

Abstract
The mechanistic links between exposure to airborne particulate matter (PM) pollution and the associated increases in cardiovascular morbidity and mortality, particularly in people with congestive heart failure (CHF), have not been identified. To advance understanding of this issue, genetically engineered mice (CREBA133) exhibiting severe dilated cardiomyopathic changes were exposed to ambient PM collected in Baltimore. CREBA133 mice, which display aberrant cardiac physiology and anatomy reminiscent of human CHF, displayed evidence of basal autonomic aberrancies (compared with wild-type mice) with PM exposure via aspiration, producing significantly reduced heart rate variability, respiratory dysynchrony, and increased ventricular arrhythmias. Carotid body afferent nerve responses to hypoxia and hyperoxia-induced respiratory depression were pronounced in PM-challenged CREBA133 mice, and denervation of the carotid bodies significantly reduced PM-mediated cardiac arrhythmias. Genome-wide expression analyses of CREBA133 left ventricular tissues demonstrated prominent Na+ and K+ channel pathway gene dysregulation. Subsequent PM challenge increased tyrosine phosphorylation and nitration of the voltage-gated type V cardiac muscle α-subunit of the Na+ channel encoded by SCN5A. Ranolazine, a Na+ channel modulator that reduces late cardiac Na+ channel currents, attenuated PM-mediated cardiac arrhythmias and shortened PM-elongated QT intervals in vivo. These observations provide mechanistic insights into the epidemiologic findings in susceptibility of human CHF populations to PM exposure. Our results suggest a multiorgan pathobiology inherent to the CHF phenotype that is exaggerated by PM exposure via heightened carotid body sensitivity and cardiac Na+ channel dysfunction.
Keywords: particulate matter, congestive heart failure, heart rate variability, electromyograms, ventricular arrhythmia score
Exposure to particulate matter (PM) air pollution increases the risk of adverse cardio-pulmonary events (1), resulting in an estimated 800,000 premature deaths annually worldwide (2). Compelling epidemiologic findings indicate that short-term increases in PM exposure are associated with a wide range of cardiovascular morbidity indicators (3). Comorbid conditions, such as age, diabetes, hypertension, chronic respiratory conditions, cardiac ischemia, and congestive heart failure (CHF), increase susceptibility to PM (4–6). For example, the mortality rate for people with CHF who have been acutely exposed to elevated ambient PM levels is estimated to be 4-fold higher when compared with healthy subjects (7). Thus, compelling evidence links PM exposure to the rapid occurrence of adverse cardiovascular effects, especially in people with preexisting cardiac disease, including CHF. This finding parallels results of observational and experimental studies of cardiovascular consequences of exposure to PM-containing secondhand tobacco smoke (8, 9).
Multiple pathogenetic mechanisms have been hypothesized to underlie the adverse cardiac effects of PM, including induction of inflammatory cascades (10), increased levels of reactive oxygen species (ROS) (11), induced coagulation cascades (10, 12), changes in blood viscosity (13), and altered autonomic function (6). In a rat model of acute myocardial infarction, PM exposure produced severe bradycardia, decreased heart rate variability (HRV), and increased ventricular arrhythmias (14). PM exposure alters the duration of the QT interval in patients with preexisting coronary disease and increases the number of discharges in patients with implanted cardiac defibrillators (15). However, despite the abundant evidence documenting the deleterious cardiac effects of PM, information remains limited regarding the mechanistic underpinnings of PM-induced cardiac pathophysiology (2).
Sympathetic nervous system hyperreactivity is a hallmark of CHF in humans and experimental models and may involve heightened carotid body reflexes (16, 17). We used a murine model of dilated cardiomyopathy to address potential mechanistic links between PM exposure and the development of life-threatening cardiac dysrhythmias in people with CHF. CREBA133 mice are engineered to express a cardiomyocyte-specific dominant/negative mutant form of CREBA133 (Ser-Ala133), a 43-kD basic leucine zipper transcription factor essential for cardiac muscle function (18), and exhibit cardiomyopathic changes, such as cardiac chamber dilation and reduced left ventricular function, that closely resemble human CHF (18). Our findings of heightened carotid body sensitivity and serious ventricular arrhythmias due to sodium channel dysregulation in PM-challenged CHF mice are relevant to understanding the risks of PM exposure for the susceptible population of older persons with CHF, who number in the millions in the United States (19).
Materials and Methods
PM Characteristics
The collection and characterization of ambient Baltimore PM has been previously described (20, 21). The PM used in our studies was collected from a sixth-floor window in urban Baltimore using a high-volume cyclone collector (theoretical cut point of 0.85 μm aerodynamic diameter) intermittently operated over a period of months with a flow rate of 0.6 m3/min. The count median diameter of the PM was 1.78 μm (from 0.1–10 μm), with a geometric standard deviation of 2.21.
Mice
CREBA133 and CD1 control mice (Jackson Laboratories, Bar Harbor, ME) were used. At 20 weeks of age, these animals were anesthetized and received a 50-μl aliquot of PBS or PM suspended in PBS (20 mg/kg) as previously described (20, 22). All measurements (transthoracic echocardiography, EKG, respiratory parameters, carotid body functional tests, and organ harvesting) were performed 36 hours after PM or PBS challenge. CREBA133 mice express a dominant-negative form of the 43-kD basic leucine zipper CREB transcription factor CREBA133 (Ser-Ala133) with a severe dilated cardiomyopathy phenotype (see Figure E1 in the online supplement), which results in 40% mortality at 20 to 25 weeks of age (23, 24).
EKG Assessment
Continuous electrocardiograms were recorded using Data Science implantable leads (Data Science, Minneapolis, MN). Electrodes were implanted subcutaneously, and animals were allowed a 24-hour recovery period before subsequent recording for 36 hours. At the completion of this recording, a single intratracheal dose of PM or PBS was administered, followed by a second 36-hour EKG recording.
Respiratory Responses to PM
Respiratory responses, including Vt, respiratory rate, and e, were measured at baseline and 36 hours after PM/PBS exposure using a whole-body plethysmograph (BUXCO chamber) as previously described (25).
Carotid Body Function and Hypoxic Responses
The effects of brief hyperoxia (Dejour's test) on diaphragmatic EMG activity (an index of neural respiration) were determined as previously described (26). Details are provided in the online supplement.
RNA Isolation and Microarray Analysis
Total RNA was isolated from the left ventricle and used for microarray analysis using Affymetrix Mouse 430_2 arrays. Chip quality was evaluated (27) and analyzed as previously described (28). Details are provided in the online supplement.
