

Abstract
The PI3K/Akt/mTOR signaling pathway is aberrantly activated in various pediatric tumors. We conducted a phase I study of the Akt inhibitor perifosine in patients with recurrent/refractory pediatric CNS and solid tumors. This was a standard 3+3 open-label dose-escalation study to assess pharmacokinetics, describe toxicities, and identify the MTD for single-agent perifosine. Five dose levels were investigated, ranging from 25 to 125 mg/m2/day for 28 days per cycle. Twenty-three patients (median age 10 years, range 4–18 years) with CNS tumors (DIPG [n = 3], high-grade glioma [n = 5], medulloblastoma [n = 2], ependymoma [n = 3]), neuroblastoma (n = 8), Wilms tumor (n = 1), and Ewing sarcoma (n = 1) were treated. Only one DLT occurred (grade 4 hyperuricemia at dose level 4). The most common grade 3 or 4 toxicity at least possibly related to perifosine was neutropenia (8.7%), with the remaining grade 3 or 4 toxicities (fatigue, hyperglycemia, fever, hyperuricemia, and catheter-related infection) occurring in one patient each. Pharmacokinetics was dose-saturable at doses above 50 mg/m2/day with significant inter-patient variability, consistent with findings reported in adult studies. One patient with DIPG (dose level 5) and 4 of 5 patients with high-grade glioma (dose levels 2 and 3) experienced stable disease for two months. Five subjects with neuroblastoma (dose levels 1 through 4) achieved stable disease which was prolonged (≥11 months) in three. No objective responses were noted. In conclusion, the use of perifosine was safe and feasible in patients with recurrent/refractory pediatric CNS and solid tumors. An MTD was not defined by the 5 dose levels investigated. Our RP2D is 50 mg/m2/day.
Introduction
Aberrant activation of the pathway defined by phosphatidylinositol 3-kinase (PI3K), Akt (protein kinase B), and mammalian target of rapamycin (mTOR) has been observed across a wide range of neoplastic diseases [1–4]. Gene mutations, rearrangements, and amplifications in this pathway lead to disordered control of cell growth and survival and are among the most frequently encountered genetic lesions in human cancers. They are estimated to be present in up to 30% of human malignancies, including pediatric solid and central nervous system (CNS) tumors [5–7]. Evidence suggests that oncogenic alterations in the PI3K/Akt/mTOR signaling cascade are associated with inferior prognoses in many pediatric cancers, including neuroblastoma, rhabdomyosarcoma, high-grade glioma, and medulloblastoma [8–11]. In normal cells, the PI3K/Akt/mTOR network is involved in processes relating to survival, cell growth, angiogenesis, glucose metabolism, and proliferation [4, 12–15]. PI3K signaling is initiated at the cell surface by growth factor receptor tyrosine kinases, activated oncogenic proteins, and/or G-protein-coupled receptors, resulting in complex downstream interactions characterized by crosstalk with other signaling cascades as well as multiple feedback loops. Once activated, PI3K recruits Akt, a serine/threonine protein kinase, to the plasma membrane where it becomes activated via phosphorylation by phosphoinositide-dependent protein kinase 1 (PDK1) and mTOR complex 2 (mTORC2). Phosphorylated Akt in turn activates a number of cellular proteins and inactivates tuberous sclerosis complex 2 (TSC2) resulting in downstream activation of mTOR complex 1 (mTORC1) with a subsequent increase in cell growth, proliferation and survival. Aberrant stimulation of the PI3K/Akt/mTOR pathway can occur via a variety of mechanisms, including activating mutations in Ras, Akt, and receptor tyrosine kinases as well as loss of function of the inhibitory regulator phosphatase and tensin homologue (PTEN) [16–18].
Perifosine (1,1-Dimethyl-4-[[(octadecyloxy)hydroxyphosphinyl]oxy]-piperidinium inner salt) is a third generation orally-available alkylphospholipid with antineoplastic activity. Alkylphospholipids differ from most conventional chemotherapy agents in that they block signal transduction pathways through cell membrane interactions rather than directly damaging DNA in the cell nucleus. The mechanism of action of alkylphospholipids is thought to be primarily mediated through targeting Akt’s pleckstrin-homology domain, resulting in inhibition of Akt membrane localization and phosphorylation. Other potential cytotoxic effects of alkylphospholipids include telomere shortening in cancer cells, induction of autophagy, inhibition of mTOR signaling, activation of cellular stress pathways leading to tumor cell apoptosis, and interference with the synthesis of phospholipids necessary for cell membrane formation, which is often rate-limiting in the setting of hyperproliferation. Of the members of the alkylphospholipid family, perifosine is the best studied and has been demonstrated to be a potent and consistent inhibitor of Akt in pre-clinical and clinical studies. In particular, perifosine has been demonstrated to be cytotoxic in murine models of glioma, medulloblastoma, and neuroblastoma [19–21]. It is further characterized by a long plasma half-life of approximately 4 days and a manageable toxicity profile that differs sufficiently from other chemotherapeutic agents to enable its use in combination regimens [22–33].
Methods
The primary aim of the study was to determine the MTD of single-agent perifosine in pediatric patients (age ≤ 21 years) with recurrent or refractory pediatric CNS and solid tumors. Secondary aims were to (1) determine whether pharmacokinetic serum levels of perifosine correlate with toxicity, (2) assess preliminary data on the efficacy of perifosine monotherapy, and (3) determine whether molecular features of the tumor were associated with likelihood of response.
Patients/guardians consented to the IRB-approved Memorial Sloan Kettering Cancer Center trial #08–091. Twenty-four pediatric patients with recurrent or refractory CNS or solid tumors were enrolled between 2008 and 2014. One enrolled subject never received study prescribed therapy and is not included in this analysis (Fig 1).
Fig 1. Diagram of study participants.

