

Abstract
Gut microbes of animals play critical roles in processes such as digestion and immunity. Therefore, identifying gut microbes will shed light on understanding the annual life of animal species, particularly those that are threatened or endangered. In the present study, we conducted nucleotide sequence analyses of the 16S rRNA genes of gut microbiome of the hooded cranes (Grus monacha) wintering at Shengjin Lake, China, by Illumina high‐throughput sequencing technology. We acquired 503,398 high‐quality sequences and 785 operational taxonomic units (OTUs) from 15 fecal samples from different cranes, representing 22 phyla that were dominated by Firmicutes, Proteobacteria, and Actinobacteria. A total of 305 genera were identified that were dominated by Clostridium, Lysinibacillus, and Enterobacter. The core gut microbiome comprised 26 genera, including many probiotic species such as Clostridium, Bacillus, Cellulosilyticum, and Cellulomonas that could catabolize cellulose. The findings reported here contribute to our knowledge of the microbiology of hooded cranes and will likely advance efforts to protect waterbirds that inhabit Shengjin Lake Reserve during winter.
Keywords: 16S rRNA gene, gut microbiome, Illumina high‐throughput sequencing, wintering hooded cranes
1. INTRODUCTION
Gut microbes play a crucial role in the physiology of their vertebrate hosts by contributing to functions such as the development of intestinal morphology, nutrition, and immunity (Leser & Mølbak, 2009; Young, 2012). Studies of the gut microbiota of avian species have focused on the effects of specific bacteria or bacterial pathogens (Collins et al., 2002; Marois et al., 2002). Recent studies of the gut microbial communities of birds are commonplace and typically employ high‐throughput sequencing technologies and bioinformatics tools to identify microbial species and their activities. An increasing number of studies have characterized the gastrointestinal microbiota of avian species, including turkeys (Meleagris gallopavo) and the kakapo (Strigops habroptilus), showing that the gut microbes of birds play an important role in digestive physiology and immunity (Lu & Domingo, 2008; Waite et al., 2012).
Numerous studies show that the genes of parasitifer as well as environmental factors alter the composition of intestinal microflora (Stanley et al., 2013). The health of the parasitifer depends on microbial metabolism, and the microbial composition of the gut depends on the dietary habits of the parasitifer. The gastrointestinal microbiota of the parasitifer may reflect their particular lifestyles. Links between the structure of microbial communities and the diet of the parasitifer have been demonstrated in humans and animals; the intestinal microflora varies among humans who engage in different lifestyles (Zhang et al., 2015). A study of the giant panda found that the intestinal microbial composition is determined by its feeding habits, which includes large numbers of clostridial species that degrade cellulose (Zhu et al., 2011). Similarly, the gastrointestinal microbiota of avian species may reflect a particular lifestyle. For example, the hoatzin (Opisthocomus hoazin) feeds mostly on leaves and carries out foregut fermentation; thus, the foregut microbiota is dominated by Bacteroidetes, Firmicutes, and Proteobacteria, similar to the gut microbiota of a bovine ruminant (Domínguez‐Bello et al., 1994; Garcia‐Amado et al., 2003; Godoyvitorino et al., 2012).
Despite the presence of numerous probiotics within the avian intestinal microflora that are associated with the avian diet, numerous studies found that potential pathogens are present as well. Identification of the latter is likely of great value to contribute a better understanding of avian health, particularly for the benefit of basic research and the conservation of endangered species. For example, the identification of the intestinal microbial composition of the kakapo detects pathogens before their numbers expand to levels that induce disease (Waite et al., 2012). Moreover, studies of the intestinal microflora of waterfowl, such as gulls (Larus audouinii), black‐winged stilts (Himantopus himantopus) (Camarda et al., 2006; Grond et al., 2014; Santos et al., 2012), identified numerous potential human pathogens.
The hooded crane (Grus monacha) is a large migratory water bird that breeds in the eastern part of Siberia and China's Lesser Khingan mountains, and winters in Japan, South Korea, the middle and lower reaches of the Yangtze River in China (Jiao et al., 2014). The hooded crane was designated a vulnerable species in the IUCN (International Union for Conservation of Nature and Natural Resources) Red List and was classified as a national grade 1 protected bird in China, and its global population is approximately 11,600 (Bird Life International, 2012). Recently, due to many human activities, such as lakes aquaculture, tourism development, and water conservancy construction, the existing wetland aquatic resources, a number of floating‐leaved plants, submerged plants were destroyed, and a sharp decline in the number of benthic fauna was found (Chen et al., 2011). Hooded crane wintering foraging habitat has been destroyed, the main food resources of plants such as Vallisneria natans and Potamogeton malaianus for wintering populations are declining (Fox et al., 2011; Liu et al., 2001; Xu et al., 2008), increasing the survival pressure of wintering hooded cranes. Because of this, researchers are paying increased attention to the demographic and behavioral ecology of migrating birds, particularly to changes in their food supplies (Chen et al., 2011; Zhao et al., 2013; Zhou et al., 2010). However, the gut microbiome of hooded cranes is unknown. Because the gut microbiome serves as an important indicator of lifestyle and health, it is important to characterize the gut microbiome of hooded cranes to contribute to efforts to conserve this endangered species. Therefore, we use multiplex pyrosequencing to identify the species that populate the gut microbiome of wintering hooded cranes in this study.
2. MATERIALS AND METHODS
2.1. Fecal sample collection
We used noninvasive techniques (Darimont et al., 2008) to collect 15 fecal samples from hooded cranes wintering at Shengjin Lake (south bank of the Yangtze River, Hefei, China; 30.25°–30.50°N, 116.92°–117.25°E) during the wintering period, and make sure that the fecal samples were only from the cranes. Before the samples were collected, we used binoculars or telescopes to observe the foraging areas of the cranes, select large groups with more than 50 individuals, and insure the absence of other cranes or geese within the range of about 50 m in the foraging areas of the cranes. After the cranes vacated their foraging grounds, samples were rapidly collected into sterile 50 ml centrifuge tubes. It is identified as the foraging areas of the cranes according to the footprints or foraging pits left by the cranes. To minimize possible contamination from the ground, we only collected the upper layer of a fecal ball. The fecal samples were transported to the laboratory on ice and stored at −80°C.
