

Abstract
By using the maximum likelihood method, we made a genome-wide comparison of the evolutionary rates in the lineages leading to the laboratory strain (S288c) and a wild strain (YJM789) of Saccharomyces cerevisiae and found that genes in the laboratory strain tend to evolve faster than in the wild strain. The pattern of elevated evolution suggests that relaxation of selection intensity is the dominant underlying reason, which is consistent with recurrent bottlenecks in the S. cerevisiae laboratory strain population. Supporting this conclusion are the following observations: (i) the increases in nonsynonymous evolutionary rate occur for genes in all functional categories; (ii) most of the synonymous evolutionary rate increases in S288c occur in genes with strong codon usage bias; (iii) genes under stronger negative selection have a larger increase in nonsynonymous evolutionary rate; and (iv) more genes with adaptive evolution were detected in the laboratory strain, but they do not account for the majority of the increased evolution. The present discoveries suggest that experimental and possible industrial manipulations of the laboratory strain of yeast could have had a strong effect on the genetic makeup of this model organism. Furthermore, they imply an evolution of laboratory model organisms away from their wild counterparts, questioning the relevancy of the models especially when extensive laboratory cultivation has occurred. In addition, these results shed light on the evolution of livestock and crop species that have been under human domestication for years.
Keywords: model organism, slightly deleterious mutation, yeast evolution
The most commonly used Saccharomyces cerevisiae haploid in the laboratory, S288c, for which the whole genome sequence is known (1), has ≈88% of its genome derived from a strain (EM93) isolated from a rotten fig ≈70 years ago (2). The origin of EM93 before fig isolation is unclear. It could be a natural fig isolate or, rather, a contaminant derived from an industrial strain (2). The domestication of S. cerevisiae therefore can be dated back to somewhere between 70 and several hundred years ago. Because of the short generation time of yeast, the experimental practices in the past several decades and the possible industrial manipulations could have left significant footprints on the evolution of the S288c strain. These activities can lead to peculiar evolutionary trajectories because frequent passages of the strains through severe bottlenecks, which may even reduce populations down to a single cell on account of common experimental practices, can result in a significant reduction in the effective population size and thus to fixation of mutations that would be too deleterious to become fixed under natural conditions. This process occurs because population genetics theory predicts that mutations with s < 1/2Ne, where s is the selection coefficient and Ne is the effective population size, behave nearly neutrally (3–8). Indeed, increased evolutionary rates of individual genes due to population size reduction were recorded for diverse organisms such as endosymbiotic bacteria and island birds (9, 10). Furthermore, in some cases more targeted evolutionary changes can occur as laboratory strains experience a loss of specific environmental selection pressure or adapt to the laboratory growth conditions. Specific laboratory adaptation for various organisms is described in refs. 11 and 12.
If such evolution did occur and formed a significant proportion of the evolutionary changes in the history leading to the laboratory strain, we would expect to see an accelerated evolution in this lineage. If relaxation of selection due to population size reduction is the most dominant factor, the increase in evolutionary rate should not be centralized on genes with specific function, because the changes of population structure should affect all genes under selection. Furthermore, this hypothesis would predict that genes under different selection pressure have different changes in evolutionary rate as the effective population size reduces. Conversely, increases in evolutionary rate due to adaptive evolution or relaxation of selection caused by loss of particular environmental selection pressure should only occur for some genes, those that are functionally relevant to the specific change. In this study, we tested these hypotheses by comparing the evolutionary rates (synonymous, nonsynonymous, and the ratio of the two) in the lineages to the laboratory strain (S288c) and a wild strain (YJM789) that was isolated from the lung of an AIDS patient (13).