Eligibility criteria included: (1) presence of any solid tumor that had failed standard therapy, (2) evidence of tumor by CT, MRI, MIBG scan, serum markers, or tissue sampling, (3) age ≤ 21 years, (4) Karnofsky or Lansky performance status ≥ 50%, (5) adequate organ function [absolute neutrophil count (ANC) ≥ 1000/μL at least 24 hours off filgrastim, platelet count ≥ 75,000/μL at least 1 week post-platelet transfusion, hemoglobin ≥ 8 g/dL at least 1 week post-red blood cell transfusion, aspartate aminotransferase (AST) and alanine aminotransferase (ALT) ≤ 3x the upper limits of normal, total bilirubin ≤ 2 mg/dL, serum creatinine ≤ 1.5x the upper limit of normal for age, or calculated creatinine clearance or nuclear glomerular filtration rate ≥ 70 ml/min/1.73 m2], (6) mandated interval since prior therapy [≥ 3 weeks since last non-nitrosourea chemotherapy, ≥ 6 weeks since last nitrosoureas, ≥ 4 weeks since last radiation therapy], (7) ability to swallow tablets whole, and (8) agreement to practice adequate contraception and not breastfeed. Exclusion criteria included (1) pregnancy, (2) uncontrolled active infection or serious medical illness, (3) use of combination anti-retroviral therapy in patients who are known to be HIV-positive, and (4) enzyme-inducing anti-convulsant usage.
Treatment protocol
This was a standard 3+3 phase I dose escalation study (NCT00776867) investigating 5 dose levels. Perifosine was only available as 50-mg tablets, complicating the phase I dose escalation scheme. A loading dose was administered on day 1 and subsequent maintenance doses were administered every 1 to 4 days, depending on dose level and BSA, with the goal of achieving dose levels of 25, 50, 75, 100, and 125 mg/m2/day (Tables 1–5). This dosing scheme, incorporating drug administration as infrequently as every 4 days, was made possible by perifosine’s long half-life. Treatment was continued until disease progression, intolerable toxicity, DLT, or death was encountered. Subjects experiencing DLT but with evidence of clinical benefit at dose level 1, 2, or 3 were eligible to remain on study with a dose level reduction.
Table 1. Perifosine dose level 1 (~25 mg/m2/day).
| BSA (m2) | Loading dose day 1 | Maintenance dose—starting on day 2 |
|---|---|---|
| 0.4 to 0.59 | 50mg | 50mg every four days |
| 0.6 to 0.79 | 50mg | 50mg every three days |
| 0.8 to 1.2 | 100mg | 50mg every other day |
| 1.21 to 1.6 | 150mg | 50mg daily five days per week |
| > 1.6 | 150mg | 50mg daily |
Table 5. Perifosine dose level 5 (~125 mg/m2/day).
| BSA (m2) | Loading dose day 1 | Maintenance dose—starting on day 2 |
|---|---|---|
| 0.4 to 0.59 | 100mg | 50mg daily five days per week and 100mg daily two days per week |
| 0.6 to 0.79 | 100mg | 100mg daily five days per week and 50mg daily two days per week |
| 0.8 to 1.2 | 100mg BID | 100mg daily alternating with 150mg daily |
| 1.21 to 1.6 | 150mg BID | 150mg daily alternating with 200mg daily |
| > 1.6 | 150mg BID | 200mg daily |
Table 2. Perifosine dose level 2 (~50 mg/m2/day).
| BSA (m2) | Loading dose day 1 | Maintenance dose—starting on day 2 |
|---|---|---|
| 0.4 to 0.59 | 100mg | 50mg every other day |
| 0.6 to 0.79 | 100mg | 50 mg daily five days per week |
| 0.8 to 1.2 | 100mg BID | 50mg daily |
| 1.21 to 1.6 | 150mg BID | 100mg daily five days per week |
| > 1.6 | 150mg BID | 100mg daily |
Table 3. Perifosine dose level 3 (~75 mg/m2/day).
| BSA (m2) | Loading dose day 1 | Maintenance dose—starting on day 2 |
|---|---|---|
| 0.4 to 0.59 | 100mg | 50mg daily five days per week |
| 0.6 to 0.79 | 100mg | 50mg daily |
| 0.8 to 1.2 | 100mg BID | 50mg daily alternating with 100mg daily |
| 1.21 to 1.6 | 150mg BID | 100mg daily |
| > 1.6 | 150mg BID | 100mg daily alternating with 150mg daily |
Table 4. Perifosine dose level 4 (~100 mg/m2/day).
| BSA (m2) | Loading dose day 1 | Maintenance dose—starting on day 2 |
|---|---|---|
| 0.4 to 0.59 | 100mg | 50mg daily |
| 0.6 to 0.79 | 100mg | 50mg daily alternating with 100mg daily |
| 0.8 to 1.2 | 100mg BID | 100mg daily |
| 1.21 to 1.6 | 150mg BID | 150mg daily six days per week and 100mg daily for one day per week |
| > 1.6 | 150mg BID | 150mg daily alternating with 200mg daily |
Subjects were seen for physical examination and laboratory assessment (complete blood count, serum chemistry) prior to study enrollment, approximately 1 week after starting treatment, and then during the first week of every 28-day treatment cycle. Tumor assessments were performed approximately every 8 weeks.
Toxicity was assessed according to the Common Toxicity Criteria (version 3.0) of the National Cancer Institute, National Institutes of Health. DLT was defined as (1) any non-hematological toxicity grade ≥ 3 [except for grade 3 nausea, vomiting, and diarrhea that could be controlled within 24 hours with supportive care measures]; (2) grade 4 neutropenia on 2 consecutive blood counts drawn at least 72 hours apart; (3) grade 4 febrile neutropenia [ANC < 1000/μL and fever ≥ 38.5°C] or grade ≥ 3 documented infection with ANC < 1000/μL, or; (4) a platelet count < 25,000/μL. Grade ≥ 3 decrease in hemoglobin that could be corrected to at least 8 g/dl (grade 2) by transfusion of red blood cells, grade ≥ 3 leukopenia in the absence of dose-limiting neutropenia, and grade ≥ 3 lymphopenia were not considered DLTs.
