

Abstract
Head and neck squamous cell carcinoma (HNSCC) is an aggressive malignancy with high morbidity and mortality. Despite advances in cytotoxic therapies and surgical techniques, overall survival (OS) has not improved over the past few decades. This emphasises the need for intense investigation into novel therapies with good tumour control and minimal toxicity. Cancer immunotherapy has led this endeavour, attempting to improve tumour recognition and expand immune responses against tumour cells. While various forms of HNSCC immunotherapy are in preclinical trials, the most promising direction thus far has been with monoclonal antibodies (mAbs), targeting growth factor and immune checkpoint receptors. Preclinical and early phase trials have shown unprecedented efficacy with minimal adverse effects. This article will review biological mechanisms of immune escape and implications for immunotherapy in HNSCC.
Keywords: Head and neck cancer, Squamous cell, carcinoma, Immunotherapy, Checkpoint receptors
1. Introduction
Head and neck squamous cell carcinoma (HNSCC) is an aggressive epithelial malignancy associated with lymph node metastasis and immunosuppression [1,2]. It is the sixth leading cancer worldwide and affects 550,000 people each year [3]. Extensive tobacco and alcohol exposure are widely accepted causes of HNSCC, while human papillomavirus (HPV)-associated HNSCC is regarded as a separate entity due to its distinct disease pathophysiology, epidemiology and improved response to conventional therapies and surgery. Unfortunately, only modest improvements in the survival rate of HNSCC have been made despite advances in chemotherapy, radiotherapy, surgical techniques and combination regimens. HNSCC remains a lethal disease, with 5-year overall survival (OS) lingering at just 40—60% for the last 50 years [4]. Treatment failures remain unacceptably common, even amongst HPV-positive cancers, calling for a new paradigm to treat HNSCC.
Since the acceptance of the immune system's role in cancer development, advances in immunotherapy are now at the forefront of cancer research. Based on restoring key signalling pathways of the host immune system to counteract immune escape by malignant cells, cancer immunotherapy shows potential for durable responses with less adverse effects than conventional treatments. The first anticancer immunotherapeutic agents were the multifunctional cytokines interleukin 2 (IL-2), interferon alpha (IFN-α), and tumour necrosis factor alpha (TNF-α) [5—7]. These agents were developed in the 1980s, yet are still widely used in various conditions. However, the real breakthrough in immunotherapy came in 2010, with the development of immune checkpoint inhibitors. The first to be approved by the Food and Drug Administration (FDA) in 2011 was ipilimumab, a cytotoxic T-lymphocyte antigen 4 (CTLA4)-blocking monoclonal antibody (mAb), approved for /metastatic melanoma. This has opened up new possibilities for anticancer immunotherapy in other malignancies, such as HNSCC.
Cetuximab, a chimeric murine/human IgG1 mAb-blocking epidermal growth factor receptor (EGFR), was the first immunotherapeutic agent approved for HNSCC. Cetuximab activity largely stays within the activation of Fc gamma receptor (FcγR), leading to antibody-dependent cell-mediated cytotoxicity (ADCC) and sub-sequent antigen processing (Fig. 1) [8—11]. This leads to dendritic cell (DC) maturation and activation of cytotoxic T lymphocytes (CTLs), linking innate and adaptive tumour immunity [12—14]. Cetuximab could therefore overcome the immunosuppressive environment in HNSCC and increase clinical response to cancer treatment. However, patients treated with cetuximab have demonstrated an increase in suppressive regulatory T cells (Tregs) that impair ADCC, translating into just 10—20% of activity when used as a monotherapy[12,15,16]. However, when combined with the inhibitory blocking agent, ipilimumab, EGFR-antagonism was able to overcome this suppression and showed enhanced ADCC [16]. This has stressed the need for further investigation into active immunomodulating agents, such as the inhibitory and costimulatory immune checkpoint receptors and their ligands. Many of these agents have been developed and are being studied clinically in HNSCC, such as the immune checkpoint receptors CTLA4, programmed cell death protein 1 (PD1)/programmed cell death-ligand 1 (PD-L1), also, lymphocyte activation gene 3 (LAG3), T-cell membrane protein 3 (TIM3), costimulatory molecule, cluster of differentiation 137 (CD137) and costimulatory molecule CD134 also known as OX40 among others. In light of the mounting evidence in its effectiveness, the PD1-blocking mAb, pembrolizumab, had an accelerated FDA approval in August 2016 for HNSCC, and there is an anticipated approval of nivolumab later this year.
Fig. 1.
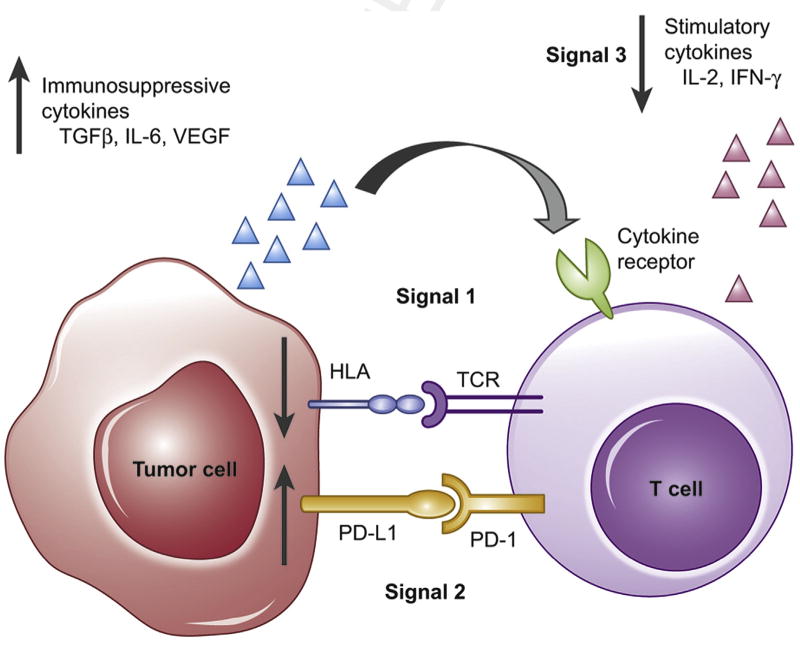
Immune surveillance and targets for evasion: T-cell activation requires three simultaneous signals at the ‘immune synapse’ in order to carry out its antitumour effects. Signal° 1 comprises the T-cell receptor (TCR)—HLA interaction, with presentation of non-self antigensfrom the tumour cell. Signal°2 is a summation of costimulatory and coinhibitory signals. These signals must occur in the presence of Signal°3, made up of immune-activating cytokines, such as IL-2 or IFN-γ. Immune evasion can occur at any of these signals (red arrows), impairing the immune system from effectively eradicating malignant cells.
Additionally, accumulating evidence demonstrates that conventional cytotoxic cancer therapies modulate the surviving tumour cells to make them more amenable to immune-mediated killing and induces a more immunostimulatory environment, termed ‘immunogenic modulation’ [17,18]. This encompasses a variety of molecular alterations to tumour cells, including the downregulation of antiapoptotic/survival genes, modulation of antigen-processing machinery (APM) components, and upregulating immune-relevant surface proteins. These consequences of cytotoxic therapy can therefore be used in synergistic regimens with immunotherapeutic agents to improve their efficacy. Indeed, these phenotypic changes translated into increased tumour sensitivity to T-cell-mediated lyses after low doses of chemotherapy or radiation [17,19—22]. While cetuximab has shown marginal benefit when added to definitive or adjuvant standard chemoradiation (cisplatin + radiation therapy) [23,24], other EGFR inhibitors show promise, such as the EGFR-targeted IgG2 mAb, panitumumab, which is FDA approved for EGFR-expressing metastatic colorectal carcinoma. In a phase II trial evaluating postoperative radiotherapy with concurrent cisplatin plus panitumumab in high risk HPV-negative patients, there was 70% progression-free survival (PFS), as compared to 50% with standard therapy [25]. While conventional cytotoxic therapies have frequent and significant acute and lifelong side-effects, immunotherapy has thus far shown to be safely tolerated with manageable side-effects. Therefore, adding immunotherapy to our clinical repertoire may be a means to not only reduce the cytotoxic load and subsequent side-effects, but also increase efficacy and durability compared to cytotoxic treatment alone. These findings have translated into promising clinical benefits for HNSCC and are the basis of many clinical trials underway.
Today, HNSCC immunotherapy encompasses a variety of diverse treatment approaches, including tumour-specific monoclonal antibodies (mAbs), immune-modulating antibodies, vaccines, oncolytic viruses and adoptive cell transfer. In this review, we will discuss biological mechanisms of immune escape and implications for immunotherapy.