Shotgun sequencing of the YJM789 genome (L.M.S., T.J., L.D., M. Miranda, D. Bruno, C. Komp, M. Nguyen, R. Tamse, J. Wilhelmy, R.W. Hyman, and R.W.D., unpublished data) shows an average difference to S288c of (9.8 ± 0.2) × 10–3 changes per synonymous site. YJM789 is a good candidate to compare with S288c, because it is not too divergent from the laboratory strain: The evolutionary changes accumulated because of laboratory and possible industrial manipulations therefore can constitute a significant part of the evolution between these two strains. Although the short separation time between the strains makes it statistically difficult to see the noteworthy effects of evolutionary forces, such as population-size reduction, on individual genes, the sequencing of the whole genome of YJM789 provides an opportunity to analyze evolutionary changes globally. By combining all genes in the genome, we can detect traces left by different evolutionary forces even if they have acted for a short time.
The genome sequence of Saccharomyces paradoxus, the closest sequenced species to S. cerevisiae (14), was used as an outgroup in a phylogenetic analysis. Altogether, 4,020 genes with good alignment among the three organisms were analyzed. As predicted from the evolution of strains that experience passage through recurrent bottlenecks, a significantly higher evolutionary rate, at both the synonymous and nonsynonymous sites, was detected in the lineage to S288c than to YJM789, and the increase of nonsynonymous evolutionary rate was shown to occur for genes in all functional categories. By dividing genes into different groups based on their codon usage bias, we demonstrate that the increase of evolutionary rate at synonymous sites occurs mostly on genes with strong codon usage bias, whereas the increase of nonsynonymous evolutionary rate is higher for genes under stronger negative selection. Furthermore, adaptive evolution was detected for more genes in the laboratory strain than in YJM789; however, the faster evolution in the laboratory strain still exists even after excluding genes under positive selection. This finding implies that adaptive evolution is not the major reason for the observed evolutionary rate increase in the laboratory strain.
Materials and Methods
Genomic Sequences and Analysis. The genome of S. cerevisiae strain YJM789 was sequenced by the Stanford Genome Technology Center. Completion of the genome is in progress (L.M.S., T.J., L.D., M. Miranda, D. Bruno, C. Komp, M. Nguyen, R. Tamse, J. Wilhelmy, R. W. Hyman, and R.W.D. unpublished data). From the shotgun assembly (Version 2, www-sequence.stanford.edu) only contigs with >10 synteny-defined orthologous genes between S288c and YJM789 were included in this study. The genome sequence of S. paradoxus was used as the outgroup in a phylogenetic analysis (Fig. 1). Orthologous genes between S. cerevisiae (S288c) and S. paradoxus were defined by Kellis et al. (14) (www.broad.mit.edu/annotation/fungi/comp_yeasts/downloads.html). The clustalw alignments of the three-way orthologous proteins were verified individually by eye. Altogether 4,020 genes were included in the analysis. Coding DNA sequences were aligned based on the protein alignments.
Fig. 1.
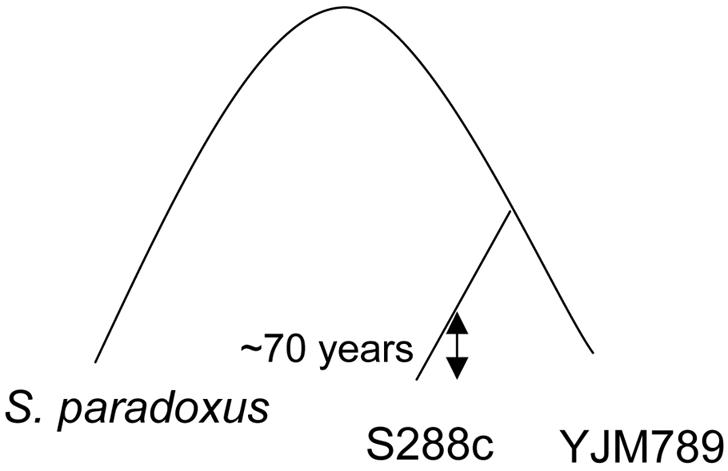
Phylogenetic relationship between the two studied S. cerevisiae strains (S288c and YJM789) and the outgroup of S. paradoxus. The arrow indicates an approximate time when EM93, an ancestor of S288c, was isolated from a rotten fig (2). The time of the most recent common ancestor of EM93 and YJM789 could not be reliably estimated.
