

Abstract
Urzymes—short, active core modules derived from enzyme superfamilies—prepared from the two aminoacyl-tRNA synthetase (aaRS) Classes contain only the modules shared by all related family members. They have been described as models for ancestral forms. Understanding them depends on inferences drawn from the crystal structures of the full-length enzymes. As aaRS Urzymes lack much of the mass of modern aaRS, retaining only a small portion of the hydrophobic cores of the full-length enzymes, it is desirable to characterize their structures. We report preliminary characterization of 15N tryptophanyl-tRNA synthetase Urzyme by heteronuclear single quantum coherence (HSQC) NMR spectroscopy supplemented by circular dichroism, thermal melting, and induced fluorescence of bound dye. The limited dispersion of 1H chemical shifts (0.5 ppm) is inconsistent with a narrow ensemble of well-packed structures in either free or substrate-bound forms, although the number of resonances from the bound state increases, indicating a modest, ligand-dependent gain in structure. Circular dichroism spectroscopy shows the presence of alpha helix and evidence of cold denaturation, and all ligation states induce Sypro Orange fluorescence at ambient temperatures. Although the term “molten globule” is difficult to define precisely, these characteristics are consistent with most such definitions. Active-site titration shows that a majority of molecules retain ~60% of the transition state stabilization free energy observed in modern synthetases. In contrast to the conventional view that enzymes require stable tertiary structures, we conclude that a highly flexible ground-state ensemble can nevertheless bind tightly to the transition state for amino acid activation.
Keywords: Amino acid activation, Thermofluor, HSQC, origin of catalysis, specificity
Graphical abstract
INTRODUCTION
Enzymes accelerate chemical reactions by binding proportionately more tightly to their transition states than to their ground states (1,2). That relative binding affinity, in turn, results from a net excess of favorable enthalpic (ΔH) versus unfavorable entropic (TΔS) contributions to the Gibbs energy of ligand binding to the transition-state substrate configuration. For this reason, enzymes are generally considered to furnish rather rigid binding sites tailored to transition-state, rather than ground-state configurations, and one expects enzymes to be properly folded.
The two structurally dissimilar Class I and Class II aminoacyl-tRNA synthetase (aaRS) superfamilies exhibit structural modularities (3,4) that may represent an archive of their evolutionary history. Like Russian matryoshka dolls, successively smaller and more broadly conserved modules are nested within each Class. We exploited these hierarchies in deconstructing both Classes into successively simpler components and testing the degrees to which successively smaller, more highly conserved modules retain the functions of the modern enzymes (3–7).
Hierarchical deconstruction produced two novel intermediates, Urzymes (5–7) and Protozymes (3). AARS Urzymes contain 120–130 amino acids, and consist of little more than is required to form intact active sites. They retain >60% of the transition-state stabilization free energy for amino acid activation and the ability to aminoacylate tRNA (5). Further, they preserve ~20% of the Gibbs energies necessary to discriminate between competing amino acid substrates and preferentially activate amino acids from within, rather than outside, their own class (4,8,9).
Protozymes are only 46 amino acids long. Although they retain only the ATP binding sites from the Urzymes, aaRS protozymes from both Class I and II aaRS significantly accelerate amino acid activation (3), but have not yet been shown either to acylate tRNA or discriminate significantly between different amino acids. Thus, Urzymes retain all necessary functions of full-length aaRS, albeit to a lesser degree, so the term “Urzyme” (Ur = primitive) is analogous to using “molecule” to define the smallest unit of matter that retains all properties of a chemical substance. Protozymes, on the other hand, are analogous to “atoms”, as they approach the smallest possible catalysts.
As Class I and II aaRS represent entirely distinct protein superfamilies, and because of the appealing notion that evolution produced complexity by stepwise accretions of simpler modules, we believe that the terms Urzyme and Protozyme may apply to the analysis of other enzyme superfamilies. They also emphasize the radical protein surgery and design necessary to make the constructs. For example, the Class I TrpRS (328 residues) Urzyme required removing and re-sealing an internal 74-residue insertion (connecting peptide 1; CP1) from the catalytic domain, as well as the 124-residue anticodon-binding domain (ABD), to yield a 130-residue fragment fusing two, discontinuous portions of the catalytic domain. Removing such masses then made it necessary to introduce 12 mutations (7) by design (10) that restored stability and solubility.
