

ABSTRACT
In patients with sepsis-induced multi-organ dysfunction syndrome, diverging patterns of oedema formation and loss of function in organs such as lung and kidney suggest that endothelial permeability-regulating molecular responses are differentially regulated. This potential differential regulation has been insufficiently studied at the level of components of adherens and tight junctions. We hypothesized that such a regulation by endothelial cells in sepsis takes place in an organ-specific manner. We addressed our hypothesis by studying by quantitative real time polymerase chain reaction the expression of a predefined subset of EC permeability-related molecules (occludin, claudin-5, PV-1, CD-31, endomucin, Angiopoietin-1, Angiopoietin-2, Tie2, VEGFA, VEGFR1, VEGFR2, and VE-cadherin) in kidney and lung after systemic lipopolysacharide injection in mice, and in kidneys of patients who died of sepsis. We showed that baseline endothelial expression of permeability-related molecules differs in mouse kidney and lung. Moreover, we showed differential regulation of these molecules after lipopolysacharide injection in the two mouse organs. In lung we found a decrease in expression levels of molecules of the adherence and tight junctions complex and related signaling systems, compatible with increased permeability. In contrast, in kidney we found expression patterns of these molecules compatible with decreased permeability. Finally, we partially corroborated our findings in mouse kidney in human kidneys from septic patients. These findings may help to understand the clinical difference in the extent of oedema formation in kidney and lung in sepsis-associated organ failure.
Keywords: Cell–cell junctions, endothelium, human, kidney, LPS, lung, MODS, mouse, sepsis, vascular permeability
INTRODUCTION
Sepsis poses a major morbidity and mortality burden since it can result in multi-organ dysfunction syndrome (1). The precise mechanisms leading to MODS are largely unknown (2). Initially, circulating humeral and cellular components of the immune and coagulation system were held responsible for the development of MODS. However, more recent studies suggest that the breakdown of the endothelial cell (EC) barrier, resulting in alterations in vascular permeability, oedema formation, and, finally, loss of organ function, plays a crucial role in the pathogenesis of MODS (3, 4).
Interestingly, oedema formation in, and loss of function of different organs in patients with MODS does not follow a uniform pattern. For example, the lung dysfunctions partially due to oedema and flooding after loss of endothelial barrier integrity and gap formation (5). On the other hand, oedema formation in the failing kidney is limited. Acute kidney injury (AKI) is marked by anuria, which could be interpreted as a result of decreased permeability of the glomerular sieving membrane. These observations suggest that differential responses of the endothelial barrier in lung and kidney might be responsible for these diverging patterns of oedema formation.
Under physiological conditions, the endothelial barrier function is maintained by junctions between EC and with the basal membrane. Vascular permeability, and thus the paracellular flow of fluids, electrolytes, proteins, and cells through the endothelial layer, is mainly controlled by molecules of the adherens and tight junctions (6–11), a subset of which is visualized in Figure 1A(8, 11–14). These EC junctions are dynamic structures that can be regulated at transcriptional, translational, and post-translational levels, resulting in permeability changes. For example, phosphorylation of adherens junctions molecules and their subsequent internalisation and degradation are key regulators of endothelial permeability (4, 7, 15). In MODS, disruption of EC junctions has been, indeed, associated with altered vascular permeability (7, 16, 17). In trauma patients there is a severity-related increase in plasma of junctional molecules (18).
Fig. 1.
Regulation of vascular permeability by microvascular endothelial cells.
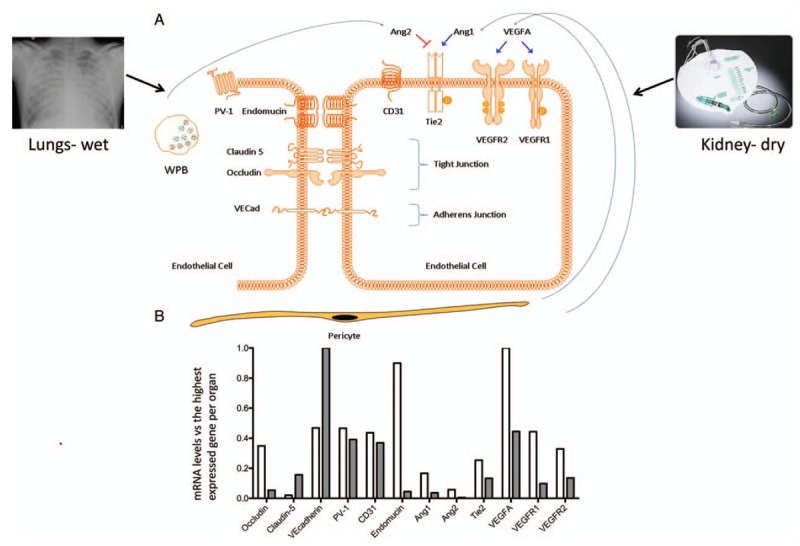
Molecules involved in vascular permeability and studied in the current study are shown schematically. A, In critically ill patients, pulmonary edema (chest X ray upper left) with increased pulmonary vascular leakage, and anuria (urine collection bag without urine, upper right) can be observed at the same time. Vascular endothelial cells (EC) cover all blood vessels in close association with supporting pericytes and play a vital role in regulating vascular leakage in health and disease, for an example in sepsis. Vascular permeability control is located in tight junctions (Claudin-5 and Occludin) and adherence junctions (VE-cadherin). Both the Ang/Tie2 (angiopoietin/Tie2 system) and the VEGFA/VEGFR1-VEGFR2 (Vascular Endothelial Growth Factor A and its receptors VEGF receptor 1, VEGFR1, also known as Flt-1) and VEGF receptor 2 (VEGFR2, also known as KDR) regulate vascular leakage. Angiopoietin1 (Ang1) is constitutively produced by pericytes and promotes EC integrity. During stress, endothelial cells release Angiopietin2 (Ang2) from intracellular Weibel Palade Bodies (WPB). Ang2 is a competitive Tie2 antagonist and promotes vascular leakage. PV1 protein (plasmalemmal vesicle associated protein-1) is required for endothelial diaphragm formation. CD31, also known as platelet endothelial cell adhesion molecule 1 (PECAM-1), and endomucin are involved in leukocyte trafficking. B, Basal gene expression levels of the molecules that are investigated in this study in healthy mouse kidney (white) and lungs (gray) normalized to the highest expressed gene (VEGFA in kidney and VE-cadherin in lungs).
It is currently unclear whether diverging outcomes in oedema formation and organ failure in MODS is attributable to differential, organ-specific regulation of endothelial barrier molecules. In this study, we hypothesized that in sepsis the molecular systems that control endothelial barrier function are differentially regulated in an organ-specific manner. To test our hypothesis, we studied the expression of mRNA of a predefined set of EC permeability-related proteins in kidney and lung after systemic exposure of lipopolysacharide (LPS) in mice. LPS in mice does not resemble human sepsis but some responses seen in sepsis can be elicited in this model (19–21). Expression of these molecules was furthermore investigated in post-mortem kidney biopsies of septic AKI patients. The obtained knowledge may aid in rational development of therapies that target vascular permeability in MODS.
MATERIALS AND METHODS
Animals
Eight- to 12-week-old C57Bl/6 male mice (20–30 g) were obtained from Harlan (Horst, The Netherlands). Mice were maintained on mouse chow and tap water ad libitum in a temperature-controlled chamber at 24 °C with a 12-h light/dark cycle. All procedures were approved by the local committee for care and use of laboratory animals and were performed according to governmental and international guidelines on animal experimentation (20, 21).
Mouse LPS model
To induce endotoxemia, mice were injected intraperitoneally (i.p.) with LPS (Escherichia coli, serotype 026:B6l; Sigma, St. Louis, Mo) at 0.5 mg/kg (1,500 EU/g) body weight. After 4, 8, or 24 h, blood was withdrawn via aortic puncture under isoflurane anesthesia, and the kidneys and lungs were excised, snap-frozen in liquid nitrogen, and stored at −80 °C until analysis. Untreated control mice were used as controls. All groups contained five mice and were previously reported.
Human kidney tissue
Kidney biopsies were obtained directly post-mortem from 19 patients aged 18 years or older, who died of sepsis; a subset of these biopsies was previously reported (22). Exclusion criteria were pre-existing chronic kidney disease, active auto-immune disorder with renal involvement, and immune-suppressive treatment. Kidneys from 12 patients diagnosed with kidney cancer who underwent a total nephrectomy of the diseased kidney, served as controls. Family was given oral and written information about the post-mortem study. Oral and written, signed consent was obtained from the family. This study was waived by the Medical Ethical Committee (METc 2011/372).
Gene expression analysis by quantitative RT-PCR
RNA was isolated from cryosections of mouse kidney and lung, and from cryosections of human kidney, using the RNeasy Mini Plus Kit (Qiagen, Venlo, The Netherlands), according to the manufacturer's instructions, and as previously described (20). cDNA was synthesized using Superscript III Reverse Transcriptase (Invitrogen, Breda, The Netherlands) and random hexamer primers (Promega, Leiden, The Netherlands). Mouse and human-specific Assay-on-Demand primers (Applied Biosystems, Nieuwekerk a/d IJssel, The Netherlands) were selected to amplify VEGFA, VEGFR1, VEGFR2, Ang1, Ang2, Tie-2, PV-1, VE-Cadherin, claudin-5, occludin, CD31, and endomucin (Table 1). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as housekeeping gene. Duplicate real-time PCR analyses were performed for each sample and the obtained threshold cycle (CT) values were averaged. Gene expression was normalized to the averaged expression of the housekeeping gene, yielding the ΔCT value. Relative gene expression levels were calculated by 2−ΔCT.
Table 1.
Mouse and human assay-on-demand primers
| Gene | Mouse assay ID | Human assay ID |
| GAPDH | Mm99999915 _g1 | Hs99999905_m1 |
| VEGFa | Mm00437304_m1 | Hs00173626_m1 |
| VEGFR-1 | Mm00438980_m1 | Hs01052936_m1 |
| VEGFR-2 | Mm00440099_m1 | Hs00176676_m1 |
| Ang1 | Mm00456503_m1 | Hs00181613_m1 |
| Ang2 | Mm00545822_m1 | Hs00169867_m1 |
| Tie2 | Mm00443242_m1 | Hs00176096_m1 |
| PV-1 | Mm00453379_m1 | Hs00229941_m1 |
| VE-cadherin | Mm00486938_m1 | Hs00174344_m1 |
| Claudin-5 | Mm00727012_s1 | Hs00533949_s1 |
| Occludin | Mm00500912_m1 | Hs00170162_m1 |
| CD31 | Mm00476702_m1 | Hs00169777_m1 |
| Endomucin | Mm00497495_m1 | Hs01038209_m1 |
Western blot
mRNA expression levels of VE-cadherin were determined at protein level by Western blot. Briefly, mouse tissue homogenates were boiled for 5 min, separated by SDS-PAGE on a 7.5% polyacrylamide gel, and blotted onto nitrocellulose membrane (Biorad, Veenendaal, The Netherlands). The membrane was blocked for 1 h in 5% (w/v) dry milk in Tris-buffered saline containing 0.1% (v/v) Tween-20 (TBS-T) and probed overnight with rat antibody to VE-cadherin in 2% (w/v) dry milk in TBS-T0.1%, and 1 h with mouse anti Actin (MAB1501, Merck Millipore, Darmstadt, Germany) diluted in 5% (w/v) BSA in TBS-T 0.1%. The membranes were subsequently washed with TBS-T0.1% and incubated with horseradish peroxidase-labeled anti-rat IgG and anti-mouse IgG (SouthernBiotech, Birmingham, Ala). After washing the membranes extensively in TBS-T0.1%, antibody binding was detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Bleiswijk, The Netherlands). Loading of protein was normalized by Actin. The blots were scanned and optical densities of the protein bands were quantified by Quantity One software (Bio-Rad, Calif).
Statistical analyses
Statistical significance of differences was calculated using the Student t test or ANOVA with post hoc comparison using Dunnet correction. Statistical analyses were performed using GraphPad Prism software (GraphPad Prism Software Inc, San Diego, Calif). Differences were considered to be significant when P < 0.05.
RESULTS
Expression of endothelial permeability-regulating molecules in LPS-injected mice
Organ expression of tight junctions molecules
Expression levels of the tight junctions molecules occludin and claudin-5 were determined in kidneys and lungs of LPS-injected and untreated (control) mice (Fig. 1B). In untreated mice, expression of occludin was 10- to 20 times higher in lungs than in kidneys (Table 2). In kidneys, LPS injection resulted in a slight increase in occludin expression at 8 h and a subsequent decrease at 24 h. In lungs, LPS injection led to a persistent decrease in occludin expression at 4, 8, and 24 h (Fig. 2, A and B).
Table 2.
Basal mRNA expression∗ of molecules under study in murine kidney and lung
| Gene | Kidney† | Lung† |
| Occludin | 0.0041 (0.0003) | 0.0660 (0.0270) |
| Claudin-5 | 0.0002 (0.00003) | 0.1906 (0.1411) |
| VE-cadherin | 0.0056 (0.0009) | 1.2187 (0.6225) |
| PV-1 | 0.0055 (0.0010) | 0.4771 (0.2179) |
| CD31 | 0.0052 (0.0006) | 0.4502 (0.2184) |
| Endomucin | 0.0107 (0.0020) | 0.0540 (0.0253) |
| Ang1 | 0.0019 (0.0002) | 0.0449 (0.0225) |
| Ang2 | 0.0006 (0.0001) | 0.0061 (0.0035) |
| Tie2 | 0.0030 (0.0003) | 0.1615 (0.0745) |
| VEGFA | 0.0119 (0.0006) | 0.5413 (0.2634) |
| VEGFR1 | 0.0053 (0.0025) | 0.1193 (0.0544) |
| VEGFR2 | 0.0039 (0.0006) | 0.165 (0.0771) |
*mRNA expression levels were determined using quantitative RT-PCR with GAPDH as reference gene.
†Data are presented as mean and SD of each group (n = 5 animals/group).
Fig. 2.
Influence of LPS challenge on expression of occludin, claudin-5, VE-cadherin, PV-1, CD31, and endomucin in mouse kidney and lung.
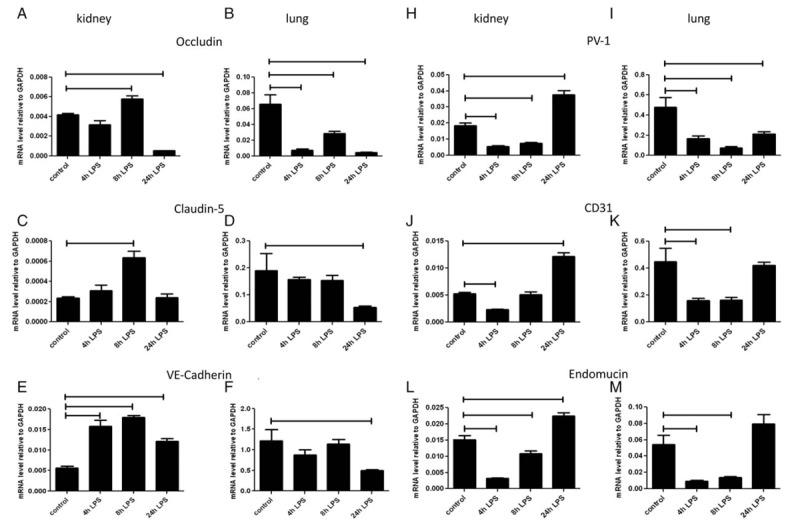
Mice were i.p. injected with LPS at 0.5 mg/kg (1,500 EU/g) bodyweight and sacrificed 4, 8, and 24 h later. Control mice were left untreated. mRNA expression of occludin (A, B), claudin-5 (C, D), VE-cadherin (E, F), PV-1 (H, I), CD31 (J, K), and endomucin (L, M) in kidney (A, C, E, H, J, L) and lungs (B, D, F, I, K, M) was determined using quantitative RT-PCR with GAPDH as housekeeping gene. Data are presented as mean ± SD (n = 5 animals/group). Significance P < 0.05.
Expression of claudin-5 in untreated mice was 100 times higher in lungs than in kidneys (Table 2). After LPS injection, an increase in claudin-5 mRNA levels was observed at 8 h in kidney. In lung there was a reduction of claudin-5 expression only at 24 h (Fig. 2, C and D).
Organ expression of adherence junction molecule VE-cadherin
In untreated mice, mRNA expression of VE-Cadherin was 200 times higher in lungs than in kidneys (Table 2). After LPS injection, VE-cadherin expression was increased 3-fold at 4 and 8 h in kidney, and although decreasing at 24 h, was still elevated at this latter time point compared to untreated mice. In the lung, LPS injection resulted in a decrease in VE-cadherin expression at 24 h (Fig. 2, E and F).
After LPS injection, VE-cadherin protein expression in the kidney did not differ from that in untreated mice, while in the lung there was a temporary decrease with the nadir at 4 h (Fig. 3, A and B).
Fig. 3.
Influence of LPS injection on VE-cadherin protein expression in mouse kidney and lung.
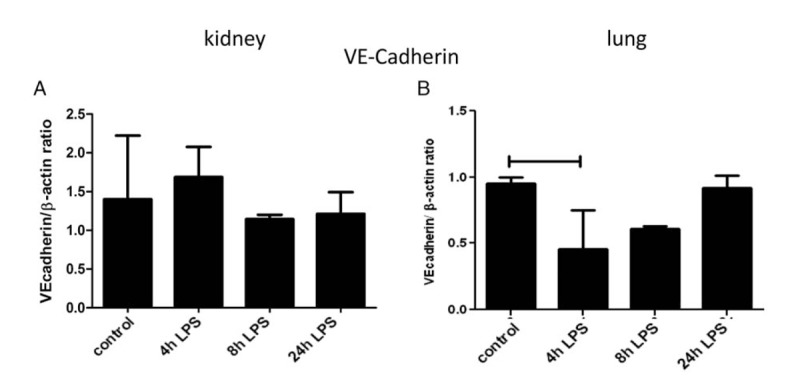
Mice were i.p. injected with LPS at 0.5 mg/kg (1,500 EU/g) bodyweight and sacrificed 4, 8, and 24 h later. VE-cadherin protein was determined using Western blot and β-actin was used as a loading control and quantified in kidney (A) and lung (B). Data are presented as mean VE-cadherin/β-actin ratio ± SD (n = 5 animals/group). Significance P < 0.05.
Expression of mediators of diapedesis and transcellular transport
Organ expression of transcellular transport molecule PV-1
In untreated mice, expression of PV-1 was 50 times higher in the lung than in the kidney (Table 2). In both kidney and lung, PV-1 expression decreased at 4 and 8 h after LPS injection, compared with untreated mice. An increase above control levels was observed at 24 h in kidney, while in lung the levels remained low (Fig. 2, H and I).
Organ expression of CD31
In untreated mice, CD31 expression was 100 times higher in the lung than in the kidney (Table 2). In kidney, CD31 expression was reduced at 4 h after LPS injection, and increased at 24 h compared with untreated mice. In lung, CD31 expression decreased at 4 and 8 h after LPS injection and returned to baseline levels at 24 h (Fig. 2, J and K).
Organ expression of endomucin
Endomucin expression in untreated mice was three times higher in the lung than in the kidney (Table 2). After LPS injection, endomucin expression decreased at 4 and 8 h in both kidney and lung. At 24 h, endomucin expression had reached baseline levels in the lung but were still higher than in controls in the kidney (Fig. 2, L and M).
Expression of vascular stability-related signaling molecules
Organ expression of molecules of the Angiopoietin/Tie2 system
In untreated mice, expression of Ang1 was 20 times higher in lungs than in kidneys (Table 2). In kidney, decrease in Ang1 expression was only observed at 8 h after LPS injection. In lung, there was a significant decrease in Ang1 expression, which persisted until 24 h (Fig. 4 A and B).
Fig. 4.
Influence of LPS injection on mRNA expression of ligands and receptors of the angiopoietin/Tie2 system and the VEGFA/VEGF-receptor system in mouse kidney and lung.
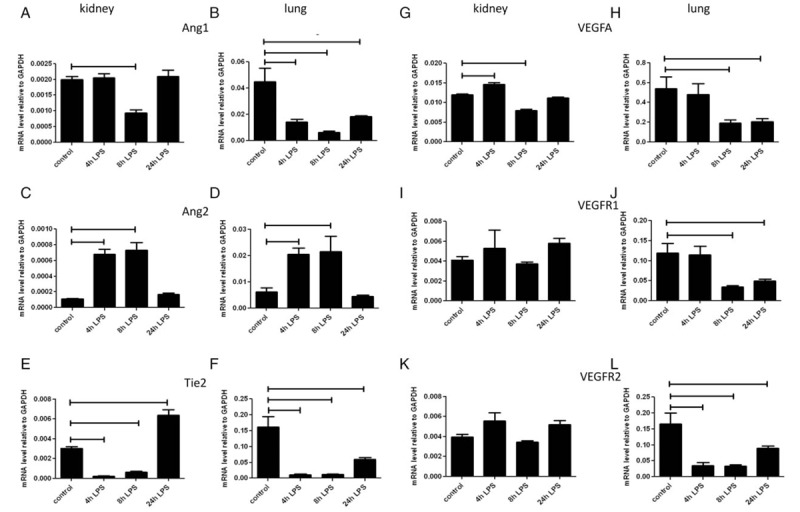
Mice were i.p. injected with LPS at 0.5 mg/kg (1,500 EU/g) bodyweight and sacrificed at 4, 8, and 24 h later. mRNA expression of Ang1 (A, B), Ang2 (C, D), Tie2 (E, F), VEGFA (G, H), VEGFR1 (I, J), and VEGFR2 (K, L) in kidney (A, C, E, G, I, K) and lungs (B, D, F, H, J, L) was determined using quantitative RT-PCR with GAPDH as housekeeping gene. Data are presented as mean ± SD (n = 5 animals/group). Significance P < 0.05.
Expression of Ang2 was 500 times higher in lungs than in kidneys of untreated mice (Table 2). After LPS injection there was an increase in Ang2 expression at 4 and 8 h in both kidney and lung, which returned to baseline at 24 h in both organs (Fig. 4, C and D).
Since Ang1 is a Tie2 agonist leading to Tie2 phosphorylation, and Ang2 is a partial Tie2 antagonist during inflammation (23), the Ang1/Ang2 ratio may be used to predict Tie2 phosphorylation in vivo. In untreated mice, the Ang1/Ang2 mRNA ratio was 3.9 in the kidney and 12.7 in the lung. After LPS injection, the Ang1/Ang2 ratio was reduced in both organs. This decrease in the Ang1/Ang2 ratio was more pronounced in lungs than in kidneys (not shown).
In untreated mice Tie2 expression was 50 times higher in the lung than in the kidney (Table 2). Tie 2 expression decreased in both organs at 4 and 8 h after LPS injection. At 24 h after LPS injection, Tie2 expression was higher in kidney and lower in lung than in their respective controls (Fig. 4, E and F).
Organ expression of VEGFA, and its receptors VEGFR1 and VEGFR2
In untreated mice, VEGFA expression was 50 times higher in the lung than in the kidney (Table 2). After LPS injection, VEGFA expression in kidney was increased at 4 h, and decreased at 8 h. In lung, VEGFA expression was decreased at 8 and 24 h (Fig. 4, G and H).
VEGFR1 expression in untreated mice was 20 times higher in the lung than in the kidney (Table 2). LPS injection did not affect expression of VEGFR1 in kidney, but led to its reduction at 8 and 24 h in lung (Fig. 4, I and J).
VEGFR2 expression in untreated mice was 40 times higher in the lung than in the kidney (Table 2). After LPS injection, VEGFR2 expression in kidney was unaffected, while it was reduced at all time points in lung (Fig. 4, K and L).
Expression of endothelial permeability regulating molecules in kidney biopsies of patients who died of sepsis
The biopsies were taken after dying with a mean time elapse of 33 min. No distinct patterns of expression of the tight- and adherence junctions molecules were present between kidneys from septic patients and controls, since expression of occludin and VE-cadherin did not differ between the two groups (Fig. 5,A), while expression of Claudin-5 expression was increased in kidneys from septic patients (Fig. 5B).
Fig. 5.
mRNA expression levels of Occludin, Claudin-5, VE-Cadherin, PV-1, CD31, endomucin, Ang1, Ang2, Tie2, VEGFA, VEGFR1, VEGFR2 in kidney of human septic patients and controls.
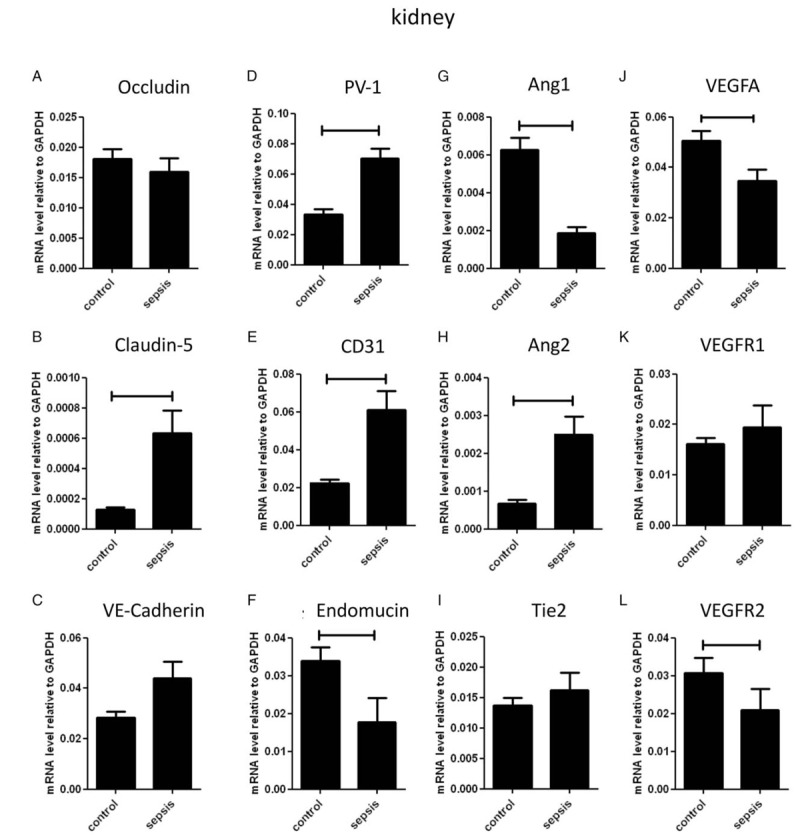
mRNA expression of Occludin (A), Claudin-5 (B), PV1 (C), CD31 (D), Endomucin (E), VE-cadherin (F), Ang1 (G), Ang2 (H), Tie2 (I), VEGFA (J), VEGFR1 (K), VEGFR2 (L) was determined using quantitative RT-PCR with GAPDH as housekeeping gene in human kidney biopsies of septic patients (n = 19) and controls (n = 12). Data are presented as mean ± SD. Significance P < 0.05.
Expression of the mediators of diapedesis and transcellular transport PV1 (Fig. 5D) and CD31 (Fig. 5E) was increased in kidneys from septic patients, while endomucin expression was decreased in this group (Fig. 5F).
As expected, Ang1 expression was decreased, whereas expression of Ang2 was increased in kidneys from septic patients (Fig. 5, G and H). No difference in expression of the angiopoietin receptor Tie2 was present in kidneys from from septic patients and controls (Fig. 5I).
Expression of VEGFA was decreased in kidneys from septic patients (Fig. 5J). Expression of VEGFR1 was similar between the two groups (Fig. 5K), whereas expression of VEGFR2 was lower in septic kidneys than in controls (Fig. 5L).
DISCUSSION
MODS in critically ill patients is associated with changes in microvascular integrity, with the clinical impression that lung capillaries become leaky while renal capillaries become less permeable. Here, we hypothesized that these organ-specific differences in permeability are attributable to differential regulation of molecular systems that control endothelial barrier function. To address this hypothesis we studied the expression of a predefined subset of EC permeability-related molecules in lung and kidney after i.p. LPS injection in mice, and in kidneys of patients who died of sepsis. We showed that baseline expression of mRNA of these molecules differs between mouse lung and kidney. Moreover, we showed differential regulation of these molecules after LPS injection in the two mouse organs. Finally, we partially corroborated the findings in mouse kidney biopsies obtained in from septic patients shortly after death.
The main molecular systems dictating vascular permeability are the tight and adherens junctions. We assessed expression of their flagship molecules occludin, claudin-5, and VE-cadherin. Reduction of occludin and claudin-5 expression is associated with breakdown of the endothelial barrier in the brain (24). Moreover, disassembly of claudin-5 from tight junctions has been shown to lead to increased vascular permeability (24, 25). Similarly, downregulation of VE-cadherin causes an increase in vascular permeability (14, 26). In our study, baseline expression levels of occludin, claudin-5, and VE-cadherin markedly differed between kidney and lung, with claudin-5 and VE-cadherin being highly expressed in lung, and occludin being highly expressed in kidney.
Interestingly, lung responses to LPS were marked by reduced expression of both tight junction molecules occludin and claudin-5, and of the adherens junction molecule VE-cadherin, albeit following different kinetics. These findings are in line with the clinical hypothesis that lung oedema may be associated with a compromised endothelial barrier. Also in a mouse model of trauma a decreased expression of junctional adhesion molecule-1 (JAM-1) protein also suggests a hampered endothelial barrier (18). Conversely, kidney responses to LPS were marked by a temporary increased expression of occludin, claudin-5, and VE-cadherin at 8 h after LPS, corroborating the notion that decreased permeability of renal microvasculature may lead to anuria. Whether the absence of a correlation between mRNA and protein expression levels of VE-cadherin in both organs reflects a dichotomy between mRNA and protein regulation per se, or a microvascular segment-specific response that was masked by total organ analysis, needs to be further investigated using more advanced techniques to quantify protein and phospho-protein expression (6, 27).
Based on these findings on renal endothelial responses to LPS in mice, we expected to find an increased expression of the studied tight- and adherens junctions in human septic kidneys as compared to healthy kidneys. The lack of such a differential expression in kidneys of sepsis and control patients may be partly attributable to the process of dying itself, and to technical issues such as the post mortem time point of biopsy sampling, which may both affect mRNA levels. But above all this may be a reflection of the huge gap between a laboratory model and clinic. Obviously, the time course of changes in these molecules cannot be studied in post mortem samples.
Next to oedema formation, an abundant influx of neutrophils is a characteristic of the lung in sepsis, while this influx is less pronounced in the kidney. Neutrophil recruitment and diapedesis is guided by selectins and cellular adhesion molecules, which have been extensively studied in mouse sepsis (15). We studied additional components of the process, i.e., PV-1, CD31, and endomucin, all located on the EC membrane and involved in multiple EC-related functions as adhesion molecules, signaling molecules, or both. PV-1 is involved in leukocyte transport, but has also a role in the repair of glomerular fenestration (28). This latter role may explain the increased expression of PV-1 expression in mouse kidney at 24 h after LPS administration and in human septic kidneys. The persistent reduction of PV-1 expression in mouse lungs is not compatible with increased neutrophil infiltration reported before. Similarly, reduced expression of CD31 in mouse lungs at 4 and 8 h after LPS injection is not in line with increased neutrophil infiltration in this organ, nor is its increased expression in human sepsis kidneys in line with a moderate renal neutrophil influx.
The sialomucin endomucin binds L-selectin and mediates leukocyte rolling. Decrease in endomucin expression reduces rolling and enhances firm leukocyte adhesion (29), a process preceding diapedesis. In our study, transiently decreased endomucin expression in lung at early time points would support increased neutrophil infiltration in this organ, while increased expression at late time points in the kidney would argue for lower neutrophil infiltration in this organ. Moreover, findings on endomucin expression in mouse kidney were not comparable to those in human sepsis kidney.
Endothelial junctional complexes are regulated by several signaling systems, among which Ang/Tie and VEGF/VEGF-R receptor signaling. The Ang/Tie system is involved in vasculogenesis, angiogenesis, permeability control, and cell adhesion (20, 23), often in conjunction with VEGF (24). Ang-1 antagonizes VEGF by sequestering intracellular Src, resulting in reduced phosphorylation and internalization of VE-cadherin, and thus decreased permeability (24). In our study, the two signaling systems displayed an organ-specific expression pattern in untreated mice. After LPS injection, a strong decline in Ang-1 expression in the lung, together with a decrease in Tie-2 and VE-cadherin protein expression, suggests that the endothelial layer may be destabilized and permeability may increase. In kidney, Ang-1 expression decreased far less and recovered more rapidly. Although Tie2 expression also decreased, it recovered quickly and increased at 24 h after LPS injection, suggesting that signaling may promote reduced permeability.
Ang1 and Ang2 are context-dependent antagonists and therefore the Ang1/Ang2 ratio is thought to be a determinant for their net signaling effect on EC (23). A low systemic Ang1/Ang2 ratio is associated with a higher permeability, decreased vascular integrity, and with higher mortality in sepsis patients. In our study, the Ang1/Ang2 ratio was lower in untreated mouse kidney compared with mouse lung and the Ang1/Ang2 ratio decreased after LPS exposure, an effect that was more pronounced in the lung than in the kidney. The kinetics of the Ang1/Ang2 ratio and Tie-2 expression are compatible with organ-specific permeability differences. Of course we only measured mRNA and this compatibility supposes a correlation between mRNA level and protein presence.
The VEGF family is involved in vascular permeability regulation, via yet incompletely understood mechanisms (7, 13, 30). The effect of VEGF on endothelial cell permeability in culture is dose dependent (31). In kidney, tight VEGF regulation is required for the development and maintenance of an intact glomerular filtration function. Studies evaluating renal VEGF in sepsis are scarce. We showed that after LPS injection, VEGFA, VEGFR1, and VEGFR2 expression was fairly stable in the kidney but declined sharply in the lung. These observations in the lung are in line with reduced pulmonary VEGF signaling in acute lung injury (32).
Our study has several limitations, among which first and foremost the fact that the complexity of endothelial permeability regulation does not allow more than a reductionist investigation in mice of a selection of molecules involved in this process. We studied mRNA but the effect of mRNA levels on the function, in this case permeability, is regulated by many post-transcriptional steps and a straightforward relationship cannot be assumed (19). We use LPS injection in young healthy mice to elicit some of the responses seen in sepsis. This is an oversimplification of the clinic where we are confronted with older patients with comorbidity, comedication, different pathogens, genetic polymorphisms, lead-time bias, and treatment differences. The strengths and weaknesses of research mice have to be balanced carefully (19). Endothelial cells respond to LPS via TLR4. We did not study TLR4 expression differences and can therefore not exclude a nonlinear effect of endothelial TLR4 signaling. There is a major difference in leukocyte influx between lung and kidney, this might also cause differences in adhesion molecule expression. The signaling pathways involved in the expression differences we found deserve further study. Functional consequences of the organ-specific, divergent expression patterns, and as yet unknown underlying regulating processes are even more cumbersome to explore and would require a full array of KO and overexpression mouse models. Moreover, the mouse LPS injection model has limited resemblance with the clinical syndrome of sepsis or MODS, even though expression of 5 of 12 molecules in the late stage of the mouse LPS model showed similar expression in human sepsis kidneys versus human control kidneys. Nevertheless, incremental improvements to the current work can be added in future studies. For example, we expect that laser microdissection of specific micro-vascular beds prior to gene (array) expression analysis will provide additional information regarding intra-organ, microvascular segment-specific endothelial responses (20, 27), which in the current study have been masked by whole organ analysis. In fact, this microvessel, segment-specific approach may give us more insight into the organ-specific vascular microenvironments, which govern endothelial permeability. Regarding human material, efforts to establish a collection of, e.g., lung and liver tissue from sepsis patients and healthy donors will greatly facilitate the evaluation of inter-organ differences in expression patterns of vascular permeability governing molecules in human sepsis. In future studies, we will try to study endothelial behavior in more organs of patients with multiple organ failure.
While major differences in the expression of adhesion molecules between organs have been reported before in endotoxaemia, caecal ligation and puncture, and animal models of haemorrhagic shock (33, 34), our study explores organ-specific responses of molecular controllers of EC permeability. The involvement of junctional molecules in MODS is supported by the increase of JAM-1 in plasma of severe trauma patients. In mice the JAM-1 is decreased in lungs but a response in the kidney was not studied (18). The observed organ-specific responses in molecular controllers of EC permeability might have therapeutic implications. For example, administration of exogenous Ang-1 or synthetic Ang-1 mimetics in animal studies led to decreased vascular leakage and survival benefit (35, 36). Furthermore, inhibition of Rho, which alters VE-cadherin function, reduces pulmonary oedema (37). Yet, our finding that expression of molecular controllers of EC permeability can strongly differ between organs also illustrates potential drawbacks of systemic interventions, which may differentially affect the function of organs such as kidney and lung.
In conclusion, our study provides insight into the occurrence of important organ-related differences in micro-vascular permeability-associated mRNA of molecules in response to LPS. Some of the differences seen in mice concur with those found in human sepsis kidneys. These differences may partly explain the striking diverging manifestations of organ permeability observed in patients with multiple organ failure. Manipulation of vascular permeability might be a target for therapy in MODS, but our study also emphasizes the challenges that such a therapy faces, since improving the vascular function of one organ might endanger the vascular function of another.
ACKNOWLEDGMENTS
Anne Marie Leliveld-Kors is acknowledged for providing human cancer kidney samples and Henk Moorlag for technical assistance.
Footnotes
The authors report no conflicts of interest.
REFERENCES
- 1.Angus DC, Linde-Zwirble WT, Lidicker J, Clermont G, Carcillo J, Pinsky MR. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit Care Med 2001; 29 7:1303–1310. [DOI] [PubMed] [Google Scholar]
- 2.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol 2008; 8 10:776–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldenberg NM, Steinberg BE, Slutsky AS, Lee WL. Broken barriers: a new take on sepsis pathogenesis. Sci Transl Med 2011; 3 88:88s25. [DOI] [PubMed] [Google Scholar]
- 4.Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY. Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Rev Mol Med 2009; 11:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Millar FR, Summers C, Griffiths MJ, Toshner MR, Proudfoot AG. The pulmonary endothelium in acute respiratory distress syndrome: insights and therapeutic opportunities. Thorax 2016; 71 5:462–473. [DOI] [PubMed] [Google Scholar]
- 6.Aman J, Weijers EM, van Nieuw Amerongen GP, Malik AB, van Hinsbergh VW. Using cultured endothelial cells to study endothelial barrier dysfunction—challenges and opportunities. Am J Physiol Lung Cell Mol Physiol 2016; 311 2:L453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci 2008; 121 13:2115–2122. [DOI] [PubMed] [Google Scholar]
- 8.Steed E, Balda MS, Matter K. Dynamics and functions of tight junctions. Trends Cell Biol 2010; 20 3:142–149. [DOI] [PubMed] [Google Scholar]
- 9.Bazzoni G. Endothelial tight junctions: permeable barriers of the vessel wall. Thromb Haemost 2006; 95 1:36–42. [PubMed] [Google Scholar]
- 10.Zihni C, Mills C, Matter K, Balda MS. Tight junctions: from simple barriers to multifunctional molecular gates. Nat Rev Mol Cell Biol 2016; 17 9:564–580. [DOI] [PubMed] [Google Scholar]
- 11.Reglero-Real N, Colom B, Bodkin JV, Nourshargh S. Endothelial cell junctional adhesion molecules: role and regulation of expression in inflammation. Arterioscler Thromb Vasc Biol 2016; 36 10:2048–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Adam A, Sharenko A, Pumiglia K, Vincent P. Src-induced tyrosine phosphorylation of VE-cadherin is not sufficient to decrease barrier function of endothelial monolayers. J Biol Chem 2010; 285 10:7045–7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eremina V, Baelde HJ, Quaggin SE. Role of the VEGF—a signaling pathway in the glomerulus: evidence for crosstalk between components of the glomerular filtration barrier. Nephron Physiol 2007; 106 2:32–37. [DOI] [PubMed] [Google Scholar]
- 14.Mochizuki N. Vascular integrity mediated by vascular endothelial cadherin and regulated by sphingosine 1-phosphate and angiopoietin-1. Circ J 2009; 73 12:2183–2191. [DOI] [PubMed] [Google Scholar]
- 15.Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol 2015; 15 11:692–704. [DOI] [PubMed] [Google Scholar]
- 16.Vestweber D, Broermann A, Schulte D. Control of endothelial barrier function by regulating vascular endothelial-cadherin. Curr Opin Hematol 2010; 17 3:230–236. [DOI] [PubMed] [Google Scholar]
- 17.Gavard J, Patel V, Gutkind JS. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev Cell 2008; 14 1:25–36. [DOI] [PubMed] [Google Scholar]
- 18.Denk S, Wiegner R, Hones FM, Messerer DA, Radermacher P, Weiss M, Kalbitz M, Ehrnthaller C, Braumuller S, McCook O, et al. Early detection of junctional adhesion molecule-1 (JAM-1) in the circulation after experimental and clinical polytrauma. Mediators Inflamm 2015; 2015:463950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osuchowski MF, Remick DG, Lederer JA, Lang CH, Aasen AO, Aibiki M, Azevedo LC, Bahrami S, Boros M, Cooney R, et al. Abandon the mouse research ship? Not just yet! Shock 2014; 41 6:463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Meurs M, Kurniati NF, Wulfert FM, Asgeirsdottir SA, de Graaf IA, Satchell SC, Mathieson PW, Jongman RM, Kümpers P, Zijlstra JG, et al. Shock-induced stress induces loss of microvascular endothelial Tie2 in the kidney which is not associated with reduced glomerular barrier function. Am J Physiol Renal Physiol 2009; 297 2:F272–F281. [DOI] [PubMed] [Google Scholar]
- 21.Kurniati NF, Jongman RM, Hagen F, Spokes KC, Moser J, Regan ER, Krenning G, Moonen JA, Harmsen MC, Struys MM, et al. The flow dependency of Tie2 expression in endotoxemia. Intensive Care Med 2013; 39 7:1262–1271. [DOI] [PubMed] [Google Scholar]
- 22.Aslan A, Jongman RM, Moser J, Stegeman CA, van GH, Diepstra A, van den Heuvel MC, Heeringa P, Molema G, Zijlstra JG, et al. The renal angiopoietin/Tie2 system in lethal human sepsis. Crit Care 2014; 18 2:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Meurs M, Kumpers P, Ligtenberg JJ, Meertens JH, Molema G, Zijlstra JG. Bench-to-bedside review: angiopoietin signalling in critical illness—a future target? Crit Care 2009; 13 2:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci USA 2009; 106 6:1977–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kondo N, Ogawa M, Wada H, Nishikawa SI. Thrombin induces rapid disassembly of claudin-5 from the tight junction of endothelial cells. Exp Cell Res 2009; 315 17:2879–2887. [DOI] [PubMed] [Google Scholar]
- 26.Vestweber D, Winderlich M, Cagna G, Nottebaum AF. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol 2009; 19 1:8–15. [DOI] [PubMed] [Google Scholar]
- 27.Langenkamp E, Kamps JA, Mrug M, Verpoorte E, Niyaz Y, Horvatovich P, Bisschoff R, Struijker-Boudier H, Molema G. Innovations in studying in vivo cell behavior and pharmacology in complex tissues—microvascular endothelial cells in the spotlight. Cell Tissue Res 2013; 354 3:647–669. [DOI] [PubMed] [Google Scholar]
- 28.Ichimura K, Stan RV, Kurihara H, Sakai T. Glomerular endothelial cells form diaphragms during development and pathologic conditions. J Am Soc Nephrol 2008; 19 8:1463–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zahr A, Alcaide P, Yang J, Jones A, Gregory M, dela Paz NG, Patel-Hett S, Nevers T, Koirala A, Luscinskas FW, et al. Endomucin prevents leukocyte-endothelial cell adhesion and has a critical role under resting and inflammatory conditions. Nat Commun 2016; 7:10363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bates DO. Vascular endothelial growth factors and vascular permeability. Cardiovasc Res 2010; 87 2:262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mirzapoiazova T, Kolosova I, Usatyuk PV, Natarajan V, Verin AD. Diverse effects of vascular endothelial growth factor on human pulmonary endothelial barrier and migration. Am J Physiol Lung Cell Mol Physiol 2006; 291 4:L718–L724. [DOI] [PubMed] [Google Scholar]
- 32.Medford AR, Millar AB. Vascular endothelial growth factor (VEGF) in acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): paradox or paradigm? Thorax 2006; 61 7:621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shapiro NI, Yano K, Sorasaki M, Fischer C, Shih SC, Aird WC. Skin biopsies demonstrate site-specific endothelial activation in mouse models of sepsis. J Vasc Res 2009; 46 5:495–502. [DOI] [PubMed] [Google Scholar]
- 34.van Meurs M, Wulfert FM, Knol AJ, de Haes A, Houwertjes M, Aarts LP, Molema G. Early organ-specific endothelial activation during hemorrhagic shock and resuscitation. Shock 2008; 29 2:291–299. [DOI] [PubMed] [Google Scholar]
- 35.Rubig E, Stypmann J, van Slyke P, Dumont DJ, Spieker T, Buscher K, Reuter S, Goerge T, Pavenstädt H, Kümpers P. The synthetic Tie2 agonist peptide vasculotide protects renal vascular barrier function in experimental acute kidney injury. Sci Rep 2016; 6:22111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.David S, Park JK, van Meurs M, Zijlstra JG, Koenecke C, Schrimpf C, Shushakova N, Gueler F, Haller H, Kümpers P. Acute administration of recombinant Angiopoietin-1 ameliorates multiple-organ dysfunction syndrome and improves survival in murine sepsis. Cytokine 2011; 55 2:251–259. [DOI] [PubMed] [Google Scholar]
- 37.Tasaka S, Koh H, Yamada W, Shimizu M, Ogawa Y, Hasegawa N, Yamaguchi K, Ishii Y, Richer SE, Doerschuk CM, et al. Attenuation of endotoxin-induced acute lung injury by the Rho-associated kinase inhibitor, Y-27632. Am J Respir Cell Mol Biol 2005; 32 6:504–510. [DOI] [PubMed] [Google Scholar]