

Abstract
We examined various histone modifications across the promoter and the coding regions of constitutively active hepatic genes in G0/G1-enriched, mitotically arrested and α-amanitin-blocked cells. Gene activation correlated with localized histone hyperacetylation, H3-K4 tri- or dimethylation and H3-K79 dimethylation and localized nucleosome remodeling at the promoter and the 5′ portion of the coding regions. Nucleosomes at more downstream locations were monomethylated at H3-K4. CBP, PCAF, Brg-1, SNF2H and FACT were recruited to the coding regions in a gene-specific manner, in a similarly restricted promoter-proximal pattern. Elongator, however, associated with the more downstream regions. While all factors were dissociated from the chromatin after transcriptional inactivation by α-amanitin, the histone modifications remained stable. In mitotic cells, histone modifications on parental nucleosomes were preserved and were regenerated in a transcription-dependent manner at the newly deposited nucleosomes, as the cells entered the next G1 phase. The findings suggest that histone modifications may function as molecular memory bookmarks for previously active locations of the genome, thus contributing to the maintenance of active chromatin states through cell division.
Keywords: active chromatin, histone modifications, transcriptional memory
Introduction
In multicellular organisms, cell type specification is achieved by the precisely regulated expression of subsets of genes. In each individual cell type, the complex pattern of gene expression is set up during cellular differentiation and is maintained through many cell generations. Dynamic changes in the structure of chromatin at gene regulatory regions have been implicated in the processes of gene repression and activation, as well as in the propagation of epigenetic information (Emerson, 2002; Lachner and Jenuwein, 2002; Fischle et al, 2003). Such structural transitions between repressed and active chromatin states are generated by the localized and, in most cases, concerted actions of histone-tail-modifying and ATP-dependent nucleosome-remodeling activities (Berger, 2002; Narlikar et al, 2002). The strong correlation between the various post-translational modifications of histones and transcriptional activity has led to the proposal of the so-called ‘histone code' hypothesis (Turner, 2000; Jenuwein and Allis, 2001; Turner, 2002). According to this concept, the combinations of different nucleosome modifications constitute a ‘code' recognized by non-histone proteins, which regulate specific chromatin functions. Histone-tail modifications may play a pivotal role in the stabilization of gene expression patterns. For example, K9-methylated histone 3 (H3) produced by Suv39h1 can recruit HP1 proteins to generate a stable repressive structure, which could spread to neighboring nucleosomes via the recruitment of additional Suv39h1-HP1 protein complexes (Bannister et al, 2001; Lachner et al, 2001). In this way, the H3-K9 methylation code has been implicated in both the formation and the epigenetic maintenance of repressive heterochromatin states (Jenuwein, 2001; Grewal and Elgin, 2002).
Active genes are also characterized by specific histone modification marks, including acetylation and H3-K4 methylation, which are known to play crucial roles in the ordered recruitment and the function of preinitiation complex components on promoters during transcriptional activation (Agalioti et al, 2000, 2002; Kouzarides, 2002; Fischle et al, 2003). Following activation, most genes remain active through many cell divisions, thus establishing a given array of stably expressed genes characteristic for a given cell type. However, unlike in the case of heterochromatin silencing, the potential role of histone modifications in the epigenetic maintenance of the active chromatin configuration is poorly understood. If such epigenetic control exists, histone methylation is predicted to serve as a molecular bookmark for active genes, mainly because, in contrast to acetylation, lysine methylation is a very stable and probably irreversible modification. Indeed, recent studies in Saccharomyces cerevisiae have shown that the Set1 histone methyltransferase is preferentially recruited to the 5′ coding regions of transcriptionally active genes, where it catalyzes trimethylation of histone 3 at the lysine 4 residue (H3-K4) (Ng et al, 2003). Hypermethylated H3-K4 within the coding regions persisted for considerable time after transcription had subsided and Set1 had dissociated from the chromatin, suggesting that the modification provides a molecular memory mark of recent transcriptional activity. Because H3-K4 hypermethylation started to decline well within the period of an individual cell cycle, the phenomenon has been termed ‘short-term transcriptional memory' (Ng et al, 2003). In higher eukaryotes, H3-K4 methylation also correlates with transcriptional activity. In contrast to yeast, where dimethylated H3-K4 displays a genome-wide distribution, in mammalian cells both di- and trimethylated H3-K4 peak at the 5′ coding regions of several active genes (Schneider et al, 2004). In addition, significant levels of H3-K4 methylation have been observed on genes within the β-globin locus and the HNF-4 gene during differentiation, before the onset of transcription, implicating this modification in both the establishment and the maintenance of a potentially active chromatin state (Hatzis and Talianidis, 2002; Schneider et al, 2004). Although, a great deal of information has accumulated for the functioning of the ‘histone code' during gene activation, direct evidence for the potential role of H3-K4 methylation or any other histone modification in the epigenetic memory of the active transcription is still missing.
To address this issue, we studied the pattern of a wide range of histone modifications at the promoter and the coding regions of constitutively active hepatic genes. Comparison of the results obtained in G0/G1-enriched, mitotically arrested and α-amanitin-blocked cells revealed a surprisingly stable pattern of H3 and H4 acetylation along with H3-K79 and H3-K4 methylation. We provide evidence for gene-specific and transcription-dependent translocation of histone acetyltransferases and chromatin-remodeling factors into the coding regions, which results in a localized pattern of histone acetylation and structural alterations of nucleosomes downstream of the promoters. The results provide new insights into the mechanism involved in the creation of a wider open chromatin domain around the promoters of active genes and into the role of histone modifications in the propagation of the active chromatin states during cell divisions.
Results
Localized histone modifications at the 5′ portion of active hepatic genes, which persist after transcriptional and mitotic inactivation
In order to investigate the histone modification states along the promoter and coding regions of active genes, we employed a modified chromatin immunoprecipitation (ChIP) assay. After crosslinking of HepG2, or HEK 293 cells, nuclei were prepared and subjected to micrococcal nuclease (MNase) cleavage. The soluble mononucleosome-sized chromatin fragments were used for immunoprecipitations (IPs) with various antibodies, followed by real-time PCR analysis of the resulting fragments with primer sets amplifying the promoter and different locations at the coding regions of the HNF-4, HNF-1 and albumin genes. All the assays were performed with G0/G1-enriched HepG2 cells, where the above genes are constitutively expressed, with HepG2 cells arrested at the G2/M phase by nocodazole treatment, with HepG2 cells treated with the RNA polymerase (pol)-II inhibitor α-amanitin and with HEK 293 cells, which do not express the studied genes.
In order to have comparable results from the different cells and to reveal transcription-dependent differences, all the values were expressed as percent of inputs and as fold increase over a single value obtained with control nucleosomes in G0/G1-enriched cells. The nucleosomes used as controls are located 4–9 kb downstream of the poly-A sites of the studied genes (see Supplementary data). In ChIP experiments studying histone modifications, the average values obtained with the control nucleosomes relative to those obtained with the nonimmune antibody were as follows: 0.95–1.05 in HEK 293 cells, 2.5–2.8 in G0/G1-enriched HepG2 cells, 1.4–1.7 in mitotic HepG2 cells and 2.4–2.8 in α-amanitin-treated cells. The difference between HepG2 and HEK 293 cells reflects different transcription-independent global levels of modifications.