It is important to summarize previously published evidence that the TrpRS Urzyme catalytic activity arises neither from tiny amounts of wild-type enzyme, nor from a separate population of folded and highly active Urzyme molecules not in equilibrium with the general population. We established the authenticity of TrpRS Urzyme catalysis by five criteria (7): (i) MBP and empty vector controls have no activity, (ii) TEV cleavage of the MBP fusion releases cryptic activity, (iii) mutations alter activity, (iv) KM values differ from WT values, and, most importantly, (v) single turnover active-site titration experiments (11,12) show pre-steady-state burst sizes implicating significant fractions of molecules (35–75%) in the rate acceleration. Thus, a major fraction of TrpRS Urzyme molecules contribute to the rate acceleration by transiently forming tight transition-state complexes.
Our parallel work with Class I and II Urzymes (4–9,13,14) and Protozymes (3) produced three lines of evidence that the two aaRS superfamilies evolved from opposite strands of the same gene. (i) Remarkably, and wholly unexpectedly, catalytic activity in both superfamilies arises largely from those modules that can be aligned sense/antisense, showing only second-order dependence on modules that cannot be so aligned (9). (ii) Coding sequences of contemporary aaRS from different Classes retain significant base-pairing complementarity of middle codon bases that increases for ancestral sequences reconstructed from the contemporary multiple sequence alignments (14). (iii) Finally, both sense and antisense peptides from a designed gene coding for Class I and II 46-residue Protozymes accelerate amino acid activation by ATP to the same extent as do the corresponding peptides with wild-type sequences (3). The sense/antisense ancestry of the Class I and II Urzymes underscores their central importance to the origins of catalytic activity in genetically-coded peptides.
Interpretations of Urzyme catalytic activities have, until now, been based on inferences drawn from the crystal structures of intact, modern enzymes. Because their active sites require vacating what in homologs forms the principal hydrophobic core, neither Class I nor Class II Urzymes can form non-polar cores as extensive as those in the full-length enzymes (7). The high rate acceleration by Urzymes —109-fold over the uncatalyzed rate (7)—is therefore surprising in light of the conventional view that catalysis by enzymes requires stable tertiary structures. It is therefore crucial to characterize their structures, in order to better understand mechanistic differences between them and the full-length enzyme (13,15–18).
We report 1H-15N NMR spectra obtained from isotopically-labeled Urzyme from Bacillus stearothermophilus tryptophanyl-tRNA synthetase (TrpRS; (5,7,19)). These data are supported by structural bioinformatic analysis, circular dichroism, and fluorescent dye-binding measurements of thermal melting behavior. These results furnish credible evidence that TrpRS Urzyme is not a properly folded protein, but is best described as a catalytically active molten globule, and is therefore unlikely to be crystallizable. Further, and importantly, the basis for transition-state stabilization by this Urzyme appears to entail a conformational change between two different unfolded ensembles. Thus, the acceleration of amino acid activation by TrpRS Urzyme most likely arises from a qualitatively different thermodynamic mechanism from the 1014-fold acceleration by the related modern, native enzyme.
RESULTS and DISCUSSION
TrpRS Urzyme prepared from inclusion bodies is an active, monomeric catalyst
We previously (5,7,20) expressed TrpRS Urzyme as a maltose-binding (MBP) fusion and cleaved with tobacco etch virus protease (TEV). For isotopically-labeled Urzyme free of signal from the MBP, the sample used in this study was necessarily prepared by a different procedure in which we renatured the Urzyme itself from inclusion bodies. Fig. 1 summarizes the purification by solubilization, initial purification using a His6 tag on Nickel-NTA, and renaturing chromatography on Superdex 75. Size exclusion profiles afford evidence that the TrpRS Urzyme is a monomer with an increased hydrodynamic radius (Fig. 1A); Fig. 1B demonstrates the purification; and Fig. 1C documents catalytic activity.
Figure 1.