Canonical Pathway and Molecular Network Modeling
Dysregulated genes were uploaded into the IPA software (http://www.ingenuity.com). Details are provided in the online supplement.
Statistical Analysis
Values are provided as the mean ± SEM. Physiological and hemodynamic results were analyzed by unpaired two-tailed Student's t test, and P < 0.05 was considered to be significant. Microarray data were bioinformatically analyzed using significance analysis of microarrays, canonical pathways analysis by Fisher's exact test, P value ≤ 0.05, and Pearson correlation for distance metric in heat maps.
Results
PM Induces Bradycardia and Cardiac Arrhythmias in CHF Mice
PM-mediated alterations in heart rate and HRV were assessed because these physiological parameters are independent risk factors for the development of cardiac arrhythmias, sudden death, and myocardial infarction (29), particularly in people with CHF and coronary artery disease (2). Continuous ECG monitoring (0–36 h) revealed marked bradycardia in CREBA133 CHF mice soon after intratracheal instillation of Baltimore PM (20 mg/kg; mean aerodynamic diameter, 1.9 μm), whereas wild-type (CD1) mice were minimally affected (Figure 1A). PM-exposed CREBA133 mice exhibited significant decreases in HRV (standard deviation of 5-min segments of normal-to-normal intervals) when compared with CD1 mice after PM exposure or with CREBA133 mice before PM exposure (24 h) (P < 0.05; Figure 1B).
Figure 1.
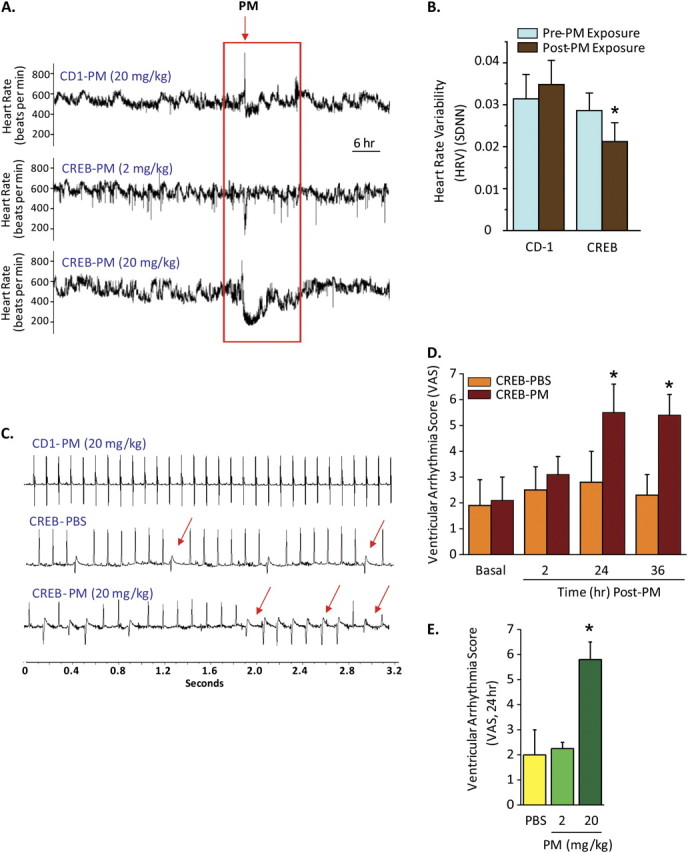
Particulate matter (PM) mediates bradycardia, decreases heart rate variability (HRV), and induces unstable electrocardiographic rhythms in CREBA133 mice. (A) Representative heart rate tracings of CREBA133 transgenic mice and wild-type CD1 mice after PM exposure via intratracheal instillation. The heart rate is obtained from continuous 10-s segments. Arrows represent the point in which Baltimore PM (2–20 mg/kg) was administered. Minimal decreases in heart rate (bradycardia) were observed in PM-exposed CD1 mice (20 mg/kg) or in CREBA133 mice receiving 2 mg/kg PM. In contrast, greater sensitivity in CREBA133 transgenic mice challenged with 20 mg/kg PM was observed with sustained and pronounced bradycardia. (B) HRV represented by the standard deviation of normal-to-normal intervals (SDNN) obtained in CREBA133 or CD1 mice 24 hours before and 24 hours after ambient Baltimore PM exposure. CD1 mice failed to exhibit significant alterations in HRV after PM exposure (P = 0.56; n = 7), whereas PM-challenged CREBA133 mice displayed significant HRV decreases (P < 0.05; n = 7). (C) Representative 3-second rhythm strips recorded with implantable EKG leads depicting ectopic beats in phosphate-buffered saline (PBS)-challenged CREBA133 mice with increased frequency of ventricular ectopy and nonsustained ventricular tachycardia sequences 36 hours after PM exposure. CD1 mice did not exhibit rhythm abnormalities even after PM exposure. (D) The bar graph depicts the average ventricular arrhythmia scores (VAS) in 20-week-old CREBA133 mice before and after PBS or PM exposure (20 mg/kg) (n = 6–8). VAS reflects a 60-minute period averaged over 2 to 36 hours after PM exposure. A VAS value of 0 reflects the absence of ectopic beats or premature ventricular contractions (PVCs); a VAS score of 1 = 1 to 30 PVCs; VAS 2 = 31 to 90 PVCs; VAS 3 = 91 to 300 PVCs; VAS 4 = 91 to 300 PVCs and an idioventricular rhythm <10 s; VAS 5 = 91 to 300 PVCs and an idioventricular rhythm >10 s; and VAS 6 = ventricular fibrillation. CREBA133 mice exhibit increased VAS scores at baseline, with further increases occurringafter PM exposure in a time-dependent manner beginning within 2 hours and reaching significance at 24 and 36 hours (*P < 0.05). CD1 mice failed to demonstrate a VAS above 0 even after 36-hour PM exposure (20 mg/kg). (E) Dose-dependent increases in VAS in CREBA133 mice before and after PBS or PM exposure (n = 4–8; *P < 0.05). VAS was not increased by exposure to 2 mg/kg PM exposure compared with PBS-exposed CREBA133 mice measured 36 hours after PM administration.
