the most common causes of acute liver injury have been excluded, consideration should be given to minor hepatitis viruses (e.g., Epstein–Barr virus, cytomegalovirus) and to autoimmune, extrahepatic and congenital causes.1,15,24,25 Autoimmune hepatitis may present with a mild increase in aminotransferase levels or with a moderate to marked increase in aminotransferase levels (up to 49% of patients) with jaundice.40,41,42 This diagnosis is further discussed in the next section. Finally, as many as 25% of the patients with AST levels greater than 10 times the upper reference limit may have acute extrahepatic biliary tract obstruction, which can be heralded by a peak in aminotransferase levels (> 50 times the upper reference value in 1%–2% of patients) that rapidly subsides once the obstruction has been removed.5,43,44,45 The patient's history, presence of symptoms such as biliary pain and the ultrasound evidence of dilated bile ducts can provide a definitive diagnosis.
Mild increase in aminotransferase levels
A minimal or mild increase in aminotransferase level is the most common biochemical alteration encountered in everyday clinical practice. In addition to considering the when and where of the alterations, there is a series of first-line tests that can be performed on all patients because of their clinical relevance and the high prevalence of the diseases screened by these tests (Fig. 4). Extrahepatic causes of aminotransferase alteration (especially in patients with isolated AST elevation) should be ruled out by considering the clinical context of enzyme abnormality. Although some reviews dealing with liver enzyme alteration suggest repeating tests as a first measure in order to rule out laboratory error, we do not feel that a second, normal result is enough to exclude the presence of disease, and we recommend that a first-line, clinically guided screening for the most prevalent causes of chronic liver disease be started together with repetition of the test (Fig. 4). In fact, chronic hepatitis C infection is characterized by a pattern of aminotransferase levels fluctuating around the upper reference value. For patients who are taking drugs known to cause liver injury or who have evidence of alcohol abuse, a second, confirmatory check of aminotransferase levels after alcohol or the medication has been stopped can be a suitable option; patients who have evidence of alcohol abuse also need to be carefully assessed for the risk of underlying chronic damage.
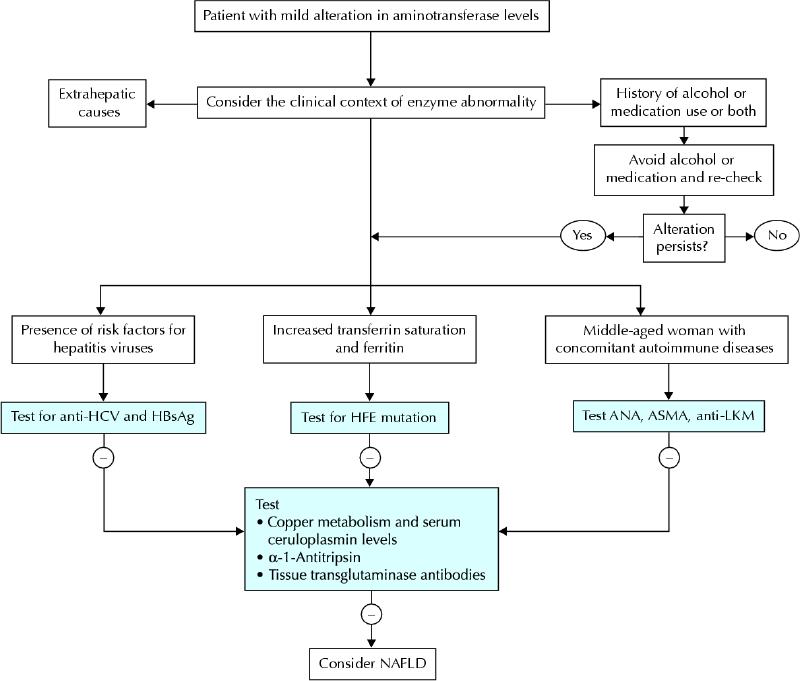
Fig. 4: Schematic, initial diagnostic algorithm for a patient presenting with mild aminotransferase abnormality. HCV = hepatitis C virus, HBsAG = hepatitis B surface antigen, ANA = antinuclear antibodies, ASMA = anti-smooth body antibodies, LKM = liver–kidney microsomes. Alcohol abuse and, to a lesser extent, drug-induced liver injury are frequently associated with mild aminotransferase abnormality, and their causality should be ruled out on a clinical basis. In the western world, chronic viral hepatitis, autoimmune hepatitis and hereditary hemochromatosis are the most common causes of mild aminotransferase alteration for which specific serological tests are available. Although nonalcoholic fatty liver disease (NAFLD) or steatohepatitis is frequently encountered in clinical practice, it remains a diagnosis of exclusion.
As in the case of acute damage, the pharmacologic history of the patient is of particular importance. All nonessential and over-the-counter medications should be discontinued, and discontinuation of an essential medication should be considered in a risk–benefit perspective. The use of dedicated scales or scores may help the clinician assess the likelihood of the hepatic drug reaction.46,47 Liver biopsy may represent a suitable diagnostic option in particular cases.