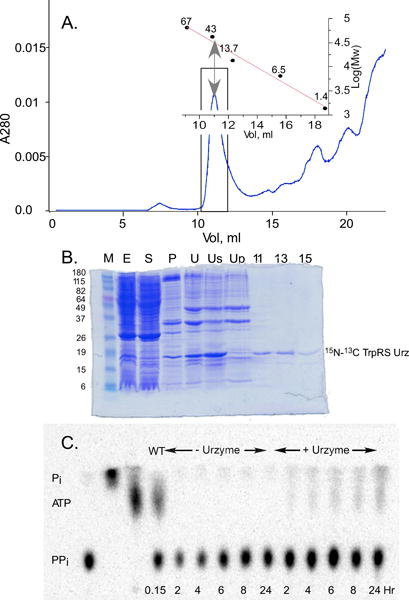
Purification and catalytic activity of TrpRS Urzyme. A. Gel filtration profile of TrpRS Urzyme purification. Boxed area includes a peak whose apparent molecular weight (inset) exceeds that of the TrpRS Urzyme monomeric molecular weight. This peak was collected and shown by PAGE (B) to contain the Urzyme. B PAGE gel analysis of the purification from inclusion bodies. M; Mw standards; E, cell extract; S, soluble fraction; P, pellet fraction; U, detergent-washed inclusion bodies; Us, solubilized inclusion bodies in 6 M urea portion, containing the Urzyme; Up, portion of inclusion bodies insoluble in 6 M urea; 11–15, SEC fractions. C Urzyme-dependent synthesis of 32ATP by the PPi exchange assay. Lane 2 is a 32PPi sample hydrolyzed with pyrophosphatase. Time points with and without Urzyme can be compared with the 32P-ATP produced by full-length enzyme in 5 minutes (WT). The time course on the right gives a rate comparable to that reported previously (35).
TrpRS Urzyme elutes earlier than expected for a folded protein of 16.3 kDa, consistent with a higher radius of gyration. At pH = 6 (Fig. 1A), the retention volume and parameters from linear fits of empirical retention volumes for model proteins in different conformational states (native, molten globule, pre-molten globule, fully denatured, for different molecular weights; (21)) suggest that the TrpRS Urzyme has a radius characteristic of a conventional molten globule. At pH = 7.4 (not shown), the radius is larger, suggesting a “pre-molten globule”.
Our conclusions require that this preparation have a catalytic proficiency comparable to that previously described. Fig. 1C illustrates that the renatured Urzyme catalyzes the appearance in 24 hours of 32P-labeled ATP comparable to that observed with full-length enzyme in 5 minutes under the same conditions. The relative specific activities of Urzyme and full-length TrpRS are in a ratio of 5.9E-5. Urzyme prepared from inclusion bodies is at least as active, if not more active in amino acid activation as reported previously for the TEV-cleaved maltose-binding protein fusion (5).
1H-15N HSQC spectra of apo- and substrate-saturated TrpRS Urzyme
The TrpRS Urzyme exhibits a poorly dispersed 1H-15N HSQC spectrum in the absence of ligands, compared to those observed for stable, well-packed proteins (Fig. 2). Depending on the threshold used, the 1H-15N spectrum of the 130-amino acid polypeptide has only between 40 and 55 resonances from individual residues, all between 7.8 ppm and 8.5 ppm in the 1H dimension.
Figure. 2.
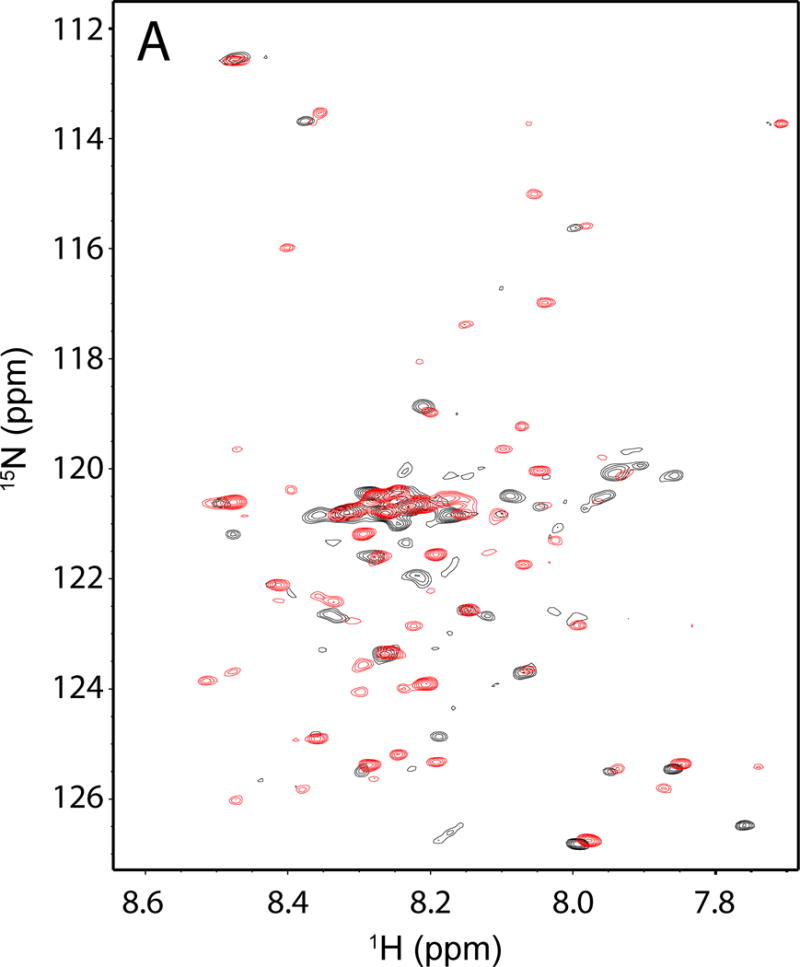
Heteronuclear NMR spectra of TrpRS Urzyme. Overlay of free (black) and ATP•TrpNH bound (red) Urzyme 1H-15N HSQCs. Note the shifts and increase in number of resonances in the bound spectrum, which are consistent, respectively, with ligand binding and modest structural ordering.