2.2. DNA extraction and PCR amplification
DNA was extracted from fecal samples according to the manufacturer's instructions by the QIAamp Fast DNA Stool Mini Kit (Qiagen, Germany). We conducted tag‐pyrosequencing analysis of the V3–V4 region of 16S rRNA gene to identify intestinal bacteria. We amplified this region using the broadly conserved primers, 338F (5′‐GGACTACHVGGGTWTCTAAT‐3′) and 806R (5′‐GGACTACHVGGGTWTCTAAT‐3′) (Dennis et al., 2013), containing the A and B sequencing adaptors. Different barcode sequences were used to tag these primers to analyze multiple samples. Each sample reaction mixture (20 μl) contained 0.5 μl of 5 U/μl Easy Taq DNA polymerase, 2 μl of 10 × Easy Taq buffer, 2 μl of 0.25 mmol/L dNTPs, 0.2 μmol/L of each primer, 10 ng of template DNA, and deionized ultrapure water (to 20 μl). An Applied Biosystems GeneAmp PCR System 9700 was used to amplify the DNA samples as follows: initial denaturation at 94°C (3 min) followed by 27 cycles at 95°C (30 s), 55°C (30 s), and 72°C (45 s), and final extension at 72°C for 10 min. We used a 2% (w/v) Tris‐Boric acid‐EDTA(TBE) agarose gel to assess the quality of the amplicons.
2.3. Illumina MiSeq sequencing
The amplicons were purified using a MiniElute PCR purification kit (Axygen) and quantified using the Applied Biosystems GeneAmp PCR System 9700. The PCR products were pooled at equal concentrations and pyrosequencing was performed using an Illumina Miseq System at Majorbio Bio‐pharm Technology Co., Ltd. (Shanghai, China). NCBI SRA database accession numbers are SAMN05207073, SAMN05207074, SAMN05207075, SAMN05207076, SAMN05207077, SAMN05207078, SAMN05207079, SAMN05211008, SAMN05211009, SAMN05211011, SAMN05211012, SAMN05211013, SAMN05211014, SAMN05211015, and SAMN05211016.
2.4. Data processing and analysis
All sequences acquired using the Illumina–MiSeq were saved in the raw fastq files. Initial processing of the raw dataset included screening to remove short and low‐quality reads; only high‐quality sequences without primer sequences were retained. Using USEARCH (version 7.1 http://drive5.com/uparse/), these high‐quality sequences were clustered into operational taxonomic units (OTUs) at a 97% identity threshold. The taxonomic classification of OTUs was performed using the Ribosomal Database Project Classifier with the QIIME bioinformatics pipeline (http://qiime.org/scripts/assign_taxonomy.html) to determine the community composition of each sample at each taxonomic level (Wang et al., 2007) at a 70% confidence level. Rarefaction curves and alpha diversity calculations were based on OTUs with >97% identity. Rarefaction analysis and alpha‐diversity indices (abundance‐based coverage estimation (ACE), Chao1, Shannon and Simpson) were calculated using Mothur. R language tools were used to generate rarefaction curves (Amato et al., 2013) and Shannon–Wiener curves (Wang et al., 2012). Heatmaps were generated using the R package (Jami et al., 2013). The core fecal microbiome of the hooded cranes was assigned if it comprised >90% of samples and represented ≤0.1% of the reads.
3. RESULTS
3.1. Barcoded 16S pyrosequencing
We acquired 503,398 valid reads from the 15 fecal samples, and among them, 96.5% (average) were classified as bacterial phyla, 3.5% could not be classified, and >90% were assigned to the levels of phylum, class, and order. The average percentages of reads assigned to bacteria at the family, genus, and species levels were 82.8%, 78.7%, and 20.8%, respectively, and 22 phyla and 305 genera were identified from the 15 fecal samples.
Alpha diversity curves of 15 samples determined using multiplex pyrosequencing with OTUs >97% identity. The rarefaction curves of each sample are shown in Figure 1 and the Shannon–Wiener curves of each sample are shown in Figure 2. The rarefaction curves for the 15 samples plateaued at 20,000 reads, indicating that the composition of the fecal microbiota was similar to that measured in this study. Table 1 indicates the diversity index of each sample. Reads ranged from 25,292 to 48,132, which accounted for the number of OTUs, ACE, Chao1, Shannon–Wiener index, and Simpson diversity index. The number of OTUs detected per sample ranged from 166 to 514 using a cutoff of 97% identity for species‐level distinctions. Furthermore, the Shannon–Wiener Index reflected the trends of detected and estimated richness, for example, diversities of samples D and H were highest and lowest, respectively.
Figure 1.
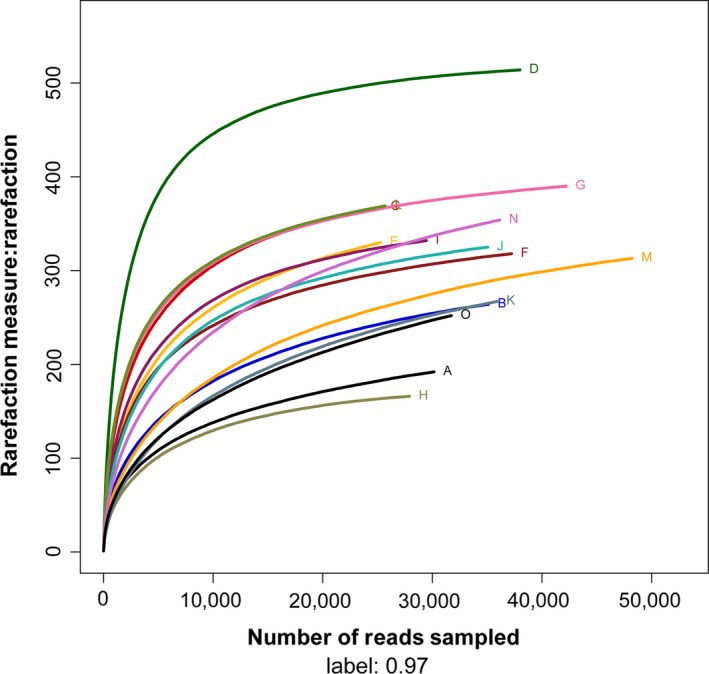
The rarefaction curves of 15 samples
Figure 2.