As expected, high levels of H3 and H4 acetylation were observed at the nucleosomes situated at the promoter-transcription start sites (Prom/TSS) of all three genes (Figure 1). With the exception of H4 hyperacetylation at the HNF-4 gene, both H3 and H4 hyperacetylated nucleosomes were detected over a considerable distance (2.5–5 kb) downstream of the promoters. Surprisingly, neither the pattern nor the absolute amounts of hyperacetylated histones changed at these regions after 5 h of α-amanitin treatment, when transcription was completely blocked (Figure 1). Similarly, in cells arrested at the G2/M phase of the cell cycle, the pattern remained the same (lines Mitotic HepG2 in Figure 1). The absolute values in these samples ranged between 50 and 65% of those obtained with G0/G1-enriched HepG2 cells. Taking into consideration a random deposition of new histone octamers during DNA replication, the above findings suggest that histone hyperacetylation at the promoters and the 5′ coding regions of these genes is a stable modification, which is not erased from the parental nucleosomes after a transcriptional block and during mitotic inactivation. Hyperacetylation of histones is clearly dependent on previous transcriptional activity, since only background signals could be observed in HEK 293 cells, which do not express the studied genes (Figure 1).
Figure 1.
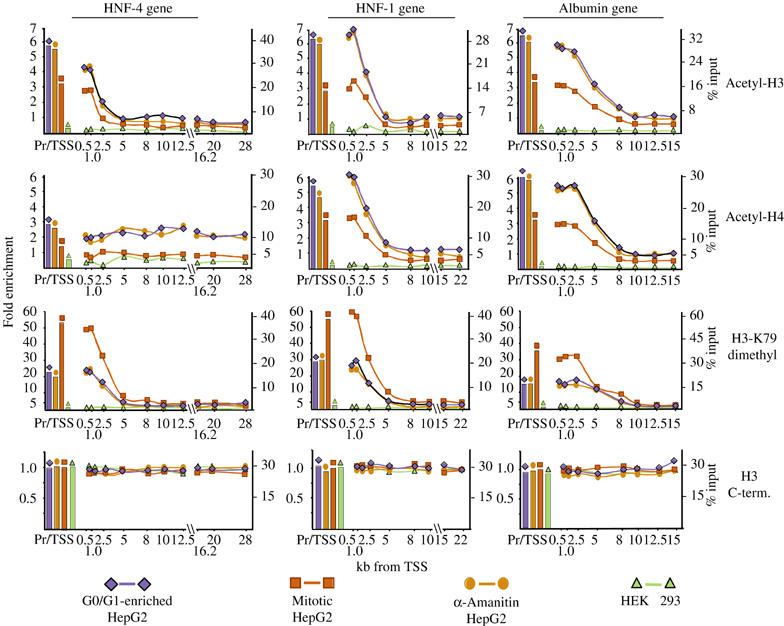
Distribution of acetylated and H3-K79-dimethylated nucleosomes over the human HNF-4, HNF-1 and albumin genes. ChIP assays were performed with G0/G1-enriched HepG2 cells, nocodazole-synchronized HepG2 cells (Mitotic HepG2), HepG2 cells that were treated with 50 μg/ml α-amanitin for 5 h (α-Amanitin HepG2) and HEK 293 cells, using antibodies recognizing K9/K14-diacetylated histone-3 (Acetyl-H3), K5/8/12/16-tetra-acetylated histone-4 (Acetyl-H4) or K79-dimethylated histone-3 (H3-K79 dimethyl), as indicated. The data represent real-time PCR measurements of the immunoprecipitated DNA at the corresponding regions and expressed as percent of inputs (Y-axis at right) and as fold enrichment over the values obtained by the amplification of control nucleosomes in G0/G1-enriched HepG2 cells (Y-axis at left). Pr/TSS corresponds to the nucleosome overlapping the transcription start site and numbers indicate nucleosome positions in kilobases relative to the transcription start site.
A similar pattern was observed for dimethyled H3-K79, another characteristic H3 modification of active genes. In mitotic cells however, the methylated H3-K79 ChIP signal was about twice higher compared to those obtained with G0/G1-enriched cells, suggesting that this modification may also be generated during or after the S phase (Figure 1). Analysis of the different methylated H3-K4 modifications revealed an interesting pattern. Similar to the pattern of modifications reported for the carbonic anhydrase and GAPDH genes (Schneider et al, 2004), we observed high levels of localized tri- and dimethylation at the coding regions of the HNF-4, HNF-1 and albumin genes (Figure 2). However, we detected similar high signals at the promoter/TSS nucleosomes, as well. In sharp contrast to this, monomethylated H3-K4 signals appeared at the nucleosomes where tri- and dimethylation declined, peaking around the middle portion of the genes (Figure 2). In all three cases, the pattern and the absolute values of methylated H3-K4 signals remained stable in α-amanitin-treated cells, while in mitotic cells the same pattern was observed with about half of the absolute values of those obtained with G0/G1-enriched cells. Similar to acetylation, no H3 methylation was detected in HEK 293 cell-derived chromatin in any of the regions studied (Figure 2). In experiments examining H3-K9 and H3-K36 methylation, only background signals were detected in all cells (data not shown).
Figure 2.
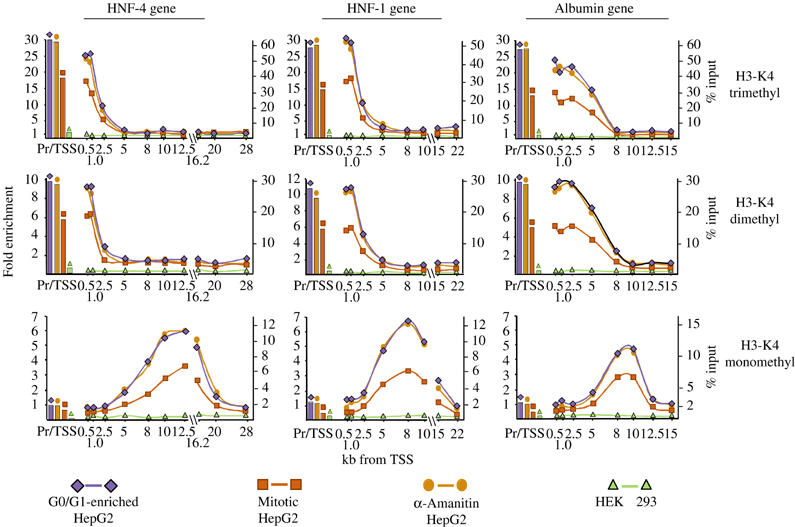
Distribution of the nucleosomes methylated at lysine-4 of histone 3 over the human HNF-4, HNF-1 and albumin genes. ChIP assays were performed using antibodies raised against the various methylated forms of H3-K4 as indicated at right. The results are presented as described in the legend of Figure 1.
The observed difference in absolute values between G0/G1-enriched and mitotic cells suggests that while the modifications remain stable in parental nucleosomes, during DNA replication, newly deposited nucleosomes are not modified by acetylation and H3-K4 methylation, up until the cells enter the next G1 phase and resume transcription of the genes. Further evidence for this was provided by time-course experiments with cells released from the mitotic block. An increase in histone acetylation, H3-K4 mono-, di- and trimethylation and a decrease in K79 dimethylation at the selected regions were observed (Figure 3). Importantly, the extent of the above changes closely correlated with the actual percentage of cells in G1 phase and RNA pol-II occupancy.