The 1H-15N spectrum changes markedly upon addition of ligands (Fig. 2). Addition of saturating ATP and non-reactive tryptophan analog tryptophanamide produces a spectrum in which peak positions shift, additional peaks appear and others disappear without, however, increasing the dispersion in the 1H dimension. The liganded 1H-15N spectrum shows ~80 resolved resonances. These ligand-dependent changes are nevertheless consistent with conformational changes associated with ligand-binding and hence with catalytic activity.
TrpRS Urzyme has substantially reduced hydrophobic stabilization
The effects of rupturing the extensive non-polar core that existed between the Urzyme and the two domain masses removed to generate it greatly reduced the extent to which hydrophobic bonding is available to stabilize its structure. The SNAPP (Simplicial Neighborhood Analysis of Protein Packing; (22,23)) algorithm is an appropriate metric for quantifying this reduction in hydrophobic stabilization free energy. The approach analytically decomposes protein tertiary structures unambiguously into tetrahedra of nearest-neighboring side chains, whose compositions can be scored quantitatively according to their frequencies in the database of protein structures, normalized by frequencies expected under a simple null hypothesis (22). These likelihood ratios span a million-fold range centered around 1.0, so their logarithms are SNAPP log-likelihood potentials. The decomposition therefore produces a comprehensive inventory of tertiary contacts.
Fig. 3A compares graphical representations of nonpolar interactions within native, full-length TrpRS (1MAU) to those from coordinates of the Urzyme alone. The total SNAPP potentials (72.4 for full-length; 12.4 for the Urzyme) furnish a metric for the relative stabilization by hydrophobic forces between the molecules. These values differ by >five-fold. Similarly, the average SNAPP log-likelihoods per residue also differ significantly, the Urzyme having only 40% of that in the full-length enzyme. Although there is no consensus minimum per-residue SNAPP score necessary to support a well-packed protein, the TrpRS Urzyme clearly appears deficient in nonpolar packing interactions that might stabilize its structure, relative to the full-length protein.
Figure. 3.