Correlative studies
Samples for pharmacokinetic analysis were obtained at baseline and during weeks 2 through 4 of cycle 1. At each time point, heparinized blood was collected into a plastic vacutainer to minimize adhesion of perifosine. Plasma was separated by centrifugation and stored in polypropylene cryovials at -70°C until assayed. Perifosine in plasma was measured by a validated reversed phase liquid chromatography/electrospray mass spectrometry method as developed and validated by Woo et al. [34].
Tumor tissue: Fifteen unstained paraffin slides and/or 100 mg of flash frozen tissue if available were obtained from the surgery closest to initiation of this clinical trial. These were used to evaluate molecular markers that may predict sensitivity to perifosine, including PI3K/Akt pathway activity as measured by phosphorylation of both Akt (AKT) and the proline-rich Akt substrate of 40 kDa (PRAS40) as well as proliferation rate as assessed by MIB-1 immunostaining.
Response criteria
For subjects with tumors other than neuroblastoma, responses were assessed by the study radiologist and principal investigator (SH, ID) via the Response Evaluation Criteria in Solid Tumors (RECIST) [35]. For subjects with neuroblastoma, responses were assessed by a single investigator (BK) using the International Neuroblastoma Response Criteria (INRC) [36].
Statistics
The DLT assessment period was the first 28-day cycle. If therapy was discontinued during the first cycle for reasons other than toxicity, an additional subject could be enrolled at that dose level to ensure adequate evaluation of toxicity. No intra-patient dose escalation was permitted.
Results
Clinical
Twenty-three subjects (median age 10 years, range 4 to 18 years) received a total of 189 cycles. Nine (39%) were male and 14 female. Thirteen subjects had CNS tumors (diffuse intrinsic pontine glioma [DIPG] n = 3, high-grade glioma n = 5, medulloblastoma n = 2, ependymoma n = 3), 8 had neuroblastoma, 1 had Wilms tumor, and 1 had Ewing sarcoma/primitive neuroectodermal tumor (PNET) (Table 6). A median of 3 cycles were initiated and a median of 2 cycles were completed per patient, ranging from <1 to 68 cycles.
Table 6. Patient characteristics and responses.
| Patient | Dose Level | Age | Sex | Diagnosis | Prior RT | Number of Prior Chemotherapy Regimens | Best Response (Duration) |
p-AKT | p-PRAS40 | MIB-1 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 18 | M | medulloblastoma | yes | 8 | PD | |||
| 2 | 1 | 17 | M | medulloblastoma | yes | 5 | PD | ++ | ++ | 30% |
| 3 | 1 | 10 | M | neuroblastoma | yes | 6 | NR (11 months) | - | - | <2% |
| 4 | 2 | 13 | F | glioblastoma | yes | 1 | PD | |||
| 5 | 2 | 12 | F | neuroblastoma | yes | 7 | NR (63 months) | - | - | <1% |
| 6 | 2 | 17 | M | anaplastic astrocytoma | yes | 2 | SD (2 months) | |||
| 7 | 3 | 9 | F | glioblastoma | yes | 3 | SD (2 months) | |||
| 8 | 3 | 5 | F | anaplastic astrocytoma | yes | 3 | SD (2 months) | - | - | 10% |
| 9 | 3 | 14 | F | anaplastic astrocytoma | yes | 1 | SD (2 months) | - | - | 10% |
| 10 | 3 | 13 | F | ependymoma | yes | 7 | PD | - | ++ | 10% |
| 11 | 3 | 4 | F | neuroblastoma | no | 5 | NR (2 months) | |||
| 12 | 4 | 8 | M | neuroblastoma | yes | 10 | PD | |||
| 13 | 4 | 18 | F | Ewing sarcoma/PNET | yes | 5 | PD | - | - | 60% |
| 14 | 4 | 13 | M | neuroblastoma | yes | 9 | NR (4 months) | |||
| 15 | 4 | 7 | M | neuroblastoma | yes | 5 | NR (42 months) | - | - | 20% |
| 16 | 4 | 7 | M | neuroblastoma | yes | 4 | PD | - | + | 70% |
| 17 | 4 | 5 | F | DIPG | yes | 0 | PD | |||
| 18 | 5 | 16 | F | Wilms tumor | yes | 6 | PD | - | + | 70% |
| 19 | 5 | 6 | M | DIPG | yes | 1 | PD | |||
| 20 | 5 | 9 | F | DIPG | yes | 0 | SD (2 months) | |||
| 21 | 5 | 16 | F | ependymoma | yes | 2 | PD | + | + | 30% |
| 22 | 5 | 7 | F | ependymoma | yes | 6 | PD | - | + | 90% |
| 23 | 5 | 7 | F | neuroblastoma | yes | 4 | PD | ++ | +++ | 90% |
RT = radiation therapy, DIPG = diffuse intrinsic pontine glioma, PNET = primitive neuroectodermal tumor, F = female, M = male, NR = no response per INRC, PD = progressive disease per RECIST or INRC, SD = stable disease per RECIST, p-AKT = phosphorylated Akt, p-PRAS40 = phosphorylated proline-rich Akt substrate of 40 kD
Toxicity
Perifosine was generally well tolerated. The most common toxicities of any grade (at least possibly related) were fatigue (65.2%), nausea (65.2%), hyperglycemia (60.9%), vomiting (56.5%), and decreased leukocytes (47.8%), with the vast majority of these toxicities ≤ grade 2. Only one DLT occurred (grade 4 hyperuricemia at dose level 4). The most common grade 3 or 4 toxicity at least possibly related to perifosine was neutropenia (8.7%), with the remaining grade 3 or 4 at least possibly related toxicities (fatigue, hyperglycemia, fever, hyperuricemia, and catheter-related infection) occurring in one patient each. Table 7 summarizes toxicities that were considered at least possibly related and observed in more than 10% of subjects or ≥ grade 3 (even if seen in fewer than 10% of subjects). Although the initial study design only included three dose levels of perifosine, the lack of DLT at the first 3 dose levels prompted the addition of dose levels 4 and 5 as permitted by protocol amendment. Enrollment on dose level 3 was concluded when this protocol amendment was approved, following enrollment of the fifth patient. The second patient (diagnosed with Ewing sarcoma/PNET) enrolled at dose level 4 experienced a grade 4 elevation in uric acid, considered to be possibly related to perifosine. That patient was therefore removed from the protocol and the dose level 4 cohort was expanded to six patients. No further grade ≥ 3 toxicities were observed in any other patient treated at dose level 4 and an additional 3 patients were enrolled at dose level 5. As dose level 5 was under consideration as the MTD, 3 final patients were enrolled at that dose level to confirm lack of toxicity. One patient at dose level 5 experienced a grade 3 hyperglycemia (possibly related) on the day of protocol removal for disease progression. There were no other grade ≥ 3 toxicities or DLTs in the patients treated at dose level 5.