In addition to heart rate alterations, PM-exposed CREBA133 mice displayed prominent cardiac arrhythmias, including multifocal premature ventricular contractions, runs of nonsustained ventricular tachycardia, and idioventricular rhythms (Figure 1C). Cardiac dysrhythmias were quantified using a ventricular arrhythmia scoring system (VAS), which incorporates the frequency and duration of premature ventricular contractions and ventricular tachycardia. CD1 mice exhibited a VAS of 0 at baseline and after PM exposure (ectopy absent; data not shown), whereas unexposed CREBA133 mice exhibited increased a basal VAS (1.8 ± 0.3), with further time-dependent VAS increases beginning 2 hours after PM instillation (3.0 ± 0.4) to a maximum VAS at 24 hours after PM exposure (5.5 ± 0.5) (Figure 1D). The VAS increase in CREBA133 mice was dose dependent, with 2 mg/kg PM failing to significantly increase VAS above baseline values (Figure 1E).
PM Mediates Heightened Carotid Body Sensitivity and Autonomic Dysfunction
Alterations in autonomic nervous system function have been implicated in the adverse effects of PM on cardiovascular mortality (30, 31); however, direct measurements of autonomic indicators, including breathing patterns, have not been previously assessed in animal models of PM exposure. CREBA133 mice demonstrated elevations in basal respiratory rates and minute ventilation compared with CD1 mice (Figure 2A), but prominent respiratory instability after PM challenge manifested as a periodic breathing pattern, a prognostic sign for the sudden cardiac decompensation and death often observed in patients with severe CHF (30, 32, 33). Respiratory dysynchrony was more pronounced during hypoxia (Figure 2B) and was abolished by carotid body denervation (Figure 2C), which also significantly reduce PM-mediated cardiac arrhythmias in CREBA133 mice (Figure 2D). In contrast, inhibition of cholinergic muscarinic autonomic neurons (atropine, 1 mg/kg, intraperitoneally) or adrenergic autonomic neurons (propranolol, 1 mg/kg, intraperitoneally) failed to prevent PM-evoked cardiac arrhythmias in CREBA133 mice (data not shown). These results strongly suggest that PM-mediated heightened carotid body sensitivity contributes to the subsequent development of cardiac arrhythmias.
Figure 2.
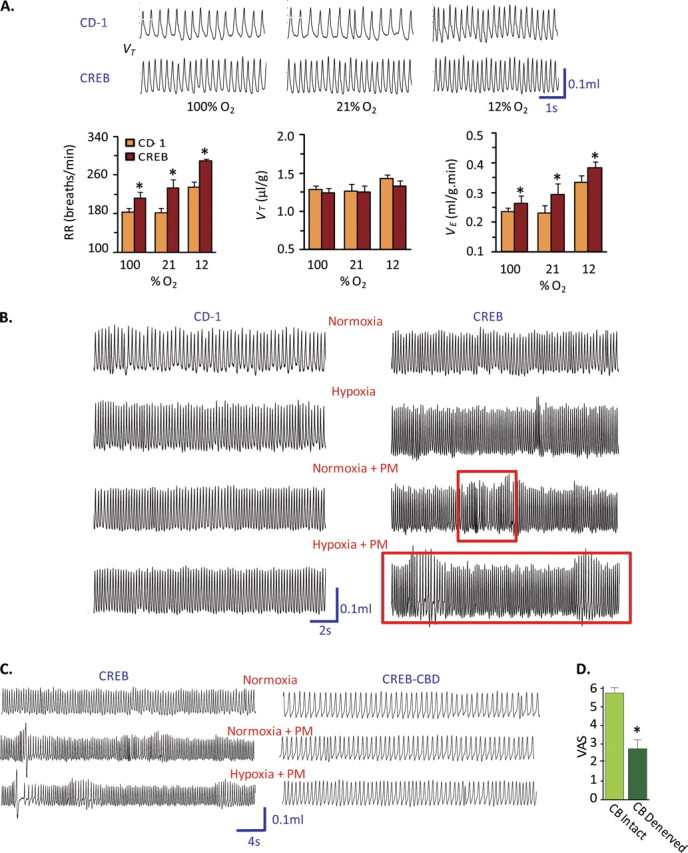
Unstable ventilation patterns in PM-exposed CREBA133 mice. (A) Respiratory tracings and bar graph summary of ventilation parameters (tidal volume or VT, respiratory rate or RR, and minute ventilation or e) in CD1 and CREBA133 mice exposed to varying levels of inspired oxygen (100, 21, and 12% O2). CREBA133 mice exhibited elevated baseline respiratory rate and minute ventilation as well as augmented ventilatory responses to acute hypoxia. The data presented are mean ± SEM from six mice per group. (B) Breathing patterns in CD1 and CREBA133 mice before and after PM exposure, during normoxia and hypoxia (12% O2). Unlike CD1 mice, which exhibit no alterations in respiratory patterns after PM exposure or hypoxic challenge, PM-exposed CREBA133 mice exhibited waxing and waning breathing patterns (red frames), which were more pronounced during hypoxia. (C) Breathing patterns in CREBA133 mice with carotid body denervation (CBD). Bilateral surgical removal of carotid bodies (CB denervation or CBD) resulted in declines in basal breathing rate and abolished PM-induced unstable ventilatory pattern in CREBA133 mice. (D) Carotid body denervation significantly reduced PM-induced cardiac arrhythmias in CHF mice.
Carotid body function in PM-challenged CREBA133 mice was further validated by monitoring the magnitude of the transient ventilatory decline by recording diaphragmatic electromyograms (EMGs) in response to brief hyperoxic exposure, an index of peripheral chemoreceptor sensitivity, especially the carotid body (34). Diaphragmatic EMGs were recorded in anesthetized mice for 5 minutes under normoxic conditions and then for 20 seconds after hyperoxic challenge. PM-challenged CREBA133 mice manifested significantly greater regulatory depression (near apnea) in response to hyperoxia compared with CD1 mice, indicating heightened carotid body sensitivity (Figures 3A and 3B). To directly demonstrate altered chemoreceptor function, “single” fiber carotid sinus nerve activities were recorded ex vivo from carotid bodies isolated from CREBA133 mice previously challenged with PM or vehicle control. The carotid body sensory response to hypoxia was markedly pronounced in PM-exposed CREBA133 mice compared with vehicle-treated CREBA133 mice or PM-challenged CD1 mice (Figures 3C and 3D), indicating that PM exposure sensitizes carotid body responses to acute hypoxia only in CHF mice, with reflexive sympathetic nervous system–mediated cardiac and respiratory arrhythmias. These findings parallel the susceptibility of patients with CHF to the adverse effects of PM exposure (14).
Figure 3.