Nonalcoholic fatty liver disease is the most common cause of mild alteration of liver enzyme levels in the western world, and, according to the National Health and Nutritional Survey, point-prevalence is about 23% among American adults.48 The biochemical picture includes mildly raised aminotransferase levels, and GGT levels can be elevated up to 3 times the upper reference value in nearly half of patients in the absence of ethanol consumption.49 As with chronic viral hepatitis, an AST/ALT ratio greater than 1, which is observed in 61% of patients with advanced fibrosis and 24% of patients with no or initial fibrosis, is highly suggestive of advanced liver disease.50 Suspicion of nonalcoholic fatty liver disease is increased by the presence of conditions linked to the metabolic syndrome and insulin resistance (increased body mass index, diabetes, hyperlipemia, hypertension), although the disease may occur in patients without these associated factors.48,49 The diagnostic approach to suspected nonalcoholic fatty liver disease is aimed at ruling out other causes of liver disease since there is no specific blood test for diagnosis. Distinguishing between simple steatosis with or without minimal inflammation and nonalcoholic steatohepatitis with fibrosis with confidence is not possible on clinical grounds alone, and therefore liver biopsy should be performed in order to confirm diagnosis and assess prognosis.7,48,49,51
All patients presenting with mild increases in aminotransferase levels should be questioned about risk factors for hepatitis B or C infection (intravenous drug use, exposure to nonsterile needles or sexual exposure to an infected person). However, since these 2 diseases have a high prevalence worldwide52,53 and infected people may lack or underreport specific risk factors for infection,54,55 testing for HCV antibodies and hepatitis B surface antigens is advisable for all patients presenting with a mild increase in aminotransferase levels. If the patient tests positive for HCV antibodies, then qualitative HCV RNA testing should follow. In subjects who are seropositive for hepatitis B, the successive diagnostic work-up may depend upon the clinical situation (e.g., e-minus v. wild-type strands). It is important to emphasize that the degree of aminotransferase alteration is a poor guide to the severity of the disease in patients with established chronic viral hepatitis (Fig. 2), unless an AST/ALT ratio greater than 1 is found.56,57,58,59 An AST/ALT ratio greater than 1 can be found in 4% of patients with chronic hepatitis C infection and in 79% of patients who also have liver cirrhosis.57 Among patients with cirrhosis of viral cause, an AST/ALT ratio greater than 1.17 was found to prognosticate 1-year survival with 87% sensitivity (69%–96%, 95% CI) and 52% specificity (40%–64%, 95% CI).59 Patients may benefit from ultrasound examination of the liver in order to evaluate the presence of signs of advanced disease or liver masses. In patients with viral hepatitis, liver biopsy is needed to assess progression, evaluate the need for therapy and establish a prognosis.
HFE-related hereditary hemochromatosis is a fairly common autosomal recessive condition (homozygote frequency 1:200-1:400)13 that is characterized by pathological deposition of iron in the liver, pancreas and heart. Serum ferritin, iron and transferrin saturation index (serum iron/total iron binding capacity) should routinely be measured in patients with altered aminotransferase levels.60 High ferritin levels and, most importantly, a transferrin saturation index greater than 45% are strongly suggestive of the disease.60 The presence of diabetes, heart disease or arthritis is also suggestive, and mutation analysis for the HFE gene may confirm the diagnosis, especially if the patient is of northern European descent.13 Nevertheless, other, rarer forms of non-HFE-related hereditary hemochromatosis are being characterized more frequently, and liver biopsy remains a fundamental diagnostic tool in the presence of strong clinical suspicion and a negative result for HFE gene mutation analysis.60
The presence of a mild elevation in aminotransferase levels in female patients with concomitant autoimmune disorders (e.g., autoimmune thyroiditis, connective tissue diseases) is suggestive of autoimmune hepatitis. The prevalence of the disease is about 1:6000 to 1:7000, and as many as 80% of patients may have hypergammaglobulinemia even in the absence of liver cirrhosis.40,61 Patients with suspected autoimmune hepatitis should have autoantibodies tested (antinuclear, anti-smooth muscle and anti-liver–kidney microsomes), although the criteria for diagnosis are complex and include liver biopsy.40,42,61 Patients may have a dramatic therapeutic response to corticosteroids, but the course of the disease may be long and can fluctuate between phases of remission and relapses that may mimic acute hepatitis.40,42,61
Wilson's disease (homozygote frequency 1:30 000– 1:300 000) should be suspected in young patients with signs of hemolysis or concomitant psychiatric or neurologic symptoms, and serum ceruloplasmin levels and copper metabolism (serum and 24-hour urinary copper) should be tested. Diagnosis in patients showing low serum ceruloplasmin levels and increased urinary copper excretion can be confirmed by slit-lamp examination for Kayser–Fleischer rings, although liver biopsy with quantitative copper measurement may be needed where no clear clinical diagnosis is possible.15,45
Although α-1-antitripsin deficiency is not a rare disease, affecting 1:1600–1:2800 newborns in Europe and the United States, it is an unusual cause of aminotransferase alteration among adults since the disease is usually identified in childhood.62 It can be suspected in adult patients with concomitant pulmonary disease (emphysema), although low serum α-1-antitripsin levels and phenotype determination provide definite diagnosis.62
Finally, it has been reported that up to 10% of patients with unexplained hypertransaminasemia actually have celiac disease, and minimal or mild alteration of aminotransferase levels may be the only visible part of the “celiac iceberg.”63 In these patients, screening by measuring tissue transglutaminase antibodies and confirmation and grading of the disease by small bowel biopsy are required for diagnosis.64
Cholestatic predominance
Presentation of liver injury with a prevalent cholestatic pattern is less frequently encountered in clinical practice than the pattern of hepatocellular damage. ALP and bilirubin levels are routinely assessed, and the level of GGT is often measured as an additional aid toward diagnosis in particular situations because of its high sensitivity but low specificity.