Analysis of the evolutionary changes in each lineage was performed by using codeml in the paml package (15, 16). The free ratio model was used in estimating the synonymous and nonsynonymous changes in each branch. Branch-Site Models (17) were used in detecting genes with positive evolution (see Results). Codon usage bias for each gene was estimated by using codonw (www.molbiol.ox.ac.uk/cu). For a gene, the smaller the effective number of codons (ENC), the stronger the codon usage bias (18). The codon preference for each amino acid was defined as in ref. 19.
To compare evolutionary rates in two branches for genes with different functions, we grouped genes based on the Gene Ontology system (www.geneontology.org). Within each category, evolutionary changes for individual genes were added together to estimate evolutionary rates for synonymous (KS) and nonsynonymous (KA) sites. The variance of the evolutionary rate ratios (or differences) between S288c and YJM789 were estimated by a bootstrap procedure (20). If there are N genes in one category, the same number of genes was sampled with replacement within the category, and the ratio (or difference) of evolutionary rate was calculated. The variance was estimated by repeating the sampling 100 times. Student's t test was used in assessing whether the ratio of evolutionary rate between S288c and YJM789 was significantly larger than 1 (or >0 for the evolutionary rate difference).
To determine the evolutionary rate changes for genes under different selection pressures, we grouped the studied genes into categories based on their codon usage bias. The stronger the codon usage bias, the higher the negative selection pressure on a gene. This assumption is valid because evolutionary rates at nonsynonymous or synonymous sites or the ratio of the two are negatively related with gene codon usage bias (21–25). To estimate the mean and variance of the evolutionary rate change between S288c and YJM789 within each ENC category, the same analysis described above for categories of gene function was performed. We report the results based on codon usage bias in S288c, but the same conclusions were reached when codon usage from orthologous genes in YJM789 or the average of S288c and YJM789 were used in the analysis. The number of categories also did not affect the conclusions (data not shown).
Excluding Genes from Different Genetic Backgrounds. Winzeler et al. (26) compared coding regions of S288c and EM93 by using Affymetrix microarrays, and polymorphisms were identified between these two strains. As performed by these authors, we clustered all polymorphic probes within 30-kb intervals and extended the boundaries of each cluster 10 kb to either side. Clusters with fewer than three polymorphic probes were dismissed. Genes located in these clusters (≈16%) were regarded as genes in highly polymorphic regions between S288c and EM93. Because it was estimated that ≈12% of the S288c genomes is derived from genetic backgrounds other than EM93 (2), to be conservative we excluded all of the genes in these highly polymorphic regions. The same analyses as those for the whole genome were carried out for the remaining genes.
Results
Increased Evolutionary Rates in the Lineage to the Laboratory Strain (S288c). The maximum likelihood method was used to estimate the evolutionary changes in each branch. R, the evolutionary rate measured by the ratio of nonsynonymous (KA) over synonymous (KS) evolutionary rates, was calculated (Fig. 1). We found that, as has been shown in other studies (27–32), R is larger for polymorphism than for divergence between species. For example, R is ≈2-fold larger in the S288 and YJM789 branches than in the S. paradoxus branch (Table 1). The salient discovery for this comparison, however, is that at the genome level R is significantly larger in the lineage to S288c than to YJM789. As shown in Table 1, when the evolutionary changes are added up for all studied genes, 21% (4400.5/3631.4 = 1.21) more nonsynonymous changes were detected in the S288c branch than in the YJM789 branch. Even after normalizing the nonsynonymous difference by the synonymous difference, a >15% (0.201/0.174 = 1.155) increase in the laboratory strain was detected. The difference is statistically significant by Fisher's exact test (P < 0.001). When the same analyses were performed separately on experimentally verified and unverified genes, similar patterns were observed (Table 5, which is published as supporting information on the PNAS web site).