2. Immune surveillance and immunoediting
Immune surveillance was first introduced by Burnet in 1970, who suggested that premalignant cells frequently develop, but are quickly eradicated by the immune system before an invasive tumour can develop [26]. Since then, mounting evidence supports the role of the immune system in preventing malignant transformation. This relationship between the immune system and malignancy can be seen clearly in immunocompromised patients who have an increased risk of cancer [27]. For example, there is an increased incidence of HNSCC in renal and bone marrow transplant patients, as well as in HIV-positive patients [28,29]. Although HNSCC patients have lower absolute lymphocyte counts than healthy subjects, as well as poor antigen presentation and impaired natural killer (NK) cell activity [27,30—35], most patients who develop HNSCC are immunocompetent. These tumours are therefore thought to evade the immune response through cancer immunoediting [36]. This theory suggests that while the immune system can eradicate some transformed cells, others that are less immunogenic and have gained a survival advantage, will allow immune evasion and subsequent tumour formation [37].
3. Mechanisms of immune escape in HNSCC and implications for therapeutic targets
In order to mount an effective immune response, effector T-cell activation requires three simultaneous signals at the ‘immune synapse’ (Fig. 2). However, there appears to be a failure in this system leading to impaired tumour antigen (TA)-specific T-cell recognition, impaired tumour cell lysis, and production of a tumour-pervasive microenvironment. This immunosuppression allows the development of clinically evident malignancies. Understanding the mechanisms of immune escape is imperative to the development of successful immunomodulating therapies.
Fig. 2.
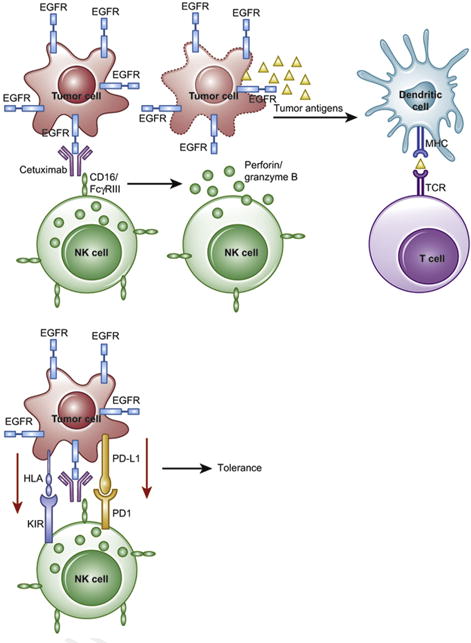
Antibody-dependent cell-mediated cytotoxicity (ADCC) is initiated by recognition of IgG-antibody-coated tumours by Fc receptors for IgG (FcγRIII), which are expressed by NK cells, macrophages and neutrophils (effector immune cells). In the absence of inhibitory signals, the effector cell releases perforin and granzymes and induces tumour cell apoptosis. This in turn releases immunogenic tumour antigens, which can be presented by an APC to T-cells, further adding to the immune response. However, in the presence of an inhibitory signal, such as PD1/PD-L1 or KIR/HLA-I, the NK cell fails to initiate ADCC and allows tumour evasion and progression.
3.1. Antigen presentation and processing
A key component for T-cell activation is the human leucocyte antigen (HLA) complex, which presents captured TAs to T cells, highlighting a potential evasion mechanism (Fig. 3). While complete loss of HLA function renders cells capable of evading T-cell recognition, it also serves as a strong stimulus for NK-cell activation and subsequent tumour cell killing [38]. Therefore, tumour cells are obligated to develop mechanisms to evade antigen recognition without total loss of HLA expression. Indeed, a significant proportion of HNSCC tumours show abnormal expression and function in HLA- and APM-encoding genes. This results in poor TA processing and presentation, providing tumour cells with a mechanism to escape T-cell-mediated lysis while avoiding NK activation [27,34,35,39–45]. Although the mechanisms behind APM component deficiency are still unclear, these abnormalities are significant as they are present in 20% of HNSCC specimens and correlate with lymph node metastasis and poor prognosis in HNSCC patients [42,46–48] and may impact the clinical response to T-cell-based immunotherapy. Ase EGFR is expressed in 80% of HNSCC tumours, EGFR-induced HLA downregulation may be an important mechanism in evading immune recognition. This escape mechanism is achieved through many oncogenic downstream pathways, including several important intracellular signalling pathways, featuring SH2 domain-containing protein-tyrosine phosphatase (SHP2)/ signal transducer and activator of transcription (STAT)1; mitogen-activated protein kinase (MAPK) and phosphatidyl inositol 3-kinase; and (PI3K)—protein kinase B, also known as AKT (AKT) [49—52]. Furthermore, HLA class I and APM component deficiency can be reversed in HNSCC by depleting SH2 domain-containing protein-tyrosine phosphatase (SHP2), or by using the signal transducer and activator of transcription (STAT)1 agonist IFN-γ, both increasing peptide: HLA class I complexes and enhancing CTL-mediated lysis of tumour cells [34,35,40,43,53—56]. Therefore, approaches that boost IFN-γ signalling and STAT1 activation, such as EGFR antagonism, may be significant in restoring antigen presentation through increased expression of HLA class I molecules and proinflammatory cytokines [54,57].
Fig. 3.
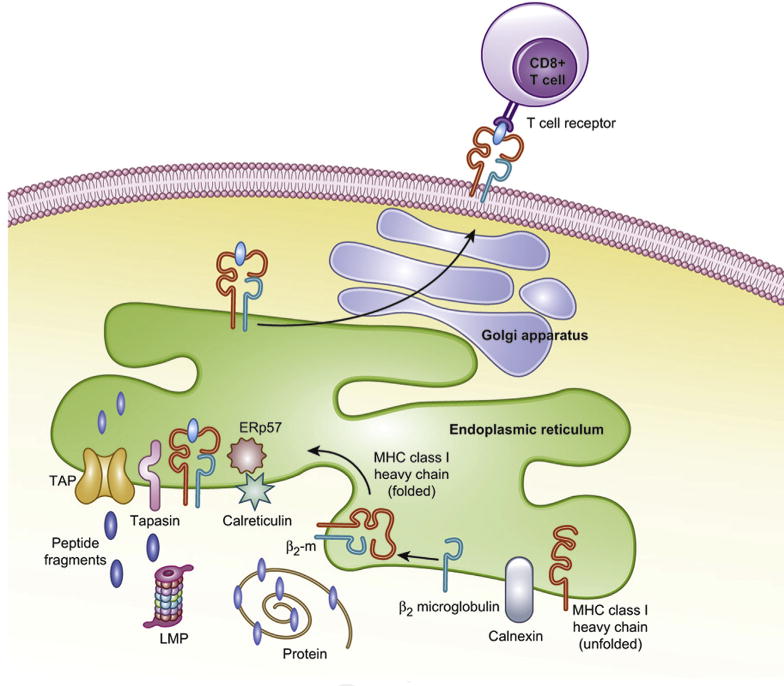
MHC class I antigen processing and presentation: Intracellular proteins are degraded via the ubiquitin-proteasome pathway. Once tagged by ubiquitin, the target protein is degraded by the proteasome. The proteasome is a multimeric protein complex formed by the 20S catalytic core and 2 regulatory 19S particles at each end. Importantly, 3 of the β-subunits of the 20S core are replaced by the IFN-U inducible subunits, LMW protein-2, 7 and 10 (LMP2, LMP7 and LMP10), forming the immunoproteasome. This structure generates different antigenic peptides with high affinity for HLA I alleles. The immunogenic peptides are then transported into the endoplasmic reticulum (ER) by the transporter of antigen processing (TAP) 1/2 complex. With the assistance of chaperones (calnexin, tapasin, calreticulin, ERp57), the peptides are loaded onto the MHC class I heavy chain/β2 microglobulin complex. The trimeric complex then traverses through the Golgi apparatus and is incorporated into the cell membrane where the peptide can interact with CD8+ T cells. Defects anywhere along this pathway can lead to impaired presentation and CTL activation.
3.2. Immune checkpoint receptors
In addition to tumour antigen presentation, T-cell activation is functionally determined by the summation of many costimulatory and coinhibitory signals (Fig. 4). The immune system employs these checkpoints to thwart an exaggerated immune response, preventing the development of autoimmune diseases, a process termed ‘adaptive immune resistance.’ However, these inhibitory checkpoints are overexpressed in the tumour microenvironment (TME), contributing to tumour-promoting immunosuppression. The two most well-studied checkpoint receptors areCTLA4 andPD1, both of which are the targets of FDA-approved inhibitory antibodies [13]. There are emerging data for the use of other novel checkpoint receptors in the treatment of HNSCC, including LAG3, TIM3 and killer immunoglobulin-like receptor (KIR). In the presence of these coinhibitory signals, the stimulatory ‘signal 2’ will fail, leading to the induction of T-cell anergy or apoptosis, weakening the immune response due to failure to induce cytotoxicity [58]. The classically described costimulatory pathway involves CD28/B7 interaction; however other costimulatory interactions between T-cell receptors and their corresponding antigen presenting cell (APC) ligands include CD137/CD137-L, OX40/OX40-L, and CD40/CD40-L. These costimulatory receptors are members of the tumour necrosis factor receptor super-family, and a decrease in their expression has been observed on T cells of HNSCC specimens, correlating with poor outcomes [59]. Therefore, blocking the coinhibitory signals and increasing the costimulatory signals may enhance tumour control. Many mAbs targeting these signalling pathways have been developed in attempts to shift the immune system towards an anti-tumour function and are currently entering the therapeutic armamentarium for HNSCC (Table 1).