Alkaline phosphatase
ALP is an enzyme that transports metabolites across cell membranes. Liver and bone diseases are the most common causes of pathological elevation of ALP levels, although ALP may originate from other tissue, such as the placenta, kidneys or intestines, or from leukocytes.65 The third trimester of pregnancy (placenta origin) and adolescence (bone origin) are associated with an isolated increase in serum ALP levels.4 Hepatic ALP is present on the surface of bile duct epithelia. Cholestasis enhances the synthesis and release of ALP, and accumulating bile salts increase its release from the cell surface.66,67 ALP half-life in the circulation is about 1 week.4 These characteristics explain why ALP levels usually rise late in bile duct obstruction and decrease slowly after resolution.
In some patients (e.g., pregnant women, adolescents) the reason for increased ALP levels may be straightforward, but in other patients it is necessary to identify the origin of the enzyme. This task can be accomplished in 2 ways: assessment of GGT levels or dosage of ALP isoenzymes (Fig. 5). From a practical point of view, measurement of GGT is preferred since it relies on automated analysis rather than on more sophisticated and expensive techniques.

Fig. 5: Suggested diagnostic algorithm for patients presenting with increased alkaline phosphatase levels. AMA = antimitochondria antibodies, PBC = primary biliary cirrhosis, ANCA = antineutrophil cytoplasmic antibodies, ERCP = endoscopic retrograde cholangiopancreatography, PSC = primary sclerosing cholangitis.
The degree and rate of enzyme alteration may provide minor and nonspecific clues to diagnosis, but the presence of symptoms and the patient's history, with particular emphasis on comorbid conditions, may provide fundamental clues (Fig. 5). Liver ultrasound may reveal the presence of bile duct dilation, demonstrate signs of chronic liver disease or even liver cirrhosis, and identify hepatic masses.
Drug-induced liver injury may present with a cholestatic pattern (preferential increase in ALP or ALT/ALP ratio < 2), although the degree of ALP alteration is variable and may be accompanied by hyperbilirubinemia.68 Commonly used drugs such as antihypertensives (e.g., angiotensin-converting enzyme inhibitors) or hormones (e.g., estrogen) may cause cholestasis and can be overlooked. Liver ultrasound in these patients is often unremarkable.
Varying degrees of ALP alteration in patients with inflammatory bowel disease (most commonly ulcerative colitis) suggest the presence of primary sclerosing cholangitis, since about 70% of these cases are associated with inflammatory bowel disease.69 The same biochemical abnormality observed in middle-aged women with a history of itching and autoimmune disease raises the suspicion of primary biliary cirrhosis.70 Further diagnostic work-up of patients with suspected autoimmune cholestatic liver disease includes testing antineutrophil cytoplasmic antibodies and cholangiography for primary sclerosing cholangitis and antimitochondria antibodies and total IgM levels for primary biliary cirrhosis. In these diseases, ALP and GGT levels may be slightly or markedly raised, whereas aminotransferase levels are often minimally altered or within the upper reference limit, unless overlapping features of autoimmune cholestatic diseases and autoimmune hepatitis occur.71 In patients with primary sclerosing cholangitis or primary biliary cirrhosis, serum bilirubin levels have prognostic meaning,72,73 and liver ultrasound may reveal an echotexture suggestive of diffuse disease or even liver cirrhosis (Fig. 5).
ALP alteration due to common bile duct obstruction may be heralded by a peak in aminotransferase levels, typical symptoms and conjugated hyperbilirubinemia, especially in the acute setting, or the ALP levels may have a fluctuating pattern (± GGT alteration) with normal serum bilirubin in “valve” choledocholithiasis.43,44 In these patients, liver ultrasound may show dilated bile ducts, and endoscopic retrograde cholangiopancreatography can be used to remove the obstacle.
Abnormal ALP levels may also be a sign of metastatic cancer of the liver, lymphoma or infiltrative diseases such as sarcoidosis. In some of these situations ALP levels may be markedly elevated and the only sign of liver involvement. In these cases, liver ultrasound examination is extremely important when the patient history is not suggestive of disease, although some patients may require liver biopsy in order to obtain a definitive diagnosis.24,25
The clinical work-up described in Fig. 5 should help to establish a diagnosis in the majority of patients with elevated ALP levels. The degree of elevation seems to have no specific diagnostic relevance. Nevertheless, diagnosis may not be evident even after thorough evaluation in asymptomatic patients with mild increases in ALP levels. As mentioned above, liver biopsy can provide a fundamental clue to some unusual and unsuspected diagnoses.