Table 7. Toxicity summary.
| Any Grade | Grade 3 or 4 | |||
|---|---|---|---|---|
| Hematologic Adverse Events | No. | % | No. | % |
| Decreased leukocytes | 11 | 47.83% | 0 | 0.00% |
| Decreased hemoglobin | 10 | 43.48% | 0 | 0.00% |
| Decreased neutrophils | 6 | 26.09% | 2 | 8.70% |
| Decreased platelets | 5 | 21.74% | 0 | 0.00% |
| Lymphopenia | 4 | 17.39% | 0 | 0.00% |
| Non-hematologic Adverse Events | ||||
| Fatigue | 15 | 65.22% | 1 | 4.35% |
| Nausea | 15 | 65.22% | 0 | 0.00% |
| Hyperglycemia | 14 | 60.87% | 1 | 4.35% |
| Vomiting | 13 | 56.52% | 0 | 0.00% |
| Diarrhea | 10 | 43.48% | 0 | 0.00% |
| ALT | 9 | 39.13% | 0 | 0.00% |
| Anorexia | 7 | 30.43% | 0 | 0.00% |
| AST | 6 | 26.09% | 0 | 0.00% |
| Fever | 5 | 21.74% | 1 | 4.35% |
| Hypocalcemia | 5 | 21.74% | 0 | 0.00% |
| Hypokalemia | 5 | 21.74% | 0 | 0.00% |
| Hyponatremia | 5 | 21.74% | 0 | 0.00% |
| Pain—abdomen NOS | 5 | 21.74% | 0 | 0.00% |
| Infection URI | 4 | 17.39% | 0 | 0.00% |
| Constipation | 3 | 13.04% | 0 | 0.00% |
| Flatulence | 3 | 13.04% | 0 | 0.00% |
| Hypomagnesemia | 3 | 13.04% | 0 | 0.00% |
| Hypophosphatemia | 3 | 13.04% | 0 | 0.00% |
| Pain—stomach | 3 | 13.04% | 0 | 0.00% |
| Hyperuricemia | 1 | 4.35% | 1 | 4.35% |
| Infection—catheter related | 1 | 4.35% | 1 | 4.35% |
Notes: Toxicities considered to be at least possibly related. Non-hematological toxicities seen in either > 10% of subjects or ≥ grade 3.
Responses
Response results are detailed in Table 6. Among 4 of 5 subjects with recurrent high-grade astrocytoma, the best responses were stable disease for 2 months in each. Three subjects with recurrent DIPG were treated with stable disease in 1 for 2 months and progressive disease in 2. All patients with ependymoma, medulloblastoma, Wilms tumor, and Ewing sarcoma/PNET had progressive disease as their best response. Five of 8 subjects with neuroblastoma experienced disease stability (labeled “no response” in the nomenclature of the INRC and defined as a less than 50% reduction of some or all measurable lesions, but no increase of greater than 25% in these lesions and no new lesions) for 2, 4, 11, 42, and 63 months, respectively. Three additional neuroblastoma patients had progressive disease, defined as a greater than 25% increase in any preexisting lesion or any new lesion.
Pharmacokinetics
The BSA grouping-based dosing scheme (Tables 1–5) was generally successful in approximating the target perifosine doses using 50-mg tablets (Table 8). Pharmacokinetic evaluation was possible with samples from 22 patients. Individual steady state plasma concentrations found are presented in Fig 2. Rather wide interpatient variability is observed in accordance with results from published adult studies [32,37]. Appreciable intra-patient variability can also be inferred in some subjects (notably patients 5, 7, 16 and 19). Average steady state levels of perifosine were calculated for each dose level (Table 8 and Fig 3). We observed saturable dose exposure at doses above 50 mg/m2/day.
Table 8. Perifosine pharmacokinetics.
| Perifosine Dose Level | Patients | Target Perifosine Dose—mg/m2/day | Given Perifosine Dose (standard deviation)—mg/m2/day | Average Plasma Level (standard deviation)—μg/mL; μM |
|---|---|---|---|---|
| 1 | 1,2,3 | 25 | 27 (1.3) | 6.8 (1.6); 18.4 (4.3) |
| 2 | 4,5,6 | 50 | 49 (2.4) | 14.4 (2.5); 39.0 (6.8) |
| 3 | 7,8,9,10,11 | 75 | 74 (5.6) | 11.5 (3.1); 31.1 (8.5) |
| 4 | 12,13,14,15,16,17 | 100 | 110 (9.1) | 12.7 (6.1); 34.4 (16.5) |
| 5 | 18,19,20,21,22 | 125 | 130 (13.1) | 12.1 (3.5); 32.8 (9.5) |
Fig 2. Steady state plasma concentration of perifosine at weeks 2, 3, and 4.

Concentrations were plotted for each study subject whose samples were available for calculation (patients 1–22) across the dose groups; enumerated individual subject trace; grey, semi-transparent thick line represents the arithmetic mean of the plasma concentration at the corresponding time-point for the given dose group. Saturable dose exposure above 50 mg/m2/day dose is evident albeit large inter-patient variability, consistent with findings reported in previous adult studies.
