

Abstract
Using poliovirus (PV) and its RNA-dependent RNA polymerase (RdRp) as our primary model system, we have advanced knowledge fundamental to the chemistry and fidelity of nucleotide addition by nucleic acid polymerase. Two fidelity checkpoints exist prior to nucleotide addition. The first toggles the enzyme between a nucleotide binding-occluded state and a nucleotide binding-competent state. The second represents an ensemble of conformational states of conserved structural motifs that permits retention of the incoming nucleotide in a state competent for phosphoryl transfer long enough for chemistry to occur. Nucleophilic attack of the alpha-phosphorous atom of the incoming nucleotide produces a pentavalent transition state, collapse of which is facilitated by protonation of the pyrophosphate leaving group by a general acid. All of the relevant conformational states of the enzyme are controlled by a network of interacting residues that permits remote-site residues to control active-site function. The current state of the art for PV RdRp enzymology is such that mechanisms governing fidelity of this enzyme can now be targeted genetically and chemically for development of attenuated viruses and antiviral agents, respectively. Application of the knowledge obtained with the PV RdRp to the development of vaccines and antivirals for emerging RNA viruses represents an important goal for the future.
1. Introduction
1.1 Four Classes of Nucleic Acid Polymerases Exist
Nucleic acid polymerases catalyze the template-dependent polymerization of (deoxy)nucleoside triphosphates and are essential for transcription, replication, and repair of the genomes of all organisms, including viruses [1,2]. Studies of nucleic acid polymerases in vitro have permitted the enzymes to be classified into four categories based upon the specificity for template and (deoxy)nucleoside triphosphate (NTP): (1) DNA-dependent DNA polymerase (DdDp); (2) RNA/DdDp (reverse transcriptase, RT); (3) DNA-dependent RNA polymerase (DdRp); and (4) RNA-dependent RNA polymerases (RdRps). It should be noted that the template and substrate specificity of the various classes of polymerases is not absolute and is very easily manipulated in vitro by subtle changes in solution conditions, for example, by changing the divalent cation cofactor employed in the reaction from Mg2+ to Mn2+ [3–8]. Model systems have emerged for the study of the kinetic, thermodynamic, and structural basis for the specificity and activity of the various classes of polymerases. We have established the RdRp from poliovirus (PV), as a model system for understanding the chemical, kinetic, thermodynamic, structural, and dynamical mechanisms employed by this class of polymerases. Studies presented in subsequent sections of this chapter will hopefully leave you convinced that the PV RdRp system is not only a useful model for the RdRp but for all classes of polymerases.
1.2 Universal and Adapted Features Revealed by Polymerase Structures
Crystal structures have been solved for single-subunit polymerases representing all four classes of polymerases [9–16]. In general, the overall topology of polymerases resembles a cupped, right hand with fingers, palm, and thumb subdomains. The RdRp contains an extension of the fingers, the so-called fingertips, that interacts with the thumb subdomain, leading to a completely encircled active site in this class of polymerases (Fig. 1A). The palm subdomain is the most conserved subdomain of polymerases and can be divided into several conserved structural motifs. In the case of the RdRp, seven conserved structural motifs exist: A–G (Fig. 1A). Motifs A and C contain conserved aspartic acid residues involved in binding the divalent cation cofactors required for nucleotidyl transfer (Fig. 1B). Motif A extends from the (deoxy)ribose-binding pocket to the catalytic center (Fig. 1B). Elements of this motif interacting with the triphosphate moiety and divalent cation superimpose exactly in different polymerases. Elements of this motif interacting with the sugar have evolved to sense the appropriate sugar configuration, thus linking the NTP-binding site to the catalytic site. Motif B contains residues that interact with the (deoxy)ribose moiety of the nucleotide substrate. Motif D was thought to provide structural integrity to the palm subdomain; however, the studies described herein have altered substantially this long-standing view by showing a role for this motif in the chemistry of nucleotidyl transfer. The remaining motifs are unique to RdRps, although some overlap with RTs also exists. Motif E provides interaction between the enzyme and primer. Motif F interacts with the triphosphate moiety of NTP and most likely functions at the earliest step of NTP binding. Motif G has no known function. Structural information is also available on polymerase elongation complexes [11,17–21]. Upon NTP binding, the fingers subdomain often moves relative to the thumb sub-domain, causing an open-to-closed transition that has been considered a rate-limiting step in the nucleotide-addition cycle. This view is now being reconsidered [22,23].
Fig. 1.
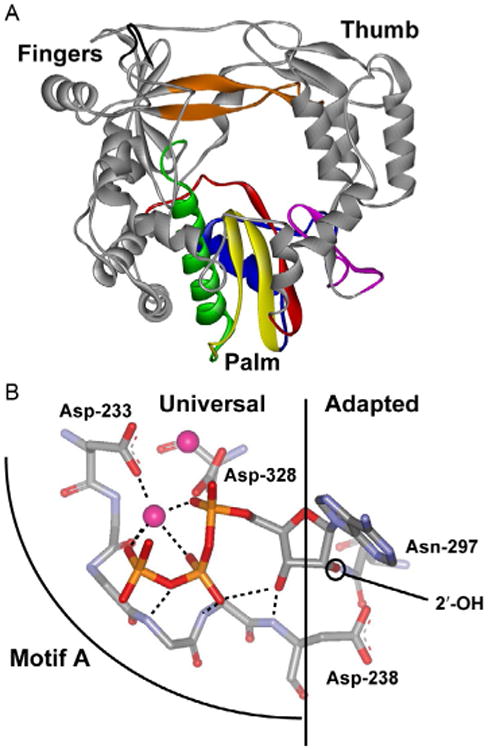
Universal and adapted features of polymerase structures. (A) PV RdRp structure. Conserved structural motifs in the palm subdomain are colored as follows: motif A, red (dark gray in the print version); motif B, green (dark gray in the print version); motif C, yellow (light gray in the print version); motif D, blue (dark gray in the print version); motif E, purple (gray in the print version); motif F, orange (gray in the print version); and motif G, black. (B) The NTP-binding site. The “universal” interactions are observed in all polymerases. The “adapted” interactions dictate the sugar configuration recognized by the polymerase. Reprinted with permission from D.W. Gohara, J.J. Arnold, C.E. Cameron, Poliovirus RNA-dependent RNA polymerase (3Dpol): kinetic, thermodynamic, and structural analysis of ribonucleotide selection, Biochemistry 43 (18) (2004) 5149–5158. Copyright (2004) American Chemical Society.
1.3 All Polymerases Employ a Two-Metal-Ion Mechanism for Catalysis
Given the conserved nature of the catalytic center of polymerases, the chemical mechanism is also conserved. The chemical mechanism is often referred to as the two-metal-ion mechanism (Fig. 2) [24,25]. Two divalent cations are required for polymerase-catalyzed nucleotidyl transfer; Mg2+ is the biologically relevant cofactor. One metal ion (metal B) enters the active site in complex with the nucleotide substrate. The other metal ion (metal A) likely enters from solution after binding of the Mg2+–NTP complex. The metals are stabilized in the active site by coordination to conserved residues in motifs A and C, oxygens from all three phosphates of NTP, and the oxygen from the primer terminus. It is generally accepted that the role of metal A is to increase the nucleophilicity of the primer 3′-OH by lowering the pKa of the hydroxyl. Metals A and B interact with oxygens of the α-phosphate so these metals could presumably increase the electrophilicity of the α-phosphorous atom. Finally, metal B may also organize the triphosphate into a conformation that is essential for catalysis—that is, permit the optimal distance between the 3′-OH and the α-phosphate to be achieved. Nucleotidyl transfer reactions likely go through a pentavalent transition state [24–27]. At least one-proton-transfer reaction must occur during the reaction: deprotonation of the 3′-OH nucleophile. As described later, we have altered this view of polymerase chemistry by revealing two proton-transfer reactions in the transition state and the use of a general acid for protonation of the PPi leaving group.
Fig. 2.
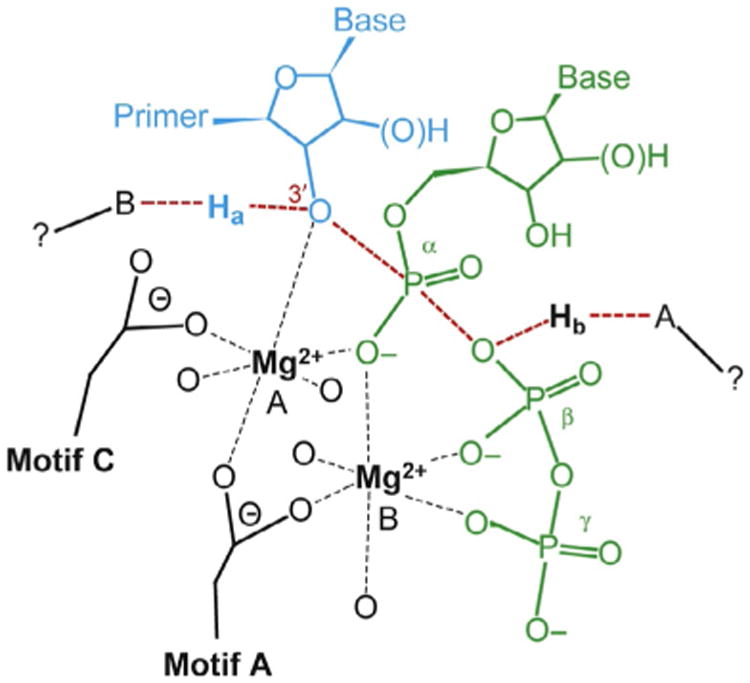
Two-metal-ion mechanism for nucleotidyl transfer. Nucleoside triphosphate (green, dark gray in the print version) enters the active site with a divalent cation (Mg2+, metal B). A second divalent cation (Mg2+, metal A) is coordinated by the 3′-OH of primer terminus (cyan, light gray in the print version), the nucleotide α-phosphate, as well as Asp residues of structural motifs A and C. The unidentified proton acceptor and proton donor are indicated as A and B, respectively. The proton from the 3′-OH is indicated as Ha, and the proton provided by the unidentified donor is indicated as Hb. Adapted from C. Castro, E.D. Smidansky, J.J. Arnold, K.R. Maksimchuk, I. Moustafa, A. Uchida, M. Gotte, W. Konigsberg, C.E. Cameron, Nucleic acid polymerases use a general acid for nucleotidyl transfer, Nat. Struct. Mol. Biol. 16 (2) (2009) 212–218.