γ-Glutamyl transpeptidase
GGT is an enzyme that is present in hepatocytes and biliary epithelial cells, renal tubules, and the pancreas and intestine. The mechanisms of alteration are similar to those described for alkaline phosphatase. GGT is a microsomal enzyme, and its activity can be induced by several drugs, such as anticonvulsants and oral contraceptives.74 Elevated GGT levels can be observed in a variety of nonhepatic diseases, including chronic obstructive pulmonary disease and renal failure, and may be present for weeks after acute myocardial infarction. Increased serum levels observed in alcoholic liver disease can be the result of enzyme induction and decreased clearance. In these patients, GGT serum levels can be markedly altered (> 10 times the upper reference value), whereas ALP levels may be normal or only slightly altered (GGT/ALP ratio > 2.5). The whole spectrum of liver diseases, regardless of cause, may be responsible for altered GGT serum levels. In particular, GGT levels may be 2–3 times greater than the upper reference value in more than 50% of the patients with nonalcoholic fatty liver disease and above the upper reference value in about 30% of patients with chronic hepatitis C infection.75,76 Furthermore, an increase in GGT levels in patients with chronic liver disease is associated with bile duct damage and fibrosis.76 Thus, because of its lack of specificity but high sensitivity for liver disease, GGT can be useful for identifying causes of altered ALP levels, or elevated levels, together with other biochemical abnormalities (AST/ALT ratio > 2),19 may support the diagnosis of alcoholic liver disease.
Bilirubin
Bilirubin is the product of hemoglobin catabolism within the reticuloendothelial system. Heme breakdown determines the formation of unconjugated bilirubin, which is then transported to the liver. In the liver, UDP-glucuronyltransferase conjugates the water-insoluble unconjugated bilirubin to glucuronic acid, and conjugated bilirubin is then excreted into the bile.77,78
Unconjugated bilirubin may increase because of augmented bilirubin production or decreased hepatic uptake or conjugation or both (Table 3). In adults, the most common conditions associated with unconjugated hyperbilirubinemia are hemolysis and Gilbert's syndrome.78 Hemolysis can be ruled out by measuring hemoglobin serum levels, reticulocyte count, and haptoglobin levels. Gilbert's syndrome is determined by a variety of genetic defects in UDP-glucuronyltransferase that affect about 5% of the population.79 In these subjects serum indirect bilirubin usually does not exceed 68 μmol/L, and the remainder of liver chemistry tests and liver ultrasound are unremarkable.80 Although a series of tests has been proposed to confirm the diagnosis, this condition is usually diagnosed on a clinical basis alone, and the patient should be reassured of the benign nature of this enzymatic alteration.80 Other, less frequent causes of unconjugated hyperbilirubinemia include reabsorption of large hematomas and ineffective erythropoiesis.24
Table 3

In healthy people, conjugated bilirubin is virtually absent from serum mainly because of the rapid process of bile secretion.1 Levels increase when the liver has lost at least half of its excretory capacity. Therefore, the presence of increased conjugated bilirubin is usually a sign of liver disease. Conjugated hyperbilirubinemia (usually < 34 μmol/L) and concomitant, markedly elevated aminotransferase levels may suggest acute viral hepatitis or toxic or ischemic liver injury. Furthermore, this biochemical picture can be the presenting feature of autoimmune hepatitis.40,41,42 On the other hand, a purely cholestatic picture, with conjugated hyperbilirubinemia, an increase in ALP levels and a negligible increase in aminotransferase levels, may be present in cholestatic drug reactions.16,68 Sometimes, the same biochemical picture may be present in the late presentation of previously unrecognized autoimmune cholestatic diseases (primary biliary cirrhosis, primary sclerosing cholangitis). In these patients, the presence of other signs of chronic liver disease may facilitate diagnosis.62,69,70
Biliary obstruction can cause various degrees of conjugated hyperbilirubinemia. The severity of alteration depends upon the degree and duraction of obstruction and the functional reserve of the liver. Biliary obstruction may have an abrupt onset and be preceded by typical symptoms (right upper quadrant pain, nausea) or may be silent and progressive. With the presence or absence of concomitant aminotransferase alteration, a liver ultrasound is essential to identify and locate the obstacle to bile flow.
Once the causal condition of conjugated hyperbilirubinemia has resolved, whatever the cause, bilirubin serum levels decrease in a bimodal fashion. There is a first, rapid decrease and then a later, slower decrease caused by the binding of bilirubin to albumin and the formation of a complex (δ-bilirubin) that has the same half-life as serum albumin.77,78,81
Albumin and prothrombin time assessment: Are we really testing liver function?
Determining serum albumin levels and assessing prothrombin time are often considered “tests of liver function.” This is mainly because hepatic synthesis of albumin tends to decrease in end-stage liver disease, and an increase in prothrombin time depends on the decreased synthesis of liver-derived coagulation factors. In fact, albumin is produced by hepatocytes, and prothrombin time depends on the activity of clotting factors I, II, V, VII and X, which are produced in the liver. However, neither test is specific for liver disease, since albumin serum levels may decrease in patients with nephrotic syndrome, malabsorption or protein-losing enteropathy, or malnutrition, and prothrombin time may be prolonged by warfarin treatment, deficiency in vitamin K (which is needed to activate clotting factors II, VII and X) and consumptive coagulopathy.1,4,5,82 The finding of hypoalbuminemia and no other alterations in liver tests virtually rules out the hepatic origin of this abnormality.