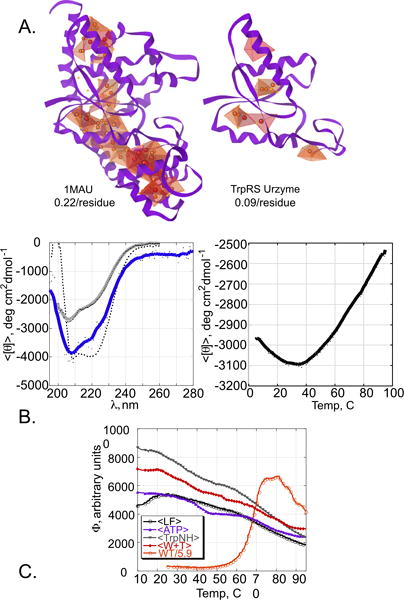
Bioinformatic and Spectroscopic data. A. Simplicial Neighborhood Analysis of Protein Packing (SNAPP) analysis of TrpRS Urzyme and native TrpRS. Tetrahedra indicate four non-polar side chains that form nearest neighbors; sphere density and color indicate the strength of hydrophobic interactions. The darkest red color indicates the highest likelihood potentials. Average SNAPP per residue is indicated for both structures. B. Circular dichroism (CD) studies. Left: spectra at 20 C. The grey line is recorded from a second sample from the same preparation renatured after thermal melting and demonstrates refolding of a significant portion of the sample heated to 95 C. Dotted line is a scaled spectrum of full length TrpRS. Right: Thermal melting profile. The temperature range over comparable to that over which the Sypro Orange fluorescence decreases in Fig. 3C. C. Temperature dependence of Sypro Orange fluorescence in native TrpRS and differently liganded TrpRS Urzymes. Fluorescence of unliganded Urzyme (black open circles) increases with temperature at lower temperatures, consistent with cold denaturation in Fig. 3B, and falls off gradually, as do those for liganded Urzyme (ATP (blue solid triangles), tryptophanamide (green solid triangles) and ATP plus tryptophanamide (red diamonds)). The melting profile of full-length TrpRS (red, open circles) has been scaled to those of the Urzymes as described in the text.
Circular dichroism (CD) analysis demonstrates reversible folding of α-helix
Native TrpRS crystal structures (Fig. 3B) suggest that 61 of 130 residues in TrpRS Urzyme are likely to assume helical structures. To secure evidence concerning the Urzyme’s helical content, we recorded the CD spectrum of a sample prepared exactly as was the 15N-labled Urzyme. At 20 C (Fig. 3B), the local CD minima at both 222 and 208 nm indicate α-helical residues. The molar ellipticity inferred from protein concentration determined by UV absorbance suggests that helical residues in the native protein are either not all helical in the Urzyme, or that they assume helical conformations only transiently. Further, unlike pure alpha helices, the ellipticity at 208 nm is appreciably greater than that at 222 nm, suggesting deviation from canonical α-helices (24,25).
Temperature-dependent CD shows non-cooperative melting and cold-denaturation
The thermal melt on the right in Fig. 3B shows first that the secondary structure disappears gradually at higher temperatures. A broad maximum stability is centered at ~35 C, suggesting that the molar ellipticity at 20 C implies a similar conformation. The linear slope in the melting at higher temperatures also suggests that the Urzyme tertiary structure is not highly cooperative. Most surprising, however, is the evidence for cold denaturation at low temperatures (26), which implies limited tertiary structure at moderate temperatures. Such behavior is unusual above 0 C for helical peptides (27–30).
Apo and liganded TrpRS Urzymes show high Sypro Orange fluorescence
To obtain another metric of Urzyme tertiary structure, we employed a temperature dependent SPYRO-orange dye binding assay originally performed with 8-analinonaphthaline sulfonic acid (ANS) (31). Like ANS, Sypro Orange fluoresces only on binding to exposed hydrophobic regions (32–34). SPYRO orange is excluded from well-packed proteins, but binds to molten globules, so fluorescence by the dye and its temperature dependence furnish a sensitive indicator of tertiary structure and the nature of folding transitions.
Temperature-dependent Sypro Orange fluorescence profiles of TrpRS Urzyme are compared to that from native TrpRS in Fig. 3C. Native TrpRS undergoes cooperative melting at 67 C, whose amplitude is substantially larger than the fluorescence seen with the Urzymes, due to the substantially larger amount of non-polar core packing (see Fig. 3A). The native melting profile determined separately was therefore scaled to those of the differently liganded Urzymes using the ratio of the total SNAPP (22) values obtained from the respective coordinate sets. The Urzymes exhibit substantial Sypro Orange fluorescence comparable to that observed only when full-length TrpRS forms a molten globule.
The Thermofluor profile of unliganded TrpRS Urzyme also suggests loss of tertiary structure at low temperature, consistent with the cold denaturation observed by CD. Addition of ligands appears to stabilize the Urzyme to cold denaturation without reducing the fluorescence at any temperature. On the contrary, addition of the tryptophan analog tryptophanamide almost doubles the fluorescence at 10 C, whereas addition of Mg2+•ATP leaves the fluorescence more or less unchanged and the addition of both ligands is intermediate between the two.