1.4 Two Rate-Limiting Conformational Changes Bracket Phosphoryl Transfer in the Kinetic Mechanism for Single-Nucleotide Incorporation
Presteady-state kinetic analysis of DdDps and RTs has produced complete kinetic mechanisms for incorporation of correct and incorrect nucleotides (Fig. 3) [28–32]. The nucleotide-addition cycle requires five steps, with the nucleotidyl-transfer step bracketed by pre and postchemistry, conformational change steps. There is a consensus on the number of steps employed during the nucleotide-addition cycle. However, the rate-limiting step differs among polymerases and a physical description of the conformational changes is debated or not known. Experiments described later for the PV model system have begun to address these issues.
Fig. 3.
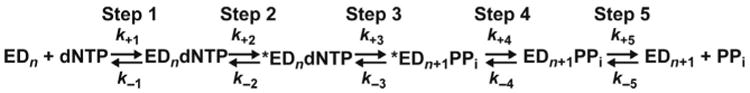
Complete kinetic mechanism for single-nucleotide incorporation for all classes of nucleic acid polymerases studied to date.
1.5 Views on the Kinetic and Structural Basis for Polymerase Fidelity Are Still Evolving
There is no debate that polymerase fidelity represents an important, and perhaps the final, frontier for the field of polymerase enzymology. From a kinetic point of view, there is no debate that much of the observed difference between correct and incorrect nucleotide incorporation is expressed in the observed rate constant for the reaction (kpol) rather than the equilibrium constant for NTP binding (Kd) [22,23,33]. The problem has been assignment of kpol to one or more of the microscopic rate constants for the elementary steps of the reaction. The reason for this problem is the absence of a universal approach to determine if or the extent to which the nucleotidyl-transfer step contributes to kpol. It was once thought that the use of the phosphorothioate elemental (thio) effect was a useful tool [33,34]. However, the absence of a clear value for the theoretical maximum that could be used to determine the extent to which chemistry is expressed in kpol has made “thio effect” a naughty pair of words in the field [23,33]. We have made two significant advances in the PV RdRp system that have begun to resolve this problem. We have uncovered solution conditions to make chemistry completely rate limiting, permitting empirical determination of the maximal thio effect [35,36]. In addition, we have shown that the solvent deuterium isotope effect can be used to probe for chemistry in kpol [37,38]. It is now becoming clear that both the prechemistry, conformational change step and chemistry are reflected in kpol. The question remains whether one or both steps contribute to fidelity. If both steps contribute, how are they coupled? Moreover, the possibility that steps after chemistry contribute to nucleotide incorporation fidelity has really not been addressed.
The structural basis for the conformational change preceding the chemical step is not known. Three hypotheses have been put forward. The first hypothesis is that this step reflects orientation of the triphosphate into the catalytically active conformation [30,39,40]. The second is that this step reflects the open-to-closed transition observed crystallographically after nucleotide binding [11,15,31,41]. The third is that this step reflects binding of metal A [19,42]. Published structures of DNA polymerase show that the open-to-closed transition can occur prior to binding of metal A, an observation interpreted to be inconsistent with this transition reflecting the conformational change observed kinetically [19].
1.6 A Role for Dynamics in Enzymic Catalysis
Conformational changes required for an enzyme to progress successfully through its reaction coordinate are now believed to be influenced by dynamics on timescales ranging from picoseconds to milliseconds [43–47]. Dynamics on the fastest timescales have been observed to be coordinated by networks that include not only residues present at the catalytic site but also at remote sites, and perhaps explain allostery [43–46]. Very little is known about polymerase dynamics. We have obtained evidence for dynamics on the nanosecond timescale as a determinant of nucleotide incorporation fidelity for the PV RdRp. We suggest that dynamics–function relationships for all polymerase will yield insight into the mechanism of nucleotide incorporation fidelity that structure–function relationships have been unable to achieve.
2. Nucleic Acid Polymerases Employ General Acid Catalysis
Nucleotidyl transfer requires, minimally, one-proton-transfer reaction. The proton from primer 3′-OH must be removed for catalysis to occur. The function of one (metal A) of the two divalent cations required for activity is to lower the pKa of this proton to facilitate catalysis. However, it was not known if the pyrophosphate leaving group needed to be protonated for efficient nucleotidyl transfer. Most structures of polymerases poised for or undergoing catalysis showed a basic amino acid near the β-phosphate, consistent with this possibility [19,48–52].
We have employed the PV RdRp to address the question of proton-transfer reactions during nucleotide addition. This system is uniquely suited to the characterization of the chemical mechanism of nucleotidyl transfer for two reasons. First, the stability of PV RdRp on our model primed template (sym/sub or s/s) is independent of pH [38,53]. Second, chemistry is the sole rate-limiting step when Mn2+ is employed as the divalent cation cofactor, thus permitting us to characterize this step without complications from conformational change steps [36].
We showed that the maximal rate constant for nucleotide incorporation (kpol) exhibited a bell-shaped dependence on pH (Fig. 4A). The pKa for the descending limb was 10.5, consistent with a Lys residue serving as a general acid. We observed a solvent deuterium kinetic isotope effect (SDKIE) on kpol, permitting us to count the number of protons transferring in the transition state for nucleotidyl transfer by performing a proton-inventory experiment [37,38,54,55]. In this experiment, the observed rate constant for nucleotide incorporation (kn) is determined at various mole fractions of D2O (n). Values for kn/kpol are plotted as function of n. If two proton-transfer reactions occur, then the data will fit best to a second-order polynomial. If one-proton-transfer reaction occurs, then the data will fit best to a line. For WT 3Dpol, the data fit best to a two-proton-transfer model (Fig. 4B). We identified Lys-359 on conserved structural motif D of 3Dpol as the putative general acid. If Lys-359 is the general acid, then by changing this residue to Leu, we should lose the descending limb of the pH rate profile and observe only one proton transferring in the transition state for nucleotidyl transfer. Both of these predictions were observed experimentally (see K359L in Fig. 4A and B).
Fig. 4.
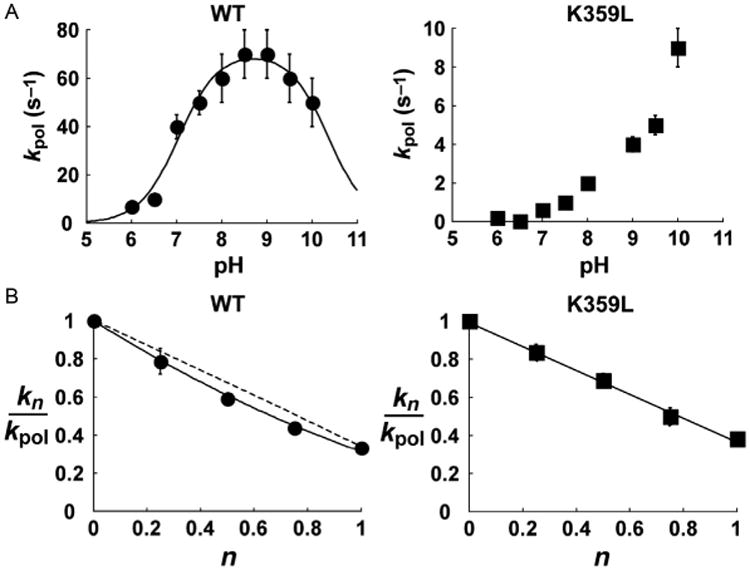
Lys-359 participates directly in nucleotidyl transfer and functions as a general acid. (A) Values for kpol plotted as a function of pH for WT and K359L PV RdRp. RdRp–sym/sub complexes were rapidly mixed with varying concentrations of ATP at different pH values using the stopped-flow assay. At each pH, time courses at a fixed nucleotide concentration were fit to a single exponential to obtain kobs. The value for kobs was then plotted as a function of ATP concentration and fit to a hyperbola to obtain kpol at a given pH value. In the case of WT PV RdRp, the solid line shows the fit of the data to a model describing two ionizable groups, yielding pKa values of 7.0±0.1 and 10.5±0.1. Error bars indicate the standard deviation. (B) Proton-inventory plots for WT and K359L PV RdRp. kn values were obtained in the presence of different mole fractions (n) of D2O.The ratio of kn/kpol (kpol is in H2O) is then plotted as a function of mole fraction of D2O. WT data (filled circles) were fit to a two-proton model (solid line) with straight, dashed line shown for comparison. K359L data (filled squares) were fit to a one-proton model. Error bars indicate the standard deviation. Adapted from C. Castro, E.D. Smidansky, J.J. Arnold, K.R. Maksimchuk, I. Moustafa, A. Uchida, M. Gotte, W. Konigsberg, C.E. Cameron, Nucleic acid polymerases use a general acid for nucleotidyl transfer, Nat. Struct. Mol. Biol. 16 (2) (2009) 212–218.
Since the first report of the two-metal-ion mechanism for nucleotidyl transfer was proposed, it has been generally accepted that active-site residues only contribute indirectly to the chemistry of nucleotidyl transfer by serving as ligands to one or more of the divalent cations required for catalysis. Our observation of a general acid in PV RdRp represents the first example of an active-site residue contributing directly to nucleotidyl transfer. This discovery is the first extension of the chemical mechanism for nucleotidyl transfer in almost three decades. Therefore, we felt obligated to determine if this observation was unique to the RdRp. We established three additional polymerase systems in the lab for this purpose [37]: the RT from human immunodeficiency virus (HIV RT), the DdDp from bacteriophage RB69 (RB69 DdDp), and the DdRp from bacteriophage T7 (T7 DdRp). T7 DdRp is a model for all A-family polymerases, and RB69 is a model for all B-family polymerases. All of these enzymes exhibited a SDKIE that was dependent on a conserved Lys residue (Table 1) [37]. Moreover, by changing this Lys residue to Leu, the proton inventory went from two to one (Table 1). These and other experiments lead us to conclude that all nucleic acid polymerases employ general acid catalysis for nucleotidyl transfer.
