

Abstract
Kinesin-1 is a heterotetramer composed of kinesin heavy chain (KHC) and kinesin light chain (KLC). The Caenorhabditis elegans genome has a single KHC, encoded by the unc-116 gene, and two KLCs, encoded by the klc-1 and klc-2 genes. We show here that UNC-116/KHC and KLC-2 form a complex orthologous to conventional kinesin-1. KLC-2 also binds UNC-16, the C. elegans JIP3/JSAP1 JNK-signaling scaffold protein, and the UNC-14 RUN domain protein. The localization of UNC-16 and UNC-14 depends on kinesin-1 (UNC-116 and KLC-2). Furthermore, mutations in unc-16, klc-2, unc-116, and unc-14 all alter the localization of cargos containing synaptic vesicle markers. Double mutant analysis is consistent with these four genes functioning in the same pathway. Our data support a model whereby UNC-16 and UNC-14 function together as kinesin-1 cargos and regulators for the transport or localization of synaptic vesicle components.
INTRODUCTION
Neurons, like all polarized cells, must regulate the transport and localization of many molecules to establish and maintain proper cellular function (Foletti et al., 1999). Intracellular transport of protein and vesicular cargos along microtubules relies on the function of ATP-dependent motor proteins, including kinesins and cytoplasmic dynein. Although kinesin-1 was the first of these motors to be biochemically purified (Brady, 1985; Vale et al., 1985), there is still much unknown about its interaction with putative cargoes in vivo.
Kinesin-1 is a heterotetramer composed of two kinesin heavy chains (KHC/KIF5) and two kinesin light chains (KLC; Goldstein and Philp, 1999; Goldstein and Yang, 2000; Vale, 2003). The KHCs contain an N-terminal motor domain, a central stalk composed of long coiled-coil domains and a C-terminal globular tail. The KLCs contain an N-terminal coiled-coil region and a stretch of tetratricopeptide (TPR) repeats. KLCs bind to the tail region of KHCs, and such binding contributes to both the activation of KHC, presumably upon binding cargo, and the full inactivation of KHC (Verhey et al., 1998; Woehlke and Schliwa, 2000; Verhey and Rapoport, 2001). Putative cargos transported by kinesin-1 include intracellular organelles, such as mitochondria, lysosomes, and endoplasmic reticulum, as well as plasma membrane receptors and vesicles that are destined to synapses (Leopold et al., 1992; Sato-Yoshitake et al., 1992).
Despite the progress in understanding the mechanics of how motor proteins move along microtubules, the in vivo functions of most kinesins remain poorly understood. A central question is how motor proteins pair with particular cargos at the right time and place. Recently, the members of the JNK interacting protein (JIP) group, which include JIP1, JIP2, and JIP3/JSAP1, have been shown to bind KLC and are molecular tethers and cargo for kinesin-1 (Bowman et al., 2000; Byrd et al., 2001; Verhey et al., 2001; Verhey and Rapoport, 2001). The localization of JIP1, JIP2, and JIP3 in cultured cells requires the function of kinesin-1, demonstrating that JIPs are kinesin-1 cargoes (Verhey et al., 2001). Binding of JIP1 and JIP2 to ApoER2 Reelin receptor may tether kinesin-1 to vesicles containing ApoER2 (Stockinger et al., 2000; Verhey et al., 2001). JIP3/JSAP1 is expressed exclusively in neurons and likely mediates the transport of synaptic components. Jip3 deletion mutant mice die shortly after birth, exhibiting severe morphological defects in telencephalon (Akechi et al., 2001; Kelkar et al., 2003). The Drosophila homolog of JIP3/JSAP1, Sunday Driver (dSYD), was identified genetically in screens for mutants with a larval sluggishness and tail flip phenotype similar to kinesin heavy chain (khc) mutants. In sunday driver mutants, axonal cargo is misaccumulated in the axons (Bowman et al., 2000).
The Caenorhabditis elegans JIP3 homolog is encoded by the unc-16 gene (Byrd et al., 2001). Previously we showed that loss-of-function mutations in unc-16 result in the improper localization of synaptic vesicle markers in multiple classes of neurons. Mutations in the kinesin heavy chain gene, unc-116, cause similar defects, and UNC-16 localization is altered in unc-116 mutants. Genetic double-mutant analysis supports the conclusion that UNC-16/JIP3 functions as a cargo adaptor for UNC-116/KHC.
What are the other components of this UNC-116 and UNC-16 motor complex? The C. elegans genome has two predicted klc genes, klc-1 and klc-2 (Koushika and Nonet, 2000). We report here that KLC-2 is a functional partner of UNC-116/KHC and that KLC-2 binds to UNC-16. We have also identified UNC-14, a novel protein with a conserved RUN domain (for RPIP8, UNC-14, and NESCA), as an UNC-16 - and KLC-2-interacting protein. Although unc-14 has previously been shown to play a role in neurite outgrowth (McIntire et al., 1992; Ogura et al., 1997), we show here that unc-14 affects both neurite extension and axonal transport. Like UNC-16, UNC-14 localization depends on kinesin-1 (UNC-116 and KLC-2). Thus, the C. elegans kinesin-1 likely utilizes at least two proteins, the UNC-16/JIP3 and the UNC-14 RUN-domain protein, for transporting cargos containing synaptic vesicle components.
MATERIALS AND METHODS
Strains and Genetics
C. elegans strains were grown on NGM plates as described and wild-type animals were Bristol strain N2 (Brenner, 1974). klc-2(km28) and klc-2(km11) were isolated by sib-selection from screens of 4 × 105 and 1 × 106 UV/4,5′,8-trimethylpsoralen (TMP) mutagenized genomes, respectively (Yandell et al., 1994). To detect the klc-2(km28) deletion, we used KLC-2-f4(5′ CGCCACGATCTCCTGTATTTCAATAGC 3′) and KLC-2-r8 (5′ CTATCAACTTTGGCGGCTTTTGCCC 3′) for external primers and KLC-2-f5 (5′ GGGCTGTTATTTAAGACGCCCTCCTC 3′) and KLC-2-r2 (5′ TGGTCTTTGCCACATTCGGG 3′) for internal primers. The klc-2(km28) lesion includes a deletion of 610 nucleotides from the genomic klc-2 locus (corresponding to nucleotides 40,117-40,726 on cosmid C18C4). To detect the klc-2(km11) deletion, we used KLC-2-f4 and KLC-2-r4 (5′ GATGACGGAGTACAATGTCGAGCAAC 3′) for external primers and KLC-2-f5 and KLC-2-r3 (5′ CATAACGGATCGTTCCATTCTTCGAG 3′) for internal primers. The klc-2(km11) lesions include both a deletion of 1479 nucleotides from the genomic klc-2 locus (corresponding to nucleotides 38,584-40,063 on cosmid C18C4) and an insertion of a second copy of klc-2 with a C-terminal deletion (after nucleotide 38,712 on cosmid C18C4) at another site on chromosome V (corresponding to 2454 nucleotides on cosmid M03E7).
unc-14(ju56) was isolated in an EMS mutagenesis screen for abnormal synapse morphology using Punc-25-SNB-1::GFP as a marker (Zhen and Jin, 1999). unc-14 genomic DNA, including all exons and intron-exon junctions, was amplified from unc-14 mutant and wild-type animals. DNA sequences were determined using 33P-labeled primers and the fmol sequencing kit (Promega, Madison, WI). The mutant lesion was confirmed on both strands from DNAs prepared in independent PCRs. Double mutants were constructed following standard procedures and confirmed by noncomplementation.
The following strains were used in the study: CZ1676 unc-14(e57); juIs1, CZ1125 unc-16(e109); juIs1, CZ936 unc-16(ju79); juIs1, CZ2017 unc-116(e2281); juIs1, CZ472 unc-14(ju56) SEM-4(n1378); juIs1, CZ4016 unc-14(ju56) SEM-4(n1378); juIs76, KU801 klc-2 (km11), KU804 klc-2(km28)/nT1[qIs51],KU807 klc-2 (km11);juIs1, KU810 klc-2(km28)/nT1[qIs51];juIs1, KU813 unc-14(ju56) SEM-4(n1378);klc-2(km28)/nT1[qIs51];juIs1, KU816 unc-16(e109);klc-2(km28)/nT1[qIs51];juIs1.
klc-2 Gene
To determine the 5′ end of the klc-2 transcript, we screened the pNVLeu C. elegans cDNA library (Kawasaki et al., 1999) using the GAL1 primer (CAAATGTAATAAAAGTATCAAC) and the KLC-2-r1 primer (TTAAGAGTTGAATCCGAGTG). The amplified fragment containing the klc-2 cDNA was subcloned into the pCR2.1-TOPO vector (Invitrogen, Carlsbad, CA) and this plasmid was sequenced. The amplified klc-2 cDNA contained the predicted second exon attached with a part of the SL1 sequence. Moreover, we searched for the 5′ end of klc-2 using the ykclone sequence database (Y. Kohara, National Institute of Genetics, Mishima, Japan). This analysis showed that klc-2 has four alternative splice forms that we have named klc-2a, klc-2b, klc-2c, and klc-2d (see Figure 1A). klc-2a, klc-2b, and klc-2d coincide with the C18C4.10b, C18C4.10c, and C18C4.10a open reading frames (ORFs), respectively.
Figure 1.