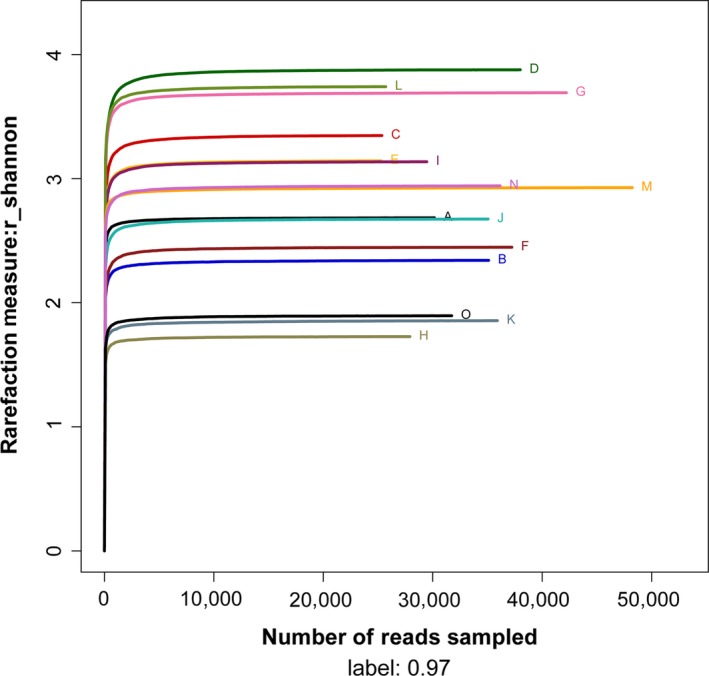
The Shannon–Wiener curves of 15 samples
Table 1.
Diversity and richness of the gut microbial communities of the wintering hooded cranes
| Samples | Reads | OTUs | ACE | Chao1 | Coverage | Shannon | Simpson |
|---|---|---|---|---|---|---|---|
| A | 30,152 | 192 | 247 | 243 | .998275 | 2.68 | .1279 |
| B | 35,109 | 264 | 313 | 301 | .998348 | 2.34 | .2159 |
| C | 25,356 | 368 | 404 | 401 | .997831 | 3.35 | .1151 |
| D | 37,999 | 514 | 525 | 523 | .999289 | 3.88 | .0994 |
| E | 25,279 | 330 | 384 | 415 | .997112 | 3.14 | .1002 |
| F | 37,227 | 318 | 351 | 364 | .998630 | 2.45 | .2432 |
| G | 42,212 | 390 | 411 | 417 | .999029 | 3.69 | .0662 |
| H | 27,913 | 166 | 181 | 176 | .999104 | 1.73 | .3894 |
| I | 29,452 | 332 | 359 | 365 | .998370 | 3.14 | .112 |
| J | 35,053 | 325 | 358 | 368 | .998459 | 2.67 | .2044 |
| K | 35,904 | 267 | 346 | 336 | .997772 | 1.86 | .3461 |
| L | 25,703 | 369 | 404 | 414 | .997821 | 3.74 | .058 |
| M | 48,235 | 313 | 378 | 374 | .998404 | 2.93 | .0886 |
| N | 36,148 | 354 | 430 | 452 | .997483 | 2.94 | .1133 |
| O | 31,728 | 252 | 441 | 360 | .997163 | 1.9 | .3181 |
OTUs, operational taxonomic units.
3.2. Microbial composition of fecal samples
We identified 22 different bacterial phyla in ≥1 sample. Figure 3 shows the relative abundance of the gut microbiome of the 15 samples. Five phyla were present in all samples as follows: Firmicutes (average 44%), Proteobacteria (average 20.4%), Actinobacteria (average 14.4%), Cyanobacteria (average 13.3%), and Fusobacteria (average 1.4%). These phyla were abundant in most samples, which represented an average of 93.5% of the microbial communities. Firmicutes were dominant in most samples.
Figure 3.
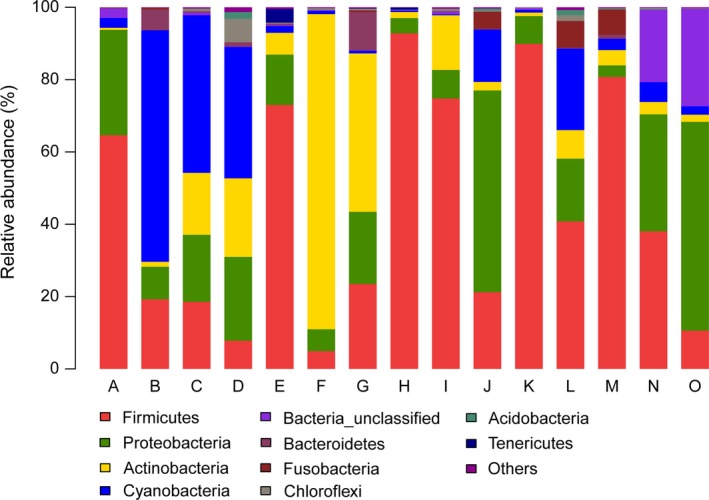
Distribution of phyla among 15 hooded crane fecal samples
We identified members of 188 families in at least one sample, and 27 families were detected in all samples. The relative distributions of the 10 most abundant core families were as follows: Peptostreptococcaceae (average 13.8%), Enterobacteriaceae (average 11%), Clostridiaceae (average 9.8%), Planococcaceae (average 6.6%), Micrococcaceae (average 4.8%), Microbacteriaceae (average 4%), Lactobacillaceae (average 3.4%), Veillonellaceae (average 2%), Paenibacillaceae (average 1.7%), and Ruminococcaceae (average 1.7%).