Fig 3. Correlation plot of perifosine steady state plasma concentration versus daily dose (actually administered).
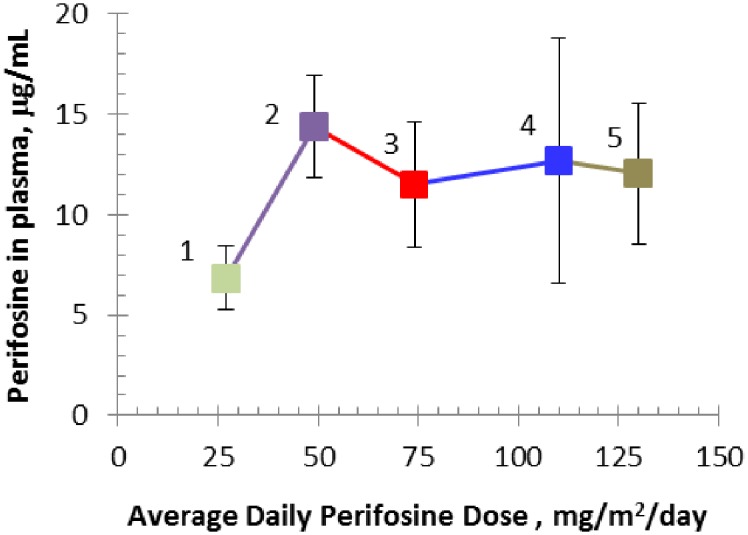
Error bars represent single standard deviation; enumerated dose groups. Saturable dose exposure above 50 mg/m2/day is observed in pediatric patients.
Tumor tissue biological assays
Biological assays were performed on 13 available tumors (Table 6). Using p-PRAS40 as a surrogate for Akt activation, we tested whether Akt activation correlated with SD/PR in this small patient cohort. Of the 13 tumors with available information regarding Akt activation status, 7 demonstrated Akt activation while 6 tumors did not. Of the 6 tumors without Akt activation, 5 had SD/NR while one had PD. Of the 7 tumors demonstrating Akt activation, all had PD. Using the Fisher’s exact test, the 2-sided p-value was 0.0047, demonstrating that lack of Akt activation is significantly associated with response.
Discussion
This phase I study represents the first evaluation of the safety and tolerability of perifosine monotherapy in pediatric patients with recurrent or refractory CNS and solid tumors. Our BSA grouping-based dosing schedule was successful in approximating perifosine doses using 50-mg tablets. As perifosine was generally well-tolerated at all investigated dose levels, an MTD was not defined. Similarly, lack of toxicity made the secondary study aim of investigating the relationship between serum levels of perifosine and toxicity less relevant. Our steady state perifosine plasma levels show saturable dose exposure (presumably due to saturable drug absorption) as observed in adult studies but were generally somewhat higher in our pediatric patients. This supports our observation that the “plateau” in pediatric patients is reached at lower maintenance doses (at 50 mg/m2/day), whereas in 2 adult studies the dose exposure plateau was reached at doses higher than 100 mg/m2/day [32, 37]. Additionally, several clinical trials have observed saturable dose-exposure to perifosine, with increasing toxicity (particularly gastrointestinal toxicity and fatigue) but no improvement in efficacy at higher doses [31, 37, 38]. The achieved plasma levels in this study compare favorably to the IC50 values published for preclinical pediatric tumor models. A wide range of IC50 values have been reported with perifosine, ranging from as low as 10 μM to as high as 30 μM in different neuroblastoma cell lines, for example [21]. Rhabdomyosarcoma cell lines were relatively sensitive to perifosine, with IC50 values around 10 μM, while medulloblastoma cell lines were comparatively more resistant, with IC50 values of 25 μM [7, 39]. Given our observation of saturable dose exposure at doses above 50 mg/m2/day that would be expected to result in anti-tumor activity, our recommended Phase 2 dose is 50 mg/m2/day.
Although we observed no objective responses within our patient cohort, a number of patients experienced disease stabilization. This is consistent with results published from previous early phase clinical trials of perifosine monotherapy in adults as well as the Children’s Oncology Group’s phase I trial of single-agent MK-2206, another Akt inhibitor, in children, which suggested that Akt inhibition resulted in anti-proliferative rather than tumoricidal effects [40, 41]. The lack of objective responses suggests that this class of agents may be more effective in combination with additional cytotoxic and/or targeted agents in future studies. For example, in early phase clinical trials, the use of Akt inhibitor-containing combination regimens in advanced adult cancers has resulted in complete or partial responses [42–44]. However, two subsequent phase 3 studies of combination therapy with perifosine in adults with refractory colorectal cancer or multiple myeloma did not demonstrate a survival benefit, prompting Aeterna Zentaris to discontinue those trials [45, 46]. Our observation that tumors with Akt activation were significantly less likely to have SD/NR and significantly more likely to have PD in response to perifosine is in concordance with the published literature which shows Akt activation to be a poor prognostic factor in a variety of pediatric tumor types. Tumors with Akt activation are more aggressive and less likely to respond to any experimental agent, particularly in the setting of recurrent disease. In support of this, the average MIB-1 of p-PRAS40 positive tumors in our cohort was 55.7% ± 32.1 while that of p-PRAS40 negative tumors was 17.2% ± 22.1. Additionally, perifosine is not a typical Akt inhibitor like MK-2206, an allosteric Akt inhibitor. As an alkylphospholipid, it has several additional proposed mechanisms of action in addition to its activity as an inhibitor of Akt. For example, it has recently been reported to inhibit telomerase activity as low micromolar doses [25]. Perifosine’s effect on telomerase activity may or may not be related to its effect on Akt activation, as Akt has been previously shown to form a complex with the protein subunit of telomerase [47]. Other putative mechanisms include activation of the stress-activated protein kinase/c-Jun NH2 terminal kinase (SAPK/JNK pathway), also at low micromolar levels, although this may be related to inhibition of Akt [48]. In addition, perifosine has also been shown to degrade components of the mTOR pathway and to induce autophagy at doses as low as 5 μM [49]. Based on our PK data with steady state plasma levels of perifosine above 10 μM observed in most patients, all of these additional mechanisms of action could have been at play in our patient cohort, including perifosine’s autophagy-promoting activity which may have resulted in diminished anti-tumor activity.