Table 1. Kinetic Analysis Of Pv Rdrp, Hiv-1 Rt, Rb69 Dddp, And T7 Ddrp Supports General Acid Catalysis In Nucleotidyl Transfer [37].
| Parameter Measured | PV RdRp | HIV RT | RB69 DdDp | T7 DdRp | ||||
|---|---|---|---|---|---|---|---|---|
| Lys-359 | Leu-359 | Lys-220 | Leu-220 | Lys-560 | Leu-560 | Lys-631 | Leu-631 | |
| kpol (s−1) | 50 ± 5 | 1 ± 0.1 | 60 ± 5 | 0.3 ± 0.1 | 200 ± 10 | 0.10 ± 0.01 | 60 ± 5 | 0.6 ± 0.1 |
| Kd,app (μM)a | 200 ± 20 | 700 ± 80 | 7 ± 1 | 5 ± 2 | 40 ± 5 | 1000 ± 100 | 300 ± 30 | 5.0 ± 1.0 × 104b |
| SDKIEc | 3.0 ± 0.3 | 2.5 ± 0.3 | 2.2 ± 0.4 | 1.8 ± 0.4 | 4.2 ± 0.2 | 1.8 ± 0.2 | 5.2 ± 0.5 | 2.6 ± 0.5b |
| PId | 2 | 1 | 2 | 1 | 2 | 1 | 2 | nde |
Kd,app is for (d)ATP.
Kd,app, kpol, and SDKIE values listed for T7 K631L were obtained by using data collected with 80 mM ATP, a subsaturating concentration.
SDKIE is solvent deuterium kinetic isotope effect, calculated as kobs in H2O/kobs in D2O at saturating [(d)ATP].
PI is proton inventory, calculated from a plot of kn/k0 as a function of n. The data were fit to a modified Gross–Butler equation for either a two-proton-transfer model or a one-proton-transfer model. The value reported is the proton-transfer model that best fits the data.
Not determined.
Together, these studies validate the PV RdRp system as a powerful model system not only for RdRps but for all classes of nucleic acid polymerases. Our studies have revealed an unexpected role for the general acid in nucleotide incorporation fidelity (Fig. 5A and B) [37,56,57]. We envision other possible roles for this residue in coupling the chemical step to other aspects of the kinetic mechanism for nucleotide incorporation.
Fig. 5.
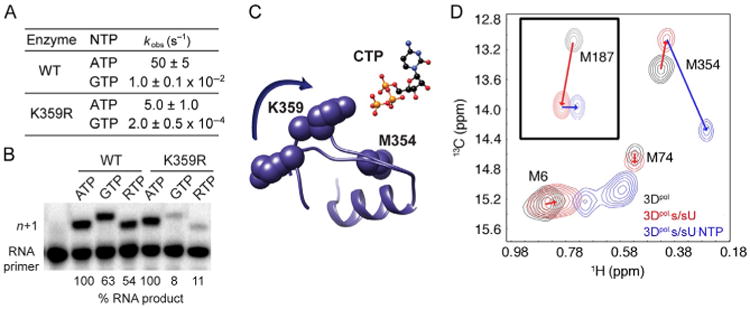
PV RdRp Lys-359 in structural motif D is a determinant of RdRp catalytic efficiency and fidelity. (A and B) K359R PV RdRp is slower and more faithful than the WT lysine-containing enzyme. RTP stands for ribavirin triphosphate. (C) Motif D changes conformation upon nucleotide binding. (D) The Met-354 resonance serves as a reporter of the catalytically competent complex. A substantial change in this resonance is only observed when the correct NTP is bound. Panels (A and B): Adapted from S.A. Weeks, C.A. Lee, Y. Zhao, E.D. Smidansky, A. August, J.J. Arnold, C.E. Cameron, A polymerase mechanism-based strategy for viral attenuation and vaccine development, J. Biol. Chem. 287 (38) (2012) 31618–31622. Panel (D): Adapted with permission from X. Yang, J.L. Welch, J.J. Arnold, D.D. Boehr, Long-range interaction networks in the function and fidelity of poliovirus RNA-dependent RNA polymerase studied by nuclear magnetic resonance, Biochemistry 49 (43) (2010) 9361–9371. Copyright (2010) American Chemical Society.
Collectively, our mechanistic studies were consistent with an active-site general acid that not only contributed to the efficiency of nucleotidyl transfer but also the fidelity of nucleotide selection. There was one problem. None of the beautiful structures of the picornaviral RdRps solved by Peersen and his group showed Lys-359 in a position consistent with hydrogen bonding to the incoming NTP [58–60]. In contrast, structures of the human norovirus RdRp in the absence and presence of NTP showed a dramatic change in the conformation of motif D, which was modeled by us for PV RdRp (Fig. 5C) [61]. This observation inspired the hypothesis that the rate-limiting conformational changes observed kinetically might actually correspond to structural changes that may not be revealed by X-ray crystallography and led to our pursuit of dynamics–function relationships of the RdRp. To this end, we established a collaboration with Dr. David Boehr in our Department of Chemistry at PSU. Together, our laboratories succeeded in applying the structural and dynamical wherewithal of multidimensional heteronuclear nuclear magnetic resonance spectroscopy to the study of PV RdRp, a 52 kDa protein [57,62,63]. Our initial experiments monitored 13C-labeled methionines in the protein. This choice was made because we hoped to use Met-354 in motif D as a reporter for any conformational changes of this motif, which contains Lys-359 (Fig. 5C). In going from unbound enzyme to the enzyme–RNA complex, a small change in the resonance for Met-354 was noted (Fig. 5D) [57,63]. Importantly, a huge change was observed for this resonance in the enzyme–RNA–NTP ternary complex [57,63], consistent with a large conformational change of motif D in the catalytically competent complex. Additional studies further implicated a hydrogen bonding interaction between the epsilon amino group of Lys-359 and the beta phosphate of the NTP [57].
3. Kinetic and Structural Determinants of Pv Rdrp Fidelity
We have solved the complete kinetic mechanism for correct and incorrect nucleotide incorporation catalyzed by PV RdRp. The kinetic mechanism for correct nucleotide incorporation is shown in Fig. 6. At least five steps are employed for a single-nucleotide-addition cycle. Nucleotide, in this case ATP, binds (step 1) followed by a conformational change step (step 2) that produces a complex competent for nucleotidyl transfer. Chemistry occurs (step 3) followed by a second conformational change step (step 4) that we currently assign to translocation. The cycle ends with release of PPi (step 5). Qualitatively, this mechanism is identical to that observed for other classes of nucleic acid polymerases [28–32]. However, this kinetic mechanism differs from others in that both the prechemistry conformational change step (step 2) and chemistry (step 3) are partially rate limiting for nucleotide addition. We have employed two methods to interrogate step 2 (Fig. 7A): isotope trapping (Fig. 7B) and EDTA vs HCl quenching (Fig. 7C and D). These experiments reveal a reduction in the equilibrium constant for step 2 when nucleotides with an incorrect sugar configuration or incorrect base are incorporated, thus revealing the use of the prechemistry, conformational change step as a kinetic determinant of nucleotide incorporation fidelity. Subsequent studies showed that fidelity was determined in large part by the stability of the ★ERnNTP complex, which is determined by the rate constant, k−2 [64].
Fig. 6.
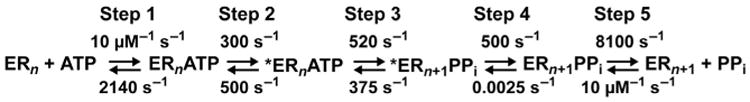
Complete kinetic mechanism for single-nucleotide incorporation catalyzed by PV RdRp. Steps 2 and 3 are both partially rate limiting for nucleotide incorporation. Adapted from J.J. Arnold, M. Vignuzzi, J.K. Stone, R. Andino, C.E. Cameron, Remote site control of an active site fidelity checkpoint in a viral RNA-dependent RNA polymerase, J. Biol. Chem. 280 (27) (2005) 25706–25716.
Fig. 7.
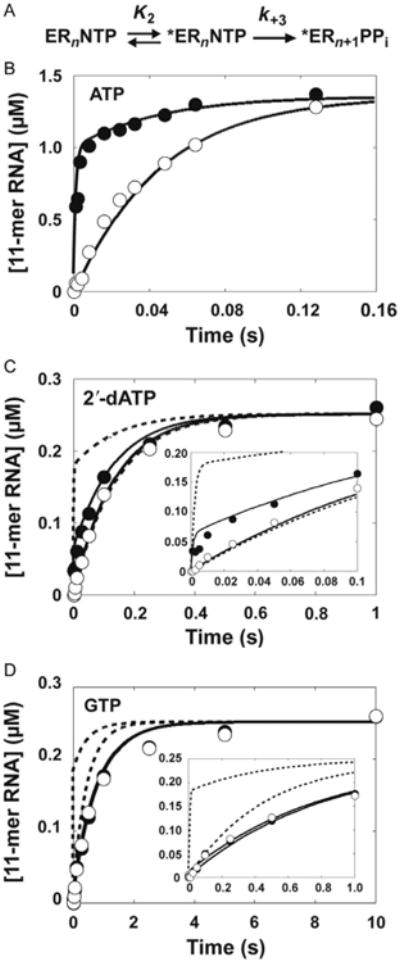
The prechemistry, conformational change step as a fidelity checkpoint for PV 3Dpol. (A) Minimal mechanism for pulse-chase analysis. (B) Kinetics of pulse-chase (•) and pulse-quench (○) using ATP. 4 μM PV RdRp was incubated with 20 μM sym/sub (10 μM duplex) and rapidly mixed with 130 μM [α-32P]ATP (3.8 Ci/mmol). At the indicated times, reactions were either chased by addition of 20 mM ATP final concentration or quenched by addition of 1 N HCl. After addition of the chase solution, the reaction was allowed to proceed for an additional 30 s at which time the reaction was quenched by addition of 1 N HCl. Immediately after addition of HCl, the solution was neutralized by addition of 1 M KOH and 300 mM Tris. The solid line represents the kinetic simulation of the data to the mechanism shown in (A) with step 2 (K2) equal to 3 and k+3 equal to 30 s−1. The simulated curve of the pulse-quench data predicts the rate of formation of ERn+1; the simulated curve of the pulse-chase data predicts the rate of formation of *ERnNTP and ERn+1. (C) 2′-dATP. A surrogate for the pulse-chase pulse-quench reaction was used for both 2′-dATP and GTP whereby the reaction was quenched by either EDTA (•) or HCl (○). The solid line represents the kinetic simulation of the data to a mechanism with step 2 (K2) equal to 0.4 and k+3 equal to 30 s−1. The dotted line represents the kinetic simulation of the data fit to a mechanism with step 2 (K2) equal to 3 and k+3 equal to 10 s−1. (D) GTP. The solid line represents the kinetic simulation of the data fit to a mechanism with step 2 (K2) equal to 0.05 and k+3 equal to 30s−1. The dotted line represents the kinetic simulation of the data to a mechanism with step 2 (K2) equal to 3 and k+3 equal to 3 s−1. Reprinted with permission from J.J. Arnold, D.W. Gohara, C.E. Cameron, Poliovirus RNA-dependent RNA polymerase (3Dpol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mn2+, Biochemistry 43 (18) (2004) 5138–5148. Copyright (2004) American Chemical Society.