However, when it is certain that the cause of their alteration is liver disease, serum albumin levels and prothrombin time are useful tests for monitoring liver synthetic activity. The half-life of albumin in circulation is long (about 20 days), and the half-life of blood clotting factors is quite short (about 1 day).4,5,82 Thus, albumin levels decrease when cirrhosis occurs, and they have prognostic meaning in these patients. On the contrary, patients with acute, massive hepatocellular necrosis (acute toxic or viral hepatitis) may have a brisk increase in prothrombin time (usually < 3 seconds) that tends to plateau, and normal albuminemia. In these patients, prothrombin time can be monitored in order to assess the risk of acute liver failure. On the other hand, prothrombin time is not useful for assessing liver function in patients with mild aminotransferase alteration, since prothrombin time can remain within normal limits for long periods unless a marked decrease in liver function occurs and patients with compensated liver cirrhosis may have normal prothrombin time.81 Thus, it is evident from these data that albumin and prothrombin time alteration should be interpreted within the clinical and biochemical context of the patient.
Obstructive jaundice may decrease the absorption of vitamin K, and therefore increase prothrombin time. In this situation, prothrombin time responds to parenteral administration of vitamin K, which is ineffective when jaundice is caused by decreased liver functioning mass. Finally, prothrombin time values are strictly dependent upon the International Sensitivity Index of the reagent that is used, and this does not allow for easy standardization of results among laboratories.4,5 Taking these limitations into account, both serum albumin and prothrombin time can be considered useful tools, alone or combined in clinical scores, for evaluating liver function.83,84,85,86 Nevertheless, tests that involve administering exogenous compounds may be used to evaluate liver function more accurately.87
Conclusions
Alterations in liver enzyme levels are one of the most common problems encountered in everyday clinical practice. Finding the way through the multiple diagnostic pathways can challenge even the experienced clinician. Knowledge of the pathophysiology of liver enzymes is an essential guide to understanding their alteration. The pattern of enzyme abnormality, interpreted in the context of the patient's characteristics, can aid in directing the subsequent diagnostic work-up. Awareness of the prevalence of determined liver disease in specific populations and of possible hepatic involvement during systemic illnesses or drug therapies may help the clinician identify the cause of alterations efficiently.
Footnotes
This article has been peer reviewed.
Competing interests: None declared.
Correspondence to: Dr. Edoardo G. Giannini, Gastroenterology Unit, Department of Internal Medicine, University of Genoa, Viale Benedetto XV, no. 6, 16132, Genoa, Italy; fax: 39 010 353 8638; egiannini@unige.it
References
- 1.Green RM, Flamm S. AGA technical review on the evaluation of liver chemistry tests. Gastroenterology 2002;123(4):1367-84. [DOI] [PubMed]
- 2.Burkitt HG, Young B, Heath JW. Wheater's functional histology: a text and colour atlas. 3rd ed. Edinburgh: Churchill Livingston, 1993.
- 3.Worobetz L, Hilsden R, Shaffer E, Simon J Pare P, Scully L, et al. The liver. In Thomson BR, Shaffer EA, editors. First Principles of Gastroenterology. 2nd ed. University of Toronto Press: Toronto; 1994.
- 4.Dufour DR, Lott JA, Nolte FS, Gretch DR, Koff RS, Seeff LB. Diagnosis and monitoring of hepatic injury. I. Performance characteristics of laboratory tests. Clin Chem 2000;46(12):2027-49. [PubMed]
- 5.Dufour DR, Lott JA, Nolte FS, Gretch DR, Koff RS, Seeff LB. Diagnosis and monitoring of hepatic injury. II. Recommendations for use of laboratory tests in screening, diagnosis, and monitoring. Clin Chem 2000;46(12):2050-68. [DOI] [PMC free article] [PubMed]
- 6.Gholson CF, Morgan K, Catinis G, Favrot D, Taylor B, Gonzalez E, et al. Chronic hepatitis C with normal aminotransferase levels: a clinical histologic study. Am J Gastroenterol 1997;92(10):1788-92. [PubMed]
- 7.Mofrad P, Contos MJ, Haque M, Sargeant C, Fisher RA, Luketic VA, et al. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology 2003;37(6):1286-92. [DOI] [PubMed]
- 8.Dufour DR. Effects of habitual exercise on routine laboratory tests [abstract]. Clin Chem 1998;44 (Suppl 6):A136.