According to high‐throughput sequencing results, we identified 305 genera among the 15 fecal samples. Peptostreptococcaceae incertae sedis was the most abundant division (M = 13.5%) followed by Chloroplast norank (M = 13.3%). The first and second most frequent genera among the 15 samples were Clostridium (M = 9.8%) and Lysinibacillus (M = 6.4%). And the heatmap (Figure 4) of the frequencies of the genera reveals three distinct groups comprising samples B, C, D, H, and J; F and G; A, E, I, M, N, K, L, and O, respectively.
Figure 4.
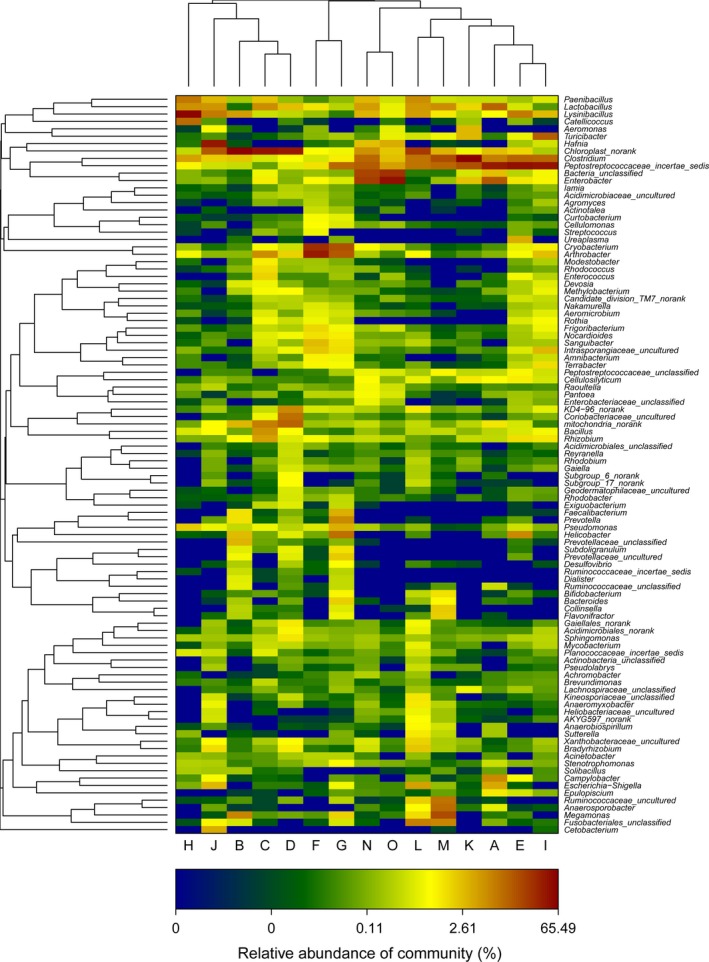
Distribution of the top 100 most abundant genera among the 15 hooded crane fecal samples
3.3. Core gut microbiome
Analysis of the frequencies of genera revealed that 37 genera were present in >90% of the samples and 26 genera were present in all samples, suggesting that approximately 12% of the genera were consistently present among most of the hooded cranes sampled, and that 9% were present at varying levels in all samples. Excluding genera present at <0.1% of the total reads, 20 genera were present in all samples, and 6 genera were present in 14 samples (Tables 2 and 3). Therefore, 9% of the gut microbiome formed the core microbiome of >90% samples. Peptostreptococcaceae genera (M = 13.5%) were not identified at genus level.
Table 2.
The relative abundance of core genera (100% core threshold) in the guts of hooded cranes
| Genus | % of total | Phylum | Class | Order | Family |
|---|---|---|---|---|---|
| Clostridium | 9.8 | Firmicutes | Clostridia | Clostridiales | Clostridiaceae |
| Enterobacter | 6.4 | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae |
| Lysinibacillus | 6.4 | Firmicutes | Bacilli | Bacillales | Bacillaceae |
| Arthrobacter | 4.5 | Actinobacteria | Actinobacteria | Actinomycetales | Micrococcaceae |
| Lactobacillus | 3.4 | Firmicutes | Bacilli | Lactobacillales | Lactobacillaceae |
| Cryobacterium | 3.3 | Actinobacteria | Actinobacteria | Actinomycetales | Microbacteriaceae |
| Paenibacillus | 1.7 | Firmicutes | Bacilli | Bacillales | Paenibacillaceae |
| Turicibacter | 1.3 | Firmicutes | Bacilli | Bacillales | Turicibacteraceae |
| Rhizobium | 0.7 | Proteobacteria | Alphaproteobacteria | Rhizobiales | Rhizobiaceae |
| Pseudomonas | 0.6 | Proteobacteria | Pseudomonadales | Pseudomonadales | Pseudomonadaceae |
| Bacillus | 0.7 | Firmicutes | Bacilli | Bacillales | Bacillaceae |
| Bradyrhizobium | 0.4 | Proteobacteria | Alphaproteobacteria | Rhizobiales | Bradyrhizobiaceae |
| Cellulosilyticum | 0.4 | Firmicutes | Clostridia | Clostridiales | Lachnospiraceae |
| Frigoribacterium | 0.3 | Actinobacteria | Actinobacteria | Actinomycetales | Microbacteriaceae |
| Nocardioides | 0.3 | Actinobacteria | Actinobacteria | Actinomycetales | Nocardioidaceae |
| Terrabacter | 0.3 | Actinobacteria | Actinobacteria | Actinomycetales | Intrasporangiaceae |
| Sphingomonas | 0.3 | Proteobacteria | Alphaproteobacteria | Sphingomonadales | Sphingomonadaceae |
| Stenotrophomonas | 0.2 | Proteobacteria | Alphaproteobacteria | Rhizobiales | Xanthomonadaceae |
| Raoultella | 0.2 | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae |
| Pantoea | 0.2 | Proteobacteria | Gammaproteobacteria | Enterobacteriales | Enterobacteriaceae |
Shown are only genera that represented >0.1% of the total reads. The Peptostreptococcaceae incertae sedis (12.6%) were not identified at the genus level.