All nucleic acid polymerases have a motif equivalent to conserved structural motif A found in RdRps and RTs. This motif extends from the (deoxy)ribose-binding pocket to the top of the catalytic site (Fig. 1B). In the ribose-binding pocket, at least one residue (Asp-238) stabilizes the ribose configuration so that the 3′-OH of incoming nucleotide can interact with the motif A backbone. In the catalytic site, both the backbone and side chains of motif A serve as ligands to metal A, the divalent cation required for catalysis. We have shown that both amino acid substitutions in motif A and the nature of the divalent cation can change the equilibrium constant for the prechemistry, conformational change step [65]. We currently assign step 2 to reorientation of the triphosphate into a position competent for nucleotidyl transfer. These and other studies link motif A to step 2 of the kinetic mechanism, thus revealing conserved structural motif A as a structural determinant of nucleotide incorporation fidelity.
4. Remote-Site Control of an Active-Site Fidelity Checkpoint
We have isolated two PV mutants: one with an antimutator phenotype [64] and the other with a mutator phenotype [66]. The antimutator phenotype was conferred by a change of Gly-64 to Ser (G64S); the mutator phenotype was conferred by a change of His-273 to Arg (H273R). Surprisingly, both Gly-64 and His-273 are located in the fingers domain of PV RdRp, not the active site (Fig. 8). We will use antimutator synonymously with high fidelity and mutator synonymously with low fidelity. As expected, G64S PV was less sensitive than WT PV to ribavirin, a mutagenic ribonucleoside [64], and H273R PV was more sensitive to ribavirin than WT PV [66]. The mutation frequency observed in cell culture was consistent with the mutation frequency observed for the corresponding PV RdRp derivatives determined biochemically [64,66]. The surprise was that the change in fidelity appeared to be no greater than threefold. Detailed kinetic analysis, in particular isotope-trapping experiments, of these PV RdRp derivatives revealed a change in the equilibrium constant for the prechemistry, conformational change step (step 2) as the kinetic basis for the change in nucleotide incorporation fidelity PV RdRp. These data provide indisputable, genetic evidence for a role of the prechemistry, conformational change step as a determinant of PV RdRp fidelity. Moreover, these data reveal remote-site control of this active-site fidelity checkpoint.
Fig. 8.
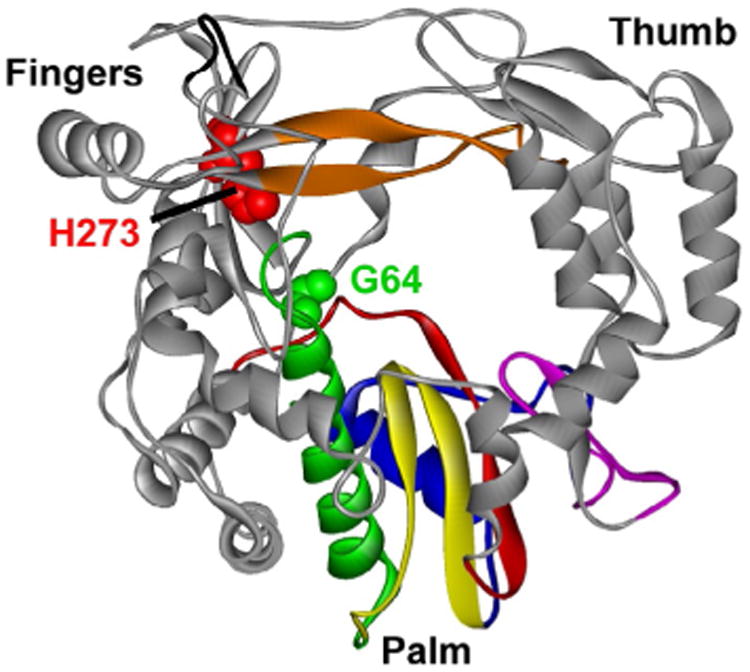
Fidelity determinants located at sites remote from the catalytic site. The location of Gly-64 (green, light gray in the print version) and His-273 (red, dark gray in the print version) is indicated. Reprinted from V.K. Korboukh, C.A. Lee, A. Acevedo, M. Vignuzzi, Y. Xiao, J.J. Arnold, S. Hemperly, J.D. Graci, A. August, R. Andino, et al., RNA virus population diversity, an optimum for maximal fitness and virulence, J. Biol. Chem. 289 (43) (2014) 29531–29544.
5. RdRp Fidelity is a Determinant of Viral Virulence
Our results suggested that PV RdRp fidelity altered viral population dynamics in cell culture. We were therefore interested in determining the impact of altered fidelity on virus multiplication in a more complex environment. For these experiments, we collaborated with Dr. Raul Andino (UCSF). Dr. Andino developed a new strain of mice expressing the PV receptor, cPVR mice [67]. Infection of these mice with WT PV at a dose of 108 plaque-forming units (pfu) kills all mice in 5 days, with 50% of the mice developing paralysis (PD50) at a dose ∼107 pfu (Table 2). The low-fidelity mutant was attenuated in this highly permissive animal model (Table 2). The PD50 was increased on the order of two logs for H273R PV (Table 2). These data suggest that RdRp fidelity is optimized. Moreover, these data show that RdRp fidelity is a determinant of virulence, a completely novel concept at the time [66]. A great deal of data exist that point to the RdRp as a virulence determinant, even in the case of the 1918 Spanish flu strain [69–71]; however, the mechanism is unknown. What an impact two mutants can have on our understanding of the chemistry and biology of a virus.
Table 2. RdRp Fidelity Is a Determinant of Viral Virulence [66,68].
| Virus | PD50a |
|---|---|
| WT | 2 × 107 |
| G64S | 1 × 107 |
| H273R | 1 × 109 |
Dose of virus in plaque-forming units that leads to paralysis in 50% of inoculated animals.
6. RdRp Fidelity Mutants are Vaccine Candidates
The practical application of any attenuated virus is vaccination. Therefore, mice were inoculated with the high-fidelity or low-fidelity virus. Four weeks or 6 months thereafter, these mice were challenged with five times the lethal dose of WT PV. In most cases, 100% of the mice were protected [56,66,68]. We conclude that an understanding of RdRp fidelity is no longer just a fundamental question of polymerase enzymology but now underpins a novel strategy for vaccine development.
The fact that the first PV fidelity mutants were caused by amino acid substitutions in the RdRp that were not only remote from the active site but were also not conserved across RdRp family of enzymes. In contrast, the general acid of PV, Lys-359, is conserved (Fig. 9A). The K359R PV mutant produced infectious virus [56]. There was a longer eclipse phase of the life cycle without a substantial change in the kinetics of the exponential phase of the life cycle and culminating with a virus yield one log lower than wild type (Fig. 9B). The ability to produce reasonable titers of virus permitted us to determine if the reduced kinetics of virus production caused attenuation of the virus in the cPVR transgenic mouse model [67]. To evaluate virus multiplication and virulence in this model, we performed an intraperitoneal inoculation as this permits the largest virus inoculum to be used (3 mL). K359R PV was attenuated relative to WT PV (Fig. 9C) [56]. The meaning of this phenotype was unclear. Was K359R PV noninfectious or just incapable of causing disease? We reasoned that if K359R PV was infectious, then these infected animals might be protected from challenge with WT PV. Indeed, at a dose of 1 × 108, K359R PV protected 100% of animals from WT PV-induced paralysis (Fig. 9D). This observation supports the conclusion that we have identified a polymerase mechanism-based strategy for PV attenuation and vaccine development [56]. We compared protection by K359R PV to the type 1 Sabin strain of PV and found that there was no more than a one-log difference in the dose required for protection (Fig. 9D). The K359R PV has one amino acid substitution, whereas Sabin 1 PV has 21 amino acid substitutions [72]. In collaboration with Dr. Andrew Macadam (National Institute of Biological Standards and Control, UK), we showed that K359R PV was as attenuated as Sabin 1 PV using an intraspinal route of inoculation, providing some evidence for the safety of this approach to viral attenuation (unpublished observations).
Fig. 9.
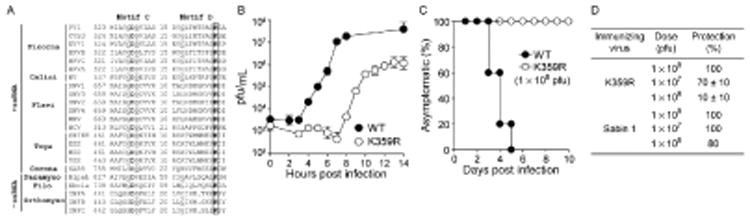
Conserved structural motif D as a target for viral attenuation and vaccine development. (A) Conservation of the motif D lysine in viral RdRps. (B) K359R PV exhibits a delayed growth phenotype. (C) K359R PV is attenuated. (D) K359R PV elicits a protective immune response. Adapted from S.A. Weeks, C.A. Lee, Y. Zhao, E.D. Smidansky, A. August, J.J. Arnold, C.E. Cameron, A polymerase mechanism-based strategy for viral attenuation and vaccine development, J. Biol. Chem. 287 (38) (2012) 31618–31622.