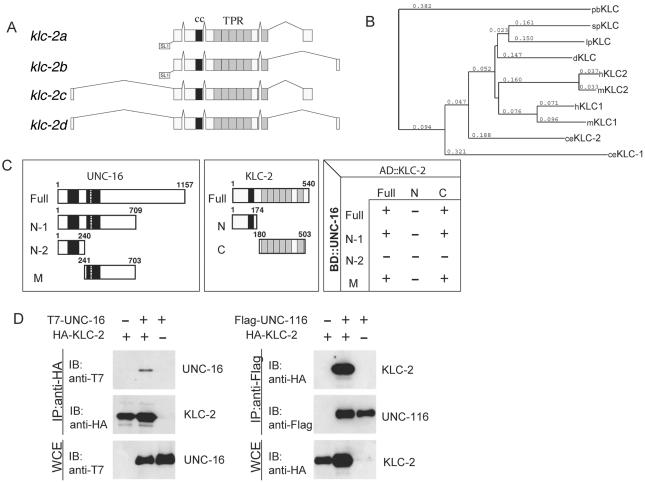
KLC-2 binds UNC-16 and the UNC-116 KHC. (A) Structures of klc-2 isoforms. The deduced four alternative splicing forms of klc-2 are shown. The black and gray boxes indicate the regions encoding the coiled-coil domain and TPR motifs, respectively. The trans-splicing sites are also indicated. (B) Alignment dendrogram generated using the neighbor-joining method within CLUSTALX. Numeric values indicate branch lengths in terms of percent divergence. pb, Plectonema boryanum (cyanobacterium); sp, Strongylocentrotus purpuratus (sea urchin); lp, Loligo pealii (squid); d, Drosophila; h, human; m, mouse; and ce, C. elegans. (C) Interaction between KLC-2 and UNC-16 in the yeast two-hybrid system. Various regions of UNC-16 fused with the LexA DNA-binding domain (BD) are shown on the left. The regions of KLC-2 fused with the GAL4 activation domain (AD) are shown in the middle. The plus (+) and minus (-) indicate positive and negative interactions between the various regions of UNC-16 and KLC-2 in the chart on the right. (D) Association of KLC-2 with UNC-16 and UNC-116. HEK 293 cells were transfected with control vector (-), HA-KLC-2, T7-UNC-16, and Flag-UNC-116 as indicated. Cell lysates were immunoprecipitated (IP) with anti-HA (left) and anti-Flag (right) antibodies. Immunoprecipitates were immunoblotted (IB) with anti-T7 (left panel) and anti-HA (right) antibodies. The amounts of immunoprecipitated HA-KLC-2 and Flag-UNC-116 were determined with anti-HA (left) and anti-Flag (right) antibodies, respectively. Whole-cell extracts (WCE) were immunoblotted with anti-T7 (left) and anti-HA (right) antibodies to determine total amounts of T7-UNC-16 and HA-KLC-2, respectively.
Rescue of klc-2(km11) movement defect was scored by thrashing assay as described (Miller et al., 1996). Briefly, young adult animals were transferred into M9 buffer. After a few minutes recovery, thrashes were counted for 10 s. A thrash was defined as a change in the direction of bending at the midbody. Ten to 30 animals from each strain were examined.
Sequence Analysis
Multiple sequence alignments for the KLCs and RUN domains were constructed using CLUSTALW (Thompson et al., 1994) and displayed using BOXSHADE version 3.2 (http://www.ch.embnet.org/software/BOX_form.html). The phylogenetic tree was constructed by the neighbor joining (N-J) method using CLUSTALX (Thompson et al., 1997) and displayed with the NJPLOT program distributed with CLUSTALX.
Yeast Two-hybrid Assays
The bait plasmid pLexA-UNC-16 was constructed by fusing the full-length unc-16 cDNA in frame with the LexA DNA-binding domain in vector pBTM116 (provided by S. Hollenberg). The truncation mutants, UNC-16N-1, UNC-16N-2, and UNC-16M, encode amino acids 1-709, 1-240, and 241-703 of UNC-16, respectively. pLexA-UNC-51 contains the full-length unc-51 cDNA derived from the plasmid pBLO (kindly provided by K. Ogura). The prey plasmid containing the full-length klc-2a cDNA (pACT-KLC-2) was isolated in yeast two-hybrid screening for UNC-16-binding proteins. The truncation mutants, KLC-2N and KLC-2C, encode amino acids 1-174 and 180-503 of KLC-2a, respectively. The unc-14 plasmids (pR4BK1, pBS1, and p14SX1) were a gift from Y. Ohshima. pR4BK1 contains the full-length unc-14 cDNA. pBS1 and p14SX1 contain the unc-14 cDNA fragment corresponding to amino acids 1-382 and 383-665 of UNC-14, respectively (Ogura et al., 1997).
The C. elegans mixed-stage cDNA library fused to the GAL4 activation domain for yeast two-hybrid screening was kindly provided by R. Barstead. The bait plasmid, pLexA-UNC-16 or pLexA-UNC-16N-1, was expressed with the C. elegans cDNA library in the yeast strain L40 (MATa his3 leu2 trp1 URA3::lexA-lacZ LYS2::lexA-HIS3). Approximately 8.9 × 104 clones of the C. elegans cDNA library were screened. Positive interacting clones were selected by growth on plates that lacked Ura, Leu, Trp, and His and contained 1-40 mM 3-aminotriazole.
Expression in 293 Cells, Immunoprecipitation, and Immunoblotting
The mammalian expression construct for T7 epitope-tagged UNC-16 (T7-UNC-16) was described previously (Byrd et al., 2001). The mammalian expression constructs for Flag-UNC-116 and HA-KLC-2 were generated by inserting full-length unc-116 cDNA and klc-2a cDNA into pFlag-CMV and pcDNA3 vectors, respectively. The expression plasmid encoding Flag-UNC-14 was a gift from Y. Ohshima.
Human embryonic kidney 293 cells were maintained in DMEM supplemented with 10% fetal calf serum, 100 μg/ml penicillin G and 100 μg/ml streptomycin at 37°C and 5% CO2. 293 cells (1 × 106) were plated in 10-cm dishes and transfected with a total of 10 μg DNA containing various expression vectors by the calcium phosphate precipitate method. After 24-36 h, cells were collected and washed once with ice-cold phosphate-buffered saline (PBS) and lysed in 0.3 ml of extraction buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 12.5 mM glycerophosphate, 1.5 mM MgCl2, 2 mM EGTA, 10 mM NaF, 2 mM dithiothreitol, 1 mM Na3 VO4, 1 mM phenylmethylsulfonyl fluoride, 20 μM aprotinin, and 0.5% Triton X-100). Cellular debris was removed by centrifugation at 10,000 × g for 4 min. Cell lysates were incubated with 1 μg of various antibodies and 15 μl protein G-Sepharose (Amersham Biosciences, Piscataway, NJ). The immune complexes were washed three times with wash buffer (20 mM HEPES, pH 7.4, 500 mM NaCl, and 10 mM MgCl2) and once with PBS and were suspended in 30 μl PBS. For immunoblotting, aliquots of immunoprecipitates or whole cell lysates were resolved by SDS-PAGE and transferred to Hybond-P membranes (Amersham Biosciences). The membranes were immunoblotted with mouse monoclonal antibodies to T7 (Novagen, Madison, WI), HA (HA.11; BAbCo, Richmond, CA), and Flag (M2; Sigma, St. Louis, MO). The bound antibodies were visualized with horseradish peroxidase (HRP)-conjugated antibodies to mouse IgG using the Enhanced Chemiluminesence (ECL) Western Blotting System (Amersham Biosciences).
Construction of GFP Expression Plasmids
Pklc-2-KLC-2ΔC::GFP was made by subcloning a 5.5-kb PstI-BamHI genomic fragment containing DNA 2.4 kb upstream of the ATG start codon and encoding the first 514 amino acids of KLC-2a from cosmid C18C4 (kindly provided by A. Coulson) into the GFP reporter vector pPD95.75 (kindly provided by A. Fire). DNA encoding amino acids 332-540 of KLC-2a was amplified as a BalI-BalI PCR product and inserted into the BalI sites of Pklc-2-KLC-2ΔC::GFP, generating plasmid Pklc-2-KLC-2::GFP. Pklc-2-KLC-2::YFP was made by using the YFP coding region from Punc-25-UNC-16::YFP. Punc-25-KLC-2 was made by inserting the full-length klc-2a cDNA into pCZ325 (Jin et al., 1999). Punc-25-KLC-2::GFP and Punc-25-KLC-2::YFP were generated by replacing the 0.9-kb BamHI-ApaI fragment of Punc-25-KLC-2 with the 1.9-kb BamHI-ApaI fragment of Pklc-2-KLC-2::GFP and Pklc-2-KLC-2::YFP, respectively. The full-length unc-14 cDNA was amplified by PCR using pR4BK1 as a template and subcloned into pBluescriptII KS-. Pjkk-1-UNC-14 was made by inserting a 0.8-kb HpaI-HincII fragment of pMK103 (Kawasaki et al., 1999) containing the jkk-1 promoter region into the GFP reporter vector pPD95.75 (provided by A. Fire), replacing the GFP coding region with a synthetic multiple cloning site to create pNHjkk1p, and inserting the KpnI-KpnI full-length unc-14 cDNA fragment into the KpnI site of pNHjkk1p. To fuse GFP or YFP to the 3′ end of the unc-14 coding region, Pjkk-1-UNC-14-1 was constructed by replacing the 0.4-kb SalI-PstI PCR product corresponding to amino acids 522-665 of UNC-14 with the 0.3-kb SalI-PstI fragment of Pjkk-1-UNC-14. The PvuII-KpnI fragment containing the jkk-1 promoter region of Pjkk-1-UNC-14-1 was replaced with the PvuII-KpnI fragment of pCZ325, generating Punc-25-UNC-14-1. To make Punc-25-UNC-14::GFP and Punc-25-UNC-14::YFP, PstI-ApaI fragments containing the GFP or YFP coding region were inserted into PstI-ApaI sites of Punc-25-UNC-14-1 in-frame, respectively.