TrpRS Urzyme is not a typical protein domain
Multi-domain enzymes can be truncated and isolated domains preserve their structures, occasionally with reduced stability, and loss of catalytic activities. For that reason, it is worth highlighting the important differences between such constructs and aaRS Urzymes. TrpRS Urzyme is not a catalytic domain, but rather a carefully engineered deconstruction of the TrpRS catalytic domain into its core—conserved throughout the Class I aaRS superfamily—the ABD, and an idiosyncratic co-domain (i.e. connecting peptide 1, CP1) that is without catalytic activity. The foremost role of CP1 appears to be to provide sufficient hydrophobic packing to sustain the amino acid binding site cavity, from which nonpolar core interactions—present in many Rossmann fold proteins—have been removed to create the amino acid binding pocket (7). CP1 is, however, strongly coupled energetically to the ABD, which was also removed to create the Urzyme (20). It is important to characterize the Urzyme structure that results from loss of these two substantial masses and their hydrophobic packing interactions.
Multiple experimental data imply molten globular ensembles
Data shown in Figs. 2–3 all argue that the TrpRS Urzyme does not behavie as a well-folded protein. The poorly dispersed HSQC spectra demonstrate unambiguously that there is a lack of tertiary structure in the Urzyme near its most stable temperature. SNAPP analysis leads us to expect a substantial quantitative loss of stabilization by hydrophobic bonding (Fig. 3A). This expectation is confirmed by the altered CD spectrum and cold denaturation (Fig. 3B). Thermal melting of secondary structure occurs over a 50-degree temperature range (Fig. 3B). Sypro orange fluorescence is observed at all temperatures (Fig. 3C) and neither CD nor dye binding have temperature dependences characteristic of cooperative phenomena. As the Thermoflour profile of unliganded Urzyme also decreases at lower temperatures, our interpretation of the cold denaturation appears is that a limited amount of tertiary structure exists in the Urzyme sample, and that much of that is lost at lower temperatures.
The ligand-dependent HSQC spectra (Fig. 2) consistently reveal a substrate-induced structural change between two different ensembles, neither of which is a folded protein. The appearance of ~30 new resonances in the HSQC spectrum and the consistency of spectroscopic indications furnish strong evidence that the TrpRS Urzyme undergoes a catalytically relevant ordering transition when bound to substrates. Liganded Thermofluor profiles indicate that ATP and the non-reactive tryptophan analog used to stabilize the pre-transition state crystal structure (35) both stabilize the Urzyme against cold denaturation, which can be confirmed by subsequent CD studies.
Molten globules remain a poorly defined structural category characterized by lack of positional stability, increased radius of gyration, and induced fluorescence of dyes like ANS and Sypro Orange that fluoresce only in hydrophobic environments. In the present case, the radius of gyration is less valuable as an indicator, because there is no corresponding “folded” state for comparison. Nevertheless, the present analysis furnishes redundant evidence that the TrpRS Urzyme lacks stable tertiary side chain packing in all ground states accessed by our experiments (unliganded, bound substrates/substrate analogs).
Potential for structural studies
The liganded HSQC spectrum resolves peaks corresponding to 62% of the 130 residues in the Urzyme, suggesting that extensive peak assignment might be possible. Prior to such effort, it is worth recording HSQC spectra at lower pH, to reduce the exchange rate of amide protons, and at higher temperature consistent with the temperature optimum. A titration series of 15N-labeled samples would help to distinguish between ligand-dependent resonances reflecting slow and/or fast exchange. Other approaches to assigning residues are suggested by our recent demonstration of the catalytic activities of the 46-residue TrpRS and HisRS protozymes, which contain the ATP binding sites (3) and are subsets of the corresponding Urzymes. Both 46-mers also catalyze amino acid activation (3). The Class I peptide corresponds to the N-terminal β-α-β crossover connection, a distant homolog of the Walker A sequence in F1-ATPase. Our work (3) reproduced for the Class I synthetases peptide titrations of ATP described by Mildvan for the F1-ATPase (36,37).
Inferences from the native crystal structures are probably useful in a limited sense. Structural characterization of a 50-residue Walker A fragment of F1-ATPase (36,37) produced a small number of NOE distances in water in the presence of ATP characteristic of a structure different from that observed in the crystal structure of the native enzyme (38). That fragment nonetheless bound ATP with an affinity (17 μM) within 15-fold of that of the full-length enzyme (1.2 μM). Mildvan’s multi-dimensional NMR studies enabled them to show that in trifluoroethanol the segment immediately following the glycine-rich region formed an α-helix, assuming a β-α-β conformation as observed in the native, full-length crystal structure (37). Corresponding residues in the TrpRS Urzyme appear to behave similarly, undergoing ligand-induced structure formation. We can use spectra of the 46-mer, perhaps stabilized by additives like trifluoroethanol and/or trimethylamine N-oxide to indentify landmarks in the Urzyme’s spectra.