- 9.Narjes H, Nehmiz G. Effect of hospitalisation on liver enzymes in healthy subjects. Eur J Clin Pharmacol 2000;56(4):329-33. [DOI] [PubMed]
- 10.Whitehead MW, Hawkes ND, Hainsworth I, Kingham JGC. A prospective study of the causes of notably raised aspartate aminotransferase of liver origin. Gut 1999;45(1):129-33. [DOI] [PMC free article] [PubMed]
- 11.Chadha MS, Walimbe AM, Chobe LP, Arankalle VA. Comparison of etiology of sporadic acute and fulminant viral hepatitis in hospitalized patients in Pune, India during 1978-81 and 1994-97. Indian J Gastroenterol 2003; 22 (1): 11 -5. [PubMed]
- 12.Feld JJ, Heathcote EJ. Epidemiology of autoimmune liver disease. J Gastroenterol Hepatol 2003;18(10):1118-28. [DOI] [PubMed]
- 13.Merryweather-Clarke AT, Pointon JJ, Shearman JD, Robson KJ. Global prevalence of putative haemochromatosis mutations. J Med Genet 1997; 34 (4): 275-8. [DOI] [PMC free article] [PubMed]
- 14.Barton JC, Acton RT. Inheritance of two HFE mutations in African Americans: cases with hemochromatosis phenotypes and estimates of hemochromatosis phenotype frequency. Genet Med 2001;3(4):294-300. [DOI] [PubMed]
- 15.Roberts EA, Schilsky ML. A practice guideline on Wilson disease. Hepatology 2003;37(6):1475-92. [DOI] [PubMed]
- 16.Lee WM. Drug-induced hepatotoxicity. N Engl J Med 2003;349(5):474-85. [DOI] [PubMed]
- 17.Fogden E, Neuberger J. Alternative medicines and the liver. Liver Int 2003; 23 (4): 213-20. [DOI] [PubMed]
- 18.Vanderlinde RE. Review of pyridoxal phosphate and the transaminases in liver disease. Ann Clin Lab Sci 1986;16(2):79-93. [PubMed]
- 19.Cohen JA, Kaplan MM. The SGOT/SGPT ratio — an indicator of alcoholic liver disease. Dig Dis Sci 1979;24(11):835-8. [DOI] [PubMed]
- 20.Diehl AM, Potter J, Boitnott J, Van Duyn MA, Herlong HF, Mezey E. Relationship between pyridoxal 5'-phosphate deficiency and aminotransferase levels in alcoholic hepatitis. Gastroenterology 1984;86(4):632-6. [PubMed]
- 21.Wroblewski F. The clinical significance of alterations in transaminase activities of serum and other body fluids. Adv Clin Chem 1958;1(2):313-51. [PubMed]
- 22.Rej R. Aminotransferases in disease. Clin Lab Med 1989;9(4):667-87. [PubMed]
- 23.Kamimoto Y, Horiuchi S, Tanase S, Morino Y. Plasma clearance of intravenously injected aspartate aminotransferase isozymes: evidence for preferential uptake by sinusoidal liver cells. Hepatology 1985;5(3):367-75. [DOI] [PubMed]
- 24.Pratt DS, Kaplan MM. Evaluation of abnormal liver-enzyme results in asymptomatic patients. N Engl J Med 2000;342(17): 1266-71. [DOI] [PubMed]
- 25.Gopal DV, Rosen HR. Abnormal findings on liver function tests. Postgrad Med 2000;107(2):100-14. [DOI] [PubMed]
- 26.Rozen P, Korn RJ, Zimmerman HJ. Computer analysis of liver function tests and their interrelationship in 347 cases of viral hepatitis. Isr Med Sci 1970; 6 (1): 67 -79. [PubMed]
- 27.Dufour DR, Teot L. Laboratory identification of ischemic hepatitis (shock liver) [abstract]. Clin Chem 1988;34:A1287.
- 28.Seeto RK, Fenn B, Rockey DC. Ischemic hepatitis: clinical presentation and pathogenesis. Am J Med 2000;109(2):109-13. [DOI] [PubMed]
- 29.Singer AJ, Carracio TR, Mofenson HC. The temporal profile of increased transaminase levels in patients with acetaminophen-induced liver dysfunction. Ann Emerg Med 1995;26(1):49-53. [DOI] [PubMed]
- 30.Fuchs S, Bogomolski-Yahalom V, Paltiel O, Ackerman Z. Ischemic hepatitis: clinical and laboratory observations of 34 patients. J Clin Gastroenterol 1998; 26 (3):183-6. [DOI] [PubMed]
- 31.Clermont RJ, Chalmers TC. The transaminase tests in liver disease. Medicine 1967; 46(2):197-207. [DOI] [PubMed]
- 32.Marcellin P. Hepatitis C: the clinical spectrum of the disease. J Hepatol 1999; 31 (Suppl 1):9-16. [DOI] [PubMed]
- 33.Geller SA. Hepatitis B and hepatitis C. Clin Liver Dis 2002;6(2):317-34. [DOI] [PubMed]
- 34.Wedemeyer H, Jackel E, Wiegand J, Cornberg M, Manns MP. Whom? When? How? Another piece of evidence for early treatment of acute hepatitis C. Hepatology 2004;39(5):1201-3. [DOI] [PubMed]
- 35.Shad JA, Chinn CG, Brann OS. Acute hepatitis after ingestion of herbs. South Med J 1999;92(11):1095-7. [DOI] [PubMed]
- 36.O'Grady JG. Acute liver failure. In: Comprehensive Clinical Hepatology. O'Grady JG, Lake JR, Howdle PD, editors. Mosby:London, 2000. p. 16.1-16.13.