There is preclinical evidence to suggest that subsets of patients may be more likely to respond to Akt inhibitors based the presence or absence of specific genetic alternations. For example, cell lines with PTEN loss or PIK3CA mutations have been shown to be significantly more sensitive to the Akt inhibitors in a variety of tumor types, while cells lines with RAS mutations are generally resistant even in the presence of concomitant PIK3CA mutations [42–44, 50]. Consideration of PI3K/Akt/mTOR pathway activation status would therefore be expected to aid in the selection of patients who would be more likely to respond to Akt inhibition. Further, a recent study of perifosine in T-cell acute lymphoblastic leukemia cell lines with constitutive activation of the PI3K/Akt/mTOR pathway showed that the combination of perifosine with two additional small molecule inhibitors of Akt resulted in synergism for the induction of cell cycle arrest as well as of apoptosis and autophagy, suggesting that multi-inhibition treatment against Akt may be a potentially advantageous pharmacologic treatment strategy in tumors with aberrant PI3K/Akt/mTOR pathway activation [51].
Prolonged disease stability in a subset of neuroblastoma patients treated at the first three dose levels prompted the creation of an expansion cohort of an additional 14 neuroblastoma patients, the results of which are reported separately [52]. One possible explanation for the lack of objective response in patients with brain tumors may relate to CNS penetration. A recent evaluation of cerebrospinal fluid pharmacokinetics in adult non-tumor bearing rhesus monkeys showed that the CNS penetration of oral perifosine was limited [53]. However, the presence of a brain tumor is known to alter drug penetration through the blood brain barrier, generally rendering it more permeable as compared with the normal brain vasculature, albeit within a spectrum of barrier integrity that may be influenced by specific CNS tumor type [54–56]. Future studies of Akt inhibitors in brain tumor patients should include assessment of whether these drugs are capable of crossing the blood brain barrier in sufficient concentrations to target CNS neoplasms. Additionally, strategies to bypass the blood brain barrier are under investigation. A recent study of convection enhanced delivery (CED) of perifosine in a mouse brainstem glioma model showed no serious acute toxicities at doses demonstrated to be effective in cell culture [57]. Further studies are needed to determine the long-term safety and potential clinical application of this drug delivery technique.
In conclusion, perifosine in pediatric patients at the dosing schedule employed is safe and well-tolerated. Evaluation of the optimal method of drug delivery in the case of CNS tumors, the rational combination of perifosine with cytotoxic chemotherapy, alternative Akt inhibitors or other targeted agents, and the potential selection of patient subsets that can be predicted to respond to Akt pathway inhibition based on molecular characteristics remain to be established.
Supporting information
(PDF)
(PDF)
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Memorial Sloan Kettering Cancer Center Core Grant P30 CA008748; Aeterna Zentaris (provision of study drug). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. 10.1038/nrd1902 [DOI] [PubMed] [Google Scholar]
- 2.Luo J, Manning BD, Cantley LC. Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell. 2003;4:257–62. [DOI] [PubMed] [Google Scholar]
- 3.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96:4240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–30. 10.1038/nature04869 [DOI] [PubMed] [Google Scholar]
- 5.Pollack IF, Hamilton RL, Burger PC, Brat DJ, Rosenblum MK, Murdoch GH, et al. Akt activation is a common event in pediatric malignant gliomas and a potential adverse prognostic marker: a report from the Children's Oncology Group. J Neurooncol. 2010;99:155–63. 10.1007/s11060-010-0297-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mueller S, Phillips J, Onar-Thomas A, Romero E, Zheng S, Wiencke JK, et al. PTEN promoter methylation and activation of the PI3K/Akt/mTOR pathway in pediatric gliomas and influence on clinical outcome. Neuro Oncol. 2012;14:1146–52. 10.1093/neuonc/nos140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar A, Fillmore HL, Kadian R, Broaddus WC, Tye GW, Van Meter TE. The alkylphospholipid perifosine induces apoptosis and p21-mediated cell cycle arrest in medulloblastoma. Mol Cancer Res. 2009;7:1813–21. 10.1158/1541-7786.MCR-09-0069 [DOI] [PubMed] [Google Scholar]
- 8.Opel D, Poremba C, Simon T, Debatin KM, Fulda S. Activation of Akt predicts poor outcome in neuroblastoma. Cancer Res. 2007;67:735–45. 10.1158/0008-5472.CAN-06-2201 [DOI] [PubMed] [Google Scholar]
- 9.Petricoin EF 3rd, Espina V, Araujo RP, Midura B, Yeung C, Wan X, et al. Phosphoprotein pathway mapping: Akt/mammalian target of rapamycin activation is negatively associated with childhood rhabdomyosarcoma survival. Cancer Res. 2007;67:3431–40. 10.1158/0008-5472.CAN-06-1344 [DOI] [PubMed] [Google Scholar]