Figure 3.
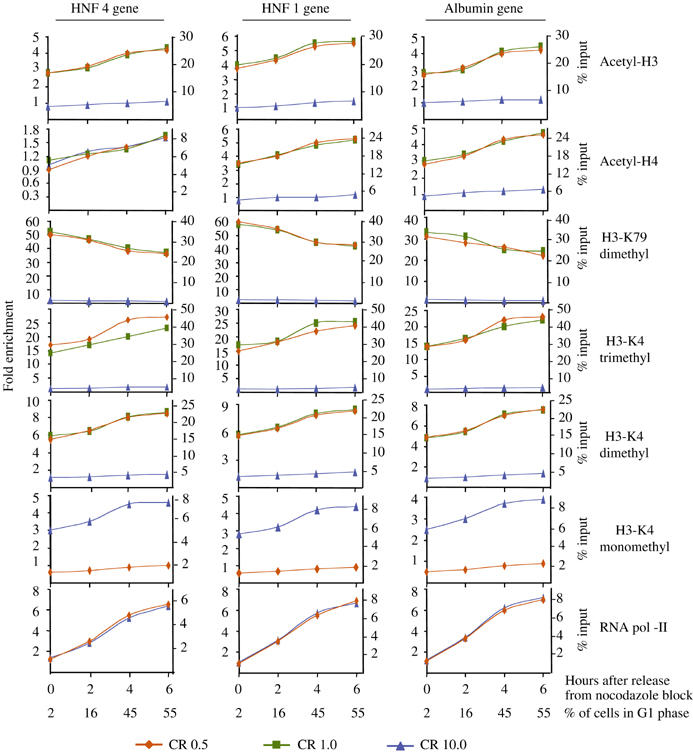
Histone modifications after releasing the cells from mitotic arrest. HepG2 cells were arrested at G2/M phase by treatment with 50 ng/ml nocodazole. Detached cells were collected and washed twice with normal growth medium (DMEM, containing 10% fetal calf serum), replated and collected at the indicated time points. ChIP assays were performed using antibodies indicated at right. The results are presented as described in the legend of Figure 1. Parallel cultures were fixed in ethanol and after staining with propidium iodine subjected to FACS analysis. Numbers at the bottom indicate the percentage of cells with 2N DNA content (% G1 cells) over the total cell population (G1+S+G2/M).
Due to the mononucleosome resolution of our ChIP assays, the detection of tri- and dimethylated H3-K4 in the same regions may be interpreted as both modifications existing on the two individual H3 molecules of the same nucleosome, as allelic variations or as signals emanating from different populations of cells containing either tri- or dimethylated H3-K4 at the corresponding nucleosome. In order to distinguish between these possibilities, we performed sequential chromatin immunoprecipitation (Re-ChIP) assays. In these assays, trimethylated H3-K4-containing nucleosomes were selected in the first IP, and after elution from the protein-G–Sepharose beads, were subjected to a second immunopurification with antibodies against dimethylated H3-K4. As shown in Figure 4A, significant amounts of DNA corresponding to the nucleosomes located at the 1 kb region of all three genes could be detected in the second immunoprecipitates, pointing to the simultaneous presence of both tri- and dimethyl H3-K4 modifications in the same nucleosome. More importantly, similar specific signals were obtained in assays when the second IP was performed with antibodies against acetylated H3 or methylated H3-K79. After normalization of the data for the Re-ChIP efficiency of the individual antibodies and performing IPs in reverse order, we detected substantial recoveries, which were in the range of 71–100% of the inputs, in all combinations of first and second IPs (Supplementary Table 1 and Figure 4A). Taking into account the experimental errors and the double H3-K79 methylation signal observed in mitotic cells, the data suggest that in G0/G1 cells within the same nucleosome one of the histone 3 polypeptide tails is trimethylated, while the other is dimethylated at lysine 4 and at least one of them is dimethylated at lysine 79 and either one or both of them are acetylated.
Figure 4.
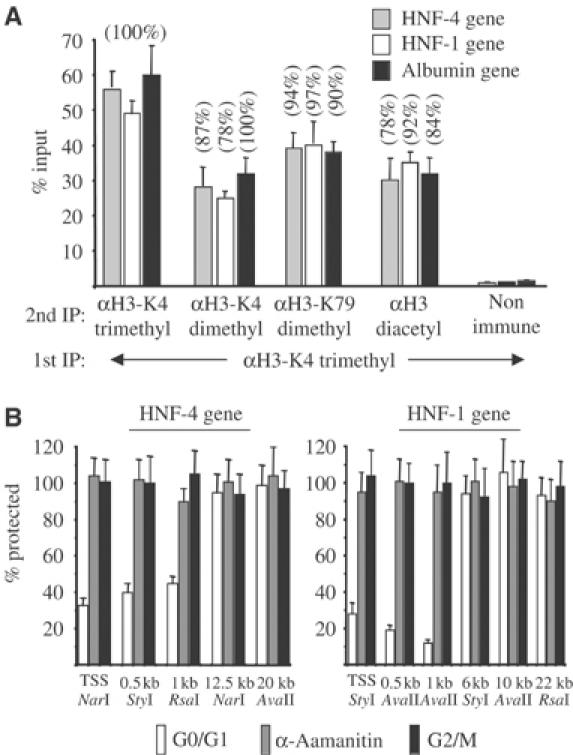
Evidence for the presence of multiple modifications in the same nucleosomes and the presence of an open chromatin domain restricted to the 5′ coding regions of the HNF-1 and HNF-4 genes. (A) Complexes immunoprecipitated with the antibody recognizing trimethylated H3-K4 (1st IP) were eluted from the protein-G–Sepharose beads and were re-immunoprecipitated with the indicated second antibodies (2nd IP). Input values correspond to the nonimmunoprecipitated eluted chromatin. Numbers in parentheses correspond to percent recoveries of the input DNA, after normalization to the Re-ChIP efficiencies of the different antibodies (see Supplementary Table 1). (B) Restriction enzyme hypersensitivity assays with the indicated enzymes were performed using nuclei prepared from G0/G1-enriched HepG2 cells, HepG2 cells that were treated with 50 μg/ml α-amanitin (α-amanitin) and nocodazole-arrested HepG2 cells (G2/M). The bars represent average real-time PCR measurements of the protected DNA segments from three experiments, relative to the values of uncut control samples, which were set at 100%.
Taken together, the results demonstrate that multiple modifications decorate most of the individual nucleosomes at the 5′ coding regions of the genes, which persist after transcriptional inactivation and can also be detected in mitotic cells specifically on the studied genes by ChIP assays (Figures 1 and 2) and globally by indirect immunofluorescence staining (Supplementary Figure 3).