We have realized a polymerase mechanism-based strategy for viral attenuation. The question now is whether or not this knowledge can be applied to other viral systems.
7. Correlated Motions of Functionally Important Motifs of The RdRp May Be a Determinant of Nucleotide Incorporation Fidelity
Our studies of incorporation fidelity revealed the ability of remote sites of PV RdRp to control conformational changes that occur at the active site. We made a similar observation in our studies of dihydrofolate reductase [73]. In that system, it turned out that remote-site control of active-site function was mediated by protein dynamics on multiple timescales [74]. Therefore, our observations with 3Dpol inspired the provocative hypothesis that polymerase incorporation fidelity might be controlled by dynamics as well. As a first step in this direction, we turned to molecular dynamics simulation (MD). MD is a powerful, computational approach to study protein dynamics [75,76]. In spite of all of the structural information available for polymerases, MD has primarily been applied to polymerases involved in DNA repair [77–80]. We have exploited the availability of structures for polymerases from multiple picornaviruses, including PV [60], Coxsackievirus B3 (CVB3) [81], human rhinovirus type 16 (HRV16) [82], and foot-and-mouth disease virus (FMDV) [83] to determine if conserved dynamical properties exist for these enzymes that could be important for function.
We performed an all-atom (or atomistic) MD study of the four picornaviral RdRps listed earlier using the protocol illustrated in Fig. 10 [84,85]. All simulations were performed without substrates or metal ions. First, we performed the simulation of PV RdRp for 25 ns. The overall structure was maintained on the timescale of this experiment. The flexibility of the enzyme structure was determined by using the MD data to calculate B-factors. These data were in good agreement with B-factors obtained from X-ray data, suggesting that the force field and the protocol employed for the simulation are reasonable. Next, we performed MD on the other picornaviral RdRps. However, in each case we evaluated an incrementally shorter timescale: FMDV 3Dpol (20 ns), CVB3 3Dpol, and HRV16 3Dpol (14 ns). The shortest timescale (14 ns) did not alter the reliability of the data, decreasing substantially the amount of computational time required to perform the experiment. All systems reached equilibrium in 2–4 ns. The temperature, energy, and density of the system were monitored throughout the simulation, and all of these parameters displayed well-behaved dynamic trajectories.
Fig. 10.
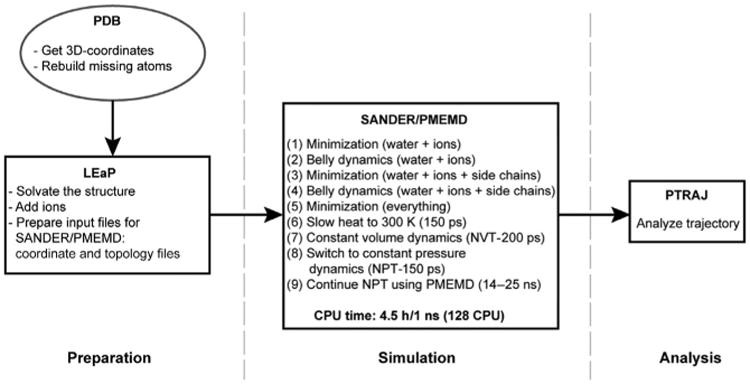
Molecular dynamics protocol. Flow chart describing the protocol used to perform the all-atom MD simulations. Programs LEaP, SANDER, PMEMD, and PTRAJ are modules in the AMBER simulation package [84].
Principal component analysis (PCA) permits us to separate large, concerted motions from the noise [86,87]. The analysis is carried out by calculating the correlation matrix for the displacements of Cα atoms from the averaged structure. Treating the matrix as an eigenvalue problem results in a set of eigenvectors (or principal components), defining a new coordinate space to describe the observed motions in the simulation. The number of PCs is equivalent to three times the number of amino acid residues in the data set. However, when the PCs are arranged in descending order, most of the variance (displacements/motion) in the data can be accounted for in a small fraction of the PCs. In the case of the simulations for the four picornaviral RdRps (∼461 residues), the first six PCs accounted for more than 50% of the global motion in the polymerases. Thus, we restricted our analysis to these PCs. In doing so, we restricted our focus to the major motions of each polymerase, facilitating comparative analysis. Our premise was that if motion is important, then it should be a major motion and should be conserved. Because the magnitude of the values for the displacements varied for each polymerase, direct visual comparison of the top six PCs for each polymerase was difficult. To address this issue, the sum of the top six PCs were normalized to the average of the sum of the five least flexible residues. This analysis revealed a remarkable conservation in the dynamics of the four polymerases [85]. In order to determine if correlated motions existed in the polymerases, we calculated dynamics cross-correlation maps (DCCMs) [88]. The DCCMs for three of the four picornaviral RdRps are shown in Fig. 11. As observed for the PCA, the DCCMs were remarkably similar. Correlated motions were observed between several conserved structural motifs, including A and D (region I in Fig. 11) and F and G (region II in Fig. 11). The amino acid sequences of CVB3 RdRp and FMDV RdRp are 74% and 30%, respectively, identical to that of PV RdRp. Interestingly, the DCCM for PV G64S RdRp exhibited substantial differences from PV RdRp [85]. The ability for MD to reveal conserved dynamics within this family of polymerases engenders confidence in our protocol. Moreover, these results provide evidence for evolution of structure and dynamics, a very exciting finding with significant implications for the existence of conserved dynamics in other polymerase systems.
Fig. 11.
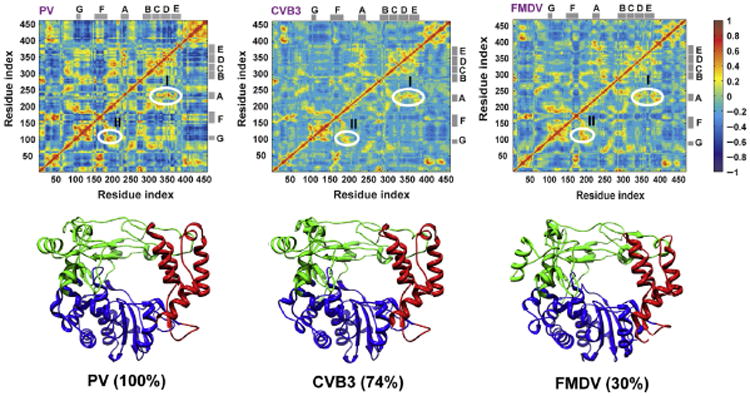
Conservation of correlated motions in picornaviral RdRps. The cross-correlation of the atomic displacement vectors of Cα atoms was examined. The pair-wise correlation of the displacements of Cα atoms relative to a reference structure was calculated and used to build the dynamic cross-correlation matrix (DCCM). Each element of the matrix is correlation between displacements of Cα atoms of a residue pair (ij). The matrix is plotted as a color-coded map, shown at the top panel for the three polymerases: PV, CVB3, and FMDV. For completely correlated motions the correlation score measures (+1), whereas for completely uncorrelated motions the score is (−1). The maps are coded with color gradient for the correlation scores (+1 more red, gray in the print version; −1 more blue, dark gray in the print version). The similarity between the correlations observed in the three simulated polymerases can be inferred from the comparable patterns of the color distributions in the three maps. Two regions I and II with positive correlations are labeled in the maps. Region I corresponds to the coupled motion of motifs A and D; region II refers to motifs F and G. The locations of the different motifs are indicated by the gray bars at the top and right of the maps. The bottom panel shows the structures of the three polymerases with the fingers, palm, and thumb subunits colored green (light gray in the print version), blue (dark gray in the print version), and red (gray in the print version), respectively. Despite the diverse sequence identity between the different picornaviral polymerases (30–74%), the similarity of the overall structure and dynamics is noteworthy. The top panel of the figure was created using MATLAB 7.6. Adapted from I.M. Moustafa, H. Shen, B. Morton, C.M. Colina, C.E. Cameron, Molecular dynamics simulations of viral RNA polymerases link conserved and correlated motions of functional elements to fidelity, J. Mol. Biol. 410 (1) (2011) 159–181. Copyright (2011), with permission from Elsevier.
In other systems in which correlated motions have been observed, the correlated motions are often coordinated by virtue of the existence of a global network of interacting amino acid residues [89,90]. We have been able to identify such a network in PV RdRp [85]. Interestingly, Gly-64 and His-273, remote-site residues that influence the prechemistry, conformational change step, are located in this network. Importantly, disruptions in the network resulting from amino acid substitutions at these positions cause changes in correlated motions observed between conserved structural motifs (unpublished observations). Together, these observations lead us to propose that this network may be responsible for this and perhaps other conformational changes required for RdRp function.
8. Nucleotide Binding-Occluded and Binding-Competent States of The RdRp–RNA Complex as Determinants of Fidelity
Our MD experiments have revealed two states of the PV RdRp–RNA complex based on the conformation and dynamics of conserved residues in conserved structural motifs A (Asp-238), B (Asn-297), D (Lys-359), E (Lys-375), and F (Arg-174) [91]. The first state occludes binding of the incoming NTP because of an ionic interaction between Asp-238 and Arg-174 (binding occluded in Fig. 12A). The second state is competent for NTP binding as the 238–174 interaction is broken, causing a rearrangement of the pocket that includes movement of Lys-359 toward the active site (binding competent in Fig. 12A). Closure of motif D to position Lys-359 to serve as the general acid would then produce the catalytically competent state (catalytic in Fig. 12A) [57]. The distance between the Asp-238 and Arg-174 side chains as a function of time is indicative of the interconversion between the NTP-binding occluded (points below the red, gray in the print version, line in Fig. 12B) and competent (points above the red, gray in the print version, line in Fig. 12B) states. In the absence of RNA and NTP, the enzyme samples both states, but the occluded state is favored 9:1 (see WT in Fig. 12B). Addition of RNA increases the sampling frequency such that the competent state is present 40% of the time (see WT-RNA in Fig. 12B). In the mutator polymerase, the competent state is present 98% of the time (see H273R-RNA in Fig. 12B). By analyzing the differences in the average structures for the WT and H273R enzymes, it is evident that accommodation of the larger side chain at position 273 leads to a chain reaction that disconnects the network of interactions stabilizing the occluded conformation (direction of red, gray in the print version, arrows in Fig. 12C indicate the change from WT, gray, to H273R, black). From these data, we suggest that at least two structural changes comprise the fidelity checkpoint observed kinetically and that the equilibrium between occluded and competent state may be a determinant of fidelity.
Fig. 12.
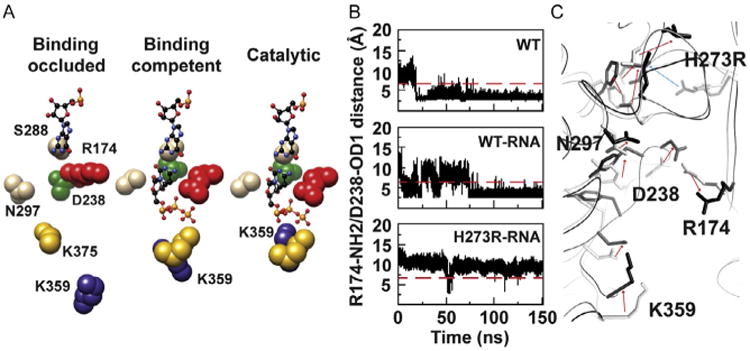
Physical mechanism of PV RdRp fidelity: an hypothesis. (A) Active-site conformational changes revealed by MD simulations of PV RdRp–RNA binary complex. (B) Time evolution of the distance between NH2 of Arg-174 and OD1 of Asp-238 atoms during 150 ns MD simulations of the free WT and the binary complexes of WT and H273R RdRps. (C) The average structures of WT-RNA (gray) and H273R-RNA (black) observed during the MD simulations are superimposed. Direction of arrows indicates the change from WT-RNA to H273R-RNA. Adapted from I.M. Moustafa, V.K. Korboukh, J.J. Arnold, E.D. Smidansky, L.L. Marcotte, D.W. Gohara, X. Yang, M.A. Sanchez-Farran, D. Filman, J.K. Maranas, et al., Structural dynamics as a contributor to error-prone replication by an RNA-dependent RNA polymerase, J. Biol. Chem. 289 (52) (2014) 36229–36248.
Worth noting, the dynamics sampled by the PV WT RdRp or complexes thereof are restricted to those of relevance to the next step in the reaction coordinate [91]. For example, PV WT RdRp samples conformations required for nucleic acid binding. Once nucleic acid is bound, however, PV WT RdRp–RNA complex samples states competent for nucleotide binding [91].
9. Conserved, Active-Site Determinants Of RdRp Incorporation Fidelity May Exist That Can Be Targeted For Viral Attenuation
One of the most exciting advances made during our studies of RdRp fidelity has been the formulation of a hypothesis linking the structural dynamics and accessibility of the NTP-binding site to fidelity [91]. WT RdRp cycles between occluded (Fig. 13A) and competent (Fig. 13B) conformations. Side chains indicated interact with one or more portions of the incoming nucleotide. In the binding-occluded conformation, all of these residues interact with each other. Therefore, a stable shift of the equilibrium to the binding-competent conformation requires binding of a correct nucleotide. When a nucleotide with an incorrect sugar configuration or base binds, one or more interactions in the binding-occluded conformation will not be disrupted. Partial disruption may preclude a stable shift to the binding-competent conformation. Therefore, an RdRp derivative that favors the binding-occluded state should exhibit a fidelity higher than WT. Conversely, one that favors the binding-competent state should exhibit a fidelity lower than WT. A central, stabilizing determinant of the occluded state is the interaction between Asp-238 (motif A) and Arg-174 (motif F) (Fig. 13A). These same residues are critical for stabilizing the bound NTP (Fig. 13B), consistent with our suggestion that binding of a correct nucleotide might diminish return to the occluded state and nucleotide release. Importantly, essentially all of these residues are conserved among the RdRps of positive-strand RNA viruses. It may be possible to identify substitutions of one or more of these residues that create fidelity variants that can serve as vaccine candidates for any RNA virus, especially those that have yet to emerge.
Fig. 13.
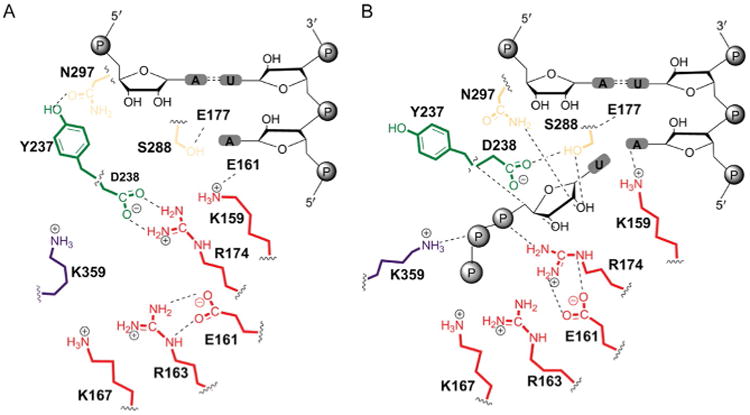
Proposed structural dynamics in the transition from NTP binding-occluded state to NTP binding-competent state employs conserved residues of the RdRp. All residues shown are conserved among RdRps of positive-strand viruses. (A) NTP binding-occluded state. (B) NTP binding-competent state. Adapted from I.M. Moustafa, V. K. Korboukh, J.J. Arnold, E.D. Smidansky, L.L. Marcotte, D.W. Gohara, X. Yang, M.A. Sanchez-Farran, D. Filman, J.K. Maranas, et al., Structural dynamics as a contributor to error-prone replication by an RNA-dependent RNA polymerase, J. Biol. Chem. 289 (52) (2014) 36229–36248.
Acknowledgments
For nearly 20 years, C.E.C. and J.J.A. have worked to elucidate the fundamental principles responsible for the specificity, chemistry, and biology of the viral RdRp. The summary presented here would not have been possible without the collaborations that we have had with graduate and postdoctoral students and colleagues both here at PSU and elsewhere. Worth special mention are Raul Andino (UCSF), David Boehr (PSU), Coray Colina (PSU) as these scientists have both enabled our technical capabilities and have enriched our research. Our studies of the RdRp have been supported by Grant AI045818 from NIAID, NIH.
References
- 1.Buck KW. Comparison of the replication of positive-stranded RNA viruses of plants and animals. Adv Virus Res. 1996;47(1):159–251. doi: 10.1016/S0065-3527(08)60736-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kornberg A, Baker T. DNA Replication. two. W. H. Freeman and Co; New York: 1991. [Google Scholar]
- 3.Arnold JJ, Ghosh SK, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3Dpol): divalent cation modulation of primer, template, and nucleotide selection. J Biol Chem. 1999;274(52):37060–37069. doi: 10.1074/jbc.274.52.37060. [DOI] [PubMed] [Google Scholar]
- 4.Beckman RA, Mildvan AS, Loeb LA. On the fidelity of DNA replication: manganese mutagenesis in vitro. Biochemistry. 1985;24(21):5810–5817. doi: 10.1021/bi00342a019. [DOI] [PubMed] [Google Scholar]
- 5.Goodman MF, Keener S, Guidotti S, Branscomb EW. On the enzymatic basis for mutagenesis by manganese. J Biol Chem. 1983;258(6):3469–3475. [PubMed] [Google Scholar]
- 6.Huang Y, Beaudry A, McSwiggen J, Sousa R. Determinants of ribose specificity in RNA polymerization: effects of Mn2+ and deoxynucleoside monophosphate incorporation into transcripts. Biochemistry. 1997;36(44):13718–13728. doi: 10.1021/bi971609o. [DOI] [PubMed] [Google Scholar]
- 7.Liu J, Tsai MD. DNA polymerase beta: pre-steady-state kinetic analyses of dATPalphaS stereoselectivity and alteration of the stereoselectivity by various metal ions and by site-directed mutagenesis. Biochemistry. 2001;40:9014–9022. doi: 10.1021/bi010646j. [DOI] [PubMed] [Google Scholar]
- 8.Tabor S, Richardson CC. Effect of manganese ions on the incorporation of dideoxynucleotides by bacteriophage T7 DNA polymerase and Escherichia coli DNA polymerase I. Proc Natl Acad Sci U S A. 1989;86(11):4076–4080. doi: 10.1073/pnas.86.11.4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beese LS, Friedman JM, Steitz TA. Crystal structures of the Klenow fragment of DNA polymerase I complexed with deoxynucleoside triphosphate and pyrophosphate. Biochemistry. 1993;32(51):14095–14101. doi: 10.1021/bi00214a004. [DOI] [PubMed] [Google Scholar]
- 10.Hansen JL, Long AM, Schultz SC. Structure of the RNA-dependent RNA poly-merase of poliovirus. Structure (London, England: 1993) 1997;5(8):1109–1122. doi: 10.1016/s0969-2126(97)00261-x. [DOI] [PubMed] [Google Scholar]
- 11.Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science (New York) 1998;282(5394):1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 12.Jacobo-Molina A, Clark AD, Williams RL, Nanni RG, Clark P, Ferris AL, Hughes SH, Arnold E. Crystals of a ternary complex of human immunodeficiency virus type 1 reverse transcriptase with a monoclonal antibody Fab fragment and doublestranded DNA diffract X-rays to 3.5-Å resolution. Proc Natl Acad Sci U S A. 1991;88(23):10895–10899. doi: 10.1073/pnas.88.23.10895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science (New York) 1992;256(5065):1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 14.Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat Struct Biol. 1999;6(10):937–943. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- 15.Ollis DL, Brick P, Hamlin R, Xuong NG, Steitz TA. Structure of large fragment of Escherichia coli DNA polymerase I complexed with dTMP. Nature. 1985;313(6005):762–766. doi: 10.1038/313762a0. [DOI] [PubMed] [Google Scholar]
- 16.Sousa R, Chung YJ, Rose JP, Wang BC. Crystal structure of bacteriophage T7 RNA polymerase at 3.3 Å resolution. Nature. 1993;364(6438):593–599. doi: 10.1038/364593a0. [DOI] [PubMed] [Google Scholar]
- 17.Cheetham GM, Jeruzalmi D, Steitz TA. Structural basis for initiation of transcription from an RNA polymerase-promoter complex. Nature. 1999;399(6731):80–83. doi: 10.1038/19999. [DOI] [PubMed] [Google Scholar]
- 18.Halvas EK, Svarovskaia ES, Freed EO, Pathak VK. Wild-type and YMDD mutant murine leukemia virus reverse transcriptases are resistant to 2′,3′-dideoxy-3′-thiacytidine. J Virol. 2000;74(14):6669–6674. doi: 10.1128/jvi.74.14.6669-6674.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnson SJ, Taylor JS, Beese LS. Processive DNA synthesis observed in a polymerase crystal suggests a mechanism for the prevention of frameshift mutations. Proc Natl Acad Sci U S A. 2003;100(7):3895–3900. doi: 10.1073/pnas.0630532100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kiefer JR, Mao C, Braman JC, Beese LS. Visualizing DNA replication in a catalytically active Bacillus DNA polymerase crystal. Nature. 1998;391(6664):304–307. doi: 10.1038/34693. [DOI] [PubMed] [Google Scholar]
- 21.Yin YW, Steitz TA. Structural basis for the transition from initiation to elongation transcription in T7 RNA polymerase. Science (New York) 2002;298(5597):1387–1395. doi: 10.1126/science.1077464. [DOI] [PubMed] [Google Scholar]
- 22.Johnson KA. Role of induced fit in enzyme specificity: a molecular forward/reverse switch. J Biol Chem. 2008;283(39):26297–26301. doi: 10.1074/jbc.R800034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joyce CM, Benkovic SJ. DNA polymerase fidelity: kinetics, structure, and checkpoints. Biochemistry. 2004;43(45):14317–14324. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 24.Joyce CM, Steitz TA. Polymerase structures and function: variations on a theme? J Bacteriol. 1995;177(22):6321–6329. doi: 10.1128/jb.177.22.6321-6329.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steitz TA. DNA polymerases: structural diversity and common mechanisms. J Biol Chem. 1999;274(25):17395–17398. doi: 10.1074/jbc.274.25.17395. [DOI] [PubMed] [Google Scholar]
- 26.Hengge AC. Isotope effects in the study of phosphoryl and sulfuryl transfer reactions. Acc Chem Res. 2002;35(2):105–112. doi: 10.1021/ar000143q. [DOI] [PubMed] [Google Scholar]
- 27.Lahiri SD, Zhang G, Dunaway-Mariano D, Allen KN. The pentacovalent phosphorus intermediate of a phosphoryl transfer reaction. Science (New York) 2003;299(5615):2067–2071. doi: 10.1126/science.1082710. [DOI] [PubMed] [Google Scholar]
- 28.Dahlberg ME, Benkovic SJ. Kinetic mechanism of DNA polymerase I (Klenow fragment): identification of a second conformational change and evaluation of the internal equilibrium constant. Biochemistry. 1991;30(20):4835–4845. doi: 10.1021/bi00234a002. [DOI] [PubMed] [Google Scholar]
- 29.Kati WM, Johnson KA, Jerva LF, Anderson KS. Mechanism and fidelity of HIV reverse transcriptase. J Biol Chem. 1992;267(36):25988–25997. [PubMed] [Google Scholar]
- 30.Kuchta RD, Mizrahi V, Benkovic PA, Johnson KA, Benkovic SJ. Kinetic mechanism of DNA polymerase I (Klenow) Biochemistry. 1987;26(25):8410–8417. doi: 10.1021/bi00399a057. [DOI] [PubMed] [Google Scholar]
- 31.Patel SS, Wong I, Johnson KA. Pre-steady-state kinetic analysis of processive DNA replication including complete characterization of an exonuclease-deficient mutant. Biochemistry. 1991;30(2):511–525. doi: 10.1021/bi00216a029. [DOI] [PubMed] [Google Scholar]
- 32.Wong I, Patel SS, Johnson KA. An induced-fit kinetic mechanism for DNA replication fidelity: direct measurement by single-turnover kinetics. Biochemistry. 1991;30(2):526–537. doi: 10.1021/bi00216a030. [DOI] [PubMed] [Google Scholar]
- 33.Showalter AK, Tsai MD. A reexamination of the nucleotide incorporation fidelity of DNA polymerases. Biochemistry. 2002;41(34):10571–10576. doi: 10.1021/bi026021i. [DOI] [PubMed] [Google Scholar]
- 34.Herschlag D, Piccirilli JA, Cech TA. Ribozyme-catalyzed and nonenzymatic reactions of phosphate diesters: rate effects upon substitution of sulfur for a nonbridging phosphoryl oxygen atom. Biochemistry. 1991;30:4844–4854. doi: 10.1021/bi00234a003. [DOI] [PubMed] [Google Scholar]
- 35.Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3Dpol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mg2+ Biochemistry. 2004;43(18):5126–5137. doi: 10.1021/bi035212y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnold JJ, Gohara DW, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3Dpol): pre-steady-state kinetic analysis of ribonucleotide incorporation in the presence of Mn2+ Biochemistry. 2004;43(18):5138–5148. doi: 10.1021/bi035213q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Castro C, Smidansky ED, Arnold JJ, Maksimchuk KR, Moustafa I, Uchida A, Gotte M, Konigsberg W, Cameron CE. Nucleic acid polymerases use a general acid for nucleotidyl transfer. Nat Struct Mol Biol. 2009;16(2):212–218. doi: 10.1038/nsmb.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Castro C, Smidansky E, Maksimchuk KR, Arnold JJ, Korneeva VS, Gotte M, Konigsberg W, Cameron CE. Two proton transfers in the transition state for nucleotidyl transfer catalyzed by RNA- and DNA-dependent RNA and DNA polymerases. Proc Natl Acad Sci U S A. 2007;104(11):4267–4272. doi: 10.1073/pnas.0608952104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferrin LJ, Mildvan AS. Nuclear Overhauser effect studies of the conformations and binding site environments of deoxynucleoside triphosphate substrates bound to DNA polymerse I and its large fragment. Biochemistry. 1985;24(24):6904–6912. doi: 10.1021/bi00345a024. [DOI] [PubMed] [Google Scholar]
- 40.Ferrin LJ, Mildvan AS. Studies of conformations and interactions of substrates and ribonucleotide templates bound to the large fragment of DNA polymerase I. Biochemistry. 1986;25:5131–5145. doi: 10.1021/bi00366a023. [DOI] [PubMed] [Google Scholar]
- 41.Doublie S, Ellenberger T. The mechanism of action of T7 DNA polymerase. Curr Opin Struct Biol. 1998;8(6):704–712. doi: 10.1016/s0959-440x(98)80089-4. [DOI] [PubMed] [Google Scholar]
- 42.Vande Berg BJ, Beard WA, Wilson SH. DNA structure and aspartate 276 influence nucleotide binding to human DNA polymerase beta. Implication for the identity of the rate-limiting conformational change. J Biol Chem. 2001;276:3408–3416. doi: 10.1074/jbc.M002884200. [DOI] [PubMed] [Google Scholar]
- 43.Benkovic SJ, Hammes GG, Hammes-Schiffer S. Free-energy landscape of enzyme catalysis. Biochemistry. 2008;47(11):3317–3321. doi: 10.1021/bi800049z. [DOI] [PubMed] [Google Scholar]
- 44.Benkovic SJ, Hammes-Schiffer S. A perspective on enzyme catalysis. Science (New York) 2003;301(5637):1196–1202. doi: 10.1126/science.1085515. [DOI] [PubMed] [Google Scholar]
- 45.Goodey NM, Benkovic SJ. Allosteric regulation and catalysis emerge via a common route. Nat Chem Biol. 2008;4(8):474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 46.Henzler-Wildman K, Kern D. Dynamic personalities of proteins. Nature. 2007;450(7172):964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 47.Ringe D, Petsko GA. Biochemistry. How enzymes work. Science (New York) 2008;320(5882):1428–1429. doi: 10.1126/science.1159747. [DOI] [PubMed] [Google Scholar]
- 48.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell. 2001;105(5):657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 49.Gillis AJ, Schuller AP, Skordalakes E. Structure of the Tribolium castaneum telomerase catalytic subunit TERT. Nature. 2008;455(7213):633–637. doi: 10.1038/nature07283. [DOI] [PubMed] [Google Scholar]
- 50.Sawaya MR, Prasad R, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 1997;36(37):11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- 51.Wang D, Bushnell DA, Westover KD, Kaplan CD, Kornberg RD. Structural basis of transcription: role of the trigger loop in substrate specificity and catalysis. Cell. 2006;127(5):941–954. doi: 10.1016/j.cell.2006.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yin YW, Steitz TA. The structural mechanism of translocation and helicase activity in T7 RNA polymerase. Cell. 2004;116(3):393–404. doi: 10.1016/s0092-8674(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 53.Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3D(pol)). Assembly of stable, elongation-competent complexes by using a symmetrical primertemplate substrate (sym/sub) J Biol Chem. 2000;275(8):5329–5336. doi: 10.1074/jbc.275.8.5329. [DOI] [PubMed] [Google Scholar]
- 54.Schowen RL. Mechanistic deductions from solvent isotope effects. Prog Phys Org Chem. 1972;9:275–332. [Google Scholar]
- 55.Venkatasubban KS, Schowen RL. The proton inventory technique. CRC Crit Rev Biochem. 1984;17(1):1–44. doi: 10.3109/10409238409110268. [DOI] [PubMed] [Google Scholar]
- 56.Weeks SA, Lee CA, Zhao Y, Smidansky ED, August A, Arnold JJ, Cameron CE. A polymerase mechanism-based strategy for viral attenuation and vaccine development. J Biol Chem. 2012;287(38):31618–31622. doi: 10.1074/jbc.C112.401471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang X, Smidansky ED, Maksimchuk KR, Lum D, Welch JL, Arnold JJ, Cameron CE, Boehr DD. Motif D of viral RNA-dependent RNA polymerases determines efficiency and fidelity of nucleotide addition. Structure (London, England: 1993) 2012;20(9):1519–1527. doi: 10.1016/j.str.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gong P, Kortus MG, Nix JC, Davis RE, Peersen OB. Structures of coxsackievirus, rhinovirus, and poliovirus polymerase elongation complexes solved by engineering RNA mediated crystal contacts. PLoS One. 2013;8(5):e60272. doi: 10.1371/journal.pone.0060272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gong P, Peersen OB. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A. 2010;107(52):22505–22510. doi: 10.1073/pnas.1007626107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thompson AA, Peersen OB. Structural basis for proteolysis-dependent activation of the poliovirus RNA-dependent RNA polymerase. EMBO J. 2004;23(17):3462–3471. doi: 10.1038/sj.emboj.7600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zamyatkin DF, Parra F, Alonso JM, Harki DA, Peterson BR, Grochulski P, Ng KK. Structural insights into mechanisms of catalysis and inhibition in Norwalk virus polymerase. J Biol Chem. 2008;283(12):7705–7712. doi: 10.1074/jbc.M709563200. [DOI] [PubMed] [Google Scholar]
- 62.Liu X, Yang X, Lee CA, Moustafa IM, Smidansky ED, Lum D, Arnold JJ, Cameron CE, Boehr DD. Vaccine-derived mutation in motif D of poliovirus RNA-dependent RNA polymerase lowers nucleotide incorporation fidelity. J Biol Chem. 2013;288(45):32753–32765. doi: 10.1074/jbc.M113.484428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yang X, Welch JL, Arnold JJ, Boehr DD. Long-range interaction networks in the function and fidelity of poliovirus RNA-dependent RNA polymerase studied by nuclear magnetic resonance. Biochemistry. 2010;49(43):9361–9371. doi: 10.1021/bi100833r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arnold JJ, Vignuzzi M, Stone JK, Andino R, Cameron CE. Remote site control of an active site fidelity checkpoint in a viral RNA-dependent RNA polymerase. J Biol Chem. 2005;280(27):25706–25716. doi: 10.1074/jbc.M503444200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gohara DW, Arnold JJ, Cameron CE. Poliovirus RNA-dependent RNA polymerase (3Dpol): kinetic, thermodynamic, and structural analysis of ribonucleotide selection. Biochemistry. 2004;43(18):5149–5158. doi: 10.1021/bi035429s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Korboukh VK, Lee CA, Acevedo A, Vignuzzi M, Xiao Y, Arnold JJ, Hemperly S, Graci JD, August A, Andino R, et al. RNA virus population diversity, an optimum for maximal fitness and virulence. J Biol Chem. 2014;289(43):29531–29544. doi: 10.1074/jbc.M114.592303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Crotty S, Hix L, Sigal LJ, Andino R. Poliovirus pathogenesis in a new poliovirus receptor transgenic mouse model: age-dependent paralysis and a mucosal route of infection. J Gen Virol. 2002;83(Pt. 7):1707–1720. doi: 10.1099/0022-1317-83-7-1707. [DOI] [PubMed] [Google Scholar]
- 68.Vignuzzi M, Stone JK, Arnold JJ, Cameron CE, Andino R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature. 2006;439(7074):344–348. doi: 10.1038/nature04388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pappas C, Aguilar PV, Basler CF, Solorzano A, Zeng H, Perrone LA, Palese P, Garcia-Sastre A, Katz JM, Tumpey TM. Single gene reassortants identify a critical role for PB1, HA, and NA in the high virulence of the 1918 pandemic influenza virus. Proc Natl Acad Sci U S A. 2008;105(8):3064–3069. doi: 10.1073/pnas.0711815105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taubenberger JK, Reid AH, Lourens RM, Wang R, Jin G, Fanning TG. Characterization of the 1918 influenza virus polymerase genes. Nature. 2005;437(7060):889–893. doi: 10.1038/nature04230. [DOI] [PubMed] [Google Scholar]
- 71.Tumpey TM, Basler CF, Aguilar PV, Zeng H, Solorzano A, Swayne DE, Cox NJ, Katz JM, Taubenberger JK, Palese P, et al. Characterization of the reconstructed 1918: Spanish influenza pandemic virus. Science (New York) 2005;310(5745):77–80. doi: 10.1126/science.1119392. [DOI] [PubMed] [Google Scholar]
- 72.Nomoto A, Omata T, Toyoda H, Kuge S, Horie H, Kataoka Y, Genba Y, Nakano Y, Imura N. Complete nucleotide sequence of the attenuated poliovirus Sabin 1 strain genome. Proc Natl Acad Sci U S A. 1982;79(19):5793–5797. doi: 10.1073/pnas.79.19.5793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cameron CE, Benkovic SJ. Evidence for a functional role of the dynamics of glycine-121 of Escherichia coli dihydrofolate reductase obtained from kinetic analysis of a site-directed mutant. Biochemistry. 1997;36:15792–15800. doi: 10.1021/bi9716231. [DOI] [PubMed] [Google Scholar]
- 74.Rajagopalan PT, Lutz S, Benkovic SJ. Coupling interactions of distal residues enhance dihydrofolate reductase catalysis: mutational effects on hydride transfer rates. Biochemistry. 2002;41(42):12618–12628. doi: 10.1021/bi026369d. [DOI] [PubMed] [Google Scholar]
- 75.Karplus M. Molecular dynamics simulations of biomolecules. Acc Chem Res. 2002;35(6):321–323. doi: 10.1021/ar020082r. [DOI] [PubMed] [Google Scholar]
- 76.Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat Struct Biol. 2002;9(9):646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 77.Sampoli Benitez BA, Arora K, Balistreri L, Schlick T. Mismatched base-pair simulations for ASFV Pol X/DNA complexes help interpret frequent G*G misincorporation. J Mol Biol. 2008;384(5):1086–1097. doi: 10.1016/j.jmb.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Woo HJ, Liu Y, Sousa R. Molecular dynamics studies of the energetics of translocation in model T7 RNA polymerase elongation complexes. Proteins. 2008;73(4):1021–1036. doi: 10.1002/prot.22134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang L, Beard W, Wilson S, Roux B, Broyde S, Schlick T. Local deformations revealed by dynamics simulations of DNA polymerase beta with DNA mismatches at the primer terminus. J Mol Biol. 2002;321(3):459–478. doi: 10.1016/s0022-2836(02)00617-4. [DOI] [PubMed] [Google Scholar]
- 80.Yang L, Beard WA, Wilson SH, Broyde S, Schlick T. Polymerase beta simulations suggest that Arg258 rotation is a slow step rather than large subdomain motions per se. J Mol Biol. 2002;317(5):651–671. doi: 10.1006/jmbi.2002.5450. [DOI] [PubMed] [Google Scholar]
- 81.Gruez A, Selisko B, Roberts M, Bricogne G, Bussetta C, Jabafi I, Coutard B, De Palma AM, Neyts J, Canard B. The crystal structure of coxsackievirus B3 RNA-dependent RNA polymerase in complex with its protein primer VPg confirms the existence of a second VPg binding site on Picornaviridae polymerases. J Virol. 2008;82(19):9577–9590. doi: 10.1128/JVI.00631-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Love RA, Maegley KA, Yu X, Ferre RA, Lingardo LK, Diehl W, Parge HE, Dragovich PS, Fuhrman SA. The crystal structure of the RNA-dependent RNA polymerase from human rhinovirus: a dual function target for common cold antiviral therapy. Structure (London, England: 1993) 2004;12(8):1533–1544. doi: 10.1016/j.str.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 83.Ferrer-Orta C, Arias A, Perez-Luque R, Escarmis C, Domingo E, Verdaguer N. Structure of foot-and-mouth disease virus RNA-dependent RNA polymerase and its complex with a template-primer RNA. J Biol Chem. 2004;279(45):47212–47221. doi: 10.1074/jbc.M405465200. [DOI] [PubMed] [Google Scholar]
- 84.Case DA, Cheatham TE, 3rd, Darden T, Gohlke H, Luo R, Merz KM, Jr, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;16:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Moustafa IM, Shen H, Morton B, Colina CM, Cameron CE. Molecular dynamics simulations of viral RNA polymerases link conserved and correlated motions of functional elements to fidelity. J Mol Biol. 2011;410(1):159–181. doi: 10.1016/j.jmb.2011.04.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Amadei A, Linssen AB, Berendsen HJ. Essential dynamics of proteins. Proteins. 1993;17(4):412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 87.Garcia AE. Large-amplitude nonlinear motions in proteins. Phys Rev Lett. 1992;68(17):2696–2699. doi: 10.1103/PhysRevLett.68.2696. [DOI] [PubMed] [Google Scholar]
- 88.Ichiye T, Karplus M. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 1991;11(3):205–217. doi: 10.1002/prot.340110305. [DOI] [PubMed] [Google Scholar]
- 89.Agarwal PK, Billeter SR, Rajagopalan PT, Benkovic SJ, Hammes-Schiffer S. Network of coupled promoting motions in enzyme catalysis. Proc Natl Acad Sci U S A. 2002;99(5):2794–2799. doi: 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Estabrook RA, Luo J, Purdy MM, Sharma V, Weakliem P, Bruice TC, Reich NO. Statistical coevolution analysis and molecular dynamics: identification of amino acid pairs essential for catalysis. Proc Natl Acad Sci U S A. 2005;102(4):994–999. doi: 10.1073/pnas.0409128102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Moustafa IM, Korboukh VK, Arnold JJ, Smidansky ED, Marcotte LL, Gohara DW, Yang X, Sanchez-Farran MA, Filman D, Maranas JK, et al. Structural dynamics as a contributor to error-prone replication by an RNA-dependent RNA polymerase. J Biol Chem. 2014;289(52):36229–36248. doi: 10.1074/jbc.M114.616193. [DOI] [PMC free article] [PubMed] [Google Scholar]