- 10.Castellino RC, Barwick BG, Schniederjan M, Buss MC, Becher O, Hambardzumyan D, et al. Heterozygosity for Pten promotes tumorigenesis in a mouse model of medulloblastoma. PLoS One. 2010;5:e10849 10.1371/journal.pone.0010849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thorarinsdottir HK, Santi M, McCarter R, Rushing EJ, Cornelison R, Jales A, et al. Protein expression of platelet-derived growth factor receptor correlates with malignant histology and PTEN with survival in childhood gliomas. Clin Cancer Res. 2008;14:3386–94. 10.1158/1078-0432.CCR-07-1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–65. 10.1038/35077225 [DOI] [PubMed] [Google Scholar]
- 13.Brazil DP, Park J, Hemmings BA. PKB binding proteins. Getting in on the Akt. Cell. 2002;111:293–303. [DOI] [PubMed] [Google Scholar]
- 14.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. 10.1038/nrg1879 [DOI] [PubMed] [Google Scholar]
- 15.Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–74. 10.1016/j.cell.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, et al. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer Cell. 2011;19:58–71. 10.1016/j.ccr.2010.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;13:1021–31. 10.1158/1535-7163.MCT-13-0639 [DOI] [PubMed] [Google Scholar]
- 18.Martini M, De Santis MC, Braccini L, Gulluni F, Hirsch E. PI3K/AKT signaling pathway and cancer: an updated review. Ann Med. 2014;46:372–83. 10.3109/07853890.2014.912836 [DOI] [PubMed] [Google Scholar]
- 19.Momota H, Nerio E, Holland EC. Perifosine inhibits multiple signaling pathways in glial progenitors and cooperates with temozolomide to arrest cell proliferation in gliomas in vivo. Cancer Res. 2005;65:7429–35. 10.1158/0008-5472.CAN-05-1042 [DOI] [PubMed] [Google Scholar]
- 20.Hambardzumyan D, Becher OJ, Rosenblum MK, Pandolfi PP, Manova-Todorova K, Holland EC. PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes Dev. 2008;22:436–48. 10.1101/gad.1627008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z, Tan F, Liewehr DJ, Steinberg SM, Thiele CJ. In vitro and in vivo inhibition of neuroblastoma tumor cell growth by AKT inhibitor perifosine. J Natl Cancer Inst. 2010;102:758–70. 10.1093/jnci/djq125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Blitterswijk WJ, Verheij M. Anticancer mechanisms and clinical application of alkylphospholipids. Biochim Biophys Acta. 2013;1831:663–74. 10.1016/j.bbalip.2012.10.008 [DOI] [PubMed] [Google Scholar]
- 23.Fensterle J, Aicher B, Seipelt I, Teifel M, Engel J. Current view on the mechanism of action of perifosine in cancer. Anticancer Agents Med Chem. 2014;14:629–35. [DOI] [PubMed] [Google Scholar]
- 24.Kondapaka SB, Singh SS, Dasmahapatra GP, Sausville EA, Roy KK. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol Cancer Ther. 2003;2:1093–103. [PubMed] [Google Scholar]
- 25.Holohan B, Hagiopian MM, Lai TP, Huang E, Friedman DR, Wright WE, et al. Perifosine as a potential novel anti-telomerase therapy. Oncotarget. 2015;6:21816–26. 10.18632/oncotarget.5200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Molife LR, Yan L, Vitfell-Rasmussen J, Zernhelt AM, Sullivan DM, Cassier PA, et al. Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors. J Hematol Oncol. 2014;7:1 10.1186/1756-8722-7-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bendell JC, Nemunaitis J, Vukelja SJ, Hagenstad C, Campos LT, Hermann RC, et al. Randomized placebo-controlled phase II trial of perifosine plus capecitabine as second- or third-line therapy in patients with metastatic colorectal cancer. J Clin Oncol. 2011;29:4394–400. 10.1200/JCO.2011.36.1980 [DOI] [PubMed] [Google Scholar]
- 28.Richardson PG, Wolf J, Jakubowiak A, Zonder J, Lonial S, Irwin D, et al. Perifosine plus bortezomib and dexamethasone in patients with relapsed/refractory multiple myeloma previously treated with bortezomib: results of a multicenter phase I/II trial. J Clin Oncol. 2011;29:4243–9. 10.1200/JCO.2010.33.9788 [DOI] [PubMed] [Google Scholar]
- 29.Friedman DR, Lanasa MC, Davis PH, Allgood SD, Matta KM, Brander DM, et al. Perifosine treatment in chronic lymphocytic leukemia: results of a phase II clinical trial and in vitro studies. Leuk Lymphoma. 2014;55:1067–75. 10.3109/10428194.2013.824080 [DOI] [PubMed] [Google Scholar]
- 30.Vink SR, Schellens JH, van Blitterswijk WJ, Verheij M. Tumor and normal tissue pharmacokinetics of perifosine, an oral anti-cancer alkylphospholipid. Invest New Drugs. 2005;23:279–86. 10.1007/s10637-005-1436-0 [DOI] [PubMed] [Google Scholar]
- 31.Crul M, Rosing H, de Klerk GJ, Dubbelman R, Traiser M, Reichert S, et al. Phase I and pharmacological study of daily oral administration of perifosine (D-21266) in patients with advanced solid tumours. Eur J Cancer. 2002;38:1615–21. [DOI] [PubMed] [Google Scholar]
- 32.Van Ummersen L, Binger K, Volkman J, Marnocha R, Tutsch K, Kolesar J, et al. A phase I trial of perifosine (NSC 639966) on a loading dose/maintenance dose schedule in patients with advanced cancer. Clin Cancer Res. 2004;10:7450–6. 10.1158/1078-0432.CCR-03-0406 [DOI] [PubMed] [Google Scholar]
- 33.Unger C, Berdel W, Hanauske AR, Sindermann H, Engel J, Mross K. First-time-in-man and pharmacokinetic study of weekly oral perifosine in patients with solid tumours. Eur J Cancer. 2010;46:920–5. 10.1016/j.ejca.2009.12.028 [DOI] [PubMed] [Google Scholar]
- 34.Woo EW, Messmann R, Sausville EA, Figg WD. Quantitative determination of perifosine, a novel alkylphosphocholine anticancer agent, in human plasma by reversed-phase liquid chromatography-electrospray mass spectrometry. J Chromatogr B Biomed Sci Appl. 2001;759:247–57. [DOI] [PubMed] [Google Scholar]
- 35.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–16. [DOI] [PubMed] [Google Scholar]