- 37.Sellers EM, Freedman F. Treatment of acetaminophen poisoning. CMAJ 1981; 125(8):827-9. [PMC free article] [PubMed]
- 38.Mendenhall CL. Alcoholic hepatitis. The VA Cooperative Study Group on Alcoholic Hepatitis. ClinGastroenterol 1981;10(2): 417-41. [PubMed]
- 39.Goldberg S, Mendenhall C, Anderson S, Garcia-Pont P, Kiernan T, Seeff L, et al. VA Cooperative Study on Alcoholic Hepatitis. IV. The significance of clinically mild alcoholic hepatitis-describing the population with minimal hyperbilirubinemia. Am J Gastroenterol 1986;81(11):1029-34. [PubMed]
- 40.Krawitt EL. Autoimmune hepatitis. N Engl J Med 1996;334(14):897-903. [DOI] [PubMed]
- 41.Kessler WR, Cummings OW, Eckert G, Chalasani N, Lumeng L, Kuo PY. Fulminant hepatic failure as the initial presentation of acute autoimmune hepatitis. Clin Gastroenterol Hepatol 2004;2(7):625-31. [DOI] [PubMed]
- 42.Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol 1999;31(5):929-38. [DOI] [PubMed]
- 43.Forston WC, Tedesco FJ, Stames EC, Shaw CT. Marked elevation of serum transaminase activity associated with extrahepatic biliary tract disease. J Clin Gastroenterol 1985;7(6):502-5. [DOI] [PubMed]
- 44.Anciaux ML, Pelletier AG, Attali P, Meduri B, Liguory C, Etienne JP. Prospective study of clinical and biochemical features of symptomatic choledocolithiasis. Dig Dis Sci 1986;31:449-53. [DOI] [PubMed]
- 45.Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, et al. Diagnosis and phenotypic classification of Wilson disease. Liver Int 2003; 23 (3):139-42. [DOI] [PubMed]
- 46.Maria VA, Victorino RM. Development and validation of a clinical scale for the diagnosis of drug-induced hepatitis. Hepatology 1997;26(3):664-9. [DOI] [PubMed]
- 47.Aithal GP, Rawlins MD, Day CP. Clinical diagnostic scale: a useful tool in the evaluation of suspected hepatotoxic adverse drug reactions. J Hepatol 2000;33(6);949-52. [DOI] [PubMed]
- 48.Harrison SA, Kadakia S, Lang KA, Schenker S. Nonalcoholic steatohepatitis: what we know in the new millenium. Am J Gastroenterol 2002;97(11):2714-24. [DOI] [PubMed]
- 49.Brunt EM. Nonalcoholic steatohepatitis. Semin Liver Dis 2004;24(1):3-20. [DOI] [PubMed]
- 50.Angulo P, Keach JC, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology 1999; 30 (6): 1356-62. [DOI] [PubMed]
- 51.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, Mc-Cullogh AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 1999;116(6):1413-9. [DOI] [PubMed]
- 52.Lavanchy D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat 2004; 11 (2): 97-107. [DOI] [PubMed]
- 53.The Global Burden Of Hepatitis C Working Group. Global burden of disease (GBD) for hepatitis C. J Clin Pharmacol 2004;44 (1):20-9. [DOI] [PubMed]
- 54.Mele A, Spada E, Sagliocca L, Ragni P, Tosti ME, Gallo G, et al. Risk of parenterally transmitted hepatitis following exposure to surgery or other invasive procedures: results from the hepatitis surveillance system in Italy. J Hepatol 2001;35(2):284-9. [DOI] [PubMed]
- 55.Memon MI, Memon MA. Hepatitis C: An epidemiological review. J Viral Hepat 2002;9(2):84-100. [DOI] [PubMed]
- 56.Williams AL, Hoofnagle JH. Ratio of serum aspartate to alanine aminotransferase in chronic hepatitis. Relationship to cirrhosis. Gastroenterology 1988; 95 (3): 734-9. [DOI] [PubMed]
- 57.Giannini E, Botta F, Fasoli A, Ceppa P, Risso D, Lantieri PB, et al. Progressive liver functional impairment is associated with an increase in AST/ALT ratio. Dig Dis Sci 1999;44(6):1249-53. [DOI] [PubMed]
- 58.Giannini E, Risso D, Botta F, Chiarbonello B, Fasoli A, Malfatti F, et al. Validity and clinical utility of the aspartate aminotransferase-alanine aminotransferase ratio in assessing disease severity and prognosis in patients with hepatitis C virus-related chronic liver disease. Arch Intern Med 2003;163(2):218-24. [DOI] [PubMed]
- 59.Giannini E, Botta F, Testa E, Romagnoli P, Polegato S, Malfatti F, et al. The 1-year and 3-month prognostic utility of the AST/ALT ratio and model for end-stage liver disease score in patients with viral liver cirrhosis. Am J Gastroenterol 2002;97(11):2855-60. [DOI] [PubMed]
- 60.Tavill AS. Diagnosis and management of Hemochromatosis. Hepatology 2001; 33 (5):1321-8. [DOI] [PubMed]
- 61.Czaja AJ. Natural history, clinical features, and treatment of autoimmune hepatitis. Semin Liver Dis 1984;4(1):1-12. [DOI] [PubMed]