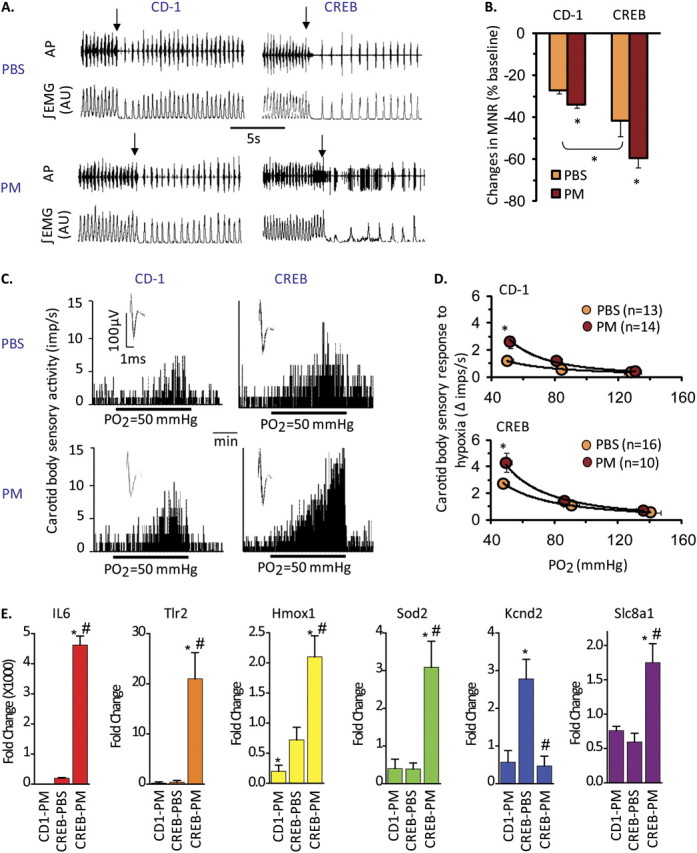
PM sensitizes carotid body function in CREBA133 mice. (A) Diaphragmatic EMG responses to brief (20 s) exposure to 100% O2 (arrows) in CREBA133 and CD1 mice exposed to vehicle (PBS) or PM. AP = action potential of the diaphragmatic EMG; AU = arbitrary units; ∫EMG = integrated diaphragmatic EMG activity. (B) Effects of brief hyperoxia (Dejour's test) on minute neural respiration (MNR). Data are mean ± SEM change in MNR (MNR100% –MNR21% O2)/MNR21%O2. n = 4 mice per group (*P < 0.05). (C and D) Integrated sensory activities of the carotid body in response to hypoxia in CD1 and CREBA133 mice exposed to PBS or PM (Po2 = partial pressures of O2 in the perfusate [Po2 = 50 mm Hg]). Black bars represent duration of the hypoxic challenge. The superimposed action potential of a “single” fiber is shown (inset). Average sensory responses to graded hypoxia are presented as changes in impulses/second (hypoxia – baseline). Values are mean ± SEM. *P < 0.05. n = Number of fibers analyzed from five mice per group. (E) PM induces dysregulation of inflammatory, oxidant, and carotid body regulatory genes in CD1 and CREBA133 mice. Pooled carotid body RNA from PBS- and PM-challenged CD1 and CREBA133 mice (36 h; n = 10–12 mice per group) was used in qPCR reactions (see Materials and Methods). Shown are expression levels for IL-6, Toll-like Receptor 2 (TLR2), heme oxygenase (Hmox1), superoxide dismutase (SOD2), the large conductance Ca2+–activated K+ channel (Kcnd2), and the Na+–Ca2+ exchanger 1 precursor protein (Slc8a1). Data represent the fold change in gene expression compared with PBS-challenged CD1 mice (*P < 0.05).
Genomic Analyses to Evaluate Gene Deregulation in CHF Mice
How might PM sensitize carotid body responses to hypoxia? Carotid bodies express receptors for proinflammatory mediators (35) that sensitize carotid body responses to acute hypoxia (31, 36, 37). Assessment of expression levels of selected genes within carotid body tissues from PM-exposed mice (by qRT-PCR) revealed profound PM-mediated gene dysregulation in CREBA133 mice (compared with CD1 mice), including genes encoding a number of proinflammatory and oxidant markers (Figure 3E and see Table E1 in the online supplement). In addition, carotid bodies from PM-challenged CREBA133 mice exhibited marked gene up-regulation of Na+ channels (scnn1b and scn8a) and Na+–Ca+ exchangers (slc8a1) with concomitant down-regulation of K+ channels (kcnd3, kcnmb2, kcnc1, kcnd1, and kcnd2), responses which potentially contribute to hyperexcitability of carotid body sensory activity (16, 38). Furthermore, significant down-regulation of glial-derived neurotrophic factor (GDNF), which is critical for maintenance of the dopaminergic phenotype of hypoxic-sensing type I carotid body cells (39), was observed in PM-challenged CREBA133 mice (Table E1). Reduced expression of GDNF likely results in down-regulation of dopamine's inhibitory effects on carotid body sensory activity.
We next evaluated genome-wide cardiac gene expression in left ventricular tissue samples obtained from the PM-exposed CREBA133 and CD-1 mice (Affymetrix Mouse 430 v2.0 array) with selected RT-PCR validation (Table E2). Unsupervised hierarchical clustering analyses demonstrated that the CREBA133 genotype was the dominant factor in left ventricular gene dysregulation (Table E2; Figure E2). Unlike PM-induced gene dysregulation in carotid body tissues, PM exposure minimally affected cardiac gene expression, although the magnitude of gene dysregulation was greater in cardiac tissues from PM-challenged CREBA133 mice than from PBS-exposed CREBA133 mice (Figure 4A). Similar to carotid body expression profiling, marked cardiac ion channel gene dysregulation was observed in CREBA133 mice, with increased expression of multiple Na+ channel subunit and Na+–Ca2+ exchanger genes accompanied by reduced K+ channel subunit gene expression (Figure 4B).
Figure 4.