Transcription-dependent localized alteration of chromatin structure at the 5′ coding regions of active genes
Because during gene activation histone acetylation and chromatin remodeling at the promoter regions are usually coupled processes, we investigated the potential structural alterations in nucleosomes located at the hyperacetylated 5′ coding regions compared to those located at the hypoacetylated and hypomethylated further downstream regions. Nucleosomal DNA is normally refractory to cleavage by restriction enzymes, but chromatin remodeling significantly increases the extent of cleavage. Therefore, we used a restriction enzyme accessibility assay to measure chromatin remodeling on selected regions of the HNF-4 and HNF-1 genes in G0/G1, α-amanitin-treated and mitotic HepG2 cells. In G0/G1-enriched HepG2 cells, significant cleavage was observed at the 0.5 and 1 kb regions, but not at the more downstream locations (Figure 4B), pointing to the existence of an extended open chromatin domain downstream of the promoters, which is limited to the 5′ portions of the coding regions. This ‘remodeling' of nucleosomes is clearly transcription dependent, since we could not observe any hypersensitivity for the same enzymes in α-amanitin-treated and mitotic HepG2 cells (Figure 4B), or the nonexpressing HEK 293 cell line (data not shown).
Gene-specific localized recruitment of histone acetyltransferase and chromatin-remodeling activities to the 5′ coding regions of active genes
In order to identify the enzymatic activities responsible for the localized hyperacetylation and open chromatin structure at the 5′ portion of the coding regions of the HNF-4, HNF-1 and albumin genes, we examined the recruitment of proteins possessing histone acetylase activities (CBP, PCAF and the Elp3 component of Elongator), the mammalian homolog of SWI/SNF remodeling complex component (Brg-1) and SNF2H and FACT/cdc68, which have been shown to function in elongation (Narlikar et al, 2002; Santos-Rosa et al, 2003; Saunders et al, 2003), along with RNA pol-II. As expected, none of these factors could be detected at any of the promoter or coding regions in the nonexpressing HEK 293 cells. RNA pol-II was uniformly distributed along the coding regions in G0/G1-enriched HepG2 cells and dissociated from the genes in α-amanitin and mitotic cells (Figure 5). Analysis of histone acetylases produced an unexpected pattern. The RNA pol-II associated histone acetyltransferase Elp3 (Wittschieben et al, 1999; Winkler et al, 2002) associated with the coding regions at positions downstream of 2.5 and 5 kb, where histone acetylation started to decline (Figure 5). The specificity of this distribution was confirmed by the similar pattern obtained with Elp1, another component of the Elongator, and by peptide competition assays (data not shown). On the other hand, CBP and PCAF, which were thought to function solely in PIC formation at gene regulatory regions, were found to associate also with the 5′ portion of the coding regions overlapping with the hyperacetylated segments. Interestingly, their association was gene specific. CBP could be detected only in the HNF-4 and HNF-1 genes, while PCAF could only be detected in the albumin gene (Figure 5). We also observed Brg-1 occupancy at all three genes, and SNF2H at the HNF-1 and albumin genes, in a similar distribution, which was restricted to the promoters and the 5′ portions of the coding regions. FACT/cdc68 occupancy was detected only at the coding region of the HNF-1 gene and was similarly limited to its 5′ segment (Figure 5). All of the above factors dissociated from the chromatin upon α-amanitin-mediated transcriptional repression and during mitosis (Figure 5 and Supplementary Figure 3). The strong correlation between the localized distribution of one or more chromatin-modifying or -remodeling activities and the altered nucleosome structure downstream of the promoters points to a continuous RNA pol-II-dependent modification and reorganization of the chromatin over an extended segment, but not the entirety of the coding regions.
Figure 5.
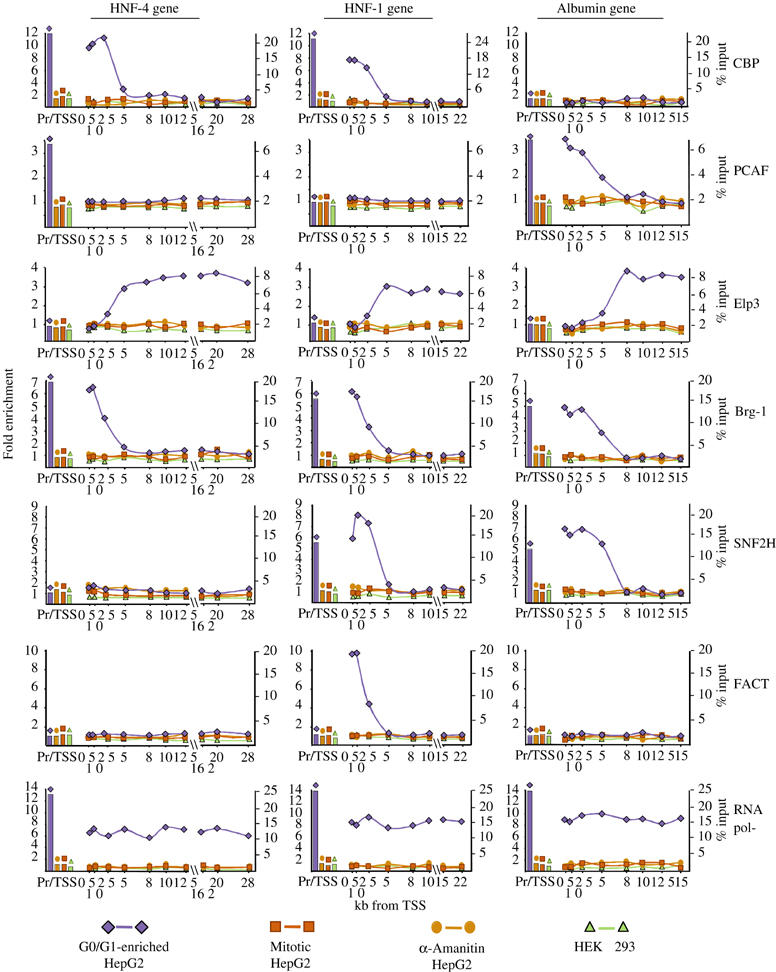
Distribution of histone acetyltransferases and chromatin-remodeling factors over the human HNF-4, HNF-1 and albumin genes. ChIP assays were performed using antibodies raised against the proteins indicated at right. The results are presented as described in the legend of Figure 1.
Using the specific CTD antibodies, Brg-1, CBP and PCAF efficiently co-immunoprecipitated with phosphorylated forms of RNA pol-II, including the Ser-2 phosphorylated, elongating form of the enzyme (Supplementary Figure 2). This provides further evidence for the notion that CBP, PCAF and Brg-1, besides functioning on promoters, are also involved in transcript elongation via association with the elongating RNA pol-II complex.
Potential role of histone-tail modifications in mediating association of chromatin with histone acetylases and chromatin-remodeling complexes
CBP and Brg-1 contain bromodomains, which can bind to acetylated histone tails with high affinity (Agalioti et al, 2002). To test whether the H3-K4 methylation mark may represent additional interaction surfaces for these factors, we performed peptide pull-down assays using HepG2 extracts. As control, we analyzed SNF2H binding, which has been shown to specifically interact with di- and trimethylated H3-K4 peptides (Santos-Rosa et al, 2003). The results presented in Figure 6 show that CBP and Brg-1 can specifically interact with the K9/K14-acetylated H3 peptides, but not with the unmodified or dimethylated H3-K4 peptide. Interestingly however, SNF2H also interacted very efficiently with the K9/K14-acetylated H3 peptides. When the binding of the FACT/cdc68 subunit was tested, we observed an interaction with the unmodified H3 peptide. Although the interaction was somewhat higher with dimethylated H3-K4, in light of the strong preference of FACT for binding to H2A/H2B dimers (Belotserkovskaya et al, 2003), the functional significance of this difference is not clear (Figure 6A). The results are in agreement with the notion that the different histone-tail modifications may stabilize interactions with individual proteins. An alternative role of the individual H3-tail modifications could be that they may create better substrates for the enzymes catalyzing another tail modification. In line with this, we found that the dimethylated H3-K4 peptide was more efficiently acetylated by CBP and PCAF than the unmodified one (Figure 6B). This finding may provide a mechanistic basis for the generation of multiple modifications of the nucleosomes located at the promoter and 5′ coding regions of active genes.