Table 1. Number of synonymous and nonsynonymous changes in the lineages to S288c, YJM789, and S. paradoxus.
| All genes
|
Genes in low polymorphic regions
|
|||||
|---|---|---|---|---|---|---|
| Strain | S* | N* | R = KA/KS* | S | N | R = KA/KS |
| S. paradoxus | 625,466.5 | 169,826.8 | 0.104 | 523,254.7 | 142,368.5 | 0.104 |
| S288c | 8,342.3 | 4,400.5 | 0.201 | 6,844.6 | 3,610.3 | 0.201 |
| YJM789 | 7,956.1 | 3,631.4 | 0.174 | 6,542.4 | 3,033.6 | 0.177 |
| S288c/YJM789 | 1.05 | 1.21 | 1.16 | 1.05 | 1.19 | 1.14 |
In the left part of the table 4,020 genes were analyzed; in the right part 663 genes that locate in the highly polymorphic regions between S288c and EM93 were excluded. Fisher's exact test shows that the nonsynonymous evolutionary rate is significantly higher in S288c than in YJM789 for both groups of genes.
S and N represent the number of synonymous and nonsynonymous changes, respectively. KS and KA represent the number of synonymous and nonsynonymous changes per synonymous and nonsynonymous site, respectively.
Previous study showed that ≈88% of the S288c genome was derived from a strain called EM93, whereas the rest of the genome originated from a mixture of other genetic backgrounds (2). Genes derived from strains other than EM93 might interfere with identifying the real evolutionary changes in the lineage to the laboratory strain. To eliminate this effect, we excluded genes located in highly polymorphic regions between S288c and EM93. As shown in Table 1, the difference in evolutionary rates between the two lineages based on the remaining 3,357 genes remains statistically significant. To exclude possible effects coming from different genetic backgrounds, we only used genes located outside of the highly polymorphic regions between S288c and EM93 in the following analyses.
Increased Evolutionary Rates Found for Genes in All Functional Categories. We investigated possible reasons for the evolutionary rate increase in the laboratory strain. If population-size reduction is the major underlying reason, the evolutionary rate should increase for all genes that experience negative selection regardless of their function. To test this prediction, the Gene Ontology system was used to group genes into different functional categories (33). We then compared the evolutionary rate in the S288c and YJM789 branches within each functional category. As shown in Fig. 2 and Fig. 5, which is published as supporting information on the PNAS web site, an increased evolutionary rate (R) in the laboratory strain was observed for all nine functional categories. The level of increase, however, was not identical for each group. For example, genes involved in transcriptional activity had the highest increase in evolutionary rate in the laboratory strain.
Fig. 2.

Relative increase of evolutionary rate in the laboratory strain for genes in different functional categories. The groups were ordered by decreasing number of genes in each category. The x-axis represents the ratio of evolutionary rate (R = KA/KS) in S288c to that in YJM789 (see Fig. 5 for the absolute increase in evolutionary rates in each functional category). ***, P < 0.001 by Student's t test for ratio >1. Genes in highly polymorphic regions between S288c and EM93 were excluded.
Greater Increase in Evolutionary Rates for Genes Under Stronger Negative Selection. To determine whether the evolutionary rate increase varies for genes with different selection pressure we grouped genes into different categories based on their codon usage bias (ENC). Indeed, evolutionary rates at nonsynonymous or synonymous sites or the ratio of the two have been shown to be negatively related with gene codon usage bias (21–25). Therefore, the more the codon usage bias for a gene, the stronger the negative selection pressure for that gene.