Punc-25-UNC-16::GFP (pCZ462) was made by first replacing the unc-16 promoter in pCZ350 (Byrd et al., 2001) with the unc-25 promoter. GFP was then added by inserting an NheI fragment containing the GFP ORF with synthetic splice donor and acceptor sites from pPD107.45 (provided by A. Fire). A similar UNC-16::GFP fusion (pCZ352) was previously shown to rescue all unc-16 mutant phenotypes (Byrd et al., 2001). Punc-25-UNC-16::CFP(pCZ460) was made by replacing the GFP fragment with the CFP DNA from pPD136.61 (provided by A. Fire).
Punc-116-UNC-116::GFP (pCZ463) was made by amplifying the unc-116 promoter and ORF from cosmid R05D3 with primers YJ658 and YJ661 (adding NotI and KpnI sites 5′ and 3′, respectively). The unc-116 PCR fragment was then cloned in frame with a C-terminal GFP ORF in pPD114.108 (provided by A. Fire).
Transgene Generation
Germline transformation was performed following standard procedures (Mello et al., 1991) using Pttx-3-GFP, Plin-15(EK), and pRF4 as coinjection markers. Transgenic arrays were generated using different concentrations as follows. Pklc-2-KLC-2::GFP and Pttx-3-GFP (20 and 50 ng/μl, respectively) were used in Ex[Pklc-2-KLC-2::GFP] (kmEx801-803, kmEx807, and kmEx808); 20 ng/μl Punc-25-KLC-2::GFP and 50 ng/μl Pttx-3-GFP were used in Ex[Punc-25-KLC-2::GFP] (kmEx811-813); 2 ng/μl Punc-25-KLC-2::GFP and 50 ng/μl Pttx-3-GFP were used in Ex[Punc-25-KLC-2::GFP] (kmEx844-846); 40 ng/μl Punc-25-UNC-14::GFP and 50 ng/μl Pttx-3-GFP were used in Ex[Punc-25-UNC-14::GFP] (kmEx820-kmEx822); 20 ng/μl Punc-25-UNC-16::GFP and 50 ng/μl Pttx-3-GFP were used in Ex[Punc-25-UNC-16::GFP] (kmEx823-825). No GFP expression was seen when Punc-25-UNC-14::GFP was expressed at 20 ng/μl or when Punc-25-KLC-2::GFP was expressed below 2 ng/μl. In Punc-25-UNC-16::GFP transgenic lines generated at 15-20 ng/μl, a range of GFP expression was seen from invisible to moderately visible, and the line that showed medium expression was chosen for further analysis. To generate Ex[Punc-25-SNB-1::CFP, Punc-25-UNC-16::YFP, lin-15(+)] lines (juEx831 and juEx832), lin-15(n765ts) animals were used as host coinjected with lin-15 DNA at concentrations of 30, 15, and 50 ng/μl respectively. juEx832 was crossed into unc-116 (e2310); lin-15(n765) mutants for further analysis.
GFP Analysis
Expression of GFP, CFP, and YFP reporters were observed and photographed in adult animals under a 100× objective of an Olympus FLUOVIEW FV500 confocal laser scanning microscope (Lake Success, NY; see Figures 2B, 5B, and 7, A and B). In Figure 2B, animals were fixed in 4% paraformaldehyde. In Figures 5B and 7, A and B, living animals were mounted on 2% agarose pads containing 0.5% 1-phenoxy 2-propanol (Sigma). Three to four independent lines were observed. Animals expressing UNC-116::GFP (see Figures 4, E-H, and 5A) and Punc-25-GFP (Figure 8A) were immobilized with 0.25 μM Levamisol. GFP was observed using an HQ-FITC filter (Chroma, Brattleboro, VT) under a 63× objective of a Zeiss Axioskop fluorescent microscope and photographed with a Hamamatsu digital camera. SNB-1::GFP (Figure 8, B and C) was observed using an HQ-FITC filter (Chroma) under a 63× objective of a Zeiss Axioplan2 fluorescence microscope. Animals fixed in 2% paraformaldehyde were photographed with a Zeiss AxioCam. The method for quantification of the L1 dorsal SNB-1::GFP phenotype was described previously (Byrd et al., 2001). Briefly, the intensity of dorsal SNB-1::GFP localization was scored manually in live, unanesthetized L1 animals on a scale from 0 to 25, where animals with no detectable SNB-1::GFP localization along the dorsal cord were scored as 0 and animals with SNB-1::GFP localization along the full length of the dorsal cord at a fluorescent intensity of ∼75% of that along the ventral cord were scored as 25.
Figure 2.
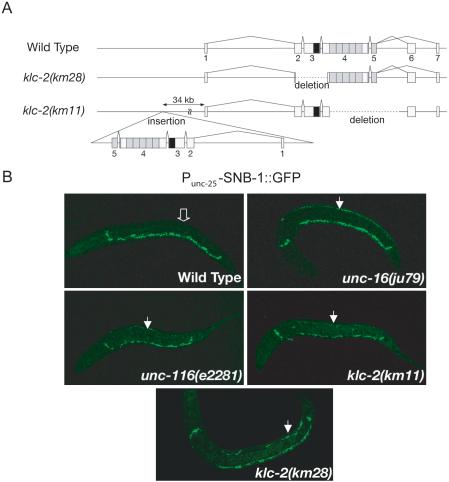
klc-2 loss-of-function mutation affects synaptic vesicle marker localization. (A) Structure of the klc-2 gene. Exons are indicated by boxes with numbers. The black and gray boxes indicate regions encoding the coiled-coil domain and TPR motifs, respectively. The klc-2(km28) mutation is a 0.6 kb deletion. The klc-2(km11) mutant has two copies of the klc-2 gene. One deletes the entire regions encoding the TPRs, and the other is an insertion of a second copy of the klc-2 gene with a deletion removing the two alternative last exons. (B) SNB-1::GFP localization. SNB-1::GFP is localized only along the ventral DD processes in wild-type L1 larvae (open arrow in top left panel indicates lack of SNB-1::GFP along dorsal side), but along both the ventral and dorsal DD processes in unc-16(ju79), unc-116(e2281), klc-2(km28), and klc-2(km11) mutant L1 larvae (closed arrows indicate dorsal SNB-1::GFP in top right and bottom panels). All animals are positioned with anterior-left, posterior-right, ventral-down, and dorsal-up.
Figure 5.
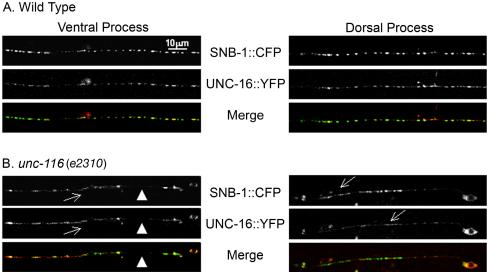
UNC-16 and SNB-1 localization is dependent on UNC-116. P unc-25-UNC-16::YFP and P unc-25-SNB-1::CFP (juEx832) were expressed in wild-type (A) and unc-116 mutant (B) animals. Shown are single plane scan confocal images, focusing on SNB-1::CFP. (A) UNC-16 is associated with SNB-1 puncta on the ventral and dorsal processes of wild-type animals (97.2 ± 0.9%). (B) In unc-116 (e2310) mutants, both UNC-16 and SNB-1 localization is altered. UNC-16 and SNB-1 are seen largely irregular and diffuse along the ventral process (arrows) and are often absent (arrowheads). In the rare case where SNB-1 expression was punctate, the incidence of colocalization with UNC-16 was decreased.
Figure 7.
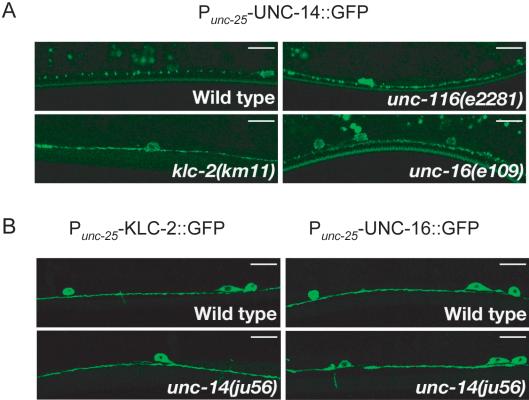
Localization of UNC-14 depends on kinesin-1. (A) Localization of UNC-14. P unc-25-UNC-14::GFP (kmEx821) was expressed in D-type neurons of wild-type, unc-116(e2281), klc-2(km11), and unc-16(e109) animals. Panels show localization of UNC-14::GFP in the ventral nerve processes of D-type neurons. UNC-14::GFP showed a punctate pattern in wild-type animals (top left). In unc-116(e2281), klc-2(km11), and unc-16(e109) mutant animals, UNC-14::GFP was diffused and irregular along the ventral nerve processes (top right and bottom panels). (B) Localization of KLC-2 and UNC-16 in unc-14 mutants. Punc-25-KLC-2::GFP (kmEx811) and Punc-25-UNC-16::GFP (kmEx823) were expressed in D-type neurons of wild-type and unc-14(ju56) mutant animals. Panels show localization of KLC-2::GFP (left panels) and UNC-16::GFP (right panels) in the ventral nerve processes of D-type neurons. Both KLC-2::GFP and UNC-16::GFP were evenly distributed in wild-type and unc-14(ju56) mutant animals. Scale, 10 μm
Figure 4.