Figure 6.
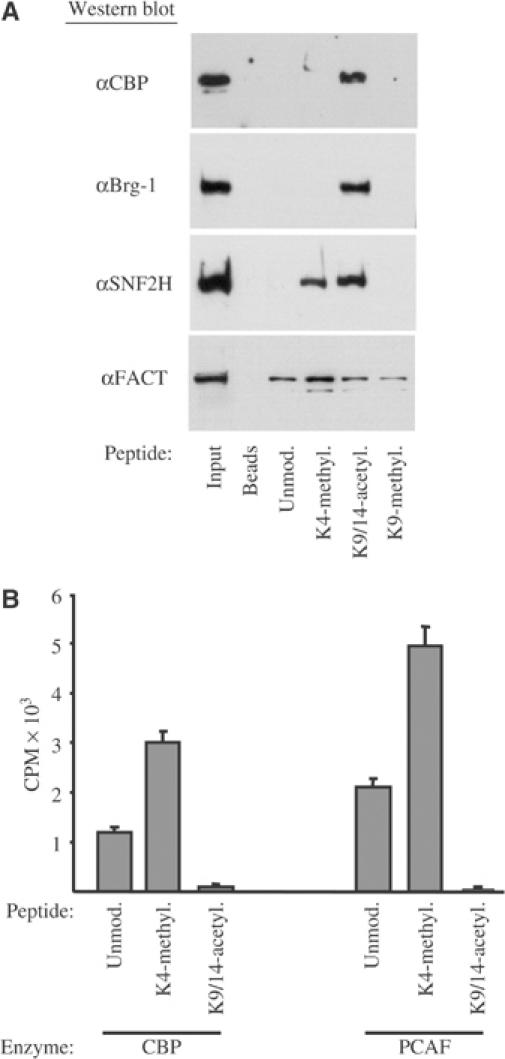
In vitro association and acetylation of modified histone-tail peptides with chromatin-remodeling factors. (A) Biotinylated histone-3-tail peptides with the indicated modifications at the specific lysine residues were immobilized to streptavidin–agarose beads and incubated with HepG2 nuclear extracts. Interacting proteins were identified by Western blot analysis using antibodies against CBP, Brg-1, SNF2H and FACT/cdc68 as indicated. (B) In vitro acetylation assays were performed using 100 ng of baculovirus-expressed recombinant CBP and PCAF and 100 ng of unmodified (Unmod.), K4-dimethylated (K4-methyl.) and K9/K14-diacetylated (K9/14-acetyl.) peptides. The reaction products were separated by 18% SDS–PAGE and the corresponding gel slices were excised and quantified by scintillation counting.
Discussion
Chromatin is dynamically restructured during each mitotic cycle, yet the information governing the expression of a particular array of genes in differentiated cells is faithfully transmitted through many cell divisions. Histone modifications are considered molecular marks for the recruitment of activator and repressor proteins into specific locations of the genome, implicating their involvement in epigenetic memory (Fischle et al, 2003). The molecular events by which specific histone modifications generate and propagate repressive chromatin structure have been comprehended in great detail (Jenuwein, 2001; Kouzarides, 2002). Although the function of various histone modifications in transcriptional activation is well established (Santos-Rosa et al, 2002; Fischle et al, 2003), their possible role in maintaining active chromatin states is less explored.
The results presented in this study demonstrate that modifications, including H3 and H4 acetylation, H3-K4 and H3-K79 methylation, which characterize active chromatin, persist over a long period of time after α-amanitin-mediated transcriptional inactivation and through mitosis on parental nucleosomes, at three differently regulated, constitutively active hepatic genes.
Localized histone modifications and open chromatin domains at the coding regions of active genes
Of particular interest is the observed highly localized pattern of histone modifications over the coding regions of the genes. Histone acetylation, H3-K4 di- and trimethylation and H3-K79 dimethylation were observed at the promoters and at the initial 2.5–5 kb of the coding regions. These segments represent a more open chromatin structure compared to the regions further downstream and coincide with the highly localized recruitment of the histone acetyltransferases CBP or PCAF, and the chromatin-remodeling complexes Brg-1, SNF2H and FACT/cdc68. Association of these factors with the promoter and 5′ portions of the coding regions is transcription dependent, mediated in part by direct or indirect interactions with the initiating and elongating forms of RNA pol-II. Unlike histone modifications, restriction enzyme hypersensitivity of the nucleosomes located at the 5′ coding regions was lost in α-amanitin-treated cells and in mitotic cells, suggesting that the continuous action of RNA pol-II-associated remodeling complexes is required to maintain the open chromatin conformation. In agreement with this scenario is the recent report demonstrating the necessity of an open chromatin structure and Brg-1 occupancy at the coding region of the heat shock-inducible Hsp70 gene for transcriptional elongation (Corey et al, 2003). Consistent with our results, it has been shown that Brg-1 is inactivated during mitosis by phosphorylation and excluded from mitotic chromatin (Muchardt et al, 1996; Sif et al, 1998). On the other hand, previous immunofluorescence studies revealed poor colocalization of Brg-1 with RNA pol-II in interphase cells (Grande et al, 1997; Reyes et al, 1997), suggesting that only a small portion of Brg-1 is involved directly in transcript elongation.
In all three genes we studied, we could observe H3-K4 monomethylation, peaking at the middle parts of the coding regions, downstream of the segments characterized by the other modifications. Although, the functional significance of this modification is not clear, its persistence through mitosis and its transcription-dependent re-establishment in G1-phase cells point to a role in transcript elongation. The components of the Elongator complex were found to associate with the coding regions downstream of the hyperacetylated, H3-K4- and H3-K79-methylated domains. Up until recently, the role of Elongator in transcription has been disputed mainly because of the reported cytoplasmic localization of some of its components (Kim et al, 2002; Pokholok et al, 2002). Using newly developed, highly specific antibodies against Elp3, we observed that it was localized mainly in the nucleus of interphase HepG2 cells (Supplementary Figure 3). This, together with our ChIP results reinforces the evidence provided by a recent study (Gilbert et al, 2004) that Elongator is involved in transcription. Its recruitment was evident from the end of the hyperacetylated segments, which indicates that, in mammalian genes, the acetyltransferase activity of Elp3 may only partially contribute to the localized histone hyperacetylation at the 5′ portion of the coding regions. The unique association of Elongator with the coding regions after the dissociation of CBP, Brg-1, SNF2H and FACT raises the possibility of novel functions of this complex in transcription. Interestingly, the occupancy of the coding regions by SNF2H and FACT was found to be gene specific, pointing to some functional redundancy. It should be noted that, in theory, the lack of FACT and SNF2H, or other ChIP signals on some genes could also be explained by gene-specific epitope masking, or a less efficient crosslinking of the proteins with certain chromatin segments. If this is the case, then the strong signals observed on other genes must reflect a more tight association, a different conformation or a more crowded presence of the proteins with the respective regions. In either case though, the results suggest differential association of these proteins with the studied genes.