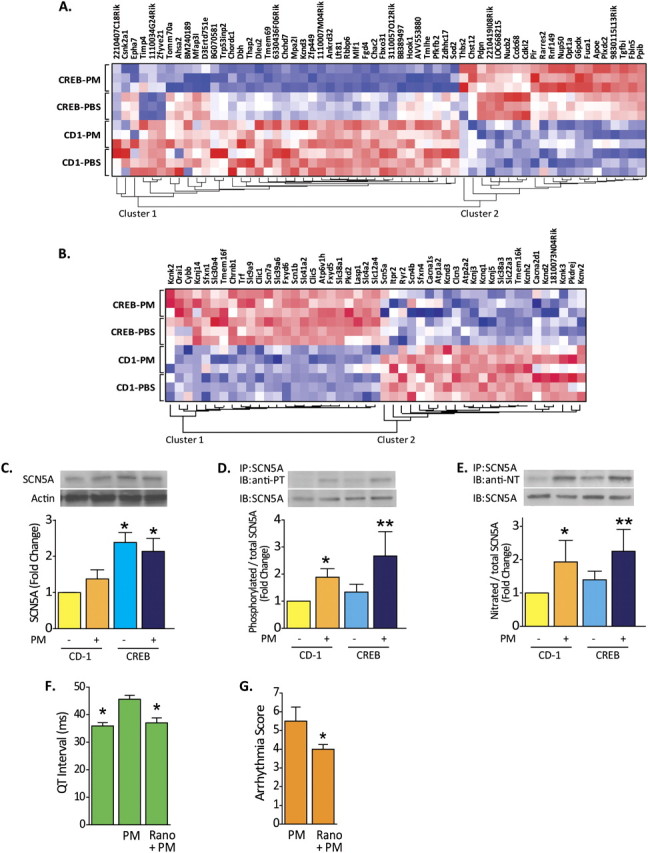
Cardiac ion channels regulation in PM-challenged CREBA133 mice. (A) Genome-wide expression profiling of PM-exposed left ventricular tissues from CREBA133 mice identified a large set of dysregulated genes with 1,927 probe sets (Table E3) when compared with PBS-challenged CD1 mice (Significance Analysis of Microarrays software). Comparison between PM- and PBS-exposed CREBA133 mice shows that 60 of the 1,927 probe sets display differential expression (Student's t test P value ≤ 0.05). The heat map (dChip software) of these 60 genes across the four groups is displayed. Red, white, or blue color represents expression level above, at, or below the mean level, respectively. (B) The expression pattern of dysregulated genes involved in ion transport (Gene Ontology ID: 0006811 and 0043269) is displayed across all left ventricle tissue samples. Noted is the up-regulation of Na+ channels and the Na+–Ca2+ exchanger with concominant down-regulation of K+ channels, events which potentially alter myocardial Na+ and Ca2+ homeostasis, resulting in increased [Ca2+]i and [Na+]i, tissue hyperexcitability, and arrhythmogenesis. Up-regulated nonchannel proteins, such as ankyrin and gelsolin, also participate in localization and regulation of pore function, including L-type Ca2+ channel activity in PM-challenged CREBA133 mice. (C) Expression and posttranslational modification of the voltage-gated, type V cardiac muscle α-subunit Na+ channel encoded by SCN5a. This Na+ channel protein was analyzed by Western blot in CD1 and CREBA133 left ventricle tissues. The late Na+ channel protein is up-regulated in CREBA133 ventricle tissues but is not altered significantly by PM exposure. (D and E) The cardiac Na+ channel subunit encoded by SCN5A was immunoprecipitated (IP) in left ventricle protein lysates and subjected to Western blotting (IB) for phosphotyrosine (PT) or nitrotyrosine (NT). Increased phosphorylation and nitration of late Na+ channel protein tyrosine residues was observed in CD1 and CREBA133 mice after PM exposure (representative blots from more than three independent experiments). Bar graphs were generated from quantification of Western blots from four independent experiments. *P < 0.05 compared with CD-1 control. **P < 0.05 compared with CREBA133 control. (F) QT interval alterations evoked by PM or by ranolazine treatment. QT intervals were quantified from ECG recordings in CREBA133 mice. PM exposure significantly prolonged QT intervals in CREBA133, mice with ranolazine pretreatment significantly reversing PM-mediated QT interval elongation. n = 4. *P < 0.05 compared with PM-alone group. (G) Inhibition of late sodium channel by ranolazine pretreatment (Rano, 50 mg/kg, intraperitoneally) attenuates PM-induced ventricular arrhythmias reflected by significantly reduced VAS in PM-treated CREBA133 mice.
The α Subunit of the Late Na+ Channel Is a Key Participant in PM-Mediated Cardiac Arrhythmias
Dysregulation of ion channel gene expression in left ventricular tissues potentially contributes to altered myocardial handling of Na+ and Ca2+ and subsequent Ca2+ overload, tissue hyperexcitability, and arrhythmogenesis in PM-challenged CREBA133 mice. Immunoprecipitation experiments were performed targeting the cardiac α subunit of the voltage-gated late Na+ channel encoded by SCN5A, an up-regulated gene in CHF mice (Figure 4C). This Na+ channel subunit facilitates the late Na+ current, which flows throughout the ventricular action potential and contributes to cardiac arrhythmia development, particularly in fibrotic tissues (40). Our results indicate that this Na+ channel subunit undergoes PM-mediated significant posttranslational modification reflected by tyrosine phosphorylation and nitration (Figures 4D and 4E), events previously observed to deregulate Na+ channel activity and to generate late-channel current leak and arrhythmogenesis (41, 42). Highly consistent with the mechanistic contribution of Na+ channel dysfunction in PM-mediated arrhythmia generation in CHF mice, the selective late Na+ channel inhibitor ranolazine (50 mg/kg, intraperitoneally) significantly attenuated PM-mediated cardiac arrhythmias in CREBA133 mice (Figure 4G) while shortening PM-mediated elongation of the QT interval (Figure 4F). These findings strongly suggest a critical role of PM-mediated dysregulation of cardiac Na+ channels in arrhythmia development in the CHF mice.
Discussion
Although the public health implications of sudden death and adverse cardiovascular outcomes after ambient PM exposure are profound, the mechanistic underpinnings for the epidemiologic observations have been incompletely understood. Using CREBA133 mice that exhibited severe cardiomyopathy, we have noted that the CHF phenotype is inherently primed to respond in exaggerated fashion to PM challenge. For example, only carotid bodies from CHF mice (but not CD1 mice) responded to PM challenge with the induction of marked inflammatory, oxidant, and ion channel gene signatures in carotid body tissues. The mechanism by which carotid bodies for CHF mice acquire this priming is not known. PM-mediated carotid body gene dysregulation may be related to the well recognized induction of lung inflammatory cascades (10) after PM exposure with accompanying increases in the levels of circulating cytokines and reactive oxygen species (ROS) (11). Proinflammatory molecules are known to augment Ca2+ flux in glomus cells of the carotid body and to sensitize carotid body responses to hypoxia via Ca2+ signaling pathways (37) and to increase carotid sinus nerve firings, leading to an increase in reflex automaticity and electromechanical cardiac coupling. Our assessment of carotid body susceptibility in CHF mice confirmed heightened carotid body sensitivity to acute hypoxia, consistent with prior observations that peripheral chemoreflex sensitivity is enhanced in rabbits with CHF (43) and plays a major role in the sympathetic activation found in human CHF (44). It is likely that these autonomic alterations in CHF mice are influenced by PM-mediated up-regulation of Na+ channels, the Na+–Ca2+ exchanger, and down-regulation of K+ channels (Figure 3B) in carotid body tissues.