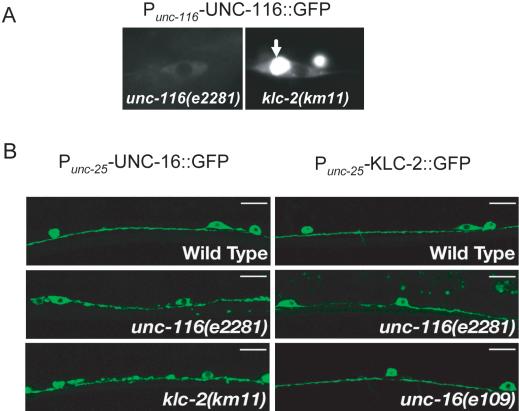
Localization dependence of UNC-16 and kinesin-1. (A) Subcellular localization of UNC-116::GFP. Panels show localization of Punc-116-UNC-116::GFP (juEx637) in an unc-116(e2281) rescued animal (left) and in a klc-2(km11) mutant animal (right). Arrow in right panel indicates perinuclear accumulation of UNC-116::GFP in the klc-2(km11) mutant cell. (B) Localization of UNC-16::GFP and KLC-2::GFP in kinesin-1 and unc-16 mutants. Punc-25-UNC-16::GFP (kmEx823) and Punc-25-KLC-2::GFP (kmEx811) were expressed in D-type neurons of wild-type, unc-116(e2281), klc-2(km11), and unc-16(e109) animals. Panels show localization of UNC-16::GFP (left panels) and KLC-2::GFP (right panels) in the ventral nerve processes of the D-type neurons. UNC-16::GFP is evenly distributed in wild-type animals (top left). In unc-116(e2281) and klc-2(km11) mutants, UNC-16::GFP is aggregated along the ventral nerve processes (middle and bottom left). KLC-2::GFP is evenly distributed in wild-type (top right) and in unc-16(e109) mutant animals (bottom right), but not uniform in unc-116(e2281) mutants (middle right). Scale, 10 μm.
Figure 8.
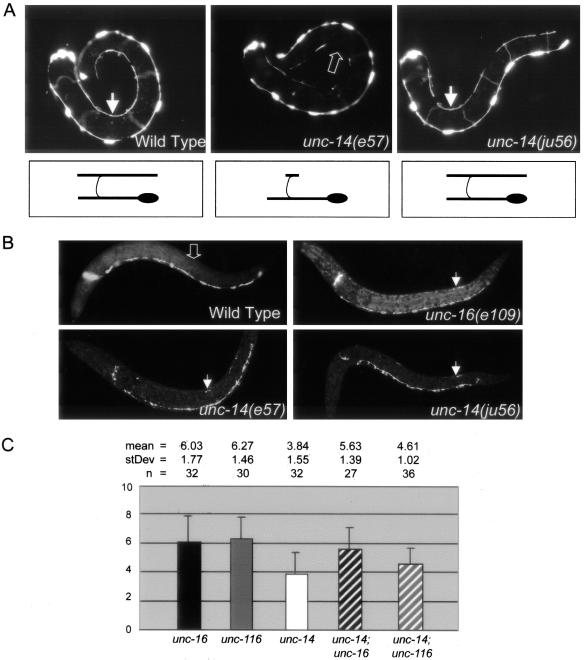
unc-14 regulates synaptic vesicle localization and functions in the same pathway as unc-16 and unc-116. (A) DD cell morphology. Panels show L1 stage animals expressing Punc-25-GFP (juIs76) to visualize the morphology of the DD motor neurons. In wild-type animals, six DD cell bodies reside along the ventral nerve cord. Each extends a process along the ventral nerve cord, around the side of the body (commissures out of the focal plane), and along the dorsal nerve cord. The arrows in the left and right panels indicate fully extended dorsal DD processes in wild-type and unc-14(ju56) L1 animals. The open arrow in the middle panel indicates missing dorsal DD processes in an unc-14(e57) L1 animal. Drawings below the animals demonstrate the morphology of an individual DD motor neuron representative of each genotype. (B) SNB-1::GFP (juIs1) localization in L1 animals. Although wild-type L1s have no SNB-1::GFP along their dorsal DD processes (open arrow in top left panel), SNB-1::GFP is misaccumulated along the dorsal DD processes of unc-16 and unc-14 mutant L1 animals (closed arrows in top right and bottom panels). (C) Quantification of L1 dorsal SNB-1::GFP phenotype. The extent of dorsal SNB-1::GFP was scored in L1 animals on a scale from 0 to 25 (see Materials and Methods) in unc-16(e109), unc-116(e2281), and unc-14(ju56) single mutants, and in unc-14(ju56); unc-16(e109) and unc-14(ju56); unc-116(e2281) double-mutant animals. The mean, SD, and n are indicated above the graph. By Student's t-tests (2 tail/unpaired/unequal variance), the L1 SNB-1::GFP phenotypes of unc-16(e109) single mutants and unc-16(e109); unc-14(ju56) double mutants are not significantly different (p = 0.333), and the L1 SNB-1::GFP phenotypes of unc-14(ju56) single mutants and unc-14(ju56); unc-116(e2281) double mutants are not significantly different (p = 0.21). Although other pair combinations have significantly different mean scores, all differences are less than additive. p-values for other pair-wise comparisons are as follows: unc-16(e109) and unc-14(ju56) (p = 1.9e-6), unc-14(ju56) and unc-14(ju56); unc-16(e109) (p = 1.9e-5), unc-116(e2281) and unc-14(ju56) (p = 3.3e-8), and unc-116(e2281) and unc-14(ju56); unc-116(e2281) (p = 3.2e-6).
For UNC-16::YFP and SNB-1::CFP analysis, images were collected using a Zeiss LSM5 PASCAL laser scanning confocal microscope using a 63× objective. Because CFP and YFP fluorescence quenched, we were unable to collect Z-stack images. Images were single plane scans with the focus on the SNB-1::CFP, from the central region of the animal and areas analyzed range from 35 to 120 μm along the length of the dorsal and ventral cords. Laser and image capture settings were optimized to avoid bleed through: the argon laser was set to 18% at 458 nm and 4% at 514 nm, with pinhole settings of 92 and 127 μm, respectively. Areas of large aggregation were avoided in our analyses because of bleed-through.
RESULTS
Direct Association of UNC-16 with the KLC-2 Kinesin Light Chain of Kinesin-1
To identify proteins that interact with UNC-16, we screened a C. elegans mixed-stage cDNA library by the yeast two-hybrid method with full-length UNC-16 (ZK1098.10b) as the bait. Five clones isolated contained cDNA inserts encoding KLC-2 (Figure 1, A-C). There are at least four klc-2 transcripts formed by inclusion or exclusion of an upstream 5′ first exon and alternative splicing of two different exons at the 3′ end, designated as klc-2a, b, c, and d (Figure 1A; see also Materials and Methods). klc-2a and klc-2d correspond to isoforms described previously as 1 and 2, respectively (Fan and Amos, 1994).
We confirmed the physical association of KLC-2 and UNC-16 by coimmunoprecipitation of HA-KLC-2 and T7-UNC-16 expressed together in mammalian 293 cells. Using anti-HA antibodies, T7-UNC-16 coimmunoprecipitated with HA-KLC-2 (Figure 1D). We also used yeast two-hybrid assays to identify the domains of each protein that mediate the binding. As shown in Figure 1C, the middle portion of UNC-16 (aa 241-703) was sufficient to bind KLC-2, and the TPR domain of KLC-2 was required for binding UNC-16. These results are consistent with the reported binding between JIP3/dSYD and KLC in mammals and in Drosophila (Bowman et al., 2000; Verhey et al., 2001).
On the basis of phenotypic resemblance and genetic interactions, we previously reported that UNC-16 functions together with the kinesin heavy chain protein, UNC-116 (Byrd et al., 2001). Although KLC-2 is closely homologous to the KLCs of other species (Figure 1B, Supplementary Figure), the UNC-116 KHC lacks a segment of rod II that is conserved in the KHCs of other species, raising some questions as to how or whether UNC-116 may interact with KLCs and cargo (Patel et al., 1993; Fan and Amos, 1994). Because the physical interactions of KLC-2 and UNC-16 suggested that KLC-2 may be a functional partner for the UNC-116 kinesin heavy chain, we tested whether epitope-tagged UNC-116 and KLC-2 expressed together in mammalian 293 cells could coimmunoprecipitate. We found that HA-KLC-2 coimmunoprecipitated with Flag-UNC-116 (Figure 1D), demonstrating that UNC-116 and KLC-2 constitute a kinesin-1 complex in C. elegans.
Although there are two kinesin light chains encoded by the C. elegans genome, KLC-1 and KLC-2 (Koushika and Nonet, 2000), KLC-2 shares more sequence homology with other metazoan KLCs, including Drosophila KLC and human KLC1 (Figure 1B). Although we cannot rule out the possibility that KLC-1 may also bind to UNC-116/KHC and/or UNC-16, KLC-1 was not identified as an UNC-16-interacting protein from the yeast two-hybrid screen.
Loss of Function in klc-2 Displays Transport Defects
To examine the in vivo function of klc-2, we isolated two deletion mutations in klc-2. The km28 mutation deletes 0.6 kb within the region that encodes the coiled-coil domain (Figure 2A). klc-2(km28) mutants are arrested as L1 larvae. A partial loss-of-function mutation klc-2(km11) mutant has two copies of a truncated klc-2: the endogenous klc-2 locus contains a ∼1.4-kb deletion that would truncate the protein product just after the coiled-coil region and eliminate the entire TPR region; the second copy of the klc-2 gene, inserted upstream of the endogenous locus, lacks the two alternative last exons, resulting in a protein truncated after the TPRs (Figure 2A). klc-2(km11) homozygous mutants are viable and exhibit uncoordinated movement.
To assess the neuronal defects in klc-2(km11) and klc-2(km28) animals, we examined the dorsal D-type (DD) motor neurons. The DD motor neurons are GABAergic neurons and express the UNC-25 glutamic acid decarboxylase (Jin et al., 1999). From first stage larvae to adult animals, the DD neurons in wild-type animals have a side-down H-shaped axon morphology with the parallel ventral and dorsal processes connecting through a single lateral extending commissure (see Figure 8A). During the first larval stage (L1), the DD neurons form synapses to the ventral body muscles (White et al., 1978). The SNB-1::GFP synaptic vesicle marker driven by the unc-25 promoter (Punc-25-SNB-1::GFP) is localized exclusively along the ventral processes of the DD neurons in L1 worms (Figure 2B; Hallam and Jin, 1998; Nonet, 1999). Mutations in unc-16 and unc-116 cause SNB-1::GFP to be improperly localized to the dorsal branches of the L1 DDs in addition to their normal ventral localization (Figure 2B). In klc-2(km11) L1 larvae, we observed similar defects. The overall cell morphology of the DD neurons was normal (unpublished data), but SNB-1::GFP was improperly localized along the dorsal branches in 30% of klc-2(km11) L1 animals (Figure 2B, Table 1). In klc-2(km28) animals, some DD neuron axons exhibited dorsal extension defects (unpublished data), but 100% of the arrested L1 animals showed mislocalization of SNB-1::GFP to the dorsal processes (Figure 2B, Table 1). The phenotypic resemblance is consistent with the conclusion that unc-116, klc-2, and unc-16 function together to regulate the transport or localization of cargo containing SNB-1::GFP.
Table 1.
Quantitation of SNB-1::GFP in L1 animals
| Genotype | All ventral (%) | Dorsal and ventral (%) | n |
|---|---|---|---|
| juIs1 | 97 | 3 | 227 |
| unc-14(ju56); juIs1 | 29 | 71 | 107 |
| unc-16(e109); juIs1 | 4 | 96 | 300 |
| klc-2(km11); juIs1 | 70 | 30 | 360 |
| klc-2(km28); juIs1a | 0 | 100 | 35 |
| unc-16(e109); klc-2(km28); juIs1b | 0 | 100 | 56 |
| unc-14(ju56);klc-2(km28); juIs1c | 0 | 100 | 3 |
n, number of animals scored.
klc-2(km28);juIs1 was born from klc-2(km28)/nT1;juIs1.
unc-16(e109);klc-2(km28);juIs1 was born from unc-16(e109);klc-2(km28)/nT1;juIs1.
unc-14(ju56);klc-2(km28);juIs1 was born from unc-14(ju56);klc-2(km28)/ nT1;juIs1.
Localization Dependence of KLC-2, UNC-116, and UNC-16
A further prediction of UNC-116, KLC-2, and UNC-16 acting in the same pathway is that they should colocalize. UNC-16 is broadly expressed in many tissues, and functional UNC-16::GFP protein is localized along nerve processes (Byrd et al., 2001). We examined the expression of UNC-116 and KLC-2 using GFP-tagged transgenes driven by their endogenous promoters (see Materials and Methods). Expression of UNC-116::GFP rescued the uncoordinated phenotype of unc-116(e2281) and expression of KLC-2::GFP rescued the lethal phenotype of klc-2(km28) and the uncoordinated phenotype of klc-2(km11). We generated a series of transgenes to determine the proper expression level of KLC-2::GFP by scoring the rescuing activity of klc-2(km11) (Supplementary Table and Figure). We chose the minimal level of expression to observe the GFP pattern, reasoning that such pattern was most likely representing the endogenous localization of UNC-116 and KLC-2. Expression of the UNC-116::GFP and KLC-2::GFP fusion proteins was seen broadly in multiple tissues including most of the neurons, muscles, and pharynx (Figure 3, A-H). The GFP expression within a given cell was in general diffuse and excluded from the nucleus. It has been suggested that the functions of the kinesin light chain may include binding cargo and regulating the heavy chain (Verhey et al., 1998). Kinesin heavy chain remains localized at the Golgi in the absence of functional kinesin light chain in vivo (Rahman et al., 1999). We addressed how the localization of UNC-116 depends on KLC-2 by examining the expression of the UNC-116::GFP transgene in klc-2(km11) mutants. We found that UNC-116::GFP was retained in perinuclear regions of the cell bodies in klc-2(km11) mutants (Figure 4A), an area where Golgi apparatus usually resides. These observations further confirm that KLC-2 and UNC-116 are orthologues of mammalian KLC and the KIF5 KHC, respectively.
Figure 3.
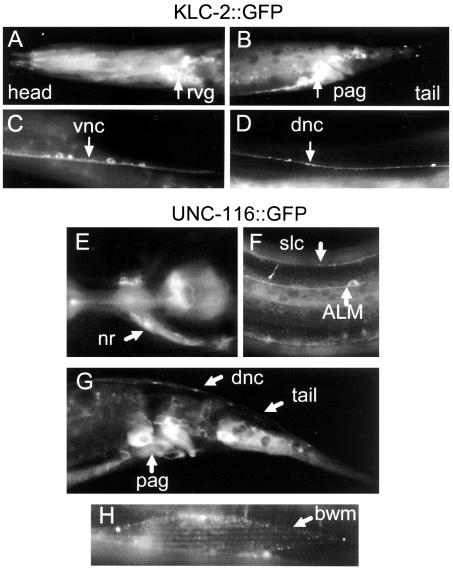
Expression of KLC-2 and UNC-116. (A-D) Expression pattern and localization of KLC-2::GFP. Panels show wild-type young adult animals expressing Pklc-2-KLC-2::GFP (kmEx801). Arrows indicate rvg, retrovesicular ganglion (A); pag, preanal ganglion (B); vnc, ventral nerve cord (C); and dnc, dorsal nerve cord (D). (E-H) Expression pattern and localization of UNC-116::GFP. Panels show unc-116(e2281) animals rescued by expression of Punc-116 UNC-116::GFP (juEx637). Arrows indicate nrn, nerve ring neuropile (E); ALM mechanosensory neuron and slnc, sublateral nerve cord (F); various cell types in the tail (G); and bwm, body wall muscle (H).
The broad tissue expression of both UNC-116 and KLC-2 impaired our ability to examine precisely how they were localized within neurons. To overcome this problem, we chose to express each protein in the type D neurons. We generated constructs to express functional GFP-tagged KLC-2 or UNC-16 fusion proteins under the control of the unc-25 promoter. KLC-2::GFP was seen diffusely throughout the axons of the D type motor neurons (Figure 4B, Supplementary Figure), whereas UNC-16::GFP showed discretely punctate pattern (Figures 4B, Supplementary Figure), although the puncta size was somewhat variable depending on the level of expression. We then introduced the transgenes expressing UNC-16::GFP or KLC-2::GFP fusion proteins into kinesin-1 (unc-116 and klc-2) and unc-16 mutants. We observed that KLC-2::GFP showed similar patterns in wild-type and unc-16(e109) mutants and was slightly irregular in unc-116(e2281) mutants (Figure 5B). In contrast, UNC-16::GFP localization in the type D neurons appeared both aggregated and diffuse in different regions of the nerve processes in unc-116(e2281) and klc-2(km11) mutants (Figure 4B), consistent with our previous report that UNC-16::GFP localization depends on UNC-116/KHC (Byrd et al., 2001).
Because UNC-16 is likely mediating the transport of SNB-1 by kinesin-1, we asked how the punctate pattern of UNC-16::GFP was correlated with SNB-1::GFP. We coexpressed UNC-16::YFP and SNB-1::CFP in the D-type neurons. We observed that UNC-16::YFP was consistently detected in regions where SNB-1::CFP puncta were prominent (97.2 ± 0.9%, n = 27 animals; Figure 5A; see Materials and Methods). We further addressed how the correlated punctate pattern of UNC-16 and SNB-1 might be affected in unc-116 mutants. We observed a range of phenotypes that we classified as 1) aggregate, 2) diffuse, 3) gaps or absent, and 4) normal/punctate (Figure 5B). Class 1-3 phenotypes were seen in most animals, although the regions affected varied along the nerve cords and between animals. Both UNC-16 and SNB-1 were largely absent from the dorsal processes and when present tend to localize as aggregates. The localization pattern of SNB-1 and UNC-16 in the ventral processes of the D neurons was largely diffuse and irregular. In the rare case where SNB-1 was punctate in unc-116 mutants (6/46 animals, length ranging between 10 and 45 μm) the incidence of detecting UNC-16 was decreased (66.0 ± 15.8%). This analysis supports our conclusion that the localization of both UNC-16 and SNB-1 is regulated by Kinesin-1. However, the limited resolution of light microscope prevents us from further concluding whether there were significant uncoupling or coupling of UNC-16 with SNB-1 in unc-116 mutants. Together, the results of protein binding, mutant phenotypes, and the localization studies demonstrate that UNC-116, KLC-2, and UNC-16 are members of a protein complex that likely transports cargos containing synaptic vesicle components in neurons.
UNC-14 Is a Binding Partner of UNC-16 and KLC-2
To identify additional proteins that interact with UNC-16, we also carried out the yeast two-hybrid screen with a bait construct encoding an N-terminal fragment of UNC-16 (aa 1-709). From this screen, we identified one clone containing a cDNA encoding UNC-14. UNC-14 is a novel protein that contains a RUN domain and has been previously shown to bind the conserved Ser/Thr kinase, UNC-51 (Ogura et al., 1997; Figure 6A). The RUN domain of UNC-14 is closely related to that of NESCA, a human protein highly expressed in the brain that likely plays a role in neurotrophin-dependent neurite outgrowth (Matsuda et al., 2000; MacDonald et al., 2004; Figure 6B). UNC-14 binds UNC-51 through the middle portion of the protein (Figure 6, A and C). By a yeast two-hybrid assay, we determined that the RUN domain-containing C-terminal region of UNC-14 was sufficient to bind the N-terminal region of UNC-16 (Figure 6C), indicating that UNC-14 binds to UNC-51 and UNC-16 through different domains. However, full-length UNC-16 did not show a positive interaction with UNC-14 in the yeast two-hybrid assay, suggesting either that the interaction might be the result of expressing a truncated UNC-16 protein or that the C-terminal region of UNC-16 might block UNC-14 binding in the absence of other factors. To discern between these two possibilities, we examined the binding interaction of UNC-14 with UNC-16 by coimmunoprecipitation in mammalian 293 cells. When full-length epitope-tagged UNC-16 and UNC-14 were coexpressed in mammalian 293 cells, T7-UNC-16 did not coimmunoprecipitate with Flag-UNC-14 (Figure 6D, lane 2). However, in the presence of HA-KLC-2, T7-UNC-16 coimmunoprecipitated with Flag-UNC-14 (Figure 6D, lane 6). Interestingly, HA-KLC-2 coimmunoprecipitated with FLAG-UNC-14 in the absence of UNC-16 (Figure 6D, lane 5). These results suggest that the interaction between full-length UNC-16 and UNC-14 may rely on KLC-2 to relieve the inhibitory effect of the C-terminal region of UNC-16.
Figure 6.
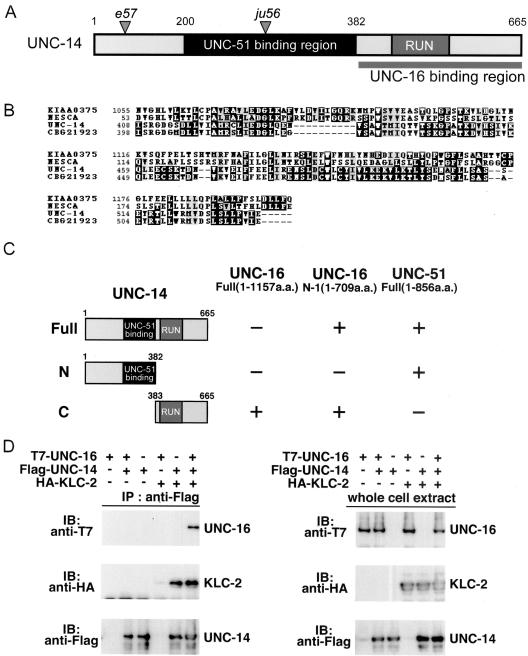
UNC-16 binds to the RUN domain region of UNC-14. (A) UNC-14 protein domain structure. The RUN domain and the UNC-16 binding region are shown; the shaded region shows the region that was previously reported to bind UNC-51 (Ogura et al., 1997). Positions of the e57 and ju56 early stop mutations are also indicated. (B) RUN domain sequence alignment. Multiple sequence alignment with human NESCA and conceptual translation of KIAA0375, C. elegans UNC-14, and C. briggsae CBG21923 RUN domains constructed with CLUSTALW and displayed by BOXSHADE. (C) Interaction of UNC-14 with UNC-16 and UNC-51 in the yeast two-hybrid system. UNC-16 N-1 and UNC-51 were constructed with the LexA DNA-binding domain (BD). Various regions of UNC-14 constructed with the GAL4 activation domain (AD) are shown on the left. Plus (+) and minus (-) indicate positive and negative interactions, respectively. (D) Coimmunopreciptation of UNC-16 and KLC-2 with UNC-14. HEK 293 cells were transfected with control vector (-), T7-UNC-16, Flag-UNC-14, and HA-KLC-2, as indicated. Cell lysates were immunoprecipitated (IP) with anti-Flag antibody (left panels). Immunoprecipitates were immunoblotted (IB) with anti-T7 (top panel) and anti-HA (middle panel) antibodies. The amounts of immunoprecipitated Flag-UNC-14 were determined with anti-Flag antibody (bottom panel). Whole-cell extracts were immunoblotted with anti-T7, anti-HA, and anti-Flag antibodies to determine total amounts of T7-UNC-16, HA-KLC-2, and Flag-UNC-14, respectively (right panels).
UNC-14 Localization Depends on Kinesin-1 and UNC-16
To address whether UNC-14 localization depends on kinesin-1 and UNC-16, we constructed functional UNC-14::GFP transgenes, expressed under the unc-25 promoter. In wild-type animals, UNC-14::GFP showed punctate expression along the nerve processes (Figure 7A). In klc-2(km11) and unc-116(e2281) mutant animals, UNC-14::GFP was diffuse and irregular (Figure 7A, Table 2), consistent with UNC-14 being a cargo for kinesin-1. UNC-14::GFP also showed a more diffuse pattern in unc-16(e109) mutants (Figure 7A, Table 2). Conversely, the localization of KLC-2::GFP and UNC-16::GFP in the D neurons of unc-14(ju56) mutants was not noticeably different from in wild-type animals (Figure 7B). These results are consistent with UNC-14 and UNC-16 being transported by the UNC-116/KLC-2 kinesin-1 and possibly functioning as adaptors for cargo recognition.
Table 2.
UNC-14::GFP expression in kinesin-1 and unc-16 mutants
| Genotype | % Punctate | % Diffuse | n |
|---|---|---|---|
| kmEx820 | 80.7 (71) | 19.3 (17) | 88 |
| kmEx821 | 75.5 (40) | 24.5 (13) | 53 |
| unc-116(e2281); kmEx820 | 23.7 (22) | 76.3 (71) | 93 |
| unc-116(e2281); kmEx821 | 23.6 (13) | 76.4 (42) | 55 |
| klc-2(km11); kmEx820 | 27.8 (25) | 72.2 (65) | 90 |
| klc-2(km11); kmEx821 | 25.5 (13) | 74.5 (38) | 51 |
| unc-16(e109); kmEx820 | 48.8 (41) | 51.2 (43) | 84 |
| unc-16(e109); kmEx821 | 50.0 (26) | 50.0 (26) | 52 |
The UNC-14::GFP localization pattern (from two independent extrachromosomal arrays) was observed by taking confocal images under the same settings. The score “punctate” refers to that UNC-14::GFP was seen as discrete and regularly spaced fluorescent puncta, and “diffuse” refers to that UNC-14::GFP appeared diffuse and were not visually separable as puncta. n, number of animals scored.
unc-14 Mutant Animals Exhibit Transport Defects
Previous work examining neurons in fixed animals has shown that neuronal differentiation is arrested at neurite extension in unc-14 mutants (McIntire et al., 1992). Using GFP markers, we further examined the development of the D-type neurons in unc-14(e57) mutants over time in live animals. We noticed that in unc-14 mutant animals, the axons of the D neurons showed dynamic axon morphology throughout the larval and adult stages. Many DD processes were able to grow and extend to the dorsal side in L1 animals (Figure 8A), but these processes collapsed and appeared to degenerate by the adult stage (n = 10, unpublished data). Some DD processes showed an initial delay in the outgrowth, as they did not reach the dorsal side by the end of the L1 stage, but later were able to continue to grow and eventually reach the dorsal cord, forming the dorsal processes in adults. For those DD neurons that had normally extended dorsal processes in unc-14(e57) mutant L1 animals, we found that the SNB-1::GFP synaptic vesicle marker was improperly localized along the dorsal processes, similar to what is observed in unc-16 mutant L1 animals (Figure 8B).