Multiplicity of stable nucleosome modifications decorates the active chromatin domains
The unexpected stability of the H3 and H4 acetylation pattern in mitotic and α-amanitin-treated cells where histone acetylases dissociate from the genes points to the lack of histone deacetylase action, probably by regulated exclusion of such activities from these regions. Analysis of global H3 and H4 acetylation levels in cells at different phases of mitosis confirmed the retention of these modifications in mitotic chromatin (Supplementary Figure 3). Notably, hyperacetylation and hypermethylation were segregated to isolated subregions of mitotic chromatin, suggesting that distinct chromatin territories exist for the residence of genes that were previously active and should imminently be reactivated upon the cells exiting mitosis. Acetylated nucleosomes were also modified by di- and trimethylation at H3-K4 and dimethylation at H3-K79, which were also retained in mitotic chromatin and after transcriptional inactivation by α-amanitin. This indicates that the simultaneous presence of multiply modified histones within the same nucleosome is important for the generation of a stable epigenetic mark for active chromatin. According to the histone code concept, the various modifications may be recognized by specific chromatin-associated proteins, which ‘translate' this code to a specific chromatin function (Turner, 2000, 2002; Jenuwein and Allis, 2001; Agalioti et al, 2002). Our sequential ChIP analysis revealed that within the same nucleosome one of the histone 3 polypeptide tails can be trimethylated at lysine 4, while the other can be dimethylated and either of them acetylated and K79-methylated. Such a multiplicity of specific modifications may be required for the stable association of several regulatory proteins possessing different interaction properties. In this regard, we demonstrate that the H3 acetylation mark can provide a specific high-affinity interaction surface for the acetyltransferases, which actually generated it, and for the chromatin-remodeling factors such as Brg-1 and SNF2H, all involved in active transcription. It is less clear, however, whether H3-K4 methylation operates in this manner. Although H3-K4 methylation provides a high-affinity surface for SNF2H binding (Santos-Rosa et al, 2003; Figure 6), the gene-specific involvement of SNF2H in transcription raised the possibility that this modification may also have a different role. At least in vitro, using H3-tail peptides as substrates, we found that dimethylated H3-K4 tails are better substrates for histone acetylases. If such preference occurs in vivo in the nucleosomal context, one may speculate that H3-K4 methylation, besides functioning as a recognition surface for some proteins, could also contribute to the generation of the acetylation code within the same nucleosome. The strict correlation between H3-K4-di- or -trimethylated and H3-K9/K14-diacetylated nucleosomes at the open chromatin domains of active genes provides an additional indication for such a mechanism.
Role of transcription in the generation of histone modification marks
Comparative examination of the absolute values of our ChIP analysis in G0/G1-phase and mitotic cells supports the view that a dynamic reorganization of nucleosomes occurs during the cell cycle. In mitotic cells, the amounts of acetylated and H3-K4-methylated histones at the different nucleosomes were approximately half compared to those in G0/G1-phase samples and gradually increased when the cells were allowed to enter the next G1 phase. This suggests that during DNA replication newly deposited histones are not modified at these sites, while parental histones remain stably modified. In agreement with this is our finding that the generation of the above modifications is transcription dependent. Previous works in yeast, studying histone methyltransferases associated with elongating RNA pol-II, established that the H3-K79 methylase Dot1p and the H3-K4 methylase Set1-containing COMPASS complex require the PAF1 complex for their function, linking histone methylation to transcript elongation (Krogan et al, 2003). It was postulated that H3-K4 and H3-K79 methylation are coupled processes and depend on active transcription. This is in agreement with our results showing that trimethylated H3-K4 nucleosomes were also methylated at K79. On the other hand, we also noticed that the H3-K79 methylation signals were always about twice higher in cells arrested in G2/M compared to G0/G1-enriched cells, indicating that this modification can also be generated independently of transcription during or after the S phase. Such transcription-independent H3-K79 methylation, however, should preferentially occur in chromatin domains defined by the other modifications, because H3-K79 hypermethylation was observed only in nucleosomes situated at the regions in which they were acetylated and K4 di- or trimethylated. If we exclude the possible action of a so far unidentified specific histone lysine demethylase, the above difference also points to a dynamic, transcription-dependent nucleosome exchange as the cells enter the G1 phase. Recent studies have identified that the H3.3 histone variant can be deposited at active loci in a replication-independent manner (Ahmad and Henikoff, 2002; Tagami et al, 2004). Consistent with such preferential targeting is the finding that H3.3 is enriched in histone modifications associated with active chromatin (McKittrick et al, 2004).
Our observations extend current models by proposing that specific histone modifications may represent memory signals involved in the propagation and maintenance of active chromatin states during cell division. According to this model, histone acetylation, H3-K4 di- and trimethylation and H3-K79 methylation marks are generated at specific nucleosomes of active genes during transcription by an interdependent action of histone-modifying enzymes. These modifications are preserved over a long period of time after transcription subsides and thus constitute a mark for a specialized chromatin domain. During DNA replication, as a result of deposition of new histones, part of these modifications is lost, although H3-K79 methylation continues to take place. Because the histone modifications at the parental nucleosomes remain stable through mitosis, they can serve as molecular bookmarks for the assembly of the transcription machinery at specific locations of the genome, as the cells enter the next G1 phase. During transcription, nucleosomes are exchanged by a displacement/replacement mechanism (Belotserkovskaya et al, 2003; Belotserkovskaya and Reinberg, 2004) and the action of promoter- and RNA pol-II-associated enzymes can regenerate the histone modifications. The concurrent action of remodeling complexes then generates an open chromatin domain, which extends several hundred bases downstream of the promoters.
Why do cells generate such highly localized histone modification marks and open chromatin domains at the coding regions of active genes? During elongation, several factors associate with RNA pol-II, destabilizing nucleosome structure and reassembling nucleosomes as they are traversed by the enzyme (Belotserkovskaya and Reinberg, 2004). This extensive remodeling of nucleosomes may facilitate passage of RNA pol-II through chromatin. However, RNA pol-II density and presumably elongation efficiency were not changed at the more downstream coding regions of the genes, pointing to distinct chromatin requirements for initiating and elongating RNA pol-II. The open chromatin environment around the promoter regions may thus have as its principal role the facilitation of the efficient release of new RNA pol-II molecules from the transcription start site during reinitiation, but could be dispensable for RNA pol-II progression through the downstream regions.
Memory of recent transcriptional activity
The histone modification pattern of the genes studied here shows similarities and some fundamental differences compared to active genes in S. cerevisiae. In yeast, only H3-K4 trimethylation is localized to the 5′ coding regions, while H3-K4 dimethylation and acetylation are uniformly distributed (Kristjuhan et al, 2002; Winkler et al, 2002; Ng et al, 2003). In contrast, at the mammalian genes of this study, H3-K4 tri- and dimethylation and also H3-K79 dimethylation and H3 and H4 acetylation were all restricted to the 5′ portions of the genes. In addition, we observed H3-K4 monomethylation at the middle parts of the coding regions, downstream of the segments characterized by the other modifications. In yeast, H3-K4 trimethylation lasts for an extended period of time after transcriptional inactivation, a phenomenon termed as ‘short-term memory' of recent transcription (Ng et al, 2003). At the mammalian genes of this study, H3-K4 methylation, H3-K79 methylation, H3 and H4 acetylation were also stably maintained for a significant period of time after α-amanitin-induced transcriptional block and persisted on the parental nucleosomes through mitosis. These transcriptional memory mechanisms may have several distinct biological functions. They may be important to prevent some active genes, which need to be expressed continuously or need to be rapidly switched on and off by environmental signals, from becoming permanently silenced. In addition, they may also provide a means for the cells to ‘remember' the locations of the genome where transcription must be resumed after the cells exit mitosis, thus contributing to the faithful maintenance of the gene expression pattern that is characteristic to the particular cell type.