The morphology of the numerous aberrantly conducted beats recorded in CREBA133 CHF mice implicates electrical activity emanating from the substantially remodeled fibrotic left ventricle previously noted in CREBA133 mice (23). Complex arrhythmogenic mechanisms resulting in sudden death due to ventricular tachycardia or ventricular fibrillation (15) are often linked to defects in myocardial intracellular Na+ and Ca2+ homeostasis (45, 46). Our unbiased network analysis of left ventricular gene expression identified marked ion channel abnormalities inherent to the CHF phenotype as well as prominent deregulation of genes encoding ryanodine receptors and cytoskeletal proteins (ankyrin, gelsolin), which participate in localization and regulation of Ca2+ channel activity and Ca2+ release in failing human heart myocytes (47, 48) (Table E2). For example, gelsolin expression is up-regulated in human and murine heart failure, and gelsolin mutations are associated with sudden cardiac death (48, 49). Together, these findings suggest that the CHF phenotype is primed by genomic alterations that contribute to aberrant Na+ and Ca2+ handling and arrhythmogenesis.
This hypothesis is supported by studies that explored the voltage-gated late Na+ channel protein, encoded by SCN5A, as a target for PM-mediated posttranslational modification. This Na+ channel protein subunit produces the sustained component of fast Na+ current of cardiac myocytes occurring in inflammatory and hypoxic conditions (45, 50). We noted that SCN5A is up-regulated in CHF mice with , inducing prominent Na+ channel subunit tyrosine phosphorylation and nitration, post-translational modifications, which presumably reflect PM-mediated increases in systemic oxidative stress (51) but which are well recognized to mediate sustained activated late Na+ currents (40, 42, 52). These events appear to directly contribute to PM-induced ventricular arrhythmias because the administration of ranolazine, the late Na+ current inhibitor, reduced PM-induced QT interval elongation and significantly attenuated PM-mediated cardiac arrhythmias (Figures 4F and 4G). These findings strongly implicate a critical contributory role for the late Na+ channel in PM-augmented cardiac arrhythmias in CHF mice.
There are several limitations of the current study, including the mode of PM exposure, intratracheal PM instillation, which is distinct from the human pattern of sustained exposure at lower dosage levels via inhalation. The instilled dose (20 mg/kg) is equivalent to 1-year exposure of a human in a highly polluted area (>200 μg/m3). In addition, we have not characterized the aspect of PM exposure that results in PM-mediated carotid body dysfunction and cardiac arrhythmias. Deleterious PM responses may be due to direct contact with PM constituents or may be evoked indirectly via lung-derived inflammatory processes that generate substantial cytokine and ROS release from lung tissues. Finally, further investigation is required to fully understand the mechanism and the functional consequences of Na+ channel protein phosphorylation and nitration induced by PM exposure, including identification of the modified tyrosine residue(s). Despite these limitations, our findings in a murine model of dilated cardiomyopathy provide novel mechanistic insights regarding complex PM pathophysiology in susceptible individuals with CHF at risk for acute cardiovascular morbidity and mortality after short-term PM exposure (7). Using complementary physiological and genomic approaches with direct measurements of carotid body neural activity, our studies significantly extend earlier in vitro (53) and in vivo studies (54, 55) and indicate that PM-mediated decreases in HRV, respiratory dysynchrony, and serious cardiac arrhythmias are driven by heightened sensitivity of carotid body function, augmented peripheral chemoreceptor reflexes, and dysregulated cardiac ion channel expression within the CREBA133 CHF phenotype. The cardiac late Na+ channel appears to be a major target for PM-mediated posttranslational modification and subsequent life-threatening cardiac arrhythmogenesis.
Acknowledgments
The authors thank Eddie T. Chiang and Carrie L. Evenoski for superb technical assistance.
Footnotes
Supported by grant RD–83241701from the United States Environmental Protection Agency.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1165/rcmb.2011-0213OC on November 22, 2011
References
- 1.Mann JK, Tager IB, Lurmann F, Segal M, Quesenberry CP, Lugg MM, Shan J, Van Den Eeden SK. Air pollution and hospital admissions for ischemic heart disease in persons with congestive heart failure or arrhythmia. Environ Health Perspect 2002;110:1247–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brook RD. Cardiovascular effects of air pollution. Clin Sci (Lond) 2008;115:175–187. [DOI] [PubMed] [Google Scholar]
- 3.Peel JL, Metzger KB, Klein M, Flanders WD, Mulholland JA, Tolbert PE. Ambient air pollution and cardiovascular emergency department visits in potentially sensitive groups. Am J Epidemiol 2007;165:625–633. [DOI] [PubMed] [Google Scholar]
- 4.Dekker JM, Schouten EG, Klootwijk P, Pool J, Swenne CA, Kromhout D. Heart rate variability from short electrocardiographic recordings predicts mortality from all causes in middle-aged and elderly men. The Zutphen Study. Am J Epidemiol 1997;145:899–908. [DOI] [PubMed] [Google Scholar]
- 5.Glantz SA. Air pollution as a cause of heart disease: time for action. J Am Coll Cardiol 2002;39:943–945. [DOI] [PubMed] [Google Scholar]
- 6.Pope CA, Burnett RT, Thurston GD, Thun MJ, Calle EE, Krewski D, Godleski JJ. Cardiovascular mortality and long-term exposure to particulate air pollution: epidemiological evidence of general pathophysiological pathways of disease. Circulation 2004;109:71–77. [DOI] [PubMed] [Google Scholar]