Table 3.
The relative abundance of the newly added core genera (90% core threshold) in the guts of hooded cranes
| Genus | % of total | Phylum | Class | Order | Family |
|---|---|---|---|---|---|
| Methylobacterium | 0.2 | Proteobacteria | Alphaproteobacteria | Rhizobiales | Methylobacteriaceae |
| Devosia | 0.2 | Proteobacteria | Alphaproteobacteri | Rhizobiales | Hyphomicrobiaceae |
| Cellulomonas | 0.1 | Actinobacteria | Actinobacteria | Actinomycetales | Cellulomonadaceae |
| Mycobacterium | 0.2 | Actinobacteria | Actinobacteria | Actinomycetales | Mycobacteriaceae |
| Gaiella | 0.1 | Actinobacteria | Actinobacteria | Gaiellales | Gaiellaceae |
| Nakamurella | 0.1 | Actinobacteria | Actinobacteria | Actinomycetales | Nakamurellaceae |
Shown are only genera that represented ≥0.1% of the total reads.
4. DISCUSSION
The present study is the first to describe the gut microbiome of hooded cranes wintering at Shengjin Lake. The feces of the hooded cranes harbored an abundant population of microbes that included 785 OTUs representing 22 phyla, 51 classes, 107 orders, 188 families, and 305 genera. Further analysis showed that a high diversity of intestinal microbial of hooded crane samples, the Simpson index and Shannon–Wiener index averaged 0.17 and 2.83, respectively. One study indicates that the location of the sampling site mainly reflects the composition of microbiota compared with taxonomy or ecology (Hird et al., 2014). This maybe because Shengjin Lake provides wintering hooded cranes with an abundant and diverse source of food.
The most abundant phyla of gut microbes of wintering hooded cranes were as follows: Firmicutes, Proteobacteria, and Actinobacteria. The microbial content was similar to wild bar‐headed geese: Firmicutes predominated (58.33%), followed by Proteobacteria (30.67%), Actinobacteria (7.33%), and Bacteroidetes (3.33%) (Wang et al., 2016a; Wang et al., 2016b). The most populous phylum was Firmicutes that includes species that catabolize complex carbohydrates, polysaccharides, sugars, and fatty acids to provide an energy source for the host (Flint et al., 2008; Tap et al., 2009). The high abundance of Firmicutes was largely accounted for by Clostridium, Lysinibacillus, and Lactobacillus, which are often found as flora of the giant panda and hoatzin (Godoyvitorino et al., 2012; Zhu et al., 2011). Proteobacteria and Actinobacteria inhabit mammals, including horse (Dougal et al., 2013) as well as avian species such as the bar‐headed goose (Anser indicus) (Wang et al., 2016a; Wang et al., 2016b). In the present study, we detected a high abundance of Proteobacteria, mainly Enterobacter. The high abundance of Actinobacteria was largely accounted for by Arthrobacter. The gut microbiomes of some seabirds such as king penguins harbor a high abundance of Leuconostocaceae, Campylobacteriaceae, Porphyromonadaceae, Helicobacteriaceae, Flavobacteriaceae, Moraxellaceae, and Streptococcaceae (Dewar et al., 2013). Furthermore, Leuconostocaceae and Streptococcaceae are the most abundant families of gut bacteria of the short‐tailed shearwater (Dewar et al., 2014), in contrast to those of hooded cranes.
The microbial composition of the gut of the hooded cranes may be related to their diet acquired from their major foraging habitats such as paddy fields and meadows (Zheng et al., 2015). Further study is required to understand the relationship between the function of “key” bacteria and the lifestyle of hooded cranes, particularly their feeding habits. In addition, some sequences were classified as Chloroplast norank, because they likely represent ingested plant material.
We show here that 9% of the genera formed the core gut microbiome of hooded cranes. These core microbes influence and determine the composition of the intestinal microbial community structure and maintain community balance. According to previous studies, we detected many probiotics among the core microbes, which are characterized by remarkable metabolic and physiologic versatility, providing nutrition for the host. For example, cellulolytic Clostridium was the most abundant genus (average 9.8% of the total) (Sabathe et al., 2002; Shoham et al., 1999; Varel & Pond, 1992; Warnick et al., 2002) as well as Bacillus, Cellulosilyticum, Cellulomonas that could convert cellulose into metabolites. Furthermore, we detected an abundance of Arthrobacter (averaged 4.5% of the total), which as nutritionally versatile bacteria existed in many animals’ gut system (Buchan et al., 2001; Lu & Domingo, 2008). Lactobacillus (averaged 3.4% of the total), which was found in all samples, can degrade starch into maltose, maltotriose, and glucose (Champ et al., 1983; Kotarski et al., 1992), and are abundant in the seed‐eating green‐rumped parrotlet (Forpus passerinus) (Pacheco et al., 2004). Further studies are required to simultaneously test and verify the diet of the host as one of the factors that influence the composition of the gut microbiota as well as studies to determine the functions of the core gut microbiome to better understand the diet and health of the hooded cranes that winter at Shengjin Lake.