Materials and methods
Cell culture and synchronization
HepG2 and HEK 293 cells were grown in Dulbecco's modified Eagle's medium, supplemented with 10% fetal calf serum. The cells were arrested at the G2/M phase by treatment with 50 ng/ml nocodazole for 16 h. Detached cells were collected by shaking and processed for further analysis. HepG2 cells were used at the time the plates became confluent to enrich for the G0/G1-phase population. FACS analysis confirmed the presence of more than 95% mitotic cells (4N DNA content) in the nocodazole-treated cultures and about 85% of G0/G1-phase cells (2N DNA content) in the confluent cultures. In certain experiments, confluent HepG2 cells were treated with 50 μg/ml α-amanitin for 5 h, before collection for analysis.
Preparation of mononucleosomes and chromatin immunoprecipitation
To crosslink chromatin, the cells were treated with 1% formaldehyde for 10 min at room temperature. Crosslinking was stopped by the addition of glycine to a final concentration of 125 mM. The cells were washed with PBS and nuclei were prepared by resuspension in buffer A, containing 0.32 M sucrose, 15 mM Hepes pH 7.9, 60 mM KCl, 2 mM EDTA, 0.5 mM EGTA, 0.5% BSA, 0.5 mM spermidine, 0.15 mM spermine and 0.5 mM DTT, followed by dounce homogenization. The nuclear suspension was layered over an equal volume of buffer B, containing 30% sucrose, 15 mM Hepes pH 7.9, 60 mM KCl, 2 mM EDTA, 0.5 mM EGTA, 0.5 mM spermidine, 0.15 mM spermine and 0.5 mM DTT and centrifuged for 15 min at 3000 r.p.m. Purified nuclei were collected from the bottom of the gradient, washed with buffer N and resuspended in buffer N, containing 0.34 mM sucrose, 15 mM Hepes pH 7.5, 60 mM KCl, 15 mM NaCl, 0.5 mM spermidine, 0.15 mM spermine and 0.15 mM β-mercaptoethanol at a 2 × 106 nuclei/ml concentration. The samples were adjusted to 3 mM CaCl2, and incubated with 30 U of micrococcal nuclease (MNase, Worthington) for 5 min at 37°C. The reactions were stopped by the addition of an equal volume of 2 × sonication buffer (90 mM Hepes pH 7.9, 220 mM NaCl, 10 mM EDTA, 2% Triton X-100, 0.2% Na-deoxycholate, 0.2% SDS, 0.5 mM PMSF, 2 μg/ml aprotinin and protease inhibitor cocktail from Roche) and, after centrifugation at 14 000 r.p.m. for 15 min, the soluble chromatin was precleared by incubation with protein-G–Sepharose beads and subjected to IP as described previously (Hatzis and Talianidis, 2002; Soutoglou and Talianidis, 2002). In each experiment, aliquots of chromatin were de-crosslinked and, after purification, the DNA was analyzed by agarose gel electrophoresis to confirm the complete digestion of chromatin to mononucleosome-sized DNA fragments. The intact nature of the nucleosomes at the different locations of the genes was confirmed by performing IP and PCR analysis with a C-terminal epitope anti-histone 3 antibody (Abcam), which gave uniform signals all over the analyzed regions (Figure 1). The nucleosome-sized resolution of the assay was confirmed by agarose gel electrophoresis of the purified DNA fragments (Supplementary Figure 1).
In Re-ChIP assays, after washing the protein-G–Sepharose beads from the primary IP, the complexes were eluted from the beads by incubation with 10 mM DTT at 37°C for 30 min. After a 50-fold dilution with 1 × sonication buffer, the eluates were subjected to IP with the second antibody. After purification, the immunoprecipitated DNA was analyzed by quantitative real-time PCR using SYBR green on MJ-Research Opticon Engine. All experimental values were normalized to those obtained with a nonimmune serum, and divided by the input. The data in the figures are presented as fold change over the signal obtained with amplifications of control nucleosomes located outside the coding regions. The antibodies and the positions of the primer sets used in this study are listed in Supplementary data.
Restriction enzyme hypersensitivity assays
Nuclei were purified as described above and resuspended in the appropriate 1 × restriction enzyme buffer at a concentration of 2 × 106 nuclei/ml. After addition of 5–20 U of the corresponding restriction enzyme, the samples were incubated at 37°C for 30 min. Reactions were stopped by the addition of an equal volume of 2 × stop buffer, containing 20 mM Tris pH 7.5, 200 mM NaCl, 8 mM EDTA and 2% SDS, followed by incubation with 0.25 mg/ml proteinase K at 42°C for 2 h. The DNA was purified by extractions with phenol/chloroform and ethanol precipitation. The samples were then analyzed by quantitative real-time PCR. The experimental values were compared to at least three control amplifications performed on mock-digested samples, and samples digested with restriction enzymes, which do not cut the corresponding segment. The data obtained with the controls were averaged and set as 100% protected.
Protein interaction and histone acetyltransferase assays
Purification of baculovirus-expressed recombinant CBP and PCAF proteins and in vitro histone acetyltransferase assays were performed as described by Soutoglou et al (2000). Preparation of nuclear extracts from HepG2 cells and co-immunoprecipitations experiments were performed essentially as described previously (Ktistaki and Talianidis, 1997; Hatzis and Talianidis, 2001). For peptide pull-down experiments, 2 μg of synthetic biotinylated histone-3-tail peptides (1–20 amino acids; Upstate Biotechnology) were coupled to streptavidin–agarose magnetic beads (Roche) and incubated with the nuclear extracts, which were precleared by incubation with plain beads. The incubation was performed in a buffer containing 25 mM Hepes pH 7.9, 0.75 mM MgCl2, 400 mM KCl, 0.1 mM EDTA, 0.5% NP-40, 0.5 mM PMSF, 2 μg/ml aprotinin, 1 mM DTT and 10 mM NaF for 5 h at 4°C. After extensive washing with the same buffer, the interacting proteins were eluted by boiling the beads in SDS sample buffer and detected by Western blot analysis.
Supplementary Material
Supplementary Information
Acknowledgments
We thank J Svejstrup for providing the Elp3 and Elp1 antibodies, N Katrakili for technical assistance and P Hatzis for critical reading of the manuscript. This work was supported by grants from HFSP (RGP-0024) and from EU (HPRN-CT-2000-00087, QLRT-2000-01513 and LSHG-CT-2004-502950).