- 7.Kwon HJ, Cho SH, Nyberg F, Pershagen G. Effects of ambient air pollution on daily mortality in a cohort of patients with congestive heart failure. Epidemiology 2001;12:413–419. [DOI] [PubMed] [Google Scholar]
- 8.Van Deusen A, Hyland A, Travers MJ, Wang C, Higbee C, King BA, Alford T, Cummings KM. Secondhand smoke and particulate matter exposure in the home. Nicotine Tob Res 2009;11:635–641. [DOI] [PubMed] [Google Scholar]
- 9.United States. Public Health Service. Office of the Surgeon General. 2006. The health consequences of involuntary exposure to tobacco smoke: a report of the Surgeon General. US Dept. of Health and Human Services, Public Health Service, Office of the Surgeon General, Rockville, MD. [Google Scholar]
- 10.Ruckerl R, Ibald-Mulli A, Koenig W, Schneider A, Woelke G, Cyrys J, Heinrich J, Marder V, Frampton M, Wichmann HE, et al. Air pollution and markers of inflammation and coagulation in patients with coronary heart disease. Am J Respir Crit Care Med 2006;173:432–441. [DOI] [PubMed] [Google Scholar]
- 11.Sorensen M, Daneshvar B, Hansen M, Dragsted LO, Hertel O, Knudsen L, Loft S. Personal PM2.5 exposure and markers of oxidative stress in blood. Environ Health Perspect 2003;111:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mutlu GM, Green D, Bellmeyer A, Baker CM, Burgess Z, Rajamannan N, Christman JW, Foiles N, Kamp DW, Ghio AJ, et al. Ambient particulate matter accelerates coagulation via an IL-6-dependent pathway. J Clin Invest 2007;117:2952–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Donaldson K, Stone V, Seaton A, MacNee W. Ambient particle inhalation and the cardiovascular system: potential mechanisms. Environ Health Perspect 2001;109:523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wellenius GA, Coull BA, Batalha JR, Diaz EA, Lawrence J, Godleski JJ. Effects of ambient particles and carbon monoxide on supraventricular arrhythmias in a rat model of myocardial infarction. Inhal Toxicol 2006;18:1077–1082. [DOI] [PubMed] [Google Scholar]
- 15.Lux RL, Pope CA. Air pollution effects on ventricular repolarization. Res Rep Health Eff Inst 2009;141:3–20, discussion 21–28. [PubMed] [Google Scholar]
- 16.Schultz HD, Li YL. Carotid body function in heart failure. Respir Physiol Neurobiol 2007;157:171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schultz HD, Li YL, Ding Y. Arterial chemoreceptors and sympathetic nerve activity: implications for hypertension and heart failure. Hypertension 2007;50:6–13. [DOI] [PubMed] [Google Scholar]
- 18.Kronke G, Bochkov VN, Huber J, Gruber F, Bluml S, Furnkranz A, Kadl A, Binder BR, Leitinger N. Oxidized phospholipids induce expression of human heme oxygenase-1 involving activation of cAMP-responsive element-binding protein. J Biol Chem 2003;278:51006–51014. [DOI] [PubMed] [Google Scholar]
- 19.Environmental-Protection-Agency. 2010. Policy assessment for the review of the particulate matter national ambient air quality standards. USEPA: Washington, DC. [Google Scholar]
- 20.Walters DM, Breysse PN, Schofield B, Wills-Karp M. Complement factor 3 mediates particulate matter-induced airway hyperresponsiveness. Am J Respir Cell Mol Biol 2002;27:413–418. [DOI] [PubMed] [Google Scholar]
- 21.Walters DM, Breysse PN, Wills-Karp M. Ambient urban Baltimore particulate-induced airway hyperresponsiveness and inflammation in mice. Am J Respir Crit Care Med 2001;164:1438–1443. [DOI] [PubMed] [Google Scholar]
- 22.Wang T, Moreno-Vinasco L, Huang Y, Lang GD, Linares JD, Goonewardena SN, Grabavoy A, Samet JM, Geyh AS, Breysse PN, et al. Murine lung responses to ambient particulate matter: genomic analysis and influence on airway hyperresponsiveness. Environ Health Perspect 2008;116:1500–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fentzke RC, Korcarz CE, Lang RM, Lin H, Leiden JM. Dilated cardiomyopathy in transgenic mice expressing a dominant-negative CREB transcription factor in the heart. J Clin Invest 1998;101:2415–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fentzke RC, Korcarz CE, Shroff SG, Lin H, Leiden JM, Lang RM. The left ventricular stress-velocity relation in transgenic mice expressing a dominant negative CREB transgene in the heart. J Am Soc Echocardiogr 2001;14:209–218. [DOI] [PubMed] [Google Scholar]
- 25.Kline DD, Overholt JL, Prabhakar NR. Mutant mice deficient in NOS-1 exhibit attenuated long-term facilitation and short-term potentiation in breathing. J Physiol 2002;539:309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peng YJ, Yuan G, Ramakrishnan D, Sharma SD, Bosch-Marce M, Kumar GK, Semenza GL, Prabhakar NR. Heterozygous HIF-1alpha deficiency impairs carotid body-mediated systemic responses and reactive oxygen species generation in mice exposed to intermittent hypoxia. J Physiol 2006;577:705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Z, Irizarry RA. Preprocessing of oligonucleotide array data. Nat Biotechnol 2004;22:656–658, author reply 658. [DOI] [PubMed] [Google Scholar]
- 28.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 2001;98:5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abrahamowicz M, Schopflocher T, Leffondre K, du Berger R, Krewski D. Flexible modeling of exposure-response relationship between long-term average levels of particulate air pollution and mortality in the American Cancer Society study. J Toxicol Environ Health A 2003;66:1625–1654. [DOI] [PubMed] [Google Scholar]
- 30.Rhoden CR, Wellenius GA, Ghelfi E, Lawrence J, Gonzalez-Flecha B. PM-induced cardiac oxidative stress and dysfunction are mediated by autonomic stimulation. Biochim Biophys Acta 2005;1725:305–313. [DOI] [PubMed] [Google Scholar]
- 31.Fitzgerald RS, Shirahata M, Chang I, Balbir A. Modulators of cat carotid body chemotransduction. Adv Exp Med Biol 2006;580:307–311; discussion 351–309. [DOI] [PubMed] [Google Scholar]