Transition state stabilization and catalytic activity from molten globules
The present experiments establish that: (i) The TrpRS Urzyme purified and renatured from inclusion bodies exhibits catalytic proficiency comparable to those reported for the maltose-binding protein fusion. (ii) The structural ensembles of native and substrate-bound Urzyme lack extensive tertiary structure. (iii) Substrates and substrate analogs significantly increase the order evident in the 15N-1H HSQC spectrum without achieving proper folding.
Two additional lines of evidence argue that catalysis by full-length TrpRS and its Urzyme occur by substantially different transition-state stabilization mechanisms: (i) We have published quite substantial evidence that full-length enzyme depends on long-range allosteric coupling during catalysis (16–18) and substrate recognition (13,15). That coupling is not possible in the Urzyme because the domains involved are entirely missing. (ii) The active-site mutation D146A reduces activity 200-fold in the full length enzyme but increases activity 25-fold in the Urzyme (20), necessarily implying significant differences in the manner in which the active-site metal assists catalysis, consistent with the allosteric effects outlined in (i). The carboxylate may bind transiently to the active-site Mg2+ ion during formation and/or decomposition of the transition state in full-length TrpRS, but actually shift the metal toward a non-productive configuration in the Urzyme because it cannot profit from dynamic effects of domain movement. Thus, although the Urzyme•transition-state complex may transiently assume a folded configuration, the material difference between the folded ground states of the full-length enzyme and the molten-globular ground states of the Urzyme necessarily imply significant differences in the catalytic trajectories themselves.
The idea that an intrinsically disordered structure can be a potent catalyst, though surprising, is not entirely unexpected. Hilvert (39) constructed a chorismate mutase variant in which the domain swapped dimer was converted by mutation to a monomer in which the swapped mass was bound to the same monomer. The catalytic proficiency of that variant was unchanged. Physically, however, the monomeric form was a molten globule in the absence of ligands.
Hu’s computational studies (40) confirmed an important inference about transition-state stabilization by the two forms of chorismate mutase: the same rate acceleration—and transition state affinity—can be achieved by very different entropic and enthalpic contributions. The molten globular form must pay a higher cost in reduced entropy, but its flexibility endows it with the ability to form tighter bonds in the transition state. The TrpRS Urzyme now confirms that high rate accelerations can be achieved by peptides that are molten globular ensembles in the absence of ligands. That realization broadly increases the manifold of polypeptides capable of natural selection for their catalytic properties, suggesting that catalysis by molten globular peptides may have been an important stage in the evolution of modern enzymes.
Finally, we should also distinguish the structural phenomena reported here from that of a “molten globular” form of other aaRS (GlnRS/GluRS; (41)). The enzymes in that work are essentially full-length, multi-domain proteins and are folded in the usual buffers. A loop involved in long-range communication between domains actually stabilizes a molten globular state at low urea concentrations. The authors propose that such an event relaxed the amino acid specificity of the ancestral GluRS and may have promoted the evolutionary generation of GlnRS.
MATERIALS AND METHODS
Purification and characterization of TrpRS Urzyme
TrpRS Urzyme itself was cloned into pET 42 vector with leading FLAG and trailing His6 tags. TrpRS Urzyme localizes in inclusion bodies when expressed in Escherichia coli. Preliminary expression experiments revealed that Bl21 cells produced only small amounts of Urzyme when grown on M9 medium, whereas HSM174 cells produced about 5-fold more. Cells (HSM174) bearing the expression plasmid were grown in M9 medium supplemented with 18 mM 15N sodium chloride and induced by addition of 0.4 mM isopropyl-βD-galactopyranoside and incubated five hours at 37 C.
Cells were harvested by centrifugation at 5,000 × g at 4 °C for 20 minutes, resuspended in 5 ml per gram wet cells of 100 mM Tris hydrocholoride, 300 mM sodium chloride, 10 mM imidazole, 5 mM ethylaminediaminetetraacetate, 5 mM diethiothreitol pH 8 and lysed by vortexing and sonication. The suspension was centrifuged at 14,000 rpm at 4 °C for 30 min. Pellets were suspended using a tissue homogenizer in the same buffer supplemented with 2 M urea and 1% Triton X-100 and washed twice with 4–6 ml of the same buffer per gram of wet cells to wash out membranous components and sparingly soluble proteins from the inclusion bodies.