- 36.Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol. 1993;11:1466–77. 10.1200/JCO.1993.11.8.1466 [DOI] [PubMed] [Google Scholar]
- 37.Figg WD, Monga M, Headlee D, Shah A, Chau CH, Peer C, et al. A phase I and pharmacokinetic study of oral perifosine with different loading schedules in patients with refractory neoplasms. Cancer Chemother Pharmacol. 2014;74:955–67. 10.1007/s00280-014-2569-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Birch R, Chawla S, Nemunaitis J, Savage P, Kaiser P, Spira A, et al. Perifosine (P) as an active agent in the treatment of patients with advanced sarcoma [abstract]. J Clin Oncol (Meeting Abstracts). 2007;25:10059. [Google Scholar]
- 39.Shen K, Hong Y, Zhou Q, Zhang JL. Preclinical evaluation of perifosine as a potential promising rhabdomyosarcoma agent. Tumor Biol. 2016;37:1025–33. [DOI] [PubMed] [Google Scholar]
- 40.Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011;29:4688–95. 10.1200/JCO.2011.35.5263 [DOI] [PubMed] [Google Scholar]
- 41.Fouladi M, Perentesis JP, Phillips CL, Leary S, Reid JM, McGovern RM, et al. A phase I trial of MK-2206 in children with refractory malignancies: a Children's Oncology Group study. Pediatr Blood Cancer. 2014;61:1246–51. 10.1002/pbc.25023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sangai T, Akcakanat A, Chen H, Tarco E, Wu Y, Do KA, et al. Biomarkers of response to Akt inhibitor MK-2206 in breast cancer. Clin Cancer Res. 2012;18:5816–28. 10.1158/1078-0432.CCR-12-1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther. 2012;11:873–87. 10.1158/1535-7163.MCT-11-0824-T [DOI] [PubMed] [Google Scholar]
- 44.Lin J, Sampath D, Nannini MA, Lee BB, Degtyarev M, Oeh J, et al. Targeting activated Akt with GDC-0068, a novel selective Akt inhibitor that is efficacious in multiple tumor models. Clin Cancer Res. 2013;19:1760–72. 10.1158/1078-0432.CCR-12-3072 [DOI] [PubMed] [Google Scholar]
- 45.Bendell JC, Ervin TJ, Senzer NN, Richards DA, Firdaus I, Lockhart AC, et al. Results of the X-PECT study: A phase III randomized double-blind, placebo-controlled study of perifosine plus capecitabine (P-CAP) versus placebo plus capecitabine (CAP) in patients (pts) with refractory metastatic colorectal cancer (mCRC) [abstract]. J Clin Oncol (Meeting Abstracts). 2012;30:LBA3501. [Google Scholar]
- 46.Vallières GB, Burroughs P. Aeterna Zentaris to Discontinue Phase 3 Trial in Multiple Myeloma with Perifosine Following Data Safety Monitoring Board Recommendation 2013 [cited 2016 February 22]; http://www.aezsinc.com/en/page.php?p=60&q=550
- 47.Haendeler J, Hoffmann J, Rahman S, Zeiher AM, Dimmeler S. Regulation of telomerase activity and anti-apoptotic function by protein-protein interaction and phosphorylation. FEBS Lett. 2003;563:180–6. [DOI] [PubMed] [Google Scholar]
- 48.Ruiter GA, Zerp SF, Bartelink H, van Blitterswijk WJ, Verheij M. Alkyl-lysophospholipids activate the SAPK/JNK pathway and enhance radiation-induced apoptosis. Cancer Res. 1999;59:2457–63. [PubMed] [Google Scholar]
- 49.Fu L, Kim YA, Wang X, Wu X, Yue P, Lonial S, Khuri FR, Sun SY. Perifosine inhibits mammalian target of rapamycin signaling through facilitating degradation of major components of the mTOR axis and induces autophagy. Cancer Res. 2009;69:8967–76. 10.1158/0008-5472.CAN-09-2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J, Davies BR, Han S, Zhou M, Bai Y, Zhang J, et al. The AKT inhibitor AZD5363 is selectively active in PI3KCA mutant gastric cancer, and sensitizes a patient-derived gastric cancer xenograft model with PTEN loss to Taxotere. J Transl Med. 2013;11:241 10.1186/1479-5876-11-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cani A, Simioni C, Martelli AM, Zauli G, Tabellini G, Ultimo S, et al. Triple Akt inhibition as a new therapeutic strategy in T-cell acute lymphoblastic leukemia. Oncotarget. 2015;6:6597–610. 10.18632/oncotarget.3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kushner BH, Cheung NKV, Modak S, Becher OJ, Basu EM, Roberts SS, et al. A phase I/Ib trial targeting the PI3K/AKT pathway using perifosine: Long-term progression-free survival of patients with resistant neuroblastoma. Int J Cancer. 2017;140(2):480–484. 10.1002/ijc.30440 Epub 2016 Sep 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cole DE, Lester-McCully CM, Widemann BC, Warren KE. Plasma and cerebrospinal fluid pharmacokinetics of the Akt inhibitor, perifosine, in a non-human primate model. Cancer Chemother Pharmacol. 2015;75:923–8. 10.1007/s00280-015-2711-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee SW, Kim WJ, Park JA, Choi YK, Kwon YW, Kim KW. Blood-brain barrier interfaces and brain tumors. Arch Pharm Res. 2006;29:265–75. [DOI] [PubMed] [Google Scholar]
- 55.Watkins S, Robel S, Kimbrough IF, Robert SM, Ellis-Davies G, Sontheimer H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat Commun. 2014;5:4196 10.1038/ncomms5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pitz MW, Desai A, Grossman SA, Blakeley JO. Tissue concentration of systemically administered antineoplastic agents in human brain tumors. J Neurooncol. 2011;104:629–38. 10.1007/s11060-011-0564-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Z, Ho SL, Singh R, Pisapia DJ, Souweidane MM. Toxicity evaluation of convection-enhanced delivery of small-molecule kinase inhibitors in naive mouse brainstem. Childs Nerv Syst. 2015;31:557–62. 10.1007/s00381-015-2640-7 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.