- 62.Morrison ED, Kowdley KV. Genetic liver disease in adults: early recognition of the three most common causes. Postgrad Med 2000;107(2):147-59. [DOI] [PubMed]
- 63.Abdo A, Meddings J, Swain M. Liver abnormalities in celiac disease. Clin Gastroenterol Hepatol 2004;2(2):107-12. [DOI] [PubMed]
- 64.Farrell RJ, Kelly CP. Diagnosis of celiac sprue. Am J Gastroenterol 2001; 96 (12): 3237-46. [DOI] [PubMed]
- 65.Fishman WH. Alkaline phosphatase isoenzymes: recent progress. Clin Biochem 1990;23(2):99-104. [DOI] [PubMed]
- 66.Moss DW. Physiochemical and pathophysiological factors in the release of membrane-bound alkaline phosphatase from cells. Clin Chim Acta 1997; 257 (1): 133-40. [DOI] [PubMed]
- 67.Schlaeger R, Haux D, Kattermann R. Studies on the mechanism of the increase in serum alkaline phosphatase activity in cholestasis: significance of the hepatic bile acid concentration for the leakage of alkaline phosphatase from rat liver. Enzyme 1982;28(1):3-13. [DOI] [PubMed]
- 68.Velayudham LS, Farrell GC. Drug-induced cholestasis. Expert Opin Drug Saf 2003;2(3):287-304. [DOI] [PubMed]
- 69.Ponsioen CI, Tytgat GN. Primary sclerosing cholangitis: a clinical review. Am J Gastroenterol 1998;93(4):515-23. [DOI] [PubMed]
- 70.Heathcote J. Update on primary biliary cirrhosis. Can J Gastroenterol 2000; 14 (1): 43-8. [DOI] [PubMed]
- 71.Poupon R. Autoimmune overlapping syndromes. Clin Liver Dis 2003; 7 (4): 865-78. [DOI] [PubMed]
- 72.Bonnand AM, Heathcote EJ, Lindor KD, Poupon RE. Clinical significance of serum bilirubin levels under ursodeoxycholic acid therapy in patients with primary biliary cirrhosis. Hepatology 1999;29(1):34-43. [DOI] [PubMed]
- 73.Kim WR, Therneau TM, Wiesner RH, Poterucha JJ, Benson JT, Malinchoc M, et al. A revised natural history model for primary sclerosing cholangitis. Mayo Clin Proc 2000;75(7):688-94. [DOI] [PubMed]
- 74.Rosalki SB, Tarlow D, Rau D. Plasma gamma-glutamyl transpeptidase elevation in patients receiving enzyme-inducing drugs. Lancet 1971;2:376-7. [DOI] [PubMed]
- 75.McCullough AJ. Update on nonalcoholic fatty liver disease. J Clin Gastroenterol 2002;34(3):255-62. [DOI] [PubMed]
- 76.Giannini E, Botta F, Fasoli A, Romagnoli P, Mastracci L, Ceppa P, et al. Increased levels of gammaGT suggest the presence of bile duct lesions in patients with chronic hepatitis C: absence of influence of HCV genotype, HCV-RNA serum levels, and HGV infection on this histological damage. Dig Dis Sci 2001;46(3):524-9. [DOI] [PubMed]
- 77.Berk PD, Noyer C. Clinical chemistry and physiology of bilirubin. Semin Liver Dis 1994; 4(4):346-55. [DOI] [PubMed]
- 78.Fevery J, Blanckaert N. What can we learn from analysis of serum bilirubin. J Hepatol 1986;2(1):113-21. [DOI] [PubMed]
- 79.Bosma PJ, Chowdury JR, Bakker C, Gantla S, de Boer A, Oostra BA, et al. The genetic bases of the reduced expression of bilirubin UDP-glucuronosyltransferase 1 in Gilbert's syndrome. N Engl J Med 1995;333(18):1171-5. [DOI] [PubMed]
- 80.Thomsen HF, Hardt F, Juhl E. Diagnosis of Gilbert's syndrome. Scand J Gastroenterol 1981;16(5):699-703. [DOI] [PubMed]
- 81.Van Hootegem P, Fevery J, Blanckaert N. Serum bilirubins in hepatobiliary disease: comparison with other liver function tests and changes in the postobstructive period. Hepatology 1985;5(1):112-7. [DOI] [PubMed]
- 82.Doumas BT, Peters T. Serum and urine albumin: a progress report on their measurement and clinical significance. Clin Chim Acta 1997;258(1):3-20. [DOI] [PubMed]
- 83.Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg 1973; 60 (8): 646-9. [DOI] [PubMed]
- 84.Bonis PA, Tong MJ, Blatt LM, Conrad A, Griffith JL. A predictive model for the development of hepatocellular carcinoma, liver failure, or liver transplantation for patients presenting to clinic with chronic hepatitis C. Am J Gastroenterol 1999;94(6):1605-12. [DOI] [PubMed]
- 85.Kamath PS, Wiesner RH, Malinchoc M, Kremers W, Therneau TM, Kosberg CL, et al. A model to predict survival in patients with end-stage liver disease. Hepatology 2001;33(2):464-70. [DOI] [PubMed]
- 86.Botta F, Giannini E, Romagnoli P, Fasoli A, Malfatti F, Chiarbonello B, et al. MELD scoring system is useful for predicting prognosis in patients with liver cirrhosis and is correlated with residual liver function: a European study. Gut 2003;52(1):134-9. [DOI] [PMC free article] [PubMed]
- 87.Jalan R, Hayes PC. Review article: quantitative tests of liver function. Aliment Pharmacol Ther 1995;9(3):263-70. [DOI] [PubMed]