Inclusion bodies purified in this way contain ~10% Urzyme (Fig. 1B), with minimal contamination by proteins of similar molecular weight. The resulting pellet was again homogenized at room temperature with a tissue homogenizer in 1.0 ml/gram of wet cells of 100 mM sodium phosphate, 6 M guanidinium· hydrochloride, pH 7.2. Solubilized proteins were collected on Nickel-NTA sepharose, eluted in a small volume with imidazole, and added to a Superdex 75 (GE Healthcare Life Sciences) gel filtration column equilibrated with 50 mM sodium phosphate buffer, pH = 7.0 plus 150 mM sodium chloride and eluted in this buffer, which removed the denaturant.
Polyacrylamide gel electrophoresis of the peak (Fig. 1A) showed that this material had a molecular weight of ~14 Kd and was free of contaminating proteins. Fractions were concentrated using an Amicon Ultra Centrifugal Filter. Activity was confirmed using a modification of the 32PPi exchange assay (15) in which 1 μl of reaction mix was spotted on cellulose-polyethyleneimine thin-layer chromatography plates and eluted with 0.75 M potassium phosphate buffer, pH = 3.5, with 4 M urea (Fig. 1B). Peaks eluted from the sizing column at different pH values were analyzed using the equations presented by Uversky (21).
NMR experiments
1H-15N HSQC spectra were acquired at 25° C and pH = 7.4 on a 600 MHz Bruker Avance III spectrometer equipped with a TCI cryogenic probehead. Datasets were recorded with (128, 1024) complex points, acquisition times of (70 ms, 85 ms) in (t1, t2), 8 scans per complex t1 point, and a recycle delay of 1 s. Conditions for the two samples were: 1) 100 μM free Urzyme. 2) 83 μM Urzyme + 1.8 mM Mg2+•ATP + 24 mM tryptophanamide. The buffer for both samples was comprised of 50 mM NaH2PO4 and 150 mM NaCl.
Simplicial Neighborhood Analysis of Protein Packing (SNAPP) analysis (22)
Delaunay tessellation analysis was performed using software provided by Stephen Cammer. SNAPP likelihood scores for tetrahedra of nearest neighbors of a given composition represent the difference between logarithms of the observed frequencies for such tetrahedra in a select subset of well-determined structures in the Protein Databank and frequencies expected under a simple null hypothesis. As only ~25% of such tetrahedra have log-likelihood gains >0.3, and as those tetrahedra contain almost exclusively hydrophobic side chains (Ala, Cys, Phe, Ile, Leu, Met, Val, Tyr, Trp), the SNAPP likelihood score is an appropriate metric for hydrophobic interactions present in a protein structure.
Temperature-dependent circular dichroism
The circular dichroism spectrum of TrpRS Urzyme was recorded from 30 μl of a 60 μM sample in a 0.1 mm path length cell using a Jasco J-815 CD spectrometer. The buffer, 50 mM potassium phosphate, pH = 7.2, 150 mM sodium chloride was identical to that used to record NMR spectra. A 180 ml sample of the same solution was used for thermal melting analysis in a 1 mm path length cell. That sample was returned to 20 C after the thermal melt and the CD spectrum from 195 nm to 260 nm was recorded again.
Thermofluor analysis (34)
Samples (30 μl) of 10 μM Urzyme were distributed using a Biomek 3000 liquid handling system onto a 384-well plate containing 6:5000 dilutions of Sypro Orange dye in 20 mM HEPES buffer, pH = 7.0 with 50 mM sodium chloride. Four groups of six wells contained unliganded Urzyme and Urzyme plus saturating concentrations of Mg2+•ATP (5 mM), trypophanamide (10 mM), and ATP plus tryptophanamide. Temperature-dependent fluorescence measurements were made using a ABI 7900 HT Real Time PCR instrument from 10 C through 95 C at intervals of 0.3 degree. The excitation and emission spectra for Sypro Orange and the bandpass filters used for Thermofluor analysis exclude stimulation of indole fluorescence of the tryptophanamide ligand. A similar melting profile for full-length native TrpRS was divided by a factor of 5.9 