Fig. 4.
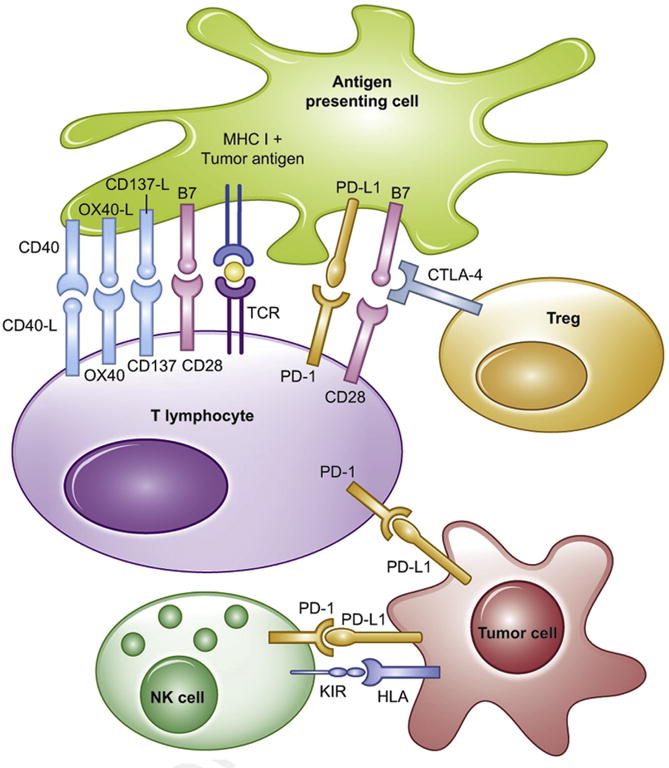
Monoclonal antibody targets. There are multiple pathways, when summated, can either trigger immune recognition and killing, or immune tolerance and evasion. The goal of immunotherapy is to induce an immune stimulatory signal that activates T effectors and NK cells, while inhibiting the immunosuppressive Treg. Red = coinhibitory signals. Green = costimulatory signals.
Table 1.
Examples of immunomodulating agents currently undergoing clinical trials.
| Coinhibitory | Binding partner | Agents in development |
|---|---|---|
| PD1 | PD-L1, PD-L2 |
|
| CTLA4 | CD80, CD86 |
|
| LAG3 | MHC class II |
|
| TIM3 | Galectin 9 |
|
| PtdSer |
|
|
| HMGB1 | ||
| KIR | MHC class I |
|
|
| ||
| Costimulatory molecules | Binding partner | Agents in development |
|
| ||
| OX40 | OX40-L |
|
| CD137/4-1BB | CD137-L |
|
| CD40 | CD40-L |
|
CTLA4 is expressed primarily on T cells in response to T-cell receptor (TCR) activation, and it counteracts CD28-mediatedcostimulatory signals [60,61]. This impairs the activation of T cells, thus limiting an exaggerated immune response [62]. Although the transient expression of CTLA4 on CD8+ T cells regulates effector/memory activity [63–65], the inhibitory function of CTLA4appears to be more importantonCD4+ Tregs, where CTLA4 is constitutively expressed at high levels [66,67]. CLTA4 and CD28 share B7 ligands, CD80 and CD86, whose expression is limited to APCs [68]. Compared to CD28, CTLA4 binds these ligands with higher affinity and avidity, competitively inhibiting ‘signal°2’ in the T-cell activation cascade [68]. Interestingly, activated CTLA4+ T cells can deplete acquired B7 ligands from APCs by a CTLA4-dependent endocytosis and degradation, further impairing the activation of T cells [69,70]. In preclinical solid tumour models, antibody-mediated blockade of CTLA4 function was found to reject transplanted cancer cells, delay growth of established tumours, and establish immune memory [71]. In less immunogenic murine tumour models, CTLA4 blockage did not demonstrate efficacy as a single agent; however, had impressive synergistic results in combination with other treatments, including tumour-antigen-based vaccines, radiation and chemotherapy [72—74]. Based on this preclinical data, two fully humanised CTLA4-blocking mAbs, ipilimumab (IgG1) and tremelimumab (IgG2), were developed and entered into human trials in advanced melanoma [62]. While ipilimumab is FDA-approved for unresectable stage III and IV metastatic melanoma, tremelimumab is currently undergoing phase III trials, with preliminary evidence of a non-significant trend towards improved OS when added to chemotherapy [75]. Although neither anti-CTLA4 mAb have been studied as a monotherapy in the HNSCC population, many CTLA4-targeted therapies combined with cytotoxic and other immunomodulating treatments are currently undergoing clinical trials.
PD1 is highly expressed on the surface of activated immune cells in the setting of chronic stimulation with minimal expression on resting cells of the immune system [76]. PD1 binds two distinct ligands, PD-L1 and PD-L2, members of the B7 family. PD-L1 is expressed constitutively on a wide range of haematopoietic cells, inhibiting T-cell cytokine production, as well as non-haematopoietic cells, including tumour cells [77]. PD-L2 on the other hand, exhibits much more restricted expression, largely limited to APCs, restraining effector T-cell function within inflamed sites and lymphoid organs [78,79]. Both ligands are induced by extrinsic proinflammatory signals, including IFN-γ, TNF-α, granulocyte macrophage colony stimulating factor (GM-CSF) and interleukin 4 (IL-4), suggesting that the inflammatory TME dictates tumour expression of programmed death-ligands 1 and 2 (PD-L1/2) in a feedback inhibitory loop frequently seen in chronic viral infection [80—82]. Additionally, intrinsic signalling pathways may lead to programmed death-ligand 1 (PD-L1) over-expression in tumour cells, including the phosphatidyl inositol 3-kinase —protein kinase B PI3K-AKT pathway [83], and the EGFR-mediated Janus kinase (JAK)2/ STAT1 pathway [84].
In the setting of chronic antigenic stimulation during viral infection, increased PD1 expression on CD8+ T cells is a marker for an exhausted phenotype, indicating a loss of effector function, including proliferation and cytokine production [85]. While PD-L1 expression by APCs has been shown to play a critical role in the induction of suppressive PD1+Tregs at the tumour periphery [86], the key function of tumour-infiltrating Tregs expressing high PD1 is not clear. In malignant glioma, these cells had impaired suppressive function, yet maintained high secretion of IFN-γ through increased PI3K/AKT activity [87]. Blocking the PD1 pathway with anti-PD1 mAb drove differentiation toward a more dysfunctional state and increased IFN-γ to further enhance tumour clearance [87]. In addition to the high expression of PD1 on CD8+ and CD4+ T cells, PD-L1 is expressed on 50—60% of HNSCC tumours[84,88]. While overexpression of PD-L1 inhibits tumour-directed T-cell cytotoxicity and permits immune evasion and tumour growth, prognostic significance of PD-L1 expression by tumours has been variable, correlating with worse outcomes in some cases [89—92] and improved outcome in others [93]. Regardless, blockade with an anti-PD-L1 mAb promotes immune-mediated tumour rejection and destruction of tumours [94]. In addition to its role in the TME, the PD1/PD-L1 pathway appears to play a significant role in modulating T-cell activity in peripheral tissues [84,88]. Thus, therapeutic blockade of the PD1/PD-L1 pathway can facilitate its effects through the simultaneous impairment of Treg function and the disinhibition of effector T cells both in the TME and in the periphery. Although less well studied, PD1 blockade as an anticancer therapy may also be mediated partially through the restoration of NK cell and B cell function [95,96].
Several antibodies that inhibit the PD1/PD-L1 pathway have been developed and show great promise as an effective anticancer therapy, with pembrolizumab, nivolumab and durvalumab being the most extensively studied in HNSCC. While pembrolizumab was recently approved by FDA for patients with platinum-refractory recurrent/metastatic (R/M) HNSCC, impressive results from phase III trials suggest other therapies will soon be available [97]. Updated results from a phase I trial of durvalumab (PD-L1-blocking mAb) in R/M HNSCC showed encouraging OS rates (62% at 6 months and 42% at 12 months) with relatively low adverse events (8% ≥ Grade 3) in this heavily pretreated population (NCT01693562) [98]. The non-redundant functions of CTLA4 and PD1 in regulating the adaptive immune system make a combined blockade an ideal target and have shown synergistic antitumour activity in preclinical mouse melanoma models [99,100]. Early phase trials combining nivolumab with ipilimumab for treatment of advanced melanoma have shown significantly improved response compared to either agent alone [99], and is now undergoing phase II and III trials in HNSCC.