We detected many potential pathogenic bacteria. For example, Corynebacterium, which was detected in 13 samples, includes several species that cause disease in mammals and birds (Hoelzle et al., 2013; Potti et al., 2002). Helicobacter, an opportunistic pathogen, were detected in 12 samples and are often present in other birds and animals (Oxley & McKay, 2005). Staphylococcus, Streptococcus, and Pasteurella were detected in 14, 8, and 3 samples, respectively. Because hooded cranes migrate annually over long distances to obtain an abundant food supply, they are potentially exposed to novel and pathogenic microbes. Moreover, pressures generated by changing lifestyles and diets associated with migration might disrupt the stable gut microbiota and diminish immunocompetence associated with a corresponding increase in susceptibility to pathogens (Owen & Moore, 2006). Therefore, we were not surprised to find these potentially pathogenic microbes. Further studies must focus on the identification of the species of these potential pathogens as well as their potential to cause disease. In addition, we have detected some zoonotic disease pathogens. For example, the Campylobacter detected in 13 samples may cause bacterial gastroenteritis in human (Whelan et al., 1988), and Escherichia–Shigella, which were detected in 13 samples, are potential human pathogens that cause diarrhea (Hermes et al., 2009). The Pseudomonas found in all samples is opportunistic human pathogens (Chan et al., 2015). There are large flock of poultry and livestock breeding at Shengjin Lake. The habitats of domestic waterfowl overlap those of the hooded cranes at Shengjin Lake, and large aggregations of migratory birds may represent a source of pathogenic microbes that can be transmitted through feces.
5. CONCLUSIONS
This study first identified the gut microbiome of hooded cranes and defines the core gut microbiome of hooded cranes wintering at Shengjin Lake. We have shown that the feces of the hooded cranes harbored an abundant population of microbes. The core gut microbiome of hooded cranes wintering at Shengjin Lake includes many probiotics. In addition, we detected many potentially pathogenic bacteria; furthermore, metagenomics and the identification of these potential pathogens should be undertaken. Our study provides a foundation for further studies aimed to characterize the normal digestive functions and health of this endangered species.
CONFLICT OF INTEREST
None declared.
ACKNOWLEDGMENTS
The work was supported by the National Natural Science Foundation of China (Grant nos. 31472020, 31172117) and the Graduate Student Innovation Research Projects of Anhui University (YQH100288).
Zhao G, Zhou L, Dong Y, Cheng Y, Song Y. The gut microbiome of hooded cranes (Grus monacha) wintering at Shengjin Lake, China. MicrobiologyOpen. 2017;6:e447 https://doi.org/10.1002/mbo3.447
REFERENCES
- Amato, K. R. , Yeoman, C. J. , Kent, A. , Righini, N. , Carbonero, F. , Estrada, A. , … Leigh, S. R. (2013). Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. The ISME Journal, 7(7), 1344–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BirdLife International . (2016). Grus monacha. The IUCN Red List of Threatened Species 2016. e.T22692151A93337861. http://dx.doi.org/10.2305/IUCN.UK.2016-3.RLTS.T22692151A93337861.en. Accessed on 16 January 2017
- Buchan, A. , Neidle, E. L. , & Moran, M. A. (2001). Diversity of the ring‐cleaving dioxygenase gene pcaH in a salt marsh bacterial community. Applied & Environmental Microbiology, 67(12), 5801–5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarda, A. , Circella, E. , Pennelli, D. , Madio, A. , Bruni, G. , Lagrasta, V. , … Campagnari, E. (2006). Wild birds as biological indicators of environmental pollution: Biotyping and antimicrobial resistance patterns of Escherichia coli isolated from Audouin's gulls (Larus audouinii) living in the Bay of Gallipoli (Italy). Italian Journal of Animal Science, 5, 287–290. [Google Scholar]
- Champ, M. , Szylit, O. , Raibaud, P. , & Aïut‐Abdelkader, N. (1983). Amylase production by three Lactobacillus strains isolated from chicken crop. Journal of Applied Bacteriology, 55, 487–493. [DOI] [PubMed] [Google Scholar]
- Chan, K. G. , Liu, Y. C. , & Chang, C. Y. (2015). Inhibiting N‐acyl‐homoserine lactone synthesis and quenching Pseudomonas quinolone quorum sensing to attenuate virulence. Frontiers in Microbiology, 6(8), 2463–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. Y. , Zhou, L. Z. , Bo, Z. , Xu, R. X. , Zhu, W. Z. , Xu, & W. B. , (2011). Seasonal dynamics of wintering waterbirds in two shallow lakes along Yangtze River in Anhui Province. Zoological Research, 32(5), 540–548. [DOI] [PubMed] [Google Scholar]
- Collins, M. D. , Hutson, R. A. , Falsen, E. , Inganas, E. , & Bisgaard, M. (2002). Streptococcus gallinaceus sp. nov from chickens. International Journal of Systematic & Evolutionary Microbiology, 52(4), 1161–1164. [DOI] [PubMed] [Google Scholar]
- Darimont, C. T. , Reimchen, T. E. , & Bryan, H. M. (2008). Faecal‐centric approaches to wildlife ecology and conservation; methods, data and ethics. Wildlife Biology in Practice, 4(2), 14. [Google Scholar]