Table 2 lists the number of increased evolutionary changes in S288c for each codon usage bias category. Of the increased synonymous changes in S228c, 76.3% (230.7 of 302.2) occurred in the first two categories (with strongest codon usage bias, P ≪ 0.001 for enrichment). This result cannot be explained by a mutation rate increase at the whole genome level or by more generations in the lineage to the laboratory strain, both of which would cause a uniform increase in KS for all groups. Conversely, population size reduction can cause this pattern because the synonymous sites for genes with strong codon usage bias are under negative selection. With decreasing population size, the selective pressure at the synonymous sites of these genes will be relaxed, and the evolutionary rate will increase.
Table 2. Increase of evolutionary changes for genes with different codon usage in the lineage to S288c.
| Codon usage bias categories
|
||||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| Synonymous | 83.9 | 146.8 | 53.9 | -6.2 | 29.4 | -5.6 |
| Nonsynonymous | 103.8 | 163.7 | 110.3 | 100.8 | 63.1 | 35 |
Categories were defined as in Fig. 3, shown in order of decreasing codon usage bias. The numbers are the difference in evolutionary changes between S288c and YJM789 branches (S288c — YJM789).
As shown in Table 2, the nonsynonymous evolutionary rate increase in S288c is also enriched in genes with strong codon usage bias [e.g., 46.4% (267.5/576.7) of increased nonsynonymous changes in S228c occurred in the first two categories; P < 0.001 for enrichment]. When R (KA/KS) was investigated, we observed the same pattern, namely that the increased evolutionary rate in the laboratory strain is highest for genes with the strongest negative selection and decreases as selection intensity on the genes, reflected here by the gene codon usage bias, decreases (Fig. 3). The same trend also was observed when the difference instead of the ratio of the evolutionary rate between the S288c and YJM789 branches was investigated (Fig. 3 Inset). Furthermore, gene groupings based on protein dispensability or R between the S288c/YJM789 ancestor and S. paradoxus, instead of ENC, led to similar conclusions (data not shown). Interestingly, as population size reduces, relatively more deleterious mutations are expected to become nearly neutral for genes under stronger negative selection (Fig. 6, which is published as supporting information on the PNAS web site), which is consistent with the above observations.
Fig. 3.

Increase of evolutionary rate in the laboratory strain for genes under different negative selection. The genes were divided into six categories basedon their ranked codon usage bias. Groups are in order of decreasing codon usage bias (increasing ENC). Each group has 559 genes (except group 6, which has 562 genes). (Inset) The absolute evolutionary rate increase in the laboratory strain. The dashed line represents the linear regression line. Grouping genes based on the average codon usage between S288c and YJM789 gave similar results (data not shown). ***, P < 0.001 by Student's t test for ratio >1. Genes in highly polymorphic regions between S288c and EM93 were excluded.
Adaptive Evolution Is Not the Major Reason for the Observed Evolutionary Rate Increase in the Laboratory Strain. Adaptive evolution of S288c to the laboratory growth conditions also can be a candidate reason for the observed increase in evolutionary rate in this strain. To examine this possibility, we applied a maximum likelihood method for identifying genes with positive selection at the codon level from both lineages (18). Two hypotheses were compared for either lineage by using the likelihood ratio test: the null hypothesis assuming neutral (KA/KS = 1 or KA/KS <1) evolution for each codon site and the alternative hypothesis that allows some sites to evolve with KA > KS. Those genes with significantly higher likelihood for the alternative hypothesis were regarded as genes under positive selection.
Table 3 lists the number of genes with evidence of adaptation under various statistical significance levels in either lineage. Although we always observed more genes with adaptive evolution in the lineage to S288c than to YJM789, we argue that adaptive evolution is not the major reason for the evolutionary rate increase in the laboratory strain for two reasons: First, we did not see more genes with evidence of adaptation than expected by random chance. For example, under P = 0.05, 138 genes with positive signals were detected in S288c, whereas we expected to see 4,020 × 0.05 = 201 genes to pass this test by random chance. Second, the evolutionary rate difference between the two lineages was still highly significant even after excluding those genes with adaptive evolution; in the latter analysis the evolutionary rate in the S288c branch still stays ≈14% higher for the remaining genes (Table 6, which is published as supporting information on the PNAS web site).
Table 3. Number of genes with adaptive evolution in the S288c and YJM789 branches.
| Threshold P value | S288c | YJM789 | Ratio |
|---|---|---|---|
| 0.05 | 138 (112) | 93 (76) | 1.48 (1.47) |
| 0.01 | 65 (54) | 42 (32) | 1.55 (1.69) |
| 0.001 | 23 (21) | 16 (11) | 1.44 (1.91) |
The values in parentheses are after excluding genes in highly polymorphic regions between S288c and EM93.
Discussion
Evolutionary rate (R) appears ≈15% faster in the laboratory strain S288c of yeast than in the wild strain YJM789. If this increase occurred because of laboratory and possible industrial manipulations, the actual rate of evolution in S288c during that period may be much higher because the observed results are averaged across the whole branch after the separation of S288c and YJM789 from their common ancestor. Several reasons, such as increased mutation rate, more generations, relaxation of selection pressure, and adaptive evolution in the laboratory lineage, can lead to elevated evolutionary rates (34, 35). The first two reasons, i.e., increased mutation rate and more generations, cannot be the major underlying reasons because the evolutionary rate difference observed above for amino acid change was normalized by synonymous change. Furthermore, as shown in Table 2, the slight increase in synonymous change in S288c occurs mostly on genes with strong codon usage bias, which is consistent with expectation from fixation of slightly deleterious mutations due to population size reduction.
A decrease in selection coefficients (s) also will lead to a relaxation of selection pressure. The evolutionary rate increase was observed for genes in all functional categories, which implies relaxation of selection due to reduction in population size (Ne) rather than selection coefficients (s), because the latter will likely occur on genes with specific functions. For the same reason we believe that adaptive evolution is not the major mechanism underlying the evolutionary rate increase in the laboratory strain. In addition, if all (or most) of the increased evolutionary rate in the laboratory strain were due to adaptive evolution, S288c should be more fit under laboratory growth conditions than YJM789. The data are not consistent with this expectation: YJM789 grows faster than S288c under laboratory growth conditions (36).
Nevertheless, genes showing adaptive evolution in the laboratory strain can provide immediate, biologically meaningful hypotheses for further investigation. For example, YBR203W (COS111), a gene involved in the response to antifungal drugs (37), shows significant positive selection in the laboratory strain (P ≪ 0.001 by likelihood ratio test). Furthermore, it will be interesting to investigate the functional consequences of the adaptation detected in genes involved in transcription, 14 of which show adaptive evolution in S288c, whereas this number is only 3 in YJM789 (Fig. 4, P < 0.05 by Fisher's exact test; see Tables 7 and 8, which are published as supporting information on the PNAS web site, for the list of all genes with adaptive evolution in either lineage).
Fig. 4.

Distribution of genes with adaptive evolution in each functional category for the lineages to S288c and YJM789. *, P < 0.05 by Fisher's exact test.
Increased evolutionary rates for individual genes as a result of population size reduction have been reported in various organisms (9, 10, 38). Several studies showed that elevated evolutionary rates are usually accompanied by relaxation of codon usage bias (9, 38). To see whether this is the case in our data, we further analyzed the direction of synonymous change in either lineage. More changes from preferred to unpreferred codons would be expected in S288c if a relaxation of codon usage bias occurred. As shown in Table 4, in the laboratory strain we observed a marginally higher rate of synonymous change from preferred to unpreferred codons for genes with strong codon usage bias. The reverse direction of synonymous change (unpreferred to preferred) is not significantly different between the S288c and YJM789 branches (Table 4).
Table 4. Direction of synonymous codon change in the S288c and YJM789 branches.
| Preferred to unpreferred*
|
Unpreferred to preferred†
|
|||
|---|---|---|---|---|
| 500 genes with the strongest codon usage bias | Rest of the genes | 500 genes with the strongest codon usage bias | Rest of the genes | |
| S288c | 236 | 1,908 | 160 | 1,767 |
| YJM789 | 203 | 1,951 | 153 | 1,682 |
| S288c/YJM789 | 1.16 | 0.98 | 1.05 | 1.05 |
Codon preference for each amino acid was taken from ref. 19. χ2 test, P = 0.086.
χ2 test, P = 0.97.
The relaxation of selection intensity due to reduction of effective population size is expected to lead to increased evolutionary rate genome-wide. Adaptive evolution and directional relaxation of selection due to growth environment changes are expected to lead to evolutionary rate increase for specific genes. The observed increase in evolutionary rate in the laboratory strain might be caused by a mixture of these factors. The observed patterns are consistent with laboratory cultivation. The laboratory growth conditions are extremely different from the ones in the wild. Streaking and picking of single clones is common during yeast experimentation and leads to a severe reduction in the effective population size of the laboratory strain. It is thus likely that laboratory domestication contributes to the observations made in this study. Nevertheless, the mysterious origin of the laboratory strain S288c makes it currently difficult to prove that all of the observed evolutionary rate increase did occur in the laboratory. If laboratory cultivation and possible industrial domestication did cause the increased evolutionary rate, the results of this study, especially the greater increase in nonsynonymous evolutionary rate for genes under stronger negative selection, imply an evolution of laboratory model organisms away from their wild counterparts. This finding has implications for all laboratory model organisms, questioning the relevancy of the models especially when extensive laboratory cultivation has occurred. Because livestock and crop species domesticated by humans went through severe bottlenecks as well, the results here also shed light on the evolution of these organisms.
Supplementary Material
Acknowledgments
We thank Tom Nagylaki, Zhiheng Yang, Martin Kreitman, Elizabeth Winzeler, Jian Lu, Jerel Davis, and Cristian Castillo-Davis for help, discussions, and comments. This work was supported by National Institutes of Health Grants HG02052, HG00205 (to R.W.D.), and GM068717 (to R.W.D and L.M.S.).
Author contributions: Z.G., L.D., D.P., and L.M.S. designed research; Z.G. performed research; R.W.D. supervised the research; T.J. and L.M.S. contributed new reagents/analytic tools; Z.G. analyzed data; and Z.G., L.D., and L.M.S. wrote the paper.
Abbreviation: ENC, effective number of codons.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AAFW00000000).
References
- 1.Goffeau, A., Barrell, B. G., Bussey, H., Davis, R. W., Dujon, B., Feldmann, H., Galibert, F., Hoheisel, J. D., Jacq, C., Johnston, M., et al. (1996) Science 274, 563–567. [DOI] [PubMed] [Google Scholar]
- 2.Mortimer, R. K. & Johnston, J. R. (1986) Genetics 113, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohta, T. (1972) J. Mol. Evol. 1, 305–314. [PubMed] [Google Scholar]
- 4.Ohta, T. & Kimura, M. (1971) J. Mol. Evol. 1, 18–25. [DOI] [PubMed] [Google Scholar]
- 5.Ohta, T. (1973) Nature 246, 96–98. [DOI] [PubMed] [Google Scholar]
- 6.Ohta, T. (1987) J. Mol. Evol. 26, 1–6. [DOI] [PubMed] [Google Scholar]
- 7.Ohta, T. (1993) Annu. Rev. Ecol. Syst. 23, 263–286. [Google Scholar]
- 8.Kimura, M. (1983) The Neutral Theory of Molecular Evolution (Cambridge Univ. Press, Cambridge, U.K.).
- 9.Moran, N. A. (1996) Proc. Natl. Acad. Sci. USA 93, 2873–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson, K. P. & Seger, J. (2001) Mol. Biol. Evol. 18, 874–881. [DOI] [PubMed] [Google Scholar]
- 11.Korona, R., Nakatsu, C. H., Forney, L. J. & Lenski, R. E. (1994) Proc. Natl. Acad. Sci. USA 91, 9037–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riehle, M. M., Bennett, A. F., Lenski, R. E. & Long, A. D. (2003) Physiol. Genomics 14, 47–58. [DOI] [PubMed] [Google Scholar]
- 13.Tawfik, O. W., Papasian, C. J., Dixon, A. Y. & Potter, L. M. (1989) J. Clin. Microbiol. 27, 1689–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kellis, M., Patterson, N., Endrizzi, M., Birren, B. & Lander, E. S. (2003) Nature 423, 241–254. [DOI] [PubMed] [Google Scholar]
- 15.Goldman, N. & Yang, Z. (1994) Mol. Biol. Evol. 11, 725–736. [DOI] [PubMed] [Google Scholar]
- 16.Yang, Z. (1996) J. Mol. Evol. 42, 294–307. [DOI] [PubMed] [Google Scholar]
- 17.Wright, F. (1990) Gene 87, 23–29. [DOI] [PubMed] [Google Scholar]
- 18.Yang, Z. & Nielsen, R. (2002) Mol. Biol. Evol. 19, 908–917. [DOI] [PubMed] [Google Scholar]
- 19.Akashi, H. (2003) Genetics 164, 1291–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Efron, B. & Tibshirani, R.J. (1998) An Introduction to the Bootstrap (Chapman & Hall/CRC, Boca Raton, FL).
- 21.Sharp, P. M. & Li, W.-H. (1987) Mol. Biol. Evol. 4, 222–230. [DOI] [PubMed] [Google Scholar]
- 22.Carulli, J. P., Krane, D. E., Hartl, D. L. & Ochman, H. (1993) Genetics 134, 837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharp, P. M. & Li, W.-H. (1989) J. Mol. Biol. 28, 398–402. [DOI] [PubMed] [Google Scholar]
- 24.Shields, D. C., Sharp, P. M., Higgins, D. G. & Wright, F. (1988) Mol. Biol. Evol. 5, 704–716. [DOI] [PubMed] [Google Scholar]
- 25.Rocha, E. P. C. & Danchin, A. (2004) Mol. Biol. Evol. 21, 108–116. [DOI] [PubMed] [Google Scholar]
- 26.Winzeler, E. A., Castillo-Davis, C. I., Oshiro, G., Liang, D., Richards, D. R., Zhou, Y. & Hartl, D. L. (2003) Genetics 163, 79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ballard, J. W. & Kreitman, M. (1994) Genetics 138, 757–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rand, D., Dorfsman, M. & Kann, L. (1994) Genetics 138, 741–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nachman, M., Brown, W., Stoneking, M. & Aquadro, C. (1996) Genetics 142, 953–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Templeton, A. (1996) Genetics 144, 1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wise, C., Sraml, M. & Easteal, S. (1998) Genetics 148, 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hasegawa, M., Cao, Y. & Yang, Z. (1998) Mol. Biol. Evol. 15, 1499–1505. [DOI] [PubMed] [Google Scholar]
- 33.Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., Davis, A. P., Dolinski, K., Dwight, S. S., Eppig, J. T., et al. (2000) Nat. Genet. 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Itoh, T., Martin, W. & Nei, M. (2002) Proc. Natl. Acad. Sci. USA 99, 12944–12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nachman, M. W., Boyer, S. N. & Aquadro, C. F. (1994) Proc. Natl. Acad. Sci. USA 91, 6364–6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Steinmetz, L. M., Sinha, H., Richards, D. R., Spiegelman, J. I., Oefner, P. J., McCusker, J. H. & Davis, R. W. (2002) Nature 416, 326–330. [DOI] [PubMed] [Google Scholar]
- 37.Leem, S. H., Park, J. E., Kim, I. S., Chae, J. Y., Sugino, A. & Sunwoo, Y. (2003) Mol. Cells 15, 55–61. [PubMed] [Google Scholar]
- 38.Akashi, H. (1996) Genetics 144, 1297–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.