Much effort has been placed in understanding tumour and/or patient characteristics that can predict who will and will not respond to particular therapies. In the nivolumab trials, there was a correlation of PD-L1 expressing tumours with clinical response to PD1-blocking treatment (36%), whereas none of the PD-L1-negativetumours displayed response to treatment [101 — 104]. However, since clinical response has been observed in PD-L1-negative tumours in other studies, investigations to identify other biomarkers are ongoing. Yearleyet al. developed a new assay testing various cancers for PD-L2 expression, and when compared to standard immunohistochemistry labelling, found a discordance between PD-L1 and PD-L2 expression. These results could possibly account for the positive clinical response seen in patients with PD-L1-negative tumours [105]. In the Keynote 012 study evaluating pembrolizumab in R/M HNSCC, those with PD-L2 expression had a trend toward higher overall response rate and longer PFS after adjusting for PD-L1 status, suggesting PD-L2 could be predictive of outcomes with pembrolizumab treatment. However, these are not perfect biomarkers, as not all PD-L1-/PD-L2-positive tumours respond to anti-PD1 treatment, and conversely, benefit has been seen in some PD-L1-/PD-L2-deficient tumours, highlighting a need to develop additional predictive biomarkers. Currently, enquiries into IFN-γexpression, major histocompatibility complex class II (MHC II) expression, CD8+ T-cell density and PD-L1 and CD8+ T-cell colocalisation at the tumour margin are demonstrating potential as predictive biomarkers for PD1/PD-L1 blockade response [106,107]. In addition to immediate resistance, long-term follow-up has now revealed a late relapse seen in 25% of patients with advanced melanoma who initially showed an objective response to PD1 blockade [108]. Acquired resistance to PD-1 blockade in these patients was associated with loss-of-function mutations in the JAK1, JAK2 and beta-2-microglobulim (B2M) genes, leading to immune resistance through impaired interferon-receptor signalling and antigen presentation [109]. Understanding the mechanisms of immediate and acquired resistance to immunotherapy will help identify the patients who are unlikely to benefit from particular treatments and help design salvage combination therapies or preventive interventions.
LAG3 (also known as CD223), is an inhibitory checkpoint receptor that enhances the function of Tregs and inhibits CD8+ effector T-cell function [110]. MHC class II molecules are the only known LAG3 ligands, which are upregulated on some epithelial cancers in response to IFN-γ but are also expressed on DCs [111]. PD1 and LAG3 are often coexpressed on exhausted or anergic T cells, and dual blockade synergistically reversed this anergy in tumour-specific CD8+ T cells [112]. Furthermore, Pd1-/- Lag3-/-double knockout mice can completely reject even poorly immunogenic tumours in a T-cell-dependent manner [113]. These developments have made LAG3 an option for anticancer therapy and have led to a phase I trial with the anti-LAG3 mAb, BMS-986016, with or without nivolumab in solid tumours, including HNSCC (NCT01968109).
TIM3 (also known as HAVcr2), is selectively expressed on IFN-γ-producing T cells, NK cells and Tregsand is closely involved in tumour-induced immune suppression. TIM3 marks the most exhausted or dysfunctional population of CD8+ T cells and NK cells in both solid and haematological malignancies [114,115], where approximately 30% of patients with advanced melanoma, non-small cell lung cancer (NSCLC) and follicular B-cell non-Hodgkin lymphoma express TIM3 [116—118]. Importantly, T-cell exhaustion can be partially reversed with TIM3-blocking antibodies in vitro, restoring up to 30—65% of NK cell function [119]. In addition to regulating CD8+ T-cell and NK-cell function, TIM3 is expressed on up to 60% of Tregs in the TME in HNSCC patients, compared to less than 20% expression on Tregs of the peripheral blood lymphocytes [120]. This is important as it leads to a more tumour-permissive environment, largely due to increased immunosuppressive cytokines and molecules (interleukin (IL)-10, perforin and granzymes) [121]. Interestingly, TIM3+ CD8+ T cells coexpress PD1 and exhibit greater deficits in both effector cytokine production (interleukin (IL)-2, TNF and IFN-γ) and cell cycle progression than with expression of either receptor alone [114].
Preclinical cancer models targeting the TIM3 pathway have shown promising results. In solid tumour models, TIM3 blockade is effective in a dose-dependent manner as a monotherapy and has similar efficacy to PD1 pathway blockade [122]. Furthermore, the combination of TIM3 and PD1 blockage is synergistic, showing more frequent and complete tumour regression than with blockade of either TIM3 or PD1 pathway alone in the poorly immunogenic and highly treatment-resistant melanoma and fibrosarcoma [114,122]. TIM3 is also an advantageous target as it is selectively expressed on T cells with an IFN-γ-producing phenotype and is primarily expressed on intratumoural T cells [117,122]. Thus, TIM3 blockade is less likely to interfere with T-cell regulation outside of tumour tissues and may not exhibit adverse autoimmune toxicities, as is frequently seen in the blockade of either CTLA4 or PD1 [122—125]. Two early phase trials are investigating TIM3-blocking mAbs (MBG453 and TSR-022) with or without a PD1-blocking mAb in advanced solid tumours (NCT02608268 and NCT02817633 respectively) and are currently recruiting patients.
KIR is expressed on NK cells and interacts with HLA molecules on target cells, playing a prominent role in modulating NK cell immune surveillance and cytotoxicity [126]. While most KIRs are inhibitory, there are a limited number of activating KIRs that bind HLA molecules with less affinity [127]. Upon binding an autologous matched HLA-C molecule, the inhibitory KIRs recruit SHP-1 and SHP-2 phosphatases, leading to subsequent suppression of activation signals [128]. However, when binding a mismatched HLA molecule or blockade by anti-KIR Ab, the NK cell lyses due to lack of an inhibitory signal. This inhibitory KIR/HLA relationship is overexpressed in patients with melanoma, breast cancer and chronic leukaemia [129], whereas activating KIRs are associated with better outcomes in leukaemia and lymphoma [130,131]. Although there have been promising results in several trials of haplo-identical allogenic stem cell transplantation in haematological malignancies [132], this method remains investigational due to its complexity and immunosuppressive morbidity. However, the anti-KIR mAb, lirilumab allows alloreactivity to all KIR+ NK cell patients and is undergoing early phase trials in solid tumours presently (NCT01714739 and NCT01750580).
In addition to blocking negative regulatory receptors on effector lymphocytes, an alternative approach has developed to enrich and activate costimulatory signals, including CD137, OX40 and CD40.
CD137 (also known as 4-1BB) is a costimulatory receptor expressed on the surface of activated T cells, DCs and NK cells [133,134]. When bound to its ligand (CD137-L), activated CD137 promotes ADCC by NK cells, differentiation of effector T cells and inhibition of immunosuppressive Tregs [135]. Administration of an agonistic CD137 mAb has been shown to induce CD8+ TA-specific T-cell-mediated eradication of established solid tumours in mice [135]. Although CD137-stimulating mAb has not been effective as a monotherapy in HNSCC models, it has been shown to synergise with other immune-stimulating therapies [136,137]. Two humanised CD137-agonist mAb have been developed, including urelumab (IgG4) and PF-05082566 (IgG2). These antibodies have been evaluated in early phase trials as a monotherapy or in combination with rituximab in melanoma, NSCLC and lymphoma [13]. A substantial proportion of HNSCC patients receiving cetuximab have shown an upregulation of CD137 on NK cells, and in preclinical models, sequential treatment with agonistic CD137mAb enhances cytolytic activity of NK cells [138,139]. This has led to clinical trials of urelumab in combination with cetuximab (NCT02110082) and in combination with tremelimumab (anti-CTLA4 mAb) in advanced and metastatic HNSCC (NCT02179918).
OX40 is expressed on the T-cell surface, and when activated by its ligand, OX40-L, promotes T-cell proliferation, antitumour cytokine secretion (IFN-γ), and enhanced memory T-cell function [13]. Thirty percent of activated Tregs isolated from HNSCC patients demonstrate OX40 expression in both tumours and tumour draining lymph node samples, compared to none of the peripheral blood mononuclear cells [140]. However, OX40-L expression is low in the tumour, and therefore fails to activateantitumour immune proliferation and cytokine production [141]. If this costimulatory pathway can be expanded and activated, it may enhance tumour-specific immunity and improve patient outcome. Indeed, OX40 stimulation has improved disease-free survival in a number of solid tumour models, including poorly immunogenic melanoma [142]. Furthermore, there is increasing evidence that agonistic OX40 mAb can synergise with other immunotherapies [143,144], and has also shown improved survival as an adjuvant following surgical resection or radiation therapy [145]. A phase I trial of an OX40 agonist, PF-04518600, as a monotherapy and in combination with a CD 137 agonist is currently recruiting patients with advanced solid tumours, including HNSCC (NCT02315066). Another phase Ib trial with the OX40 agonistic mAb, MEDI6469, prior to definitive surgical resection is currently recruiting patients with locoregionally advanced HNSCC (NCT02274155).