References
- Agalioti T, Chen G, Thanos D (2002) Deciphering the transcriptional histone acetylation code for a human gene. Cell 111: 381–392 [DOI] [PubMed] [Google Scholar]
- Agalioti T, Lomvardas S, Parekh B, Yie J, Maniatis T, Thanos D (2000) Ordered recruitment of chromatin modifying and general transcription factors to the IFN-beta promoter. Cell 103: 667–678 [DOI] [PubMed] [Google Scholar]
- Ahmad K, Henikoff S (2002) The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol Cell 9: 1191–1200 [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410: 120–124 [DOI] [PubMed] [Google Scholar]
- Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D (2003) FACT facilitates transcription-dependent nucleosome alteration. Science 301: 1090–1093 [DOI] [PubMed] [Google Scholar]
- Belotserkovskaya R, Reinberg D (2004) Facts about FACT and transcript elongation through chromatin. Curr Opin Genet Dev 14: 139–146 [DOI] [PubMed] [Google Scholar]
- Berger SL (2002) Histone modifications in transcriptional regulation. Curr Opin Genet Dev 12: 142–148 [DOI] [PubMed] [Google Scholar]
- Corey LL, Weirich CS, Benjamin IJ, Kingston RE (2003) Localized recruitment of a chromatin-remodeling activity by an activator in vivo drives transcriptional elongation. Genes Dev 17: 1392–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emerson BM (2002) Specificity of gene regulation. Cell 109: 267–270 [DOI] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Allis CD (2003) Histone and chromatin cross-talk. Curr Opin Cell Biol 15: 172–183 [DOI] [PubMed] [Google Scholar]
- Gilbert C, Kristjuhan A, Winkler G, Svejstrup JQ (2004) Elongator interactions with nascent mRNA revealed by RNA immunoprecipitation. Mol Cell 14: 457–464 [DOI] [PubMed] [Google Scholar]
- Grande MA, Van der Kraan I, De Jong L, Van Driel R (1997) Nuclear distribution of transcription factors in relation to sites of transcription and RNA polymerase II. J Cell Sci 110: 1781–1791 [DOI] [PubMed] [Google Scholar]
- Grewal SI, Elgin SC (2002) Heterochromatin: new possibilities for the inheritance of structure. Curr Opin Genet Dev 12: 178–187 [DOI] [PubMed] [Google Scholar]
- Hatzis P, Talianidis I (2001) Regulatory mechanisms controlling human hepatocyte nuclear factor 4alpha gene expression. Mol Cell Biol 21: 7320–7330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzis P, Talianidis I (2002) Dynamics of enhancer–promoter communication during differentiation-induced gene activation. Mol Cell 10: 1467–1477 [DOI] [PubMed] [Google Scholar]
- Jenuwein T (2001) Re-SET-ting heterochromatin by histone methyltransferases. Trends Cell Biol 11: 266–273 [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074–1080 [DOI] [PubMed] [Google Scholar]
- Kim JH, Lane WS, Reinberg D (2002) Human Elongator facilitates RNA polymerase II transcription through chromatin. Proc Natl Acad Sci USA 99: 1241–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T (2002) Histone methylation in transcriptional control. Curr Opin Genet Dev 12: 198–209 [DOI] [PubMed] [Google Scholar]
- Kristjuhan A, Walker J, Suka N, Grunstein M, Roberts D, Cairns BR, Svejstrup JQ (2002) Transcriptional inhibition of genes with severe histone h3 hypoacetylation in the coding region. Mol Cell 10: 925–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Dover J, Wood A, Schneider J, Heidt J, Boateng MA, Dean K, Ryan OW, Golshani A, Johnston M, Greenblatt JF, Shilatifard A (2003) The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: linking transcriptional elongation to histone methylation. Mol Cell 11: 721–729 [DOI] [PubMed] [Google Scholar]
- Ktistaki E, Talianidis I (1997) Modulation of hepatic gene expression by hepatocyte nuclear factor 1. Science 277: 109–112 [DOI] [PubMed] [Google Scholar]
- Lachner M, Jenuwein T (2002) The many faces of histone lysine methylation. Curr Opin Cell Biol 14: 286–298 [DOI] [PubMed] [Google Scholar]
- Lachner M, O'Carroll D, Rea S, Mechtler K, Jenuwein T (2001) Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410: 116–120 [DOI] [PubMed] [Google Scholar]
- McKittrick E, Gafken PR, Ahmad K, Henikoff S (2004) Histone H3.3 is enriched in covalent modifications associated with active chromatin. Proc Natl Acad Sci USA 101: 1525–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchardt C, Reyes JC, Bourachot B, Legouy E, Yaniv M (1996) The hbrm and BRG-1 proteins, components of the human SNF/SWI complex, are phosphorylated and excluded from the condensed chromosomes during mitosis. EMBO J 15: 3394–3402 [PMC free article] [PubMed] [Google Scholar]
- Narlikar GJ, Fan HY, Kingston RE (2002) Cooperation between complexes that regulate chromatin structure and transcription. Cell 108: 475–487 [DOI] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K (2003) Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell 11: 709–719 [DOI] [PubMed] [Google Scholar]
- Pokholok DK, Hannett NM, Young RA (2002) Exchange of RNA polymerase II initiation and elongation factors during gene expression in vivo. Mol Cell 9: 799–809 [DOI] [PubMed] [Google Scholar]
- Reyes JC, Muchardt C, Yaniv M (1997) Components of the human SWI/SNF complex are enriched in active chromatin and are associated with the nuclear matrix. J Cell Biol 137: 263–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419: 407–411 [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bernstein BE, Karabetsou N, Morillon A, Weise C, Schreiber SL, Mellor J, Kouzarides T 2003. Methylation of histone H3 K4 mediates association of the Isw1p ATPase with chromatin. Mol Cell 12: 1325–1332 [DOI] [PubMed] [Google Scholar]
- Saunders A, Werner J, Andrulis ED, Nakayama T, Hirose S, Reinberg D, Lis JT (2003) Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science 301: 1094–1096 [DOI] [PubMed] [Google Scholar]
- Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T (2004) Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol 6: 73–77 [DOI] [PubMed] [Google Scholar]
- Sif S, Stukenberg PT, Kirchner MW, Kingston RE (1998) Mitotic inactivation of human SWI/SNF chromatin remodeling complex. Genes Dev 12: 2842–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soutoglou E, Katrakili N, Talianidis I (2000) Acetylation regulates transcription factor activity at multiple levels. Mol Cell 5: 745–751 [DOI] [PubMed] [Google Scholar]
- Soutoglou E, Talianidis I (2002) Coordination of PIC assembly and chromatin remodeling during differentiation-induced gene activation. Science 295: 1901–1904 [DOI] [PubMed] [Google Scholar]
- Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y (2004) Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell 116: 51–61 [DOI] [PubMed] [Google Scholar]
- Turner BM (2000) Histone acetylation and an epigenetic code. BioEssays 22: 836–845 [DOI] [PubMed] [Google Scholar]
- Turner BM (2002) Cellular memory and the histone code. Cell 111: 285–291 [DOI] [PubMed] [Google Scholar]
- Winkler GS, Kristjuhan A, Erdjument-Bromage H, Tempst P, Svejstrup JQ (2002) Elongator is a histone H3 and H4 acetyltransferase important for normal histone acetylation levels in vivo. Proc Natl Acad Sci USA 99: 3517–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittschieben BO, Otero G, de Bizemont T, Fellows J, Erdjument-Bromage H, Ohba R, Li Y, Allis CD, Tempst P, Svejstrup JQ (1999) A novel histone acetyltransferase is an integral subunit of elongating RNA polymerase II holoenzyme. Mol Cell 4: 123–128 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information