- Dennis, K. L. , Wang, Y. , Blatner, N. R. , Wang, S. , Saadalla, A. , & Trudeau, E. (2013). Adenomatous polyps are driven by microbe‐instigated focal inflammation and are controlled by IL‐10‐producing T cells. Cancer Research, 73(19), 5905–5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar, M. L. , Arnould, J. P. Y. , Dann, P. , Trathan, P. , Groscolas, R. , & Smith, S. (2013). Interspecific variations in the gastrointestinal microbiota in penguins. MicrobiologyOpen, 2(1), 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar, M. L. , Arnould, J. P. Y. , Krause, L. , Dann, P. , & Smith, S. C. (2014). Interspecific variations in the faecal microbiota of Procellariiform, seabirds. Fems Microbiology Ecology, 89(1), 47–55. [DOI] [PubMed] [Google Scholar]
- Domínguez‐Bello, M. G. , Michelangeli, F. , Ruiz, M. C. , García, A. , & Rodríguez, E. (1994). Ecology of the folivorous hoatzin (Opisthocomus hoazin) on the Venezuelan plains. The Auk, 111, 643–651. [Google Scholar]
- Dougal, K. , Fuente, G. , Harris, P. A. , Girdwood, S. E. , Pinloche, E. , et al. (2013). Identification of a core bacterial community within the large intestine of the horse. PLoS ONE, 8(10), e77660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint, H. J. , Bayer, E. A. , Rincon, M. T. , Lamed, R. , & White, B. A. (2008). Polysaccharide utilization by gut bacteria: Potential for new insights from genomic analysis. Nature Reviews Microbiology, 6(2), 121–131. [DOI] [PubMed] [Google Scholar]
- Fox, A. D. , Cao, L. , Zhang, Y. , Barter, M. , Zhao, M. J. , Meng, F. J. , & Wang, S. L. (2011). Declines in the tuber—feeding waterbird guild at Shengjin Lake National Nature Reserve, China—a barometer of submerged macrophyte collapse. Aquatic Conservation: Marine and Freshwater Ecosystems, 21(1), 82–91. [Google Scholar]
- Garcia‐Amado, M. A. , Gueneau, P. , Michelangeli, F. , & Domínguez‐Bello, M. G. (2003). Rate of detoxification of Quillaja saponins by crop bacteria from Opisthocomus hoazin is increased in the presence of methanogenic bacteria. Tropical and Subtropical Agroecosystems, 3, 595–598. [Google Scholar]
- Godoyvitorino, F. , Goldfarb, K. C. , Karaoz, U. , Leal, S. , Garcia‐Amado, M. A. , Hugenholtz, P. , … Dominguez‐Bello, M. G. (2012). Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. The ISME Journal, 6(3), 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grond, K. , Ryu, H. , Baker, A. J. , Domingo, J. W. S. , & Buehler, D. M. (2014). Gastro‐intestinal microbiota of two migratory shorebird species during spring migration staging in Delaware Bay. USA. Journal of Ornithology, 155(4), 969–977. [Google Scholar]
- Hermes, R. G. , Molist, F. , Ywazaki, M. , Nofrarías, M. , Gomez de Segura, A. , Gasa, J. , & Pérez, J. F. (2009). Effect of dietary level of protein and fiber on the productive performance and health status of piglets. Journal of Animal Science, 87(11), 3569–3577. [DOI] [PubMed] [Google Scholar]
- Hird, S. M. , Carstens, B. C. , Cardiff, S. W. , Dittmann, D. L. , & Brumfield, R. T. (2014). Sampling locality is more detectable than taxonomy or ecology in the gut microbiota of the brood‐parasitic brown‐headed cowbird (Molothrus ater). PeerJ, 2(10), e321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoelzle, L. E. , Scherrer, T. , Muntwyler, J. , Wittenbrink, M. M. , Philipp, W. , & Hoelzle, K. (2013). Differences in the antigen structures of Corynebacterium pseudotuberculosis and the induced humoral immune response in sheep and goats. Veterinary Microbiology, 164, 359–365. [DOI] [PubMed] [Google Scholar]
- Jami, E. , Israel, A. , Kotser, A. , & Mizrahi, I. (2013). Exploring the bovine rumen bacterial community from birth to adulthood. Isme Journal, 7(6), 1069–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao, S. W. , Guo, Y. M. , Falk, H. , & Lei, G. C. (2014). Nest‐site selection analysis of hooded crane (Grus monacha) in Northeastern China based on a multivariate ensemble model. Zoological Science, 31, 430–437. [DOI] [PubMed] [Google Scholar]
- Kotarski, S. F. , Waniska, R. D. , & Thurn, K. K. (1992). Starch hydrolysis by the ruminal microflora. Journal of Nutrition, 122(1), 178–190. [DOI] [PubMed] [Google Scholar]
- Leser, T. D. , & Mølbak, L. (2009). Better living through microbial action: The benefits of the mammalian gastrointestinal microbiota on the host. Environmental Microbiology, 11(9), 2194–2206. [DOI] [PubMed] [Google Scholar]
- Liu, Z. Y. , Xu, W. B. , Wang, Q. S. , Shi, K. C. , Xu, J. S. , & Yu, G. Q. (2001). Environmental carrying capacity for over‐wintering hooded cranes in Shengjin Lake. Resources & Environment in the Yangtze Basin, 10(5), 454–459. [Google Scholar]
- Lu, J. , & Domingo, J. S. (2008). Turkey fecal microbial community structure and functional gene diversity revealed by 16 S rRNA gene and metagenomic sequences. The Journal of Microbiology, 46(5), 469–477. [DOI] [PubMed] [Google Scholar]
- Marois, C. , Savoye, C. , Kobisch, M. , & Kempf, I. (2002). A reverse transcription‐PCR assay to detect viable mycoplasma synoviae in poultry environmental samples. Veterinary Microbiology, 89(1), 17–28. [DOI] [PubMed] [Google Scholar]
- Owen, J. C. , & Moore, F. R. (2006). Seasonal differences in immunological condition of three species of thrushes. Condor, 108, 389–398. [Google Scholar]
- Oxley, A. P. A. , & McKay, D. B. (2005). Comparison of Helicobacter spp. genetic sequences in wild and captive seals, and gulls. Diseases of Aquatic Organisms, 65(2), 99–105. [DOI] [PubMed] [Google Scholar]