CD40 is expressed on APCs, as well as non-immunogenic cells and tumour cells [146,147]. Upon binding with its trimeric ligand (CD40-L), the downstream effects are multifaceted and depend on the type of cell expressing CD40 and the TME in which it is expressed [147]. Ligand binding on activated CD40+ T cells causes the induction of adaptive immunity and cytokine release. Ligand binding induces CD40+ macrophages to become tumouricidal, and can promote the induction of apoptosis in CD40+tumour cells, including HNSCC [146,148—151]. Expression of both CD40 and CD40-L decrease with increasing HNSCC stage and surgical resection results in increased APC expression of CD40 [152]. Although ligation with agonistic CD40 mAb has been shown to inhibit HNSCC cell growth in some cases, CD40 activation can also support tumour growth and angiogenesis due to expression on tumour endothelial cells [148,149]. Many agonistic CD40 mAbs and recombinant ligands have been developed to target this signalling pathway. Although not extensively studied in HNSCC, these antibodies have been evaluated in early phase trials, showing efficacy as a monotherapy in melanoma [153], and show synergistic promise in combination with other cytotoxic therapies and immunotherapies. However, response rates remain at 20% or less [154,155]. Specific concerns limiting CD40 use include thromboembolic syndromes, cytokine release syndromes, and tumour angiogenesis and growth [156,157].
4. Future directions
In addition to tumour- or immune-targeted mAbs, various novel techniques that boost the antitumour response are being developed. For instance, intratumoural oncolytic viral therapy uses naturally occurring or modified viruses to infect and kill tumour cells. Talimogene laherparepvec (T-VEC), tradename Imlygic, is an attenuated herpes simplex virus engineered to express GM-CSF in replicating tumour cells to induce host immunity. T-VEC is the first oncolytic virus to be approved for melanoma in the USA, Europe and Australia. With increased IFN-γ and TNF-α production, T-VEC can increase PD-L1 expression, promote effector T-cell influx and reduce Treg suppression [158]. This has led to early phase trials in combination with pembrolizumab in HNSCC (NCT02626000), and with ipilimumab in melanoma (NCT01740297). Vaccines are also making their way into the clinical arena. Early clinical trials are investigating various vaccines for HNSCC, including dendritic cell p53 peptide vaccine and HPV vaccines, with promising results thus far [159,160] (NCT02002182). Other novel agents showing promise are targeting immunosuppression, yet multimodality therapeutic strategies are beginning to gain momentum. Surgery can be combined with immunomodulating therapies as a means to eliminate the immunosuppressive environment, as well as use the surgical biopsy to guide adjuvant therapy decisions in a personalised manner. Some current trials investigating neoadjuvant immunotherapy includes pembrolizumab and nivolumab (NCT02296684, NCT02124850) and as adjuvant therapy with pembrolizumab either as a monotherapy or in conjunction with chemoradiation (NCT02841748, NCT02641093) in surgically resectable HNSCC tumours.
5. Conclusion
Cancer immunology is a rapidly evolving field, and we have only just begun to understand the relationship between the immune system and cancer development. Understanding the complex pathways of tumour immune evasion is imperative to develop novel therapies and to continue to improve clinical effectiveness through appropriate therapeutic combinations and identifying appropriate patient populations that will benefit from immunotherapy. Although monoclonal antibody therapies are still in early clinical development and just starting to make their way into the clinical therapeutic repertoire for HNSCC, the data thus far suggests these agents targeting immunomodulating pathways may play a substantial role in boosting antitumour responses. In most cases, these antibodies demonstrate synergistic responses with surgery, cytotoxic therapies and other immunomodulating therapies, leading to improved tumour control and reduced adverse side-effects.
Acknowledgments
Funding: Bristol-Myers Squibb (Inst), AstraZeneca (Inst), VentiRx Pharmaceuticals (Inst), Amgen (Inst).
Footnotes
Conflict of interest statement: We wish to draw the attention of the Editor to the following facts which may be considered as potential conflicts of interest.
Jennifer Moy: None.
Jessica Moskovitz: None.
Robert L. Ferris: Consulting or Advisory Role: Bristol-Myers Squibb, Merck, AstraZeneca, CureVac, ONO Pharmaceutical, Celgene.
References
- 1.Haddad RI, Shin DM. Recent advances in head and neck cancer. N Engl J Med. 2008;359(11):1143–54. doi: 10.1056/NEJMra0707975. [DOI] [PubMed] [Google Scholar]
- 2.Shah JP. Patterns of cervical lymph node metastasis from squamous carcinomas of the upper aerodigestive tract. Am J Surg. 1990;160(4):405–9. doi: 10.1016/s0002-9610(05)80554-9. [DOI] [PubMed] [Google Scholar]
- 3.Siegel R, et al. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 4.De Costa AM, Young MR. Immunotherapy for head and neck cancer: advances and deficiencies. Anticancer Drugs. 2011;22(7):674–81. doi: 10.1097/CAD.0b013e328340fd18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bindon C, et al. Clearance rates and systemic effects of intravenously administered interleukin 2 (IL-2) containing preparations in human subjects. Br J Cancer. 1983;47(1):123–33. doi: 10.1038/bjc.1983.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirkwood JM, et al. Comparison of intramuscular and intravenous recombinant alpha-2 interferon in melanoma and other cancers. Ann Intern Med. 1985;103(1):32–6. doi: 10.7326/0003-4819-103-1-32. [DOI] [PubMed] [Google Scholar]
- 7.Lienard D, et al. High-dose recombinant tumor necrosis factor alpha in combination with interferon gamma and melphalan in isolation perfusion of the limbs for melanoma and sarcoma. J Clin Oncol. 1992;10(1):52–60. doi: 10.1200/JCO.1992.10.1.52. [DOI] [PubMed] [Google Scholar]
- 8.Schoppy DW, Sunwoo JB. Immunotherapy for head and neck squamous cell carcinoma. Hematol Oncol Clin North Am. 2015;29(6):1033–43. doi: 10.1016/j.hoc.2015.07.009. [DOI] [PubMed] [Google Scholar]
- 9.Ferris RL, Jaffee EM, Ferrone S. Tumor antigen-targeted, monoclonal antibody-based immunotherapy: clinical response, cellular immunity, and immunoescape. J Clin Oncol. 2010;28(28):4390–9. doi: 10.1200/JCO.2009.27.6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee SC, et al. Natural killer (NK): dendritic cell (DC) cross talk induced by therapeutic monoclonal antibody triggers tumor antigen-specific T cell immunity. Immunol Res. 2011;50(2—3):248–54. doi: 10.1007/s12026-011-8231-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kondadasula SV, et al. Colocalization of the IL-12 receptor and FcgammaRIIIa to natural killer cell lipid rafts leads to activation of ERK and enhanced production of interferon-gamma. Blood. 2008;111(8):4183–83. doi: 10.1182/blood-2007-01-068908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Srivastava RM, et al. Cetuximab-activated natural killer and dendritic cells collaborate to trigger tumor antigen-specific T-cell immunity in head and neck cancer patients. Clin Cancer Res. 2013;19(7):1858–72. doi: 10.1158/1078-0432.CCR-12-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bauman JE, Ferris RL. Integrating novel therapeutic monoclonal antibodies into the management of head and neck cancer. Cancer. 2014;120(5):624–32. doi: 10.1002/cncr.28380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trivedi S, Jie HB, Ferris RL. Tumor antigen-specific monoclonal antibodies and induction of T-cell immunity. Semin Oncol. 2014;41(5):678–84. doi: 10.1053/j.seminoncol.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vermorken JB, et al. Overview of the efficacy of cetuximab in recurrent and/or metastatic squamous cell carcinoma of the head and neck in patients who previously failed platinum-based therapies. Cancer. 2008;112(12):2710–9. doi: 10.1002/cncr.23442. [DOI] [PubMed] [Google Scholar]