- 32.Lanfranchi PA, Braghiroli A, Bosimini E, Mazzuero G, Colombo R, Donner CF, Giannuzzi P. Prognostic value of nocturnal Cheyne-Stokes respiration in chronic heart failure. Circulation 1999;99:1435–1440. [DOI] [PubMed] [Google Scholar]
- 33.Poletti R, Passino C, Giannoni A, Zyw L, Prontera C, Bramanti F, Clerico A, Piepoli M, Emdin M. Risk factors and prognostic value of daytime Cheyne-Stokes respiration in chronic heart failure patients. Int J Cardiol 2009;137:47–53. [DOI] [PubMed] [Google Scholar]
- 34.Dejours P. Chemoreflexes in breathing. Physiol Rev 1962;42:335–358. [DOI] [PubMed] [Google Scholar]
- 35.Zhang XJ, Wang X, Xiong LZ, Fan J, Duan XL, Wang BR. Up-regulation of IL-1 receptor type I and tyrosine hydroxylase in the rat carotid body following intraperitoneal injection of IL-1beta. Histochem Cell Biol 2007;128:533–540. [DOI] [PubMed] [Google Scholar]
- 36.Liu X, He L, Stensaas L, Dinger B, Fidone S. Adaptation to chronic hypoxia involves immune cell invasion and increased expression of inflammatory cytokines in rat carotid body. Am J Physiol Lung Cell Mol Physiol 2009;296:L158–L166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shu HF, Wang BR, Wang SR, Yao W, Huang HP, Zhou Z, Wang X, Fan J, Wang T, Ju G. IL-1beta inhibits IK and increases [Ca2+]i in the carotid body glomus cells and increases carotid sinus nerve firings in the rat. Eur J Neurosci 2007;25:3638–3647. [DOI] [PubMed] [Google Scholar]
- 38.Balbir A, Lee H, Okumura M, Biswal S, Fitzgerald RS, Shirahata M. A search for genes that may confer divergent morphology and function in the carotid body between two strains of mice. Am J Physiol Lung Cell Mol Physiol 2007;292:L704–L715. [DOI] [PubMed] [Google Scholar]
- 39.Villadiego J, Mendez-Ferrer S, Valdes-Sanchez T, Silos-Santiago I, Farinas I, Lopez-Barneo J, Toledo-Aral JJ. Selective glial cell line-derived neurotrophic factor production in adult dopaminergic carotid body cells in situ and after intrastriatal transplantation. J Neurosci 2005;25:4091–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruan Y, Liu N, Priori SG. Sodium channel mutations and arrhythmias. Natl Rev 2009;6:337–348. [DOI] [PubMed] [Google Scholar]
- 41.Ahern CA, Zhang JF, Wookalis MJ, Horn R. Modulation of the cardiac sodium channel NaV1.5 by Fyn, a Src family tyrosine kinase. Circ Res 2005;96:991–998. [DOI] [PubMed] [Google Scholar]
- 42.Ashki N, Hayes KC, Bao F. The peroxynitrite donor 3-morpholinosydnonimine induces reversible changes in electrophysiological properties of neurons of the guinea-pig spinal cord. Neuroscience 2008;156:107–117. [DOI] [PubMed] [Google Scholar]
- 43.Sun SY, Wang W, Zucker IH, Schultz HD. Enhanced activity of carotid body chemoreceptors in rabbits with heart failure: role of nitric oxide. J Appl Physiol 1999;86:1273–1282. [DOI] [PubMed] [Google Scholar]
- 44.Farrell TG, Bashir Y, Cripps T, Malik M, Poloniecki J, Bennett ED, Ward DE, Camm AJ. Risk stratification for arrhythmic events in postinfarction patients based on heart rate variability, ambulatory electrocardiographic variables and the signal-averaged electrocardiogram. J Am Coll Cardiol 1991;18:687–697. [DOI] [PubMed] [Google Scholar]
- 45.Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart 2006;92:iv6–iv14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scoote M, Williams AJ. Myocardial calcium signalling and arrhythmia pathogenesis. Biochem Biophys Res Commun 2004;322:1286–1309. [DOI] [PubMed] [Google Scholar]
- 47.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, duBell WH, Song LS, Haurogne K, Kyndt F, Ali ME, et al. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature 2003;421:634–639. [DOI] [PubMed] [Google Scholar]
- 48.Yang J, Moravec CS, Sussman MA, DiPaola NR, Fu D, Hawthorn L, Mitchell CA, Young JB, Francis GS, McCarthy PM, et al. Decreased SLIM1 expression and increased gelsolin expression in failing human hearts measured by high-density oligonucleotide arrays. Circulation 2000;102:3046–3052. [DOI] [PubMed] [Google Scholar]
- 49.Danieli GA. Sudden arrhythmic death: which genetic determinants? In: A.Raviele, editor. Cardiac arrhythmias 2005. Milan: Springer; 2005. pp. 385–392. [Google Scholar]
- 50.Lader AS, Kwiatkowski DJ, Cantiello HF. Role of gelsolin in the actin filament regulation of cardiac L-type calcium channels. Am J Physiol 1999;277:C1277–C1283. [DOI] [PubMed] [Google Scholar]
- 51.Ying Z, Kampfrath T, Thurston G, Farrar B, Lippmann M, Wang A, Sun Q, Chen LC, Rajagopalan S. Ambient particulates alter vascular function through induction of reactive oxygen and nitrogen species. Toxicol Sci 2009;111:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Bemmelen MX, Rougier JS, Gavillet B, Apotheloz F, Daidie D, Tateyama M, Rivolta I, Thomas MA, Kass RS, Staub O, et al. Cardiac voltage-gated sodium channel Nav1.5 is regulated by Nedd4-2 mediated ubiquitination. Circ Res 2004;95:284–291. [DOI] [PubMed] [Google Scholar]
- 53.Wold LE, Simkhovich BZ, Kleinman MT, Nordlie MA, Dow JS, Sioutas C, Kloner RA. In vivo and in vitro models to test the hypothesis of particle-induced effects on cardiac function and arrhythmias. Cardiovasc Toxicol 2006;6:69–78. [DOI] [PubMed] [Google Scholar]
- 54.Anselme F, Loriot S, Henry JP, Dionnet F, Napoleoni JG, Thuillez C, Morin JP. Inhalation of diluted diesel engine emission impacts heart rate variability and arrhythmia occurrence in a rat model of chronic ischemic heart failure. Arch Toxicol 2007;81:299–307. [DOI] [PubMed] [Google Scholar]
- 55.Ghelfi E, Rhoden CR, Wellenius GA, Lawrence J, Gonzalez-Flecha B. Cardiac oxidative stress and electrophysiological changes in rats exposed to concentrated ambient particles are mediated by TRP-dependent pulmonary reflexes. Toxicol Sci 2008;102:328–336. [DOI] [PubMed] [Google Scholar]