- Pacheco, M. A. , García‐Amado, M. A. , Bosque, C. , & Domínguez‐Bello, M. G. (2004). Bacteria in the crop of the seed‐eating green‐rumped parrotlet. Condor, 106(1), 139–143. [Google Scholar]
- Potti, J. , Moreno, J. , Yorio, P. , Briones, V. , Garcia‐Borboroglu, P. , & Villar, S. (2002). Bacteria divert resources from growth for magellanic penguin chicks. Ecology Letters, 5(6), 709–714. [Google Scholar]
- Sabathe, F. , Belaich, A. , & Soucaille, P. (2002). Characterization of the cellulolytic complex (cellulosome) of Clostridium acetobutylicum . Fems Microbiology Letters, 217, 15–22. [DOI] [PubMed] [Google Scholar]
- Santos, S. S. , Pardal, S. , Proenca, D. N. , Lopes, R. J. , Ramos, J. A. , Mendes, L. , & Morais, P. V. (2012). Diversity of cloacal microbial community in migratory shorebirds that use the Tagus estuary as stopover habitat and their potential to harbor and disperse pathogenic microorganisms. Fems Microbiology Ecology, 82(1), 63–74. [DOI] [PubMed] [Google Scholar]
- Shoham, Y. , Lamed, R. , & Bayer, E. A. (1999). The cellulosome concept as an efficient microbial strategy for the degradation of insoluble polysaccharides. Trends in Microbiology, 7(7), 275–281. [DOI] [PubMed] [Google Scholar]
- Stanley, D. , Geier, M. S. , Hughes, R. J. , Denman, S. E. , & Moore, R. J. (2013). Highly variable microbiota development in the chicken gastrointestinal tract. PLoS ONE, 8(12), e84290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tap, J. , Mondot, S. , Levenez, F. , Pelletier, E. , Caron, C. , Furet, J. P. , … Leclerc, M. (2009). Towards the human intestinal microbiota phylogenetic core. Environmental Microbiology, 11(10), 2574–2584. [DOI] [PubMed] [Google Scholar]
- Varel, V. H. , & Pond, W. G. (1992). Characteristics of a new cellulolytic Clostridium sp. isolated from pig intestinal tract. Applied & Environmental Microbiology, 58(5), 1645–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite, D. W. , Deines, P. , & Taylor, M. W. (2012). Gut microbiome of the critically endangered New Zealand Parrot, the Kakapo (Strigops habroptilus). PLoS ONE, 7(4), 410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Cao, J. , Yang, F. , Wang, X. L. , Zheng, S. S. , Sharshov, K. , & Li, L. X. (2016. a). High‐ throughput sequencing reveals the core gut microbiome of Bar‐headed goose (Anser indicus) in different wintering areas in Tibet. MicrobiologyOpen, 5(2), 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Garrity, G. M. , Tiedje, J. M. , & Cole, J. R. (2007). Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied & Environmental Microbiology, 73(16), 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Sheng, H. F. , He, Y. , Wu, J. Y. , Jiang, Y. X. , Tam, N. F. Y. , & Zhou, H. W. (2012). Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of Illumina tags. Applied & Environmental Microbiology, 78(23), 8264–8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Zheng, S. , Sharshov, K. , Cao, J. , Sun, H. , Yang, F. , … Li, L. (2016. b). Distinctive gut microbial community structure in both the wild and farmed Swan goose (Anser cygnoides). Journal of Basic Microbiology, 56(11), 1299–1307. [DOI] [PubMed] [Google Scholar]
- Warnick, T. A. , Methe, B. A. , & Leschine, S. B. (2002). Clostridium phytofermentans sp. nov. a cellulolytic mesophile from forest soil. International Journal of Systematic & Evolutionary Microbiology, 52, 1155–1160. [DOI] [PubMed] [Google Scholar]
- Whelan, C. D. , Monaghan, P. , Girdwood, R. W. , & Fricker, C. R. (1988). The significance of wild birds (Larus sp.) in the epidemiology of Campylobacter infections in humans. Epidemiology & Infection, 101(2), 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L. L. , Xu, W. B. , Sun, Q. Y. , Zhou, Z. Z. , Shen, J. , & Zhao, X. X. (2008). Flora and vegetation in Shengjin Lake. Journal of Wuhan Botanical Research, 27(3), 264–270. [Google Scholar]
- Young, V. B. (2012). The intestinal microbiota in health and disease. Current Opinion in Gastroenterology, 28, 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. C. , Guo, Z. , Xue, Z. S. , Sun, Z. H. , Zhang, M. H. , Wang, L. , … Zhang, H. (2015). A phylo‐functional core of gut microbiota in healthy young Chinese cohorts across lifestyles, geography and ethnicities. The ISME Journal, 9(9), 1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, F. , Zhou, L. , Xu, W. (2013). Habitat utilization and resource partitioning of wintering hooded cranes and three goose species at Shengjin Lake. Chinese Birds, 4(4), 281–290. [Google Scholar]
- Zheng, M. , Zhou, L. Z. , Zhao, N. N. , & Xu, W. B. (2015). Effects of variation in food resources on foraging habitat use by wintering hooded cranes (Grus monacha). Avian Research, 6, 11. [Google Scholar]
- Zhou, B. , Zhou, L. Z. , Chen, J. Y. , Cheng, Y. Q. , & Xu, W. B. (2010). Diurnal time‐activity budgets of wintering hooded cranes (Grus monacha) in Shengjin Lake, China. Waterbirds, 33(1), 110–115. [Google Scholar]
- Zhu, L. F. , Wu, Q. , Dai, J. Y. , Zhang, S. N. , & Wei, F. W. (2011). Evidence of cellulose metabolism by the giant panda gut microbiome. Proceedings of the National Academy of Sciences of the United States of America, 108(43), 17714–17719. [DOI] [PMC free article] [PubMed] [Google Scholar]