- 16.Jie HB, et al. CTLA-4(+) regulatory T cells increased in cetuximab-treated head and neck cancer patients suppress NK cell cytotoxicity and correlate with poor prognosis. Cancer Res. 2015;75(11):2200–10. doi: 10.1158/0008-5472.CAN-14-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hodge JW, et al. The tipping point for combination therapy: cancer vaccines with radiation, chemotherapy, or targeted small molecule inhibitors. Semin Oncol. 2012;39(3):323–39. doi: 10.1053/j.seminoncol.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hodge JW, et al. Attacking malignant cells that survive therapy: exploiting immunogenic modulation. Oncoimmunology. 2013;2(12):e26937. doi: 10.4161/onci.26937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodge JW, et al. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int J Cancer. 2013;133(3):624–36. doi: 10.1002/ijc.28070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Biasi AR, Villena-Vargas J, Adusumilli PS. Cisplatin-induced antitumor immunomodulation: a review of preclinical and clinical evidence. Clin Cancer Res. 2014;20(21):5384–91. doi: 10.1158/1078-0432.CCR-14-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gameiro SR, et al. Exploitation of differential homeostatic proliferation of T-cell subsets following chemotherapy to enhance the efficacy of vaccine-mediated antitumor responses. Cancer Immunol Immunother. 2011;60(9):1227–42. doi: 10.1007/s00262-011-1020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gameiro SR, et al. Radiation-induced immunogenic modulation of tumor enhances antigen processing and calreticulin exposure, resulting in enhanced T-cell killing. Oncotarget. 2014;5(2):403–16. doi: 10.18632/oncotarget.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ang KK, et al. Randomized phase III trial of concurrent accelerated radiation plus cisplatin with or without cetuximab for stage III to IV head and neck carcinoma: RTOG 0522. J Clin Oncol. 2014;32(27):2940–50. doi: 10.1200/JCO.2013.53.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harari PM, et al. Postoperative chemoradiotherapy and cetuximab for high-risk squamous cell carcinoma of the head and neck: radiation Therapy Oncology Group RTOG-0234. J ClinOncol. 2014;32(23):2486–95. doi: 10.1200/JCO.2013.53.9163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferris RL, et al. Phase II trial of post-operative radiotherapy with concurrent cisplatin plus panitumumab in patients with high-risk, resected head and neck cancer. Ann Oncol. 2016 doi: 10.1093/annonc/mdw428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res. 1970;13:1–27. doi: 10.1159/000386035. [DOI] [PubMed] [Google Scholar]
- 27.Badoual C, et al. Better understanding tumor-host interaction in head and neck cancer to improve the design and development of immunotherapeutic strategies. Head Neck. 2010;32(7):946–58. doi: 10.1002/hed.21346. [DOI] [PubMed] [Google Scholar]
- 28.Bhatia S, et al. Solid cancers after bone marrow transplantation. J Clin Oncol. 2001;19(2):464–71. doi: 10.1200/JCO.2001.19.2.464. [DOI] [PubMed] [Google Scholar]
- 29.Haigentz M., Jr Aerodigestive cancers in HIV infection. Curr Opin Oncol. 2005;17(5):474–8. doi: 10.1097/01.cco.0000174036.46785.8f. [DOI] [PubMed] [Google Scholar]
- 30.Allen CT, et al. The clinical implications of antitumor immunity in head and neck cancer. Laryngoscope. 2012;122(1):144–57. doi: 10.1002/lary.21913. [DOI] [PubMed] [Google Scholar]
- 31.Kuss I, et al. Decreased absolute counts of T lymphocyte subsets and their relation to disease in squamous cell carcinoma of the head and neck. Clin Cancer Res. 2004;10(11):3755–62. doi: 10.1158/1078-0432.CCR-04-0054. [DOI] [PubMed] [Google Scholar]
- 32.Dasgupta S, et al. Inhibition of NK cell activity through TGF-beta 1 by down-regulation of NKG2D in a murine model of head and neck cancer. J Immunol. 2005;175(8):5541–50. doi: 10.4049/jimmunol.175.8.5541. [DOI] [PubMed] [Google Scholar]
- 33.Young MR, et al. Mechanisms of immune suppression in patients with head and neck cancer: influence on the immune infiltrate of the cancer. Int J Cancer. 1996;67(3):333–8. doi: 10.1002/(SICI)1097-0215(19960729)67:3<333::AID-IJC5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 34.Lopez-Albaitero A, et al. Role of antigen-processing machinery in the in vitro resistance of squamous cell carcinoma of the head and neck cells to recognition by CTL. J Immunol. 2006;176(6):3402–9. doi: 10.4049/jimmunol.176.6.3402. [DOI] [PubMed] [Google Scholar]
- 35.Ferris RL, Whiteside TL, Ferrone S. Immune escape associated with functional defects in antigen-processing machinery in head and neck cancer. Clin Cancer Res. 2006;12(13):3890–5. doi: 10.1158/1078-0432.CCR-05-2750. [DOI] [PubMed] [Google Scholar]
- 36.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331(6024):1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 37.Dunn GP, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–8. doi: 10.1038/ni1102-991. [DOI] [PubMed] [Google Scholar]
- 38.Ljunggren HG, Karre K. In search of the ‘missing self: MHC molecules and NK cell recognition. Immunol Today. 1990;11(7):237–44. doi: 10.1016/0167-5699(90)90097-s. [DOI] [PubMed] [Google Scholar]
- 39.Duray A, et al. Immune suppression in head and neck cancers: a review. ClinDevImmunol. 2010;2010:701657. doi: 10.1155/2010/701657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campoli M, Ferrone S. HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene. 2008;27(45):5869–85. doi: 10.1038/onc.2008.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seliger B. Different regulation of MHC class I antigen processing components in human tumors. J Immunotoxicol. 2008;5(4):361–7. doi: 10.1080/15476910802482870. [DOI] [PubMed] [Google Scholar]
- 42.Concha-Benavente F, et al. Immunological and clinical significance of HLA class I antigen processing machinery component defects in malignant cells. Oral Oncol. 2016;58:52–8. doi: 10.1016/j.oraloncology.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leibowitz MS, et al. Deficiency of activated STAT1 in head and neck cancer cells mediates TAP1-dependent escape from cytotoxic T lymphocytes. Cancer Immunol Immunother. 2011;60(4):525–35. doi: 10.1007/s00262-010-0961-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferris RL, Hunt JL, Ferrone S. Human leukocyte antigen (HLA) class I defects in head and neck cancer: molecular mechanisms and clinical significance. Immunol Res. 2005;33(2):113–33. doi: 10.1385/IR:33:2:113. [DOI] [PubMed] [Google Scholar]
- 45.Matsui M, Ikeda M, Akatsuka T. High expression of HLA-A2 on an oral squamous cell carcinoma with down-regulated transporter for antigen presentation. BiochemBiophys Res Commun. 2001;280(4):1008–14. doi: 10.1006/bbrc.2000.4234. [DOI] [PubMed] [Google Scholar]
- 46.Meissner M, et al. Defects in the human leukocyte antigen class I antigen processing machinery in head and neck squamous cell carcinoma: association with clinical outcome. Clin Cancer Res. 2005;11(7):2552–60. doi: 10.1158/1078-0432.CCR-04-2146. [DOI] [PubMed] [Google Scholar]
- 47.Ogino T, et al. HLA class I antigen down-regulation in primary laryngeal squamous cell carcinoma lesions as a poor prognostic marker. Cancer Res. 2006;66(18):9281–9. doi: 10.1158/0008-5472.CAN-06-0488. [DOI] [PubMed] [Google Scholar]
- 48.Ogino T, et al. Association of tapasin and HLA class I antigen down-regulation in primary maxillary sinus squamous cell carcinoma lesions with reduced survival of patients. Clin Cancer Res. 2003;9(11):4043–51. [PubMed] [Google Scholar]
- 49.Concha-Benavente F, et al. EGFR-mediated tumor immunoescape: the imbalance between phosphorylated STAT1 and phosphorylated STAT3. Oncoimmunology. 2013;2(12):e27215. doi: 10.4161/onci.27215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leibowitz MS, et al. SHP2 is overexpressed and inhibits pSTAT1-mediated APM component expression, T-cell attracting chemokine secretion, and CTL recognition in head and neck cancer cells. Clin Cancer Res. 2013;19(4):798–808. doi: 10.1158/1078-0432.CCR-12-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Agazie YM, Hayman MJ. Molecular mechanism for a role of SHP2 in epidermal growth factor receptor signaling. Mol Cell Biol. 2003;23(21):7875–86. doi: 10.1128/MCB.23.21.7875-7886.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mimura K, et al. The MAPK pathway is a predominant regulator of HLA-A expression in esophageal and gastric cancer. J Immunol. 2013;191(12):6261–72. doi: 10.4049/jimmunol.1301597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trivedi S, et al. Immune biomarkers of anti-EGFR monoclonal antibody therapy. Ann Oncol. 2015;26(1):40–7. doi: 10.1093/annonc/mdu156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pollack BP, Sapkota B, Cartee TV. Epidermal growth factor receptor inhibition augments the expression of MHC class I and II genes. Clin Cancer Res. 2011;17(13):4400–13. doi: 10.1158/1078-0432.CCR-10-3283. [DOI] [PubMed] [Google Scholar]
- 55.Srivastava RM, et al. STAT1-induced HLA class I upregulation enhances immunogenicity and clinical response to anti-EGFR mAb cetuximab therapy in HNC patients. Cancer Immunol Res. 2015;3(8):936–45. doi: 10.1158/2326-6066.CIR-15-0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chang CC, Campoli M, Ferrone S. Classical and nonclassical HLA class I antigen and NK Cell-activating ligand changes in malignant cells: current challenges and future directions. Adv Cancer Res. 2005;93:189–234. doi: 10.1016/S0065-230X(05)93006-6. [DOI] [PubMed] [Google Scholar]
- 57.Fletcher EV, et al. EGFR inhibition induces proinflammatory cytokines via NOX4 in HNSCC. Mol Cancer Res. 2013;11(12):1574–84. doi: 10.1158/1541-7786.MCR-13-0187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ferris RL. Immunology and immunotherapy of head and neck cancer. J Clin Oncol. 2015;33(29):3293–304. doi: 10.1200/JCO.2015.61.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baruah P, et al. Decreased levels of alternative co-stimulatory receptors OX40 and 4-1BB characterise T cells from head and neck cancer patients. Immunobiology. 2012;217(7):669–75. doi: 10.1016/j.imbio.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 60.Rudd CE, Taylor A, Schneider H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol Rev. 2009;229(1):12–26. doi: 10.1111/j.1600-065X.2009.00770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell. 1992;71(7):1065–8. doi: 10.1016/s0092-8674(05)80055-8. [DOI] [PubMed] [Google Scholar]