Axon outgrowth depends on membrane addition. The above observations indicate that unc-14 may be involved in both membrane addition and synaptic vesicle transport. To further decipher the functions of unc-14 in these two processes, we examined a partial loss-of-function mutation of unc-14. The unc-14(ju56) mutation generates a stop codon at amino acid Lys277 (Figure 6A). This lesion removes the UNC-16-binding region of UNC-14, but leaves much of the UNC-51-binding region intact. unc-14(ju56) animals showed largely normal movement (unpublished data). In unc-14(ju56), DD neurons had normal axon morphology throughout larval development and in adults (Figure 8A for L1 stage). However, the SNB-1::GFP marker was found in the dorsal processes of L1 DD neurons (Figure 8B). These observations suggest that the interaction between UNC-14 and UNC-51 may be required for proper axon outgrowth and that the interaction between UNC-14 and UNC-16 may be required for the proper transport or localization of synaptic vesicles. Consistent with this interpretation, expression of a truncated UNC-14 containing the UNC-51-binding region has been shown to rescue the movement and axon outgrowth phenotypes of unc-14(e57) mutants (Ogura et al., 1997). Our data do not exclude the possibility that the N-terminal of UNC-14 may contribute to both axon outgrowth and SNB-1::GFP trafficking.
We further used genetic double-mutant analysis to address whether unc-14, unc-16, unc-116, and klc-2 function in the same pathway. Null mutations in unc-116 and klc-2 result in embryonic and early larval lethality (Patel et al., 1993; see above), therefore impairing our ability to obtain viable homozygous double mutants (see Table 1). We constructed unc-14; unc-16, unc-14; unc-116, and unc-14; klc-2 double mutants using strong loss-of-function alleles available. We quantified the L1 SNB-1::GFP synaptic vesicle marker phenotype in unc-16(e109), unc-116(e2281), and unc-14(ju56) single mutants and unc-14(ju56); unc-16(e109) and unc-14(ju56); unc-116(e2281) double mutants (Figure 8C). Similar to unc-116 (Byrd et al., 2001), unc-14 does not enhance the L1 SNB-1::GFP phenotype of unc-16 mutants. The mean score for L1 dorsal SNB-1::GFP in unc-14(ju56); unc-16(e109) double mutants is not significantly different from the mean score for unc-16(e109) single mutants (p = 0.333). The mean scores for L1 dorsal SNB-1::GFP in unc-14(ju56); unc-116(e2281) double mutants and unc-14(ju56) are also not significantly different (p = 0.21). If unc-14 functions in a pathway parallel to that of unc-16 and unc-116 rather than in the same pathway, we would expect the phenotypes of the double mutants to be additive. Although the mean L1 SNB-1::GFP scores are significantly different in other pairwise comparisons (Figure 8C), the scores of the unc-14; unc-16 and unc-14; unc-116 double mutants do not reflect an additive effect indicative of two genes functioning in parallel pathways. Similar observations were seen in unc-14; klc-2 animals (Table 1). This analysis supports the model that unc-16 and unc-14 function in the same pathway to regulate the localization or transport of synaptic vesicle precursors.
DISCUSSION
In this report, we provide direct evidence demonstrating that UNC-116 and KLC-2 constitute a functional kinesin-1 complex in C. elegans. This kinesin-1 complex physically interacts with the UNC-16 JNK-signaling scaffold and UNC-14 RUN-domain proteins through their binding to KLC-2. UNC-16, UNC-14, and the components of kinesin-1, UNC-116 and KLC-2, are all required for the proper localization of a synaptic vesicle marker, perhaps by directly transporting cargos containing synaptic vesicle components. Our results provide in vivo evidence that neurite extension and vesicular transport are functionally separable processes that involve common components, including UNC-14.
UNC-116 and KLC-2 Are Functional Components of Kinesin-1
Although UNC-116 is the only KIF5 homolog in C. elegans, there are two predicted KLCs (Koushika and Nonet, 2000). KLC-1 and KLC-2 share 50% identity. We show here that KLC-2 and UNC-116 constitute the C. elegans kinesin-1 complex. First, KLC-2 and UNC-116 physically interact. Second, they are detected in the same set of cells. Third, reduction of their activity by mutations results in similar transport defects. Our finding is in agreement with the composition of C. elegans kinesin-1 purified biochemically (Signor et al., 1999). Furthermore, the data reported here support our previous conclusion that UNC-16 is a cargo adaptor or regulator of kinesin-1 through its binding to KLC-2. The domains that mediate such binding are consistent with those reported for the vertebrate and fly orthologues (Bowman et al., 2000; Verhey et al., 2001), indicating that the kinesin-1 and JIP3/SYD/UNC-16 protein complex formation is evolutionarily conserved.
UNC-14 May Bridge Synaptic Transport by Kinesin-1 and Membrane Addition during Axon Outgrowth
All previously isolated unc-14 mutations are nonsense mutations that truncate the protein within the first 200 amino acids (Ogura et al., 1997). In unc-14 null mutants, neuronal differentiation is arrested at neurite outgrowth, and the arrested neurites contain varicosities and atypical vesicles (McIntire et al., 1992), suggesting that unc-14 function may be required for transport of membranous vesicles. Our analysis of the unc-14(ju56) mutant has led us to resolve the two functions of unc-14 in neurite extension and neuronal transport. In unc-14(ju56) animals, neuronal morphology is normal, but SNB-1::GFP is localized improperly. The ju56 lesion likely leads to the production of a truncated UNC-14 protein that would lack the domain interacting with UNC-16, but would leave its interaction with UNC-51 intact. Consistently, expression of a truncated UNC-14 containing the UNC-51-binding region rescued the movement and axon outgrowth phenotypes of unc-14(e57) mutants (Ogura et al., 1997).
UNC-16 and UNC-14 Function in Cargo Recognition of Kinesin-1
UNC-16 and UNC-14 show physical binding with KLC-2. Loss of function in both genes affects the localization of SNB-1::GFP marker to a similar degree. Our double-mutant analysis is consistent with their functioning in a linear pathway, although definitive proof is impaired by the embryonic and early larval lethality associated with null mutations of kinesin-1. We have further explored the cellular interaction between UNC-16, UNC-14, and kinesin-1 using GFP-tagged functional transgenes in a specific type of neurons. Our rationale is that the broad tissue expression of these genes imposes technical ambiguity in detecting subcellular changes by light microscope analysis. Such an approach is confounded by potential artifactural effects associated with transgene overexpression. We have tried to express the GFP reporter genes at minimal detection level, with the caveat that the expression pattern and level of the endogenous proteins studied here are currently not known. Our analysis shows that both UNC-14 and UNC-16 subcellular localization is altered in kinesin-1 mutants, consistent with them being carried by kinesin-1. The observation that UNC-14 localization is subtly altered in unc-16 mutants suggests that UNC-16 may have a regulatory role in either facilitating the binding of UNC-14 to kinesin-1 or fine-tuning UNC-14 localization at the destination.
How Might UNC-14 and UNC-16 Regulate Synaptic Vesicle Transport/Localization?
The UNC-14 protein binds the UNC-51 serine/threonine kinase and is thought to play a role in oligomerizing UNC-51, thereby regulating its function in axon outgrowth (Ogura et al., 1997). Mammalian UNC-51 (mUnc51) is also required for axon outgrowth, interacts with the endosome protein syntenin, and localizes to vesicular membranes (Tomoda et al., 2004). The UNC-51-binding region in UNC-14 is in the central region, spatially distinct from the RUN domain. Although the role of the RUN domain in UNC-14 is not yet known, RUN domains appear to be linked to the functions of Rap and Rab family GTPases and may be required for localization to detergent-insoluble endosomal microdomains (Callebaut et al., 2001; Mari et al., 2001). It is possible that UNC-16 and UNC-14 may regulate the spatial or temporal dynamics of cargo-motor interactions. These kinesin-1-binding proteins could respond to positional or temporal cues within their intracellular microenvironment, such as the structural ends of microtubules or hormonal/developmental signals, and direct cargo-motor interactions accordingly. Intriguingly, the dynein light IC, DLI-1, was also identified as a potential binding partner of UNC-16 (our unpublished data), suggesting that UNC-16 could signal changes in cargo direction through a physical interaction with both the kinesin-1 plus-end-directed microtubule motor and the cytoplasmic dynein minus-end-directed microtubule motor. UNC-16 can also bind JNK and its activator kinases (Byrd et al., 2001). Another possibility is that UNC-14 may link the UNC-51 Ser/Thr kinase and the UNC-16 JNK-signaling scaffold to complete a JNK-signaling module, thereby regulating kinesin-1 transport activity through cargo recognition or attachment.
The identification of UNC-14 as an associated protein of UNC-16 and kinesin-1 complex begins to fill the gap in understanding how motor proteins pair with particular cargos at the right time and place. Because UNC-14 is required for both proper axon outgrowth and localization of synaptic material, an UNC-14/UNC-16 signaling complex could then possibly regulate the balance between the transport of plasma membrane or synaptic materials by regulating different cargo interactions with the kinesin-1 motor. Future experiments will be necessary to address whether UNC-16 interacts separately with its interacting partners or in one large complex. Moreover, determining the dynamics and spatial regulation of protein binding of UNC-16 and UNC-14 with their partners in vivo will potentially provide a key to understanding the mechanisms of vesicular genesis and transport.
Supplementary Material
Acknowledgments
We thank A. Coulson, A. Fire, Y. Ohshima, K. Ogura, and Caenorhabditis Genetics Center for reagents and strains; M. Taniguchi for the yeast two-hybrid screen using UNC-16 N-1 as a bait; K. Maki for isolation of the klc-2(km28) mutant; J. Kimble for her generous support; L. Lamont and A. Kidd for critical reading of the manuscript; and our lab members for helpful discussions and suggestions. This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture and Science of Japan (N.H.), by special grants for CREST and Advanced Research on Cancer from the Ministry of Education, Culture, and Science of Japan (K.M.), and by grants from the National Science Foundation and National Institutes of Health to Y. J. Y.J. is an assistant investigator of Howard Hughes Medical Institute. D.B. was partially supported by a UC president's dissertation-year fellowship.
Article published online ahead of print in MBC in Press on November 24, 2004 (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-07-0553).
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Akechi, M., Ito, M., Uemura, K., Takamatsu, N., Yamashita, S., Uchiyama, K., Yoshioka, K., and Shiba, T. (2001). Expression of JNK cascade scaffold protein JSAP1 in the mouse nervous system. Neurosci. Res. 39, 391-400. [DOI] [PubMed] [Google Scholar]
- Bowman, A. B., Kamal, A., Ritchings, B. W., Philp, A. V., McGrail, M., Gindhart, J. G., and Goldstein, L.S. (2000). Kinesin-dependent axonal transport is mediated by the sunday driver (SYD) protein. Cell 103, 583-594. [DOI] [PubMed] [Google Scholar]
- Brady, S. T. (1985). A novel brain ATPase with properties expected for the fast axonal transport motor. Nature 317, 73-75. [DOI] [PubMed] [Google Scholar]
- Brenner, S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd, D. T., Kawasaki, M., Walcoff, M., Hisamoto, N., Matsumoto, K., and Jin, Y. (2001). UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans. Neuron 32, 787-800. [DOI] [PubMed] [Google Scholar]
- Callebaut, I., de Gunzburg, J., Goud, B., and Mornon, J. P. (2001). RUN domains: a new family of domains involved in Ras-like GTPase signaling. Trends Biochem. Sci. 26, 79-83. [DOI] [PubMed] [Google Scholar]
- Fan, J., and Amos, L. A. (1994). Kinesin light chain isoforms in Caenorhabditis elegans. J. Mol. Biol. 240, 507-512. [DOI] [PubMed] [Google Scholar]
- Foletti, D. L., Prekeris, R., and Scheller, R. H. (1999). Generation and maintenance of neuronal polarity: mechanisms of transport and targeting. Neuron 23, 641-644. [DOI] [PubMed] [Google Scholar]
- Goldstein, L. S., and Philp, A. V. (1999). The road less traveled: emerging principles of kinesin motor utilization. Annu. Rev. Cell. Dev. Biol. 15, 141-183. [DOI] [PubMed] [Google Scholar]
- Goldstein, L. S., and Yang, Z. (2000). Microtubule-based transport systems in neurons: the roles of kinesins and dyneins. Annu. Rev. Neurosci. 23, 39-71. [DOI] [PubMed] [Google Scholar]
- Hallam, S. J., and Jin, Y. (1998). lin-14 regulates the timing of synaptic remodelling in Caenorhabditis elegans. Nature 395, 78-82. [DOI] [PubMed] [Google Scholar]
- Jin, Y., Jorgensen, E., Hartwieg, E., and Horvitz, H. R. (1999). The Caenorhabditis elegans gene unc-25 encodes glutamic acid decarboxylase and is required for synaptic transmission but not synaptic development. J. Neurosci. 19, 539-548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki, M., Hisamoto, N., Iino, Y., Yamamoto, M., Ninomiya-Tsuji, J., and Matsumoto, K. (1999). A Caenorhabditis elegans JNK signal transduction pathway regulates coordinated movement via type-D GABAergic motor neurons. EMBO J. 18, 3604-3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelkar, N., Delmotte, M. H., Weston, C. R., Barrett, T., Sheppard, B. J., Flavell, R. A., and Davis, R. J. (2003). Morphogenesis of the telencephalic commissure requires scaffold protein JNK-interacting protein 3 (JIP3). Proc. Natl. Acad. Sci. USA 100, 9843-9848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koushika, S. P., and Nonet, M. L. (2000). Sorting and transport in C. elegans: a model system with a sequenced genome. Curr. Opin. Cell Biol. 12, 517-523. [DOI] [PubMed] [Google Scholar]
- Leopold, P. L., McDowall, A. W., Pfister, K. K., Bloom, G. S., and Brady, S. T. (1992). Association of kinesin with characterized membrane-bounded organelles. Cell Motil. Cytoskelet. 23, 19-33. [DOI] [PubMed] [Google Scholar]
- MacDonald, J. I., Kubu, C. J., and Meakin, S. O. (2004). Nesca, a novel adapter, translocates to the nuclear envelope and regulates neurotrophin-induced neurite outgrowth. J. Cell Biol. 164, 851-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari, M., Macia, E., Le Marchand-Brustel, Y., and Cormont, M. (2001). Role of the FYVE finger and the RUN domain for the subcellular localization of Rabip4. J. Biol. Chem. 276, 42501-42508. [DOI] [PubMed] [Google Scholar]
- Matsuda, S., Miyazaki, K., Ichigotani, Y., Kurata, H., Takenouchi, Y., Yamamoto, T., Nimura, Y., Irimura, T., Nakatsugawa, S., and Hamaguchi, M. (2000). Molecular cloning and characterization of a novel human gene (NESCA) which encodes a putative adapter protein containing SH3. Biochim. Biophys. Acta 1491, 321-326. [DOI] [PubMed] [Google Scholar]
- McIntire, S. L., Garriga, G., White, J., Jacobson, D., and Horvitz, H. R. (1992). Genes necessary for directed axonal elongation or fasciculation in C. elegans. Neuron 8, 307-322. [DOI] [PubMed] [Google Scholar]
- Mello, C. C., Kramer, J. M., Stinchcomb, D., Ambros, V. (1991). Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J. 10, 3959-3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, K. G., Alfonso, A., Nguyen, M., Crowell, J. A., Johnson, C. D., and Rand, J. B. (1996). A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc. Natl. Acad. Sci. USA 93, 12593-12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet, M. L. (1999). Visualization of synaptic specializations in live C. elegans with synaptic vesicle protein-GFP fusions. J. Neurosci. Methods 89, 33-40. [DOI] [PubMed] [Google Scholar]
- Ogura, K., Shirakawa, M., Barnes, T. M., Hekimi, S., and Ohshima, Y. (1997). The UNC-14 protein required for axonal elongation and guidance in Caenorhabditis elegans interacts with the serine/threonine kinase UNC-51. Genes Dev. 11, 1801-1811. [DOI] [PubMed] [Google Scholar]
- Patel, N., Thierry-Mieg, D., and Mancillas, J. R. (1993). Cloning by insertional mutagenesis of a cDNA encoding Caenorhabditis elegans kinesin heavy chain. Proc. Natl. Acad. Sci. USA 90, 9181-9185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman, A., Kamal, A., Roberts, E. A., and Goldstein, L. S. (1999). Defective kinesin heavy chain behavior in mouse kinesin light chain mutants. J. Cell Biol. 146, 1277-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato-Yoshitake, R., Yorifuji, H., Inagaki, M., and Hirokawa, N. (1992). The phosphorylation of kinesin regulates its binding to synaptic vesicles. J. Biol. Chem. 267, 23930-23936. [PubMed] [Google Scholar]
- Signor, D., Wedaman, K. P., Rose, L. S., Scholey, J. M. (1999). Two heteromeric kinesin complexes in chemosensory neurons and sensory cilia of Caenorhabditis elegans. Mol. Biol. Cell 10, 345-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockinger, W., Brandes, C., Fasching, D., Hermann, M., Gotthardt, M., Herz, J., Schneider, W. J., and Nimpf, J. (2000). The reelin receptor ApoER2 recruits JNK-interacting proteins-1 and -2. J. Biol. Chem. 275, 25625-25632. [DOI] [PubMed] [Google Scholar]
- Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F., and Higgins, D. G. (1997). The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoda, T., Kim, J. H., Zhan, C., and Hatten, M. E. (2004). Role of Unc51.1 and its binding partners in CNS axon outgrowth. Genes Dev. 18, 541-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale, R. D. (2003). The molecular motor toolbox for intracellular transport. Cell 112, 467-480. [DOI] [PubMed] [Google Scholar]
- Vale, R. D., Reese, T. S., and Sheetz, M. P. (1985). Identification of a novel forcegenerating protein, kinesin, involved in microtubule-based motility. Cell 42, 39-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey, K. J., Lizotte, D. L., Abramson, T., Barenboim, L., Schnapp, B. J., and Rapoport, T. A. (1998). Light chain-dependent regulation of Kinesin's interaction with microtubules. J. Cell Biol. 143, 1053-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey, K. J., Meyer, D., Deehan, R., Blenis, J., Schnapp, B. J., Rapoport, T. A., and Margolis, B. (2001). Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J. Cell Biol. 152, 959-970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey, K. J., and Rapoport, T. A. (2001). Kinesin carries the signal. Trends Biochem. Sci. 26, 545-550. [DOI] [PubMed] [Google Scholar]
- White, J. G., Albertson, D. G., and Anness, M. A. (1978). Connectivity changes in a class of motoneurone during the development of a nematode. Nature 271, 764-766. [DOI] [PubMed] [Google Scholar]
- Woehlke, G., and Schliwa, M. (2000). Walking on two heads: the many talents of kinesin. Nat. Rev. Mol. Cell Biol. 1, 50-58. [DOI] [PubMed] [Google Scholar]
- Yandell, M. D., Edgar, L. G., and Wood, W. B. (1994). Trimethylpsoralen induces small deletion mutations in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 91, 1381-1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen, M., and Jin, Y. (1999). The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C. elegans. Nature 401, 371-375. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.