

Abstract
ATP-sensitive potassium (KATP) channels couple cell metabolism to electrical activity by regulating K+ flux across the plasma membrane. Channel closure is mediated by ATP, which binds to the pore-forming subunit (Kir6.2). Here we use homology modelling and ligand docking to construct a model of the Kir6.2 tetramer and identify the ATP-binding site. The model is consistent with a large amount of functional data and was further tested by mutagenesis. Ligand binding occurs at the interface between two subunits. The phosphate tail of ATP interacts with R201 and K185 in the C-terminus of one subunit, and with R50 in the N-terminus of another; the N6 atom of the adenine ring interacts with E179 and R301 in the same subunit. Mutation of residues lining the binding pocket reduced ATP-dependent channel inhibition. The model also suggests that interactions between the C-terminus of one subunit and the ‘slide helix' of the adjacent subunit may be involved in ATP-dependent gating. Consistent with a role in gating, mutations in the slide helix bias the intrinsic channel conformation towards the open state.
Keywords: ATP binding, KATP channel, Kir6.2
Introduction
ATP-sensitive potassium (KATP) channels play key roles in many tissues by linking cell metabolism to electrical activity. They are involved in glucose sensing in pancreatic islets, gut L cells and the brain; in the regulation of vascular smooth muscle tone; in the response to ischaemic stress in heart and brain; and in seizure protection (Seino and Miki, 2003).
KATP channels are octameric complexes of two different proteins (Clement et al, 1997). Four inwardly rectifying K+ channel subunits form a tetrameric pore: in almost all tissues except vascular smooth muscle, Kir6.2 serves this role. Each Kir subunit is associated with a regulatory sulphonylurea receptor (SUR) subunit, which belongs to the ATP-binding cassette family of transporter proteins. Metabolic regulation of KATP channel activity is mediated by changes in the intracellular concentrations of adenine nucleotides, which interact with both Kir6.2 and SUR subunits. Binding of ATP or ADP to Kir6.2 produces channel inhibition (Tucker et al, 1998; Tanabe et al, 2000), whereas interaction of Mg nucleotides with the nucleotide-binding domains (NBDs) of SUR stimulates channel activity (Nichols et al, 1996; Gribble et al, 1997). Loss-of-function mutations in both Kir6.2 and SUR1 genes cause congenital hyperinsulinism (Dunne et al, 2004). Conversely, mutations in Kir6.2 that lead to reduced ATP sensitivity of the channel cause permanent neonatal diabetes, which in some cases is associated with muscle weakness, developmental delay and epilepsy (Gloyn et al, 2004).
To understand how ATP inhibits the KATP channel and how mutations in Kir6.2 impair this process, it is necessary to identify the location and structure of the ATP-binding site. The structure of this inhibitory site is unknown, but is likely to differ from that of classical ATP-binding sites, because it has several unusual properties. For example, Mg2+ is not required for the inhibitory action of the nucleotide (Ashcroft and Kakei, 1989); the site is extremely selective for the adenine base (Tucker et al, 1998), and addition of bulky groups to the end of the phosphate chain does not abolish the inhibitory effect of ATP (Ämmälä et al, 1991; Tanabe et al, 2000). Many experiments suggest that the ATP-binding site involves residues from both the N- and C-terminal domains (N and C domains) of Kir6.2 (Drain et al, 1998; Proks et al, 1999; Tucker et al, 1998; Reimann et al, 1999; Cukras et al, 2002; John et al, 2003; Ribalet et al, 2003; Tsuboi et al, 2004). In addition, although the primary site at which ATP mediates channel inhibition lies on Kir6.2, coexpression with SUR enhances the potency of ATP block about 10-fold (Tucker et al, 1997).
Attempts to crystallize Kir6.2, or its C domains, have proved unsuccessful. However, the crystal structures of a putative bacterial Kir channel, KirBac1.1 (Kuo et al, 2003), and of the N and C domains of Kir3.1 (Nishida and MacKinnon, 2002), have recently been solved. We have therefore used these structures to construct a molecular model of the Kir6.2 tetramer, and ligand docking to identify residues interacting with ATP. The model is consistent with a large amount of functional data and was further tested by additional site-directed mutagenesis. Our results indicate the location and structure of the four ATP-binding sites, and suggest a possible model for how binding of ATP is transduced to transmembrane elements of the channel and thus to closure of the pore.
Results
Construction of a molecular model of Kir6.2
A homology model of Kir6.2 (GenBank D50581) was constructed based on the X-ray crystal structures of KirBac1.1 (Kuo et al, 2003) and the intracellular (IC) domains of Kir3.1 (Nishida and MacKinnon, 2002). KirBac1.1 was used as a template for the proximal N domain and the transmembrane (TM) domains of Kir6.2, whereas the distal N domain and the C domain were modelled on the IC domains of Kir3.1 (Figure 1A). This was because, over the region modelled, the C domains of Kir3.1 and Kir6.2 exhibit a greater sequence identity (48%) than those of Kir6.2 and KirBac1.1 (27%). Further, the crystal structure of the IC domain of Kir3.1 was determined at a resolution higher (2.0 Å) than that of KirBac1.1 (3.6 Å). Each of the three segments of the model (TMs, N and C domains) were constructed separately, and then joined together. The spatial orientation of the IC domain, with respect to the TMs, was determined from the location of conserved residues in the IC domain of KirBac1.1.
Figure 1.
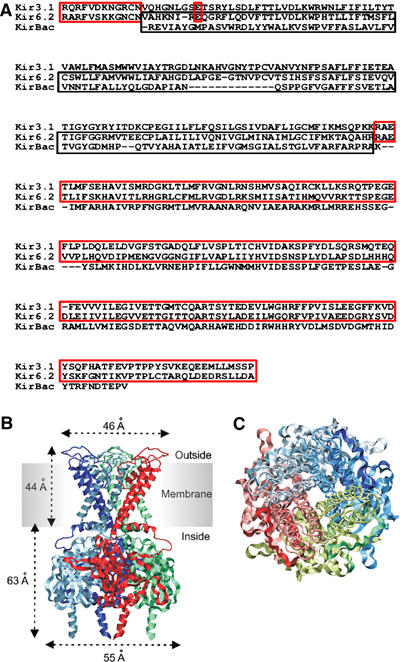
(A) Alignment of Kir3.1, Kir6.2 and KirBac1.1 sequences. Regions boxed in red and black show residues in Kir3.1 and KirBac1.1, respectively, used to construct the model. (B, C) Model of the Kir6.2 tetramer viewed from the side (B) or above (C). (B) Residues are shown in ribbon format, with different colours representing individual subunits. (C) TMs are shown in backbone format and IC domains in ribbon format. Different colours indicate individual subunits, with dark and light shades representing the N and C domains, respectively.
The structure of the N domain of Kir3.1 was resolved between residues 43 and 57 (Nishida and MacKinnon, 2002), which correspond to residues 32–46 of Kir6.2 (Figure 1A). The position of these residues with respect to the C domain of Kir6.2 is fixed by the crystal structure of the IC domain of Kir3.1. Residue 51 in Kir6.2 is also fixed, by the position of residue 63 in the Kir3.1 crystal structure (Figure 1A). The residues corresponding to positions 47–50 of Kir6.2 were not resolved in the crystal structure of Kir3.1: therefore, they were modelled as a loop and energy minimized. The minimization procedure did not significantly alter the position of the backbone of residue 50 in Kir6.2. The Cα r.m.s.d. between the N domain of Kir3.1 (template) and the N domain of Kir6.2 was 0.4 Å. The C domain of Kir6.2 was built as described previously (Trapp et al, 2003). The Cα r.m.s.d. between the Kir3.1 C-domain (template) and the Kir6.2 C-domain (model) was 1.3 Å and that between the TMs of KirBac (template) and Kir6.2 (model) was 1.7 Å. These values lie within the range expected for proteins sharing 25% sequence identity (0.7–2.3 Å) (Russel et al, 1997).
The position of the ATP-binding site was predicted using ligand docking. ATP was randomly positioned on the C-domain model and 25 dockings were carried out (Trapp et al, 2003). No information about the potential binding site obtained from mutagenesis studies was specified in the docking procedure. In 19/25 runs (76%), the β-phosphate of ATP interacted with K185 (which is suggestive of an energy minimum on the potential energy landscape). The rest of the molecule lay in an identical position in five runs, and close to one or more of a consensus set of residues in all cases. In similar studies, ADP found the same set of residues as ATP 16/25 times (64%), but GTP, azido-ATP, ADP-ribose and ATP-ribose never localized to the same position.
The ligand-docking program used did not allow for reorientation of side chains in response to the presence of ATP. It is clear that this would occur, however, because of the strong negative charge of the phosphate tail. Thus, the side chains of R201 and K185 were manually oriented to point towards the ATP molecule and then minimized. However, the backbone was not altered. The positions of the side chains of other residues, including R50, were not adjusted. Automated docking on the modified model revealed that the β-phosphate of ATP found K185 in all 25/25 runs. The Kir6.2–ATP complexes were then evaluated based on interaction energies between ATP and the protein, in order to determine the conformation of the docked ATP and the residues that contribute to its binding site. ATP was then superimposed in the same spatial orientation in the other subunits within the tetrameric model.
It is likely that the crystal structures of both KirBac1.1 and Kir3.1 represent the closed state of the channel. First, KirBac1.1 is clearly closed, as bulky side-chain residues occlude access of hydrated K+ to the intracellular mouth of the pore (Kuo et al, 2003). Second, Kir3.1 is activated by binding Gβγ proteins, and the IC domains were crystallized in the absence of ligand. Thus, we assume that these structures form a reasonable template for the closed state of Kir6.2, in which ATP is bound.
The KATP channel comprises both Kir6.2 and SUR subunits, and it is possible that SUR imposes conformational changes on Kir6.2. Thus, our model corresponds most closely to Kir6.2ΔC, a truncated form of Kir6.2 that expresses in the absence of SUR1 (Tucker et al, 1997). Thus, in our discussions, we distinguish functional data obtained for Kir6.2ΔC from that measured for Kir6.2/SUR1 channels.
Overall structure of Kir6.2
Figure 1B and C give the overall structure of the tetrameric model of Kir6.2. It can be divided into two main regions, with the ‘slide helix' (residues 54–66) lying at their interface. The TM region is 44 Å long (measured as the Cα–Cα distance between residues P109 and F60) and the cytosolic (IC) domain is 63 Å long (D65–A358). When viewed from above, the TM tetramer forms a square with dimensions of 46 Å × 46 Å between the furthest points on adjacent subunits (E104–P102). It sits on a pedestal-like IC domain, which measures 55 Å × 55 Å between D323 and D323 of adjacent subunits. The cytosolic pore that permeates the IC domain ranges from 13 to 50 Å in diameter.
The TM domains. Kir6.2 has two membrane-spanning α-helices (TM1 and TM2) per subunit. TM1 and TM2 are connected via loop segments and a short α-helix (the pore helix) that loops partway into the membrane. The selectivity filter, which lies within the loop linking the pore helix to TM2, exhibits the characteristic signature sequence of K+ channels, except that GYG is replaced by GFG. The r.m.s.d. between the TMs of the present model and that of an earlier model of the TMs alone based on KcsA (Capener et al, 2003) was 2.2 Å. Most of the variation resides in the extracellular loops that connect TM1 with the pore helix (residues 92–116). If these are omitted, the r.m.s.d. between the two models is excellent: ∼0.8–1.1 Å.
The IC domain. The IC domain shows extensive interactions between subunits: thus, the C domain of one subunit interacts both with the N domain of the adjacent subunit on one side, and with the C domain of the adjacent subunit on the other side (Figure 1C). The region of the N domain involved is consistent with protein–protein interaction studies, which implicate residues 30–46 as binding to the C domain (Tucker and Ashcroft, 1999).
The model predicts several interactions within the IC domain of Kir6.2, which may help to stabilize the tetrameric structure. These include a π-stacking interaction between F35 in the N domain of one subunit and Y326 in the C domain of the adjacent subunit (Figure 2). Two intersubunit ion pairs, R32–E321 and R34–E308, also connect these domains (Figure 2). Several electrostatic interactions are found between the C domains of adjacent subunits (Figure 2). These include three ion pairs: E229 with R314, R301 with E292 and R192 with E227.
Figure 2.
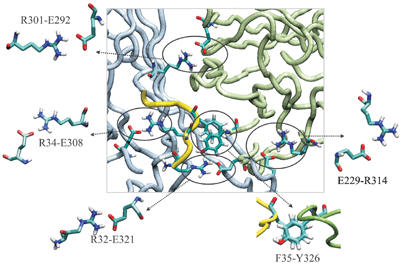
Interactions between adjacent subunits. The C domain of one subunit is shown in grey and its N domain in yellow. The C domain of the adjacent subunit is shown in green. Residues are shown in cpk colours.
Interactions between the IC and TM domains. There are three main sites of interaction between the IC and TM domains. The N domain is linked directly to the slide helix (Figures 1B and 3). The C domain connects directly to TM2, but also appears to be linked to the slide helix of the adjacent subunit by two loops that span residues 204–209 (loop 1) and 289–299 (loop 2) (Figure 3A and B). A web of interactions link loops 1 and 2 both to the slide helix and to the ATP-binding pocket (Figure 3C and D).
Figure 3.
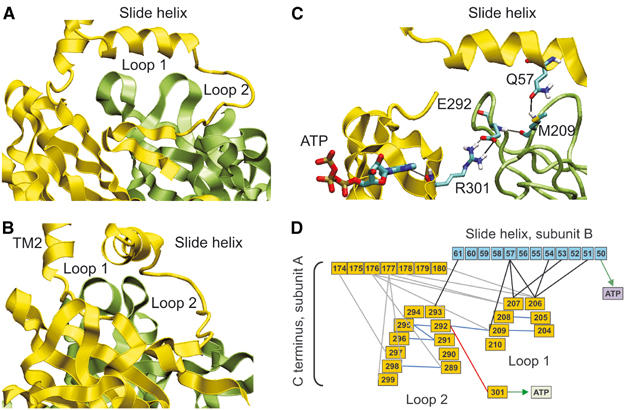
(A, B) Relationship between the N domain of one subunit (yellow) and the C domain of another (green), showing the relation of loops 1 and 2 to the slide helix. (C) Interactions between loops 1 and 2 and the C domain of one subunit and the slide helix of the adjacent subunit. (D) Web of predicted interactions between the C linker, loops 1 and 2 of the C domain of one subunit (yellow) and the slide helix of the adjacent subunit (blue). Coloured lines connect interacting residues, or indicate interactions with ATP.
As homology models of the TMs of Kirs have been constructed and evaluated previously (Loussouarn et al, 2001; Capener et al, 2003), in this paper we focus on the IC domain, and the ATP-binding site in particular. We also consider the slide helix which links the TM and IC domains, as its position suggests that it may be involved in gating of the pore, both in the absence and presence of ATP.
The ATP-binding site
There are four ATP-binding pockets, one per subunit, as predicted by functional studies (Markworth et al, 2000). Each is located in the upper part of the IC domain, about 17 Å below the plane of the membrane. The position of this pocket on the outer face of the protein (Figure 4A and B) may facilitate access of cytosolic ATP to its binding site.
Figure 4.
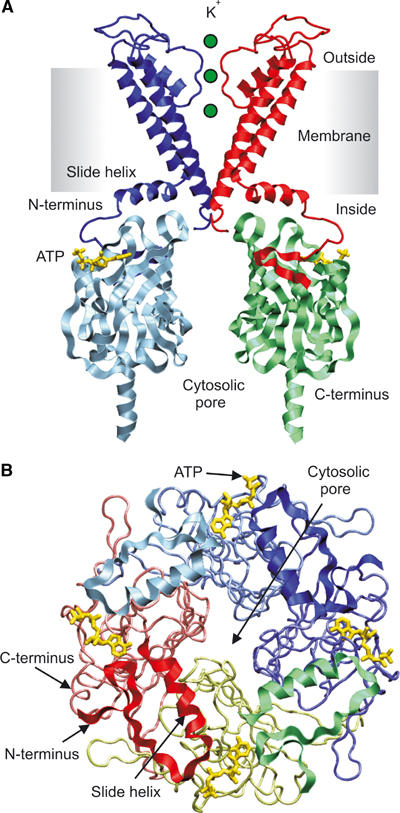
(A) Side view of the ATP-binding site. For clarity, the TMs of only two subunits and the IC domains of two separate subunits are illustrated. ATP (yellow) is docked into its binding sites. (B) Kir6.2 tetramer, viewed from above, with the TMs removed (residues 64–177). ATP (yellow) is docked into its binding sites. The N domain is shown in ribbon format and the C domain in backbone format. Different colours represent individual subunits.
The main binding pocket lies at the interface between the N and C domains of the same subunit. In addition, the N domain of the adjacent subunit loops across the C-terminal part of the binding pocket, such that the side chain of R50 lies to one side of the entrance to the pocket, while K185 from the other subunit lies at the opposite side (Figure 5A and B). Docking of ATP was conducted in the absence of the N-terminal residues, but it is clear that the location of R50 in the complete, tetrameric, homology model would restrict access of ATP to its binding site. Thus, either the position of R50 is incorrect, or the N-domain must move to enable ATP to enter the binding pocket.
Figure 5.
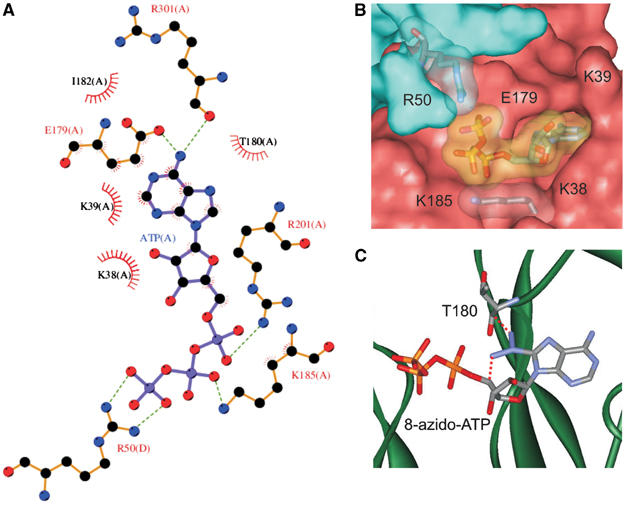
(A) Interactions between ATP and residues lining the ATP-binding pocket. The subunit origin (A or D) of the residue is indicated. Predicted hydrogen bonds are indicated by dashed lines and hydrophobic interactions by sunbursts. Residues are shown in cpk colours. (B) Space-filling model of the ATP-binding pocket. Different subunits are indicated in blue and pink. The electron shells of R50 and K185, and of ATP, are shown in transparency. (C) Close-up of the binding pocket with 8-azido-ATP placed in the same position as ATP. The dashed lines indicate where the azido group makes a steric clash with T180 and the ATP molecule itself.
The overall size of the ATP-binding pocket is sufficient to accommodate the ATP molecule (volume ∼1070 Å3). The Cα–Cα distance from A300, which forms the innermost residue of the hydrophobic pocket, to G334, which forms the outer boundary, is about 17 Å. The width of the pocket, measured from K39 to T180, is ∼10 Å and its height, measured between residues E179 and F183, is ∼8 Å.
In all, 17 residues lie within 4.5 Å of ATP in the model: K38, K39, G40 in the N domain, E179, T180, L181, I182, F183, S184, K185, R201, A300, R301, T302, F333 and G334 in the C domain, and R50 from the N domain of the adjacent subunit. The adenine ring and the ribose sugar of the nucleotide are positioned on one side of a β-sheet and the phosphate tail on the other, which effectively separates the charged and uncharged groups of ATP.
The adenine ring of ATP lies within a mainly hydrophobic pocket, lined by the side chains of E179, T180, L181, I182, and the backbone atoms of K38, K39, G40 and R301. The side chain of E179 and the carbonyl oxygen atom of the backbone of R301 make hydrogen bonds with the N6 atom in the adenine ring (Figure 5A). The adenine ring is accommodated tightly within the ATP-binding pocket (Figure 5B), and addition of an azido group at the 8′ position would cause a steric clash with the ribose moiety of ATP and with the side chain of T180 (Figure 5C). This explains why 8′-azido-ATP fails to dock into the ATP-binding site of the model or to inhibit the channel (Tanabe et al, 1999).
No interactions are predicted with the ribose moiety of ATP, but S184 lies under the ribose and K38 alongside it.
The phosphate tail of ATP makes putative electrostatic interactions with three positively charged residues: R201 with the α-phosphate, K185 with the β-phosphate and R50 (from the adjacent subunit) with the γ-phosphate (Figure 5A). Although the residue equivalent to R50 is absent in the crystal structure of the IC domains of Kir3.1, on which this model is based, the adjacent residue (equivalent to E51) is not. Thus, the position of E51 is fixed, which also restrains the predicted position of R50 and brings it close enough to interact with the γ-phosphate of ATP, as predicted by ATP-binding (Tanabe et al, 1999) and thiol modification (Trapp et al, 2003) studies. The γ-phosphate is not buried in the protein, as in many ATP-binding sites, but points out into the extracellular solution (Figure 4).
Predictive power of the model
The model accounts for a large amount of published data and enables previously puzzling findings to be explained (see Discussion). This provides significant validation of the model, as none of this information was used in its construction. In addition, we made new mutations to test the predictive power of the model.
ATP-binding site. Of the 17 residues that lie within 4.5 Å of ATP in the model, most have already been mutated in Kir6.2ΔC or Kir6.2/SUR1. The effects of these mutations are discussed below. However, residues A300 and F333 have not been mutated in either construct, nor have L181, F183, R201, A300, R301, T302 and F333 been mutated in Kir6.2ΔC. To test the model, we examined the effects of mutating (in Kir6.ΔC) residue R201, which is predicted to make an electrostatic interaction with the α-phosphate of ATP, residues A300 and F333, which lie close to the β-phosphate of ATP, and residue E179. In addition to measuring concentration-inhibition curves for ATP, we also examined the channel open probability (Po) in the absence of ATP (which we define as the intrinsic Po) by single-channel recording. This is because mutations that increase intrinsic Po indirectly reduce KATP channel ATP sensitivity (Shyng et al, 1997; Trapp et al, 1998; Cukras et al, 2002). Thus, we only considered mutations that alter ATP sensitivity without affecting intrinsic Po as influencing ATP binding and/or transduction.
The side chain of E179 is predicted to make a hydrogen bond with the N6 atom in the adenine ring of ATP (Figure 5A). To test this, we mutated E179 to a range of residues, some of which are capable of forming H bonds (Q, N), and some of which are not (M, L). Consistent with this idea, mutation of E179 to glutamine has no effect, whereas mutation to methionine reduced the IC50 two-fold (Figure 6, Table I). However, mutation to leucine caused a small but insignificant shift in IC50, which does not support an important role for H bonding. None of these mutations altered the intrinsic Po (Table I). Mutation of E179 to asparagine caused an ∼7-fold reduction in the IC50 for ATP inhibition, but also increased the intrinsic Po (Figure 6, Table I). This change in Po could account for the reduced ATP sensitivity (Shyng et al, 1997; Trapp et al, 1998; Cukras et al, 2002) and this mutation therefore cannot be used to address the role of H bonding. The relatively small effect of mutating E179 may explained by the fact that the N6 atom is also stabilized by H bonding to the backbone of R301 (Figure 5A), and further suggests that R301 may be more important in co-ordinating the adenine moiety. That E179 and ATP are slightly out of plane, and so likely to form only a weak H bond, whereas R301 and ATP lie in the same plane, is harmonious with this idea.
Figure 6.
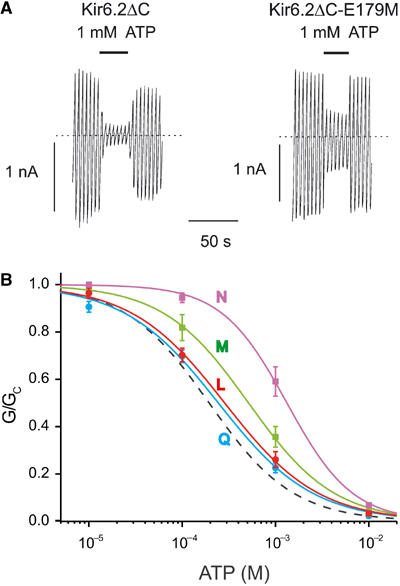
(A) KATP currents elicited by voltage ramps from −110 to +100 mV to an inside-out patch excised from a Xenopus oocyte expressing Kir6.2ΔC or Kir6.2ΔC-E179M. The dotted line indicates the zero current level. (B) Mean relationship between [ATP] and KATP conductance (G), expressed relative to the conductance in the absence of nucleotide (Gc) for Kir6.2ΔC with E179 mutated to N ( , n=5), M (
, n=5), M ( , n=5), L (
, n=5), L ( , n=5) and Q (
, n=5) and Q ( , n=7). The curves are the best fit to equation (1), using values for IC50 and h given in Table I. The dashed line indicates the wild-type data.
, n=7). The curves are the best fit to equation (1), using values for IC50 and h given in Table I. The dashed line indicates the wild-type data.
Table 1.
Mean parameters for ATP inhibition and open probability of wild-type and mutant channels
| Mutation | IC50(n) | h | Po |
|---|---|---|---|
| Kir6.2ΔC (wt) | 194±10 μM (n=5) | 0.96±0.04 | 0.08±0.02 (n=6) |
| Kir6.2ΔC-V59G | 1.7±0.1 mM*** (n=5) | 1.1±0.1 | 0.78±0.03*** (n=6) |
| Kir6.2ΔC-E179Q | 240±36 μM (n=7) | 0.88±0.5 | 0.10±0.01 (n=7)a |
| Kir6.2ΔC-E179L | 299±47 μM (n=5) | 0.89± 0.06 | 0.14±0.01 (n=5) |
| Kir6.2ΔC-E179M | 515±51 μM** (n=5) | 1.08±0.17 | 0.09±0.02 (n=5) |
| Kir6.2ΔC-E179N | 1.4±0.23 mM*** (n=6) | 1.3±0.1 | 0.58±0.03*** (n=8) |
| Kir6.2ΔC-R201C |
14±l mMb*** (n=6) |
0.81±0.05b |
0.12±0.02 (n=6) |
| IC50=ATP concentration at which inhibition is half-maximal. h, Hill coefficient. Values indicate the means of the individual fits to equation (1), for n patches. aFrom Tucker et al (1998). bEstimated by fitting line through data points that extend to 10 mM. Statistical significance was determined using Student's t-test. **P<0.01; ***P<0.001. | |||
The α-phosphate of ATP is predicted to form an electrostatic interaction with the side chain of R201 (Figure 5A). Consistent with this prediction, mutation of R201 to cysteine results in permanent neonatal diabetes in humans (Gloyn et al, 2004), and causes a marked reduction in the ATP sensitivity of Kir6.2ΔC (Figure 7): the IC50 could not be measured accurately because even as much as 10 mM ATP only inhibited the channel by 43±5% (n=6), compared with 96±1% (n=5) for the wild-type channel. The IC50 was estimated to be 14 mM, 70-fold larger than the wild-type channel (Table I).
Figure 7.
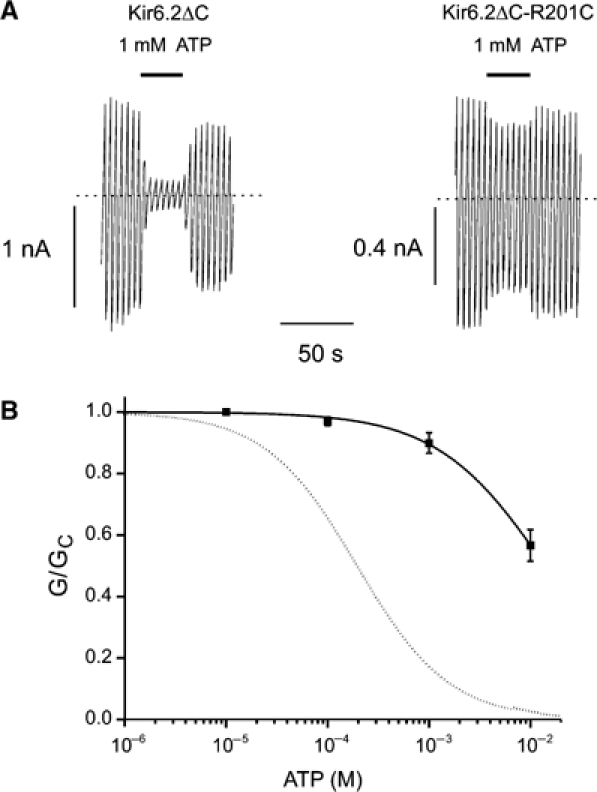
(A) Kir6.2ΔC and Kir6.2ΔC-R201H currents evoked by voltage ramps from −110 to +100mV. (B) Mean relationship between [ATP] and KATP conductance (G), expressed relative to the conductance in the absence of nucleotide (Gc) for Kir6.2ΔC-R201C (n=5). The curve is the best fit to equation (1) using the values given in Table I. The dotted line indicates the wild-type data.
We next tested the effect of mutating F333, which is predicted to lie within 3 Å of the α-phosphate of ATP (Figure 8A). In this experiment, we used full-length Kir6.2. As full-length Kir6.2 does not express by itself, it was coexpressed with SUR1: to avoid the stimulatory effects of Mg nucleotides on SUR1, we used a mutant SUR1 (SUR1-KA/KM) that is not modulated by adenine nucleotides (Gribble et al, 1997). This ensures that only ATP binding to Kir6.2 influences channel activity. The F333L mutation markedly increased the IC50 for channel inhibition—from 17 to 514 μM (Figure 8B). We also tested the effect of mutating Y330, which lies at some distance from ATP in the model, to leucine. This had no effect (Figure 8B). The model predicts that A300 lies within 4.5 Å of the adenine ring of ATP (Figure 8A). Unfortunately, mutation of A300 to either D or L failed to yield functional currents.
Figure 8.
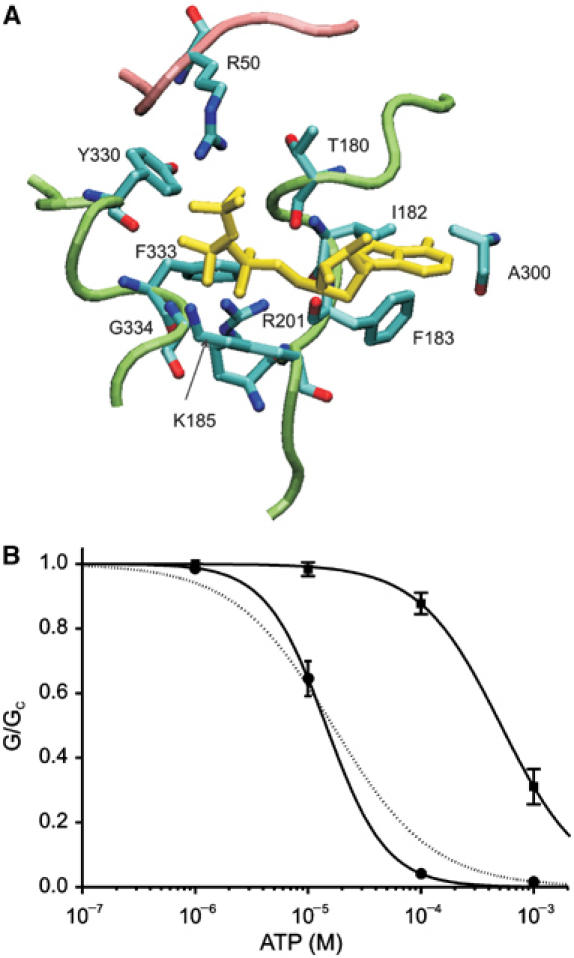
(A) Model of the ATP-binding pocket of Kir6.2 illustrating the proximity of F333 and Y330 to the phosphate tail of ATP. (B) Mean relationship between [ATP] and KATP conductance (G), expressed relative to the conductance in the absence of nucleotide (Gc) for Kir6.2-Y330L/SUR1-KA/KM (•, n=5), and Kir6.2-F333L/SUR1-KAKM (▪, n=6) channels. The smooth curves are the best fit to equation (1). For Kir6.2-Y330L, IC50=16.9±1.9 μM, h=1.3±0.1. For Kir6.2-F333L, IC50=514±93 μM, h=1.2±0.1. The dashed line indicates the control data (IC50=17 μM, h=0.99; Gribble et al, 1997).
The model predicts that addition of an azido group at the 2′ position on the adenine ring will impair ATP binding, due to a steric clash with the backbone of K38 and K39. Consistent with this prediction, 2-azido-ATP blocked Kir6.2ΔC currents by 6±4% at 100 μM and by 47±2% at 1 mM (n=6). Assuming a Hill coefficient of 1, the IC50 of was 1.2 mM, ∼10-fold greater than for ATP.
The ribose moiety of ATP does not make a direct interaction with the channel (Figure 5A). However, it lies within 4 Å of K185, which may explain why substitution of ribose with 2′-deoxyribose or ribose 2′,3′-dialdehyde reduces the ATP sensitivity of native skeletal muscle KATP channels (Spruce et al, 1987). At a concentration of 1 mM, 2′-deoxyribose ATP inhibited Kir6.2ΔC by 61±2% (n=11) as compared with 82±3% for ATP (n=16).
Slide helix mutations The position of the slide helix suggests that it may be involved in channel gating (Kuo et al, 2003). With the exception of R54 at the N-terminal end (Cukras et al, 2002; Schulze et al, 2003), no mutations in the Kir6.2 slide helix have yet been studied: thus, we tested this idea by mutagenesis of Kir6.2ΔC. We chose to mutate V59 to glycine (V59G), because this mutation produces neonatal diabetes with developmental delay, muscle weakness and epilepsy (Gloyn et al, 2004). Figure 9 shows that the V59G mutation caused a marked increase in the intrinsic Po, which was accompanied by a 10-fold reduction in ATP-sensitivity (Table I).
Figure 9.
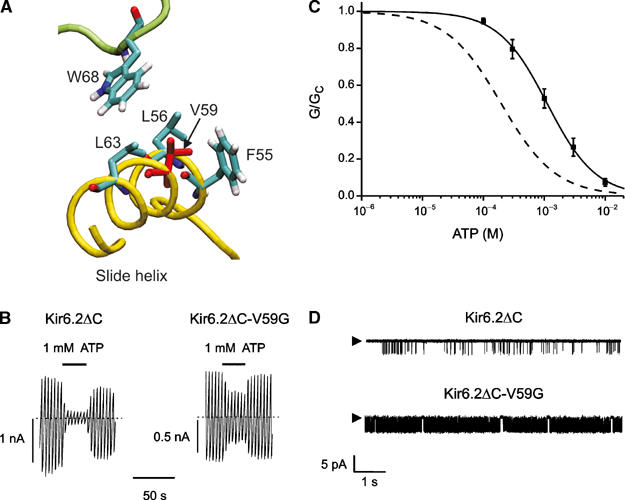
(A) Slide helix of Kir6.2, showing the location of V59. The slide helix of one subunit is shown in yellow and the TM domain of the adjacent subunit in green. (B) Kir6.2ΔC and Kir6.2ΔC-V59G currents evoked by voltage ramps from −110 to +100 mV in inside-out patches. (C) Mean relationship between [ATP] and KATP conductance (G), expressed relative to the conductance in the absence of nucleotide (Gc) for Kir6.2ΔC-V59G (n=6). The curve is the best fit to equation (1) using the values given in Table I. The dashed line indicates the wild-type data. (D) Single Kir6.2ΔC and Kir6.2ΔC-V59G channel currents recorded at –60 mV. The arrow indicates the zero current level.
Discussion
Subunit interactions in the IC domains
Evidence for a physical interaction between the N and C domains of Kir6.2 has been obtained from protein–protein interaction studies (Tucker and Ashcroft, 1999; Jones et al, 2001), which showed that residues 30–46 in the N domain interact with residues in three separate regions of the C domain of Kir6.2. The same N-terminal region contributes to the interaction of the N and C domains of our model (R32, R34 and F35). Furthermore, the π-stacking interaction predicted between F35 in the N domain of one subunit and Y326 in the C domain of the adjacent subunit is also observed in Kir3.1 (Nishida and MacKinnon, 2002). Indeed, hydrophobic residues at both these positions are conserved across Kir6.x, 2.x and 3.x channels. Naturally occurring mutations at residues F35 (to V or A) and E322 (to K, which would be expected to disrupt the adjacent E321-R32 ion pair) cause permanent neonatal diabetes mellitus (PNDM) in humans (Sagen et al, 2004; Vaxillaire et al, 2004). This supports the idea that interactions between the N and C domains of adjacent subunits are important for KATP channel function.
A number of electrostatic interactions occur between adjacent C domains. Mutagenesis studies suggest that residues E229 and R314 form an ion pair, since a single charge neutralization at either position induces channel ‘inactivation' after patch excision, which can be rescued by a double charge reversal (E229R/R314E) (Lin et al, 2003). Furthermore, if E229 and R314 are both mutated to cysteine, they can be reversibly crosslinked in the presence of an oxidizing agent, indicating that the residues must lie <4 Å apart. Tandem dimer constructs showed that this ion pair is likely to be an intersubunit interaction.
Two residues previously suggested as candidates for ion pair formation (R192, R301), on the basis of the fact that their mutation to alanine enhances channel inactivation (Lin et al, 2003), are also predicted to form ion pairs in our model: residue 192 with E227, and R301 with E292, in adjacent subunits. Both ion pairs lie in close proximity and are highly conserved. A glutamate is found at the position equivalent to E227, an arginine at the equivalent position to R301 and a negatively charged residue at the E292 position, in all Kir channels. R192 is also highly conserved. This conservation of sequence identity suggests an important role. Further, the cluster of acidic and basic residues in this region suggests that different combinations of electrostatic interactions may occur during channel gating.
Slide helix
Our results provide evidence that the slide helix plays a role in gating, since the V59G stabilizes the open state. In contrast, mutation of R54 reduces the intrinsic Po of Kir6.2/SUR channels; however, this may be secondary to changes in PIP2 or acyl CoA modulation (Cukras et al, 2002; Schulze et al, 2003). Its amphipathic nature suggests that the slide helix slides along the membrane interface during channel opening, without substantial rotational movement.
ATP-binding pocket
The ATP-binding site suggested by the model involves contributions from the N and C domains of adjacent subunits. This is supported by a substantial amount of data. First, mutations in both these regions impair channel inhibition by ATP (Drain et al, 1998; Proks et al, 1999; Tucker et al, 1998; Reimann et al, 1999; Cukras et al, 2002; John et al, 2003; Ribalet et al, 2003). Further, when mutations in the N and C domains that affect ATP sensitivity are combined, the ATP sensitivity is further reduced, as expected if the N and C domains cooperate to influence channel closure by ATP (Proks et al, 1999). Second, protein–protein interaction studies demonstrate a physical interaction between the N and C domains of Kir6.2 (see above). Importantly, the model predicts that each N domain contributes to two ATP-binding sites: that of the same and the adjacent subunit (Figure 4). This is harmonious with FRET experiments which show that the physical relationship of the N and C domains of adjacent subunits is influenced by ATP binding (Tsuboi et al, 2004).
SUR enhances the ATP sensitivity of Kir6.2 about 10-fold (Tucker et al, 1997). The exposed position of the ATP-binding site in Kir6.2 suggests that one way this might be achieved would be if SUR embraced the outer part of the binding site, perhaps by contributing additional residues that stabilize ATP binding, or by allosterically altering the structure of the binding site.
Our studies demonstrate that mutation of R201 causes a marked reduction in the ATP sensitivity of Kir6.2ΔC. Similar results have been found for Kir6.2/SUR1 channels (Shyng et al, 2000; John et al, 2003; Ribalet et al, 2003). These studies reveal that mutation of R201 to a negatively charged, or uncharged, residue weakens ATP inhibition, and in the case of Kir6.2-R201E/SUR1 the current could not be completely blocked even at millimolar ATP levels. Furthermore, inhibition by ATP, ADP and AMP are all affected, consistent with the idea that R201 interacts with the α-phosphate of ATP. Mutation of R201 to histidine also reduces the ATP sensitivity of the Kir6.2/SUR1 channel, and causes PNDM (Gloyn et al, 2004). It is interesting that the shift in ATP sensitivity produced by the R201C mutation is greater in the absence of SUR1: the IC50 increased ∼120-fold for Kir6.2ΔC (Table I) and 15-fold for Kir6.2/SUR1 (John et al, 2003). This suggests that SUR1 may modify the binding pocket at the level of R201, consistent with previous work (Dabrowski et al, 2004).
There is considerable experimental evidence to support the interaction of the β-phosphate of ATP with K185 predicted by the model (Figure 5A). First, mutation of K185 to a negative charge strongly impairs ATP inhibition, whereas mutation to arginine has only a small effect, in both Kir6.2ΔC and Kir6.2/SUR1 (Tucker et al, 1997, 1998; Reimann et al, 1999; John et al, 2003). Similar results are found when K185C is modified by charged thiol regents (Trapp et al, 2003). This supports an electrostatic interaction between K185 and ATP. Furthermore, these modifications impair ATP and ADP inhibition, but not that by AMP, consistent with an interaction with the β-phosphate of ATP (John et al, 2003; Trapp et al, 2003). In addition, binding of 8-γ-32P-azido ATP to Kir6.2 is impaired by the K186Q mutation (Tanabe et al, 1999). Mutation of residues S184 and H186, which flank K185, reduces the ATP sensitivity of Kir6.2ΔC (Tucker et al, 1998); mutation of S184 also impairs the ATP sensitivity of Kir6.2/SUR1, and introduction of charged thiol reagents at either position in Kir6.2/SUR1 shifts the channel ATP sensitivity as expected if the residue lies close to the phosphate tail of ATP (Trapp et al, 2003).
The γ-phosphate is not buried in the protein, as in many ATP-binding sites, but points out into the external solution (Figure 4). This may explain why Mg2+ is not required for ATP binding to Kir6.2 (Tanabe et al, 1999, 2000), and why large groups can be added to the phosphate tail without substantially reducing ATP block. For example, AP4A and ATP block both Kir6.2ΔC (Dabrowski et al, 2004) and native KATP channels (Jovanovic et al, 1997) with similar affinity, as does the photo-activatable ATP analogue DMNPE-ATP (Ämmälä et al, 1991). Likewise, azido-anilino ATP blocks Kir6.2ΔC channels with an IC50 comparable to that for ATP (Tanabe et al, 2000). All these data are consistent with the open-ended structure of the ATP-binding site predicted by the model. It is worth noting that this is in contrast to most ATP-binding sites, which are closed.
Our model predicts that R50 lies close to the γ-phosphate of ATP. This is in good agreement with the fact that the ATP sensitivity of Kir6.2/SUR1 channels is reduced by substitution of a cysteine at R50, and that this effect is reversed by modification with a positively charged thiol reagent but is further decreased by modification with a negatively charged thiol agent (Trapp et al, 2003). This suggests that ATP is able to sense the charge at residue 50 electrostatically, and must therefore lie in close proximity. As ADP block was unaffected by thiol modification of R50, it appears that the γ-phosphate of ATP is involved. However, mutation of R50 in Kir6.2ΔC or Kir6.2/SUR1 (Proks et al, 1999; John et al, 2003) is not consistent with the interaction of R50 and ATP being entirely electrostatic: for example, the IC50 for ATP block of Kir6.2ΔC is greater for R50G, than for R50E (although there is no change in intrinsic Po). Thus, mutations at R50 may simply perturb the protein structure, and thereby access of ATP to its binding site.
G334, which is located far from K185 in the primary sequence, lies close to both the β-phosphate of ATP (3.3 Å) and to K185 in the three-dimensional model structure (Figure 8). This rationalizes the previously puzzling finding that mutation of G334 to aspartate markedly decreases ATP sensitivity (Drain et al, 1998). As our model shows, this mutation would be expected to impede nucleotide binding both sterically and electrostatically. Residues F333 and Y330 also lie close to the phosphate tail of the ATP molecule. That PNDM is caused by mutations at either of these residues (F333I, Y330C) is indicative of their importance for channel function (Sagen et al, 2004; Vaxillaire et al, 2004).
The model prediction that the N6 atom in the adenine ring of ATP hydrogen bonds with residues lining the binding pocket is harmonious with the known efficacy of trinucleotide phosphates at inhibiting KATP currents: GTP, ITP, UTP and purine triphosphate (PTP) are all ineffective (Tucker et al, 1998; Dabrowski et al, 2004). A common feature of these other nucleotide triphosphates is that, unlike ATP, they lack an NH2 group at the 6′ position of the adenine ring and are therefore unable to form a hydrogen bond. In support of this idea, methyl ATP, which has an NCH3 group and is therefore capable of hydrogen bonding, is able to block the channel (Dabrowski et al, 2004).
Residues T180, L181 and I182 provide a hydrophobic lining to the binding pocket. Extensive mutagenesis has been carried out on I182 in Kir6.2ΔC (Li et al, 2000). Substitution with a variety of amino acids markedly reduces channel inhibition by ATP, the IC50's increasing from ∼200 μM (I) to ∼5 mM (E, M, Y, P) and >10 mM (K, R, S, W), without altering intrinsic gating. I182 is accessible to cysteine-modifying agents, but modification with a negatively charged thiol reagent was without effect on the ATP sensitivity of I182C, indicating that this residue does not lie close to the phosphate tail of the nucleotide (Trapp et al, 2003). This is consistent with the predicted location of I182 adjacent to the adenine ring. The threonine at position 180 is interesting as mutations at this position affect the intrinsic Po of the channel (Light et al, 2000), as observed for the asparagine mutation at E179 (Table I). Kir6.2/SUR1 containing either L181C or F183C mutations did not express functional channels (Trapp et al, 2003).
The model predicts that the binding pocket is lined by backbone atoms of K38, K39 and G40 in the N domain (Figure 5A and B). Consistent with this structure, mutation of K39 to alanine in Kir6.2/SUR1 reduced ATP sensitivity indirectly, by altering the open state stability rather than impairing ATP binding (Cukras et al, 2002).
How does ATP binding lead to closure of the pore?
ATP binding occurs at the interface between two subunits, as is common in ligand-gated channels (Brejc et al, 2001). Moreover, subunit interactions indicate that each monomer is connected to not one, but two other subunits (Figure 4). This suggests that ATP binding to one subunit will influence the relative conformations of the two adjacent subunits, and may help to explain why binding of a single ATP molecule is sufficient to close the pore. As the binding lies at the interface between subunits, it also seems possible that the overall conformation of each monomer will not undergo a dramatic change upon ATP binding: rather, nucleotide binding may primarily influence the relationship between the monomers.
Conclusions
In conclusion, by constructing a molecular model of the Kir6.2 tetramer, we have identified the putative location of the ATP-binding site. Residues that form the ATP-binding site are not well conserved across Kir channel subfamilies, in contrast to those implicated in intra-subunit interactions. This suggests that, while the ATP-binding site is unique, Kir channels may share a common structural basis for subunit assembly and that the mechanism by which ligand binding is linked to opening and closing of the pore may also be similar.
The model is in good agreement with a large body of mutagenesis data, offers an explanation for the relatively low affinity of Kir6.2 for other nucleotide triphosphates, and suggests that SUR1 might enhance ATP sensitivity by embracing the outer part of Kir6.2. Our results leads us to suggest that gating of Kir6.2 by ATP probably involves changes in inter-subunit interactions and, further, to speculate that flexible loops in the C-domain (204–209 and 289–299) may help translate conformational changes induced by ATP binding into movement of the slide helix and thereby closing of the channel. The model also makes specific suggestions for subunit–subunit interactions and ligand–protein interactions that provide a solid basis for further mutagenesis and simulation studies.
Materials and methods
Molecular modelling
All sequence alignments were carried out using ClustalX. Comparative modelling was used to construct the molecular models using MODELLER v6.2 (Sali and Blundell, 1993; Fiser et al, 2000). Several models were constructed, with the final one being selected on the basis of low energy function and Cα r.m.s.d. values after superimposition on the template. The models were constructed in three segments.
The percent identity between the IC domains of Kir3.1 and Kir6.2 is 48%, and that between the KirBac1.1 and Kir6.2 is 32% for the TMs and slide helices, and 27% for the IC domains. The slide helix and TMs of Kir6.2 (residues 55–172) were modelled as a tetramer, using the crystal structure of KirBac1.1 as a template (Kuo et al, 2003). Residues that connect the TMs with the C domain (residues 173–176) were modelled as loops. The consensus secondary structure predictions for these loops were obtained from the programs PHD (www.cubic.bioc.columbia.edu) and JPRED (www.compbio.dundee.ac.uk). The C domain of Kir6.2 (residues 177–358) was modelled as a tetramer, based on the 2.0 Å crystal structure of the C domain of Kir3.1 (Trapp et al, 2003). The orientation of the C domain with respect to the TMs was obtained by superimposing the conserved stretches of residues in Kir6.2 on KirBac1.1. Once the orientation of the segments had been established, they were joined using the modelled loops. The loops were then subjected to energy minimization, while keeping the position of the atoms of the core model frozen.
The N domain of Kir6.2 (residues 32–46) was modelled based on the N-terminal (residues 43–57) structure of Kir3.1 (Nishida and MacKinnon, 2002). The remaining residues that contribute to the N domain of Kir3.1 are not resolved in the crystal structure, except for E63. This residue is conserved in both Kir3.1 and Kir6.2 (E51). The remainder of the N-domain residues in Kir6.2 (47–54) were modelled using KirBac1.1 as a template. As this region is a loop in KirBac1.1, we also confirmed that residues 47–50 and 52–54 are loops, based on the predictions of the programs above. The position of E63 in the crystal structure of Kir3.1 was taken as a reference to obtain the spatial position and orientation for the N-terminal loops. The equivalent residue in Kir6.2 (E51) was fixed and the loops on either side were subjected to restrained energy minimization procedures using GROMACS v3.1.4 (www.gromacs.org). The N domain was modelled as a monomer, and docked onto the tetrameric model of the C domain, using the fixed points described above.
The Cα–Cα r.m.s.d. between the templates and the model was 0.4 Å for the N domain, 1.7 Å for the slide helix and the TMs, and 1.3 Å for the C domain.
Kir6.2 is a homotetramer with four potential ATP-binding sites (one per subunit) that do not interact (Markworth et al, 2000): for simplicity, only a monomer was used to dock ATP to the C domain (Trapp et al, 2003). Docking was carried out using AUTODOCK 3.0.5 (Goodsell et al, 1996), which allows for ligand flexibility. Monte Carlo simulated annealing was used to perform the dockings. The hydrogen atoms were generated and partial charges assigned using InsightII (www.accelrys.com). A grid box of 100 × 120 × 100 points was created with a grid spacing of 0.375 Å around the protein to define the interaction space between the ligand and the protein. In all, 11 rotatable bonds in the ligand were allowed to be flexible. A total of 25 dockings were carried out with 5000 cycles per run. Interaction energies were calculated for favourable ligand–protein-binding sites. Dockings were ranked according to the interaction energies between the protein and the ligand. The final conformation of the docked ATP was based on a combination of interaction energies, functional data obtained from physiological studies and chemical intuition. Once residues contributing to the binding site were identified, ATP was superimposed in the same spatial orientation in the remaining subunits. The quality and the stereochemical properties of the model were assessed using PROCHECK v3.4.4. (Morris et al, 1992).
Functional studies
Mouse Kir6.2 (Genbank D50581) and rat SUR1 (Genbank L40624) were used in this study. Site-directed mutagenesis of Kir6.2 and preparation of mRNAs was as described (Trapp et al, 1998). Xenopus laevis oocytes were injected with 0.1 ng full-length wild-type or mutant Kir6.2 and ∼2 ng of SUR1 mRNAs (giving a 1:20 ratio), and studied 1–4 days later (Trapp et al, 1998).
Macroscopic currents were recorded from giant inside-out patches at a holding potential of 0 mV and 20–24°C. Currents were evoked by 3 s voltage ramps from −110 to +100 mV, recorded with an Axopatch 200B patch-clamp amplifier (Axon Instruments, Foster City, CA, USA), filtered at 0.15 kHz, digitized at 0.5 kHz using a Digidata 1200 Interface and analysed using pClamp software (Axon Instruments). The pipette solution contained (mM): 140 KCl, 1.2 MgCl2, 2.6 CaCl2 and 10 HEPES (pH 7.4 with KOH). The internal solution contained (mM): 107 KCl, 1 MgCl2, 1 CaCl2, 10 EGTA (free Mg2+∼600 nM), 10 HEPES (pH 7.2 with KOH) and nucleotides as indicated. Rapid exchange of internal solutions was achieved using a sewer-pipe system. Single-channel currents were recorded from small inside-out membrane patches at −60 mV, filtered at 5 kHz and sampled at 20 kHz. Open probability was determined from records of ∼1 min duration as I/iN, where I is the macroscopic current, i is the single-channel current amplitude and N is the number of channels (maximum number of open levels).
The macroscopic slope conductance was obtained by fitting a straight line to the I–V relation between −20 and −100 mV (Trapp et al, 1998). To control for possible rundown, the control conductance (Gc) was taken as the mean of that in control solution before and after ATP application. ATP concentration–response curves were fitted by the Hill equation:
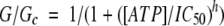 |
1 |
where [ATP] is the ATP concentration, IC50 is the concentration at which inhibition is half-maximal and h is the Hill coefficient. All data are given as mean±s.e.m. Statistical significance was tested by Student's t-test or ANOVA, as appropriate.
Acknowledgments
We thank the Wellcome Trust and the Royal Society for support. JA is a Scholar of Christ Church College, Oxford. FMA is the Royal Society GlaxoSmithKline Research Professor.
References
- Ämmälä C, Bokvist K, Galt S, Rorsman P (1991) Inhibition of ATP-regulated K+-channels by a photoactivatable ATP-analogue in mouse pancreatic beta-cells. Biochim Biophys Acta 1092: 347–349 [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Kakei M (1989) ATP-sensitive K-channels: modulation by ATP and Mg2+ ions. J Physiol 416: 349–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK (2001) Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411: 269–276 [DOI] [PubMed] [Google Scholar]
- Capener CE, Proks P, Ashcroft FM, Sansom MS (2003) Filter flexibility in a mammalian K channel: models and simulations of Kir6.2 mutants. Biophys J 84: 2345–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement JP, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar Bryan L, Bryan J (1997) Association and stoichiometry of KATP channel subunits. Neuron 18: 827–838 [DOI] [PubMed] [Google Scholar]
- Cukras CA, Jeliazkova I, Nichols CG (2002) The role of NH2-terminal positive charges in the activity of inward rectifier KATP channels. J Gen Physiol 120: 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski M, Tarasov A, Ashcroft FM (2004) Mapping the architecture of the ATP-binding site of the KATP channel subunit Kir6.2. J Physiol 557: 347–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drain P, Li L, Wang J (1998) KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc Natl Acad Sci USA 95: 13953–13958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunne MJ, Cosgrove KE, Shepherd RM, Aynsley-Green A, Lindley KJ (2004) Hyperinsulinism in infancy: from basic science to clinical disease. Physiol Rev 84: 239–275 [DOI] [PubMed] [Google Scholar]
- Fiser A, Do R, Sali A (2000) Modeling of loops in protein structures. Prot Sci 9: 1753–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, Edghill EL, Frayling TM, Temple IK, Mackay D, Shield JP, Sumnik Z, van Rhijn A, Wales JK, Clark P, Gorman S, Aisenberg J, Ellard S, Njolstad PR, Ashcroft FM, Hattersley AT (2004) Activating mutations in the ATP-sensitive potassium channel subunit Kir6.2 gene are associated with permanent neonatal diabetes. New Engl J Med 350: 1838–1849 [DOI] [PubMed] [Google Scholar]
- Goodsell DS, Morris GM, Olson AJ (1996) Automated docking of flexible ligands: applications of AutoDock. J Mol Recog 9: 1–5 [DOI] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Ashcroft FM (1997) The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by MgADP and diazoxide. EMBO J 16: 1145–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John SA, Weiss JN, Xie LH, Ribalet B (2003) Molecular mechanism for ATP-dependent closure of the K+ channel Kir6.2. J Physiol 552: 23–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, Tucker S, Ashcroft FM (2001) Multiple sites of interaction between the intracellular domains of an inwardly-rectifying potassium channel, Kir6.2. FEBS Lett 508: 85–89 [DOI] [PubMed] [Google Scholar]
- Jovanovic A, Alexseev AE, Terzic A (1997) Intracellular diadenosine phosphates. A novel family of inhibitory ligands of the ATP-sensitive K+ channel. Biochem Pharm 54: 219–225 [DOI] [PubMed] [Google Scholar]
- Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA (2003) Crystal structure of the potassium channel KirBac1.1 in the closed state. Science 300: 1922–1926 [DOI] [PubMed] [Google Scholar]
- Li L, Wang J, Drain P (2000) The I182 region of K(ir)6.2 is closely associated with ligand binding in KATP channel inhibition by ATP. Biophys J 79: 841–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Light PE, Bladen C, Winkfein RJ, Walsh MP, French RJ (2000) Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. Proc Natl Acad Sci USA 97: 9058–9063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YW, Jia T, Weinsoft AM, Shyng SL (2003) Stabilization of the activity of ATP-sensitive potassium channels by ion pairs formed between adjacent Kir6.2 subunits. J Gen Physiol 122: 225–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loussouarn G, Phillips LR, Masia R, Rose T, Nichols CG (2001) Flexibility of the Kir6.2 inward rectifier K+ channel pore. Proc Natl Acad Sci USA 98: 4227–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markworth E, Schwanstecher C, Schwanstecher M (2000) ATP4− mediates closure of pancreatic beta-cell ATP-sensitive potassium channels by interaction with 1 of 4 identical sites. Diabetes 49: 1413–1418 [DOI] [PubMed] [Google Scholar]
- Morris AL, MacArthur MW, Hutchinson EG, Thornton JM (1992) Stereochemical quality of protein structure coordinates. Proteins 12: 345–364 [DOI] [PubMed] [Google Scholar]
- Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JP IV, Gonzalez G, Aguilar-Bryan L, Permutt MA, Bryan J (1996) Adenosine diphosphate as an intracellular regulator of insulin secretion. Science 272: 1785–1787 [DOI] [PubMed] [Google Scholar]
- Nishida M, MacKinnon R (2002) Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 Å resolution. Cell 111: 957–965 [DOI] [PubMed] [Google Scholar]
- Proks P, Gribble FM, Adhikari R, Tucker SJ, Ashcroft FM (1999) Involvement of the N-terminus of Kir6.2 in the inhibition of the KATP channel by ATP. J Physiol 514: 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann F, Ryder TJ, Tucker SJ, Ashcroft FM (1999) An investigation of the role of lysine 185 in Kir6.2 in inhibition of the KATP channel by ATP. J Physiol 520: 661–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribalet B, John SA, Weiss JN (2003) Molecular basis for Kir6.2 channel inhibition by adenine nucleotides. Biophys J 84: 266–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russel RB, Saqi MA, Sayle RA, Bates PA, Sternberg MJ (1997) Recognition of analogous and homologous protein folds: analysis of sequence and structure conservation. J Mol Biol 269: 423–439 [DOI] [PubMed] [Google Scholar]
- Sagen JV, Raeder H, Hathout E, Shehadeh N, Gudmundsson K, Baevre H, Abuelo D, Phornphutkul C, Molnes J, Bell GI, Gloyn AL, Hattersley AT, Molven A, Sovik O, Njolstad PR (2004) Permanent neonatal diabetes due to mutations in KCNJ11 encoding Kir6.2: patient characteristics and initial response to sulfonylurea therapy. Diabetes 53: 2713–2718 [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 234: 779–815 [DOI] [PubMed] [Google Scholar]
- Schulze D, Krauter T, Fritzenschaft H, Soom M, Baukrowitz T (2003) Phosphatidylinositol 4,5-bisphosphate (PIP2) modulation of ATP and pH sensitivity in Kir channels. A tale of an active and a silent PIP2 site in the N terminus. J Biol Chem 278: 10500–10505 [DOI] [PubMed] [Google Scholar]
- Seino S, Miki T (2003) Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol 81: 133–176 [DOI] [PubMed] [Google Scholar]
- Shyng S, Ferrigni T, Nichols CG. (1997) Control of rectification and gating of cloned KATP channels by the Kir6.2 subunit. J Gen Physiol 110: 141–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Cukras CA, Harwood J, Nichols CG (2000) Structural determinants of PIP2 regulation of inward rectifier KATP channels. J Gen Physiol 116: 599–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruce AE, Standen NB, Stanfield PR (1987) Studies of the unitary properties of adenosine-5′-triphosphate-regulated potassium channels of frog skeletal muscle. J Physiol 382: 213–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Tucker SJ, Ashcroft FM, Proks P, Kioka N, Amachi T, Ueda K (2000) Direct photoaffinity labeling of Kir6.2 by [γ-32P]ATP-[γ]4-azidoanilide. Biochim Biophys Res Commun 272: 316–319 [DOI] [PubMed] [Google Scholar]
- Tanabe K, Tucker SJ, Matsuo M, Proks P, Ashcroft FM, Seino S, Amachi T, Ueda K (1999) Direct photoaffinity labeling of the Kir6.2 subunit of the ATP-sensitive K+ channel by 8-Azido-ATP. J Biol Chem 274: 3931–3933 [DOI] [PubMed] [Google Scholar]
- Trapp S, Haider S, Jones P, Sansom MSP, Ashcroft FM (2003) Identification of residues contributing to the ATP-binding site of Kir6.2. EMBO J 22: 2903–2912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Proks P, Tucker SJ, Ashcroft FM (1998) Molecular analysis of KATP channel gating and implications for channel inhibition by ATP. J Gen Physiol 112: 333–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi T, Lippiat JD, Ashcroft FM, Rutter GA (2004) ATP-dependent interaction of the cytosolic domains of the inwardly rectifying K+ channel Kir6.2 revealed by fluorescence resonance energy transfer. Proc Natl Acad Sci USA 101: 76–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Ashcroft FM (1999) Mapping of the physical interaction between the intracellular domains of an inwardly rectifying potassium channel, Kir6.2. J Biol Chem 274: 33393–33397 [DOI] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Proks P, Trapp S, Ryder TJ, Haug T, Reimann F, Ashcroft FM (1998) Molecular determinants of KATP channel inhibition by ATP. EMBO J 17: 3290–3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM (1997) Truncation of Kir6.2 produces ATP-sensitive K-channels in the absence of the sulphonylurea receptor. Nature 387: 179–183 [DOI] [PubMed] [Google Scholar]
- Vaxillaire M, Populaire C, Busiah K, Cave H, Gloyn AL, Hattersley AT, Czernichow P, Froguel P, Polak M (2004) Kir6.2 mutations are a common cause of permanent neonatal diabetes in a large cohort of French patients. Diabetes 53: 2719–2722 [DOI] [PubMed] [Google Scholar]