- 62.Honeychurch J, et al. Immuno-regulatory antibodies for the treatment of cancer. Expert Opin Biol Ther. 2015;15(6):787–801. doi: 10.1517/14712598.2015.1036737. [DOI] [PubMed] [Google Scholar]
- 63.Alegre ML, et al. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J Immunol. 1996;157(11):4762–70. [PubMed] [Google Scholar]
- 64.Chambers CA, Kuhns MS, Allison JP. Cytotoxic T lymphocyte antigen-4 (CTLA-4) regulates primary and secondary peptide-specific CD4(+) T cell responses. Proc Natl Acad Sci USA. 1999;96(15):8603–8. doi: 10.1073/pnas.96.15.8603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 2002;16(1):23–35. doi: 10.1016/s1074-7613(01)00259-x. [DOI] [PubMed] [Google Scholar]
- 66.Takahashi T, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192(2):303–10. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wing K, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–5. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 68.Linsley PS, et al. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994;1(9):793–801. doi: 10.1016/s1074-7613(94)80021-9. [DOI] [PubMed] [Google Scholar]
- 69.Qureshi OS, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–3. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Oderup C, et al. Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology. 2006;118(2):240–9. doi: 10.1111/j.1365-2567.2006.02362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–6. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 72.Hurwitz AA, et al. Combination immunotherapy of primary prostate cancer in a transgenic mouse model using CTLA-4 blockade. Cancer Res. 2000;60(9):2444–8. [PubMed] [Google Scholar]
- 73.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190(3):355–66. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hurwitz AA, et al. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. ProcNatlAcadSci USA. 1998;95(17):10067–71. doi: 10.1073/pnas.95.17.10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robert C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–26. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]
- 77.Ribas A, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J ClinOncol. 2013;31(5):616–22. doi: 10.1200/JCO.2012.44.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Keir ME, et al. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Keir ME, et al. Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med. 2006;203(4):883–95. doi: 10.1084/jem.20051776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liang SC, et al. Regulation of PD-1, PD-L1, and PD-L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33(10):2706–16. doi: 10.1002/eji.200324228. [DOI] [PubMed] [Google Scholar]
- 81.Latchman Y, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2(3):261–8. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 82.Freeman GJ, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–34. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Eppihimer MJ, et al. Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation. 2002;9(2):133–45. doi: 10.1038/sj/mn/7800123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Butte MJ, et al. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27(1):111–22. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Parsa AT, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med. 2007;13(1):84–8. doi: 10.1038/nm1517. [DOI] [PubMed] [Google Scholar]
- 86.Concha-Benavente F, et al. Identification of the cell-intrinsic and -extrinsic pathways downstream of EGFR and IFNgamma that induce PD-L1 expression in head and neck cancer. Cancer Res. 2016;76(5):1031–43. doi: 10.1158/0008-5472.CAN-15-2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–9. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 88.Francisco LM, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206(13):3015–29. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lowther DE, et al. PD-1 marks dysfunctional regulatory T cells in malignant gliomas. JCI Insight. 2016;1(5) doi: 10.1172/jci.insight.85935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Seiwert TY, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-012): an open-label, multicentre, phase 1b trial. Lancet Oncol. 2016;17(7):956–65. doi: 10.1016/S1470-2045(16)30066-3. [DOI] [PubMed] [Google Scholar]
- 91.Hamanishi J, et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc Natl Acad Sci USA. 2007;104(9):3360–5. doi: 10.1073/pnas.0611533104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Inman BA, et al. PD-L1 (B7-H1) expression by urothelial carcinoma of the bladder and BCG-induced granulomata: associations with localized stage progression. Cancer. 2007;109(8):1499–505. doi: 10.1002/cncr.22588. [DOI] [PubMed] [Google Scholar]
- 93.Ohigashi Y, et al. Clinical significance of programmed death-1 ligand-1 and programmed death-1 ligand-2 expression in human esophageal cancer. Clin Cancer Res. 2005;11(8):2947–53. doi: 10.1158/1078-0432.CCR-04-1469. [DOI] [PubMed] [Google Scholar]
- 94.Iwai Y, et al. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. 2002;99(19):12293–7. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Taube JM, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hirano F, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65(3):1089–96. [PubMed] [Google Scholar]
- 97.Terme M, et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res. 2011;71(16):5393–9. doi: 10.1158/0008-5472.CAN-11-0993. [DOI] [PubMed] [Google Scholar]
- 98.Okazaki T, et al. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-con-taining tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci USA. 2001;98(24):13866–71. doi: 10.1073/pnas.231486598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pai SI, Zandberg DP, Strome SE. The role of antagonists of the PD-1:PD-L1/PD-L2 axis in head and neck cancer treatment. Oral Oncol. 2016 doi: 10.1016/j.oraloncology.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Segal N, Ou SHI, Balmanoukian AS, Massarelli E, Brahmer JR, Weiss J, et al. Updated safety and efficacy of durvalumab (MEDI4736), an anti-PD-L1 antibody, in patients from squamous cell carcinoma of the head and neck (SCCHN) expansion cohort. Ann Oncol. 2016;27(Suppl. 6):vi328. [Google Scholar]
- 101.Wolchok JD, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14(8):561–84. doi: 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- 103.Topalian SL, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gillison ML, Blumenschein G, Fayette J, Guigay J, Colevas AD, Licitra L, et al. Nivolumab (nivo) vs investigator's choice (IC) for recurrent or metastatic (R/M) head and neck squamous cell carcinoma (HNSCC): Checkmate 141. AACR oral presentation [Google Scholar]
- 105.Segal NH, Balmanoukian AS, Brahmer JR, Maio M, Blake-Haskins A, Li X, et al. Safety and efficacy of MEDI4736, an anti-PD-L1 antibody, in patients from a squamous call carcinoma of the head and neck (SCCHN) expansion cohort. ACSO meeting. 2015:33. [Google Scholar]
- 106.Seiwert TY, Burtness B, Weiss J, Gluck I, Elder JP, Pai SI, et al. A phase Ib study of MK-3475 in patients with human papillomavirus (HPV)-associated and non-HPV-associated head and neck (H/N) cancer. ASCO meeting. 2014:32. [Google Scholar]
- 107.Yearley J. PD-L2 expression in human tumors: relevance to anti-D1 therapy in cancer. European Cancer Congress. doi: 10.1158/1078-0432.CCR-16-1761. [DOI] [PubMed] [Google Scholar]
- 108.Tumeh PC, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Johnson DB, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun. 2016;7:10582. doi: 10.1038/ncomms10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ribas A, et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA. 2016;315(15):1600–9. doi: 10.1001/jama.2016.4059. [DOI] [PubMed] [Google Scholar]
- 111.Zaretsky JM, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375(9):819–29. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Goldberg MV, Drake CG. LAG-3 in cancer immunotherapy. Curr Top Microbiol Immunol. 2011;344:269–78. doi: 10.1007/82_2010_114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Camisaschi C, et al. Alternative activation of human plasmacytoid DCs in vitro and in melanoma lesions: involvement of LAG-3. J Invest Dermatol. 2014;134(7):1893–902. doi: 10.1038/jid.2014.29. [DOI] [PubMed] [Google Scholar]
- 114.Grosso JF, et al. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J Immunol. 2009;182(11):6659–69. doi: 10.4049/jimmunol.0804211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Woo SR, et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012;72(4):917–27. doi: 10.1158/0008-5472.CAN-11-1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sakuishi K, et al. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–94. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhou Q, et al. Coexpression of Tim-3 and PD-1 identifies a CD8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011;117(17):4501–10. doi: 10.1182/blood-2010-10-310425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fourcade J, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. 2010;207(10):2175–86. doi: 10.1084/jem.20100637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gao X, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS One. 2012;7(2):e30676. doi: 10.1371/journal.pone.0030676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yang ZZ, et al. IL-12 upregulates TIM-3 expression and induces T cell exhaustion in patients with follicular B cell non-Hodgkin lymphoma. J Clin Invest. 2012;122(4):1271–82. doi: 10.1172/JCI59806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.da Silva IP, et al. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol Res. 2014;2(5):410–22. doi: 10.1158/2326-6066.CIR-13-0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jie HB, et al. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br J Cancer. 2013;109(10):2629–35. doi: 10.1038/bjc.2013.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sakuishi K, et al. TIM3+FOXP3+ regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology. 2013;2(4):e23849. doi: 10.4161/onci.23849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ngiow SF, et al. Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71(10):3540–51. doi: 10.1158/0008-5472.CAN-11-0096. [DOI] [PubMed] [Google Scholar]
- 125.Sabatos CA, et al. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat Immunol. 2003;4(11):1102–10. doi: 10.1038/ni988. [DOI] [PubMed] [Google Scholar]
- 126.Tivol EA, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3(5):541–7. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 127.Nishimura H, et al. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motifcarryingi mmunoreceptor. Immunity. 1999;11(2):141–51. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 128.Thielens A, Vivier E, Romagne F. NK cell MHC class I specific receptors (KIR): from biology to clinical intervention. CurrOpinImmunol. 2012;24(2):239–45. doi: 10.1016/j.coi.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 129.Moesta AK, Parham P. Diverse functionality among human NK cell receptors for the C1 epitope of HLA-C: KIR2DS2, KIR2DL2, and KIR2DL3. Front Immunol. 2012;3:336. doi: 10.3389/fimmu.2012.00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Yusa S, Campbell KS. Src homology region 2-containing protein tyrosine phosphatase-2 (SHP-2) can play a direct role in the inhibitory function of killer cell Ig-like receptors in human NK cells. J Immunol. 2003;170(9):4539–47. doi: 10.4049/jimmunol.170.9.4539. [DOI] [PubMed] [Google Scholar]
- 131.Purdy AK, Campbell KS. Natural killer cells and cancer: regulation by the killer cell Ig-like receptors (KIR) Cancer Biol Ther. 2009;8(23):2211–20. doi: 10.4161/cbt.8.23.10455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Besson C, et al. Association of killer cell immunoglobulin-like receptor genes with Hodgkin's lymphoma in a familial study. PLoS One. 2007;2(5):e406. doi: 10.1371/journal.pone.0000406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Verheyden S, Bernier M, Demanet C. Identification of natural killer cell receptor phenotypes associated with leukemia. Leukemia. 2004;18(12):2002–7. doi: 10.1038/sj.leu.2403525. [DOI] [PubMed] [Google Scholar]
- 134.Ruggeri L, et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood. 2007;110(1):433–40. doi: 10.1182/blood-2006-07-038687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pollok KE, et al. Inducible T cell antigen 4-1BB. Analysis of expression and function. J Immunol. 1993;150(3):771–81. [PubMed] [Google Scholar]
- 136.Melero I, et al. NK1.1 cells express 4-1BB (CDw137) costimulatory molecule and are required for tumor immunity elicited by anti-4-1BB monoclonal antibodies. Cell Immunol. 1998;190(2):167–72. doi: 10.1006/cimm.1998.1396. [DOI] [PubMed] [Google Scholar]
- 137.Lynch DH. The promise of 4-1BB (CD137)-mediated immunomodulation and the immunotherapy of cancer. Immunol Rev. 2008;222:277–86. doi: 10.1111/j.1600-065X.2008.00621.x. [DOI] [PubMed] [Google Scholar]
- 138.Lucido CT, et al. CD137 enhancement of HPV positive head and neck squamous cell carcinoma tumor clearance. Vaccines (Basel) 2014;2(4):841–53. doi: 10.3390/vaccines2040841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bauman JE, Grandis JR. Targeting secondary immune responses to cetuximab: CD137 and the outside story. J Clin Invest. 2014;124(6):2371–5. doi: 10.1172/JCI76264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kohrt HE, et al. Targeting CD137 enhances the efficacy of cetuximab. J Clin Invest. 2014;124(6):2668–82. doi: 10.1172/JCI73014. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 141.Srivastava RM, et al. CD137 stimulation enhances cetuximab induced natural killer (NK): dendritic cell (DC) priming of antitumor T cell immunity in head and neck cancer patients. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Vetto JT, et al. Presence of the T-cell activation marker OX-40 on tumor infiltrating lymphocytes and draining lymph node cells from patients with melanoma and head and neck cancers. Am J Surg. 1997;174(3):258–65. doi: 10.1016/s0002-9610(97)00139-6. [DOI] [PubMed] [Google Scholar]
- 143.Bell RB, et al. OX40 signaling in head and neck squamous cell carcinoma: overcoming immunosuppression in the tumor microenvironment. Oral Oncol. 2016;52:1–10. doi: 10.1016/j.oraloncology.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 144.Weinberg AD, et al. Blocking OX-40/OX-40 ligand interaction in vitro and in vivo leads to decreased T cell function and amelioration of experimental allergic encephalomyelitis. J Immunol. 1999;162(3):1818–26. [PubMed] [Google Scholar]
- 145.Redmond WL, Linch SN, Kasiewicz MJ. Combined targeting of costimulatory (OX40) and coinhibitory (CTLA-4) pathways elicits potent effector T cells capable of driving robust antitumor immunity. Cancer Immunol Res. 2014;2(2):142–53. doi: 10.1158/2326-6066.CIR-13-0031-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Guo Z, et al. PD-1 blockade and OX40 triggering synergistically protects against tumor growth in a murine model of ovarian cancer. PLoS One. 2014;9(2):e89350. doi: 10.1371/journal.pone.0089350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gough MJ, et al. Adjuvant therapy with agonistic antibodies to CD134 (OX40) increases local control after surgical or radiation therapy of cancer in mice. J Immunother. 2010;33(8):798–809. doi: 10.1097/CJI.0b013e3181ee7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Eliopoulos AG, Young LS. The role of the CD40 pathway in the pathogenesis and treatment of cancer. CurrOpinPharmacol. 2004;4(4):360–7. doi: 10.1016/j.coph.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 149.van Kooten C, Banchereau J. CD40-CD40 ligand. J Leukoc Biol. 2000;67(1):2–17. doi: 10.1002/jlb.67.1.2. [DOI] [PubMed] [Google Scholar]
- 150.Cao W, et al. CD40 function in squamous cell cancer of the head and neck. Oral Oncol. 2005;41(5):462–9. doi: 10.1016/j.oraloncology.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 151.Posner MR, et al. Surface membrane-expressed CD40 is present on tumor cells from squamous cell cancer of the head and neck in vitro and in vivo and regulates cell growth in tumor cell lines. Clin Cancer Res. 1999;5(8):2261–70. [PubMed] [Google Scholar]
- 152.Funakoshi S, et al. Inhibition of human B-cell lymphoma growth by CD40 stimulation. Blood. 1994;83(10):2787–94. [PubMed] [Google Scholar]
- 153.von Leoprechting A, et al. Stimulation of CD40 on immunogenic human malignant melanomas augments their cytotoxic T lymphocyte-mediated lysis and induces apoptosis. Cancer Res. 1999;59(6):1287–94. [PubMed] [Google Scholar]
- 154.Sathawane D, et al. Monocyte CD40 expression in head and neck squamous cell carcinoma (HNSCC) Hum Immunol. 2013;74(1):1–5. doi: 10.1016/j.humimm.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 155.Vonderheide RH, et al. Clinical activity and immune modulation in cancer patients treated with CP-870,893, a novel CD40 agonist monoclonal antibody. J ClinOncol. 2007;25(7):876–83. doi: 10.1200/JCO.2006.08.3311. [DOI] [PubMed] [Google Scholar]
- 156.Beatty GL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–6. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Vonderheide RH, et al. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology. 2013;2(1):e23033. doi: 10.4161/onci.23033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tong AW, Stone MJ. Prospects for CD40-directed experimental therapy of human cancer. Cancer Gene Ther. 2003;10(1):1–13. doi: 10.1038/sj.cgt.7700527. [DOI] [PubMed] [Google Scholar]
- 159.Henn V, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391(6667):591–4. doi: 10.1038/35393. [DOI] [PubMed] [Google Scholar]
- 160.Curran MA, et al. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. 2010;107(9):4275–80. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Schuler PJ, et al. Phase I dendritic cell p53 peptide vaccine for head and neck cancer. Clin Cancer Res. 2014;20(9):2433–44. doi: 10.1158/1078-0432.CCR-13-2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Yoshitake Y, et al. Phase II clinical trial of multiple peptide vaccination for advanced head and neck cancer patients revealed induction of immune responses and improved OS. Clin Cancer Res. 2015;21(2):312–21. doi: 10.1158/1078-0432.CCR-14-0202. [DOI] [PubMed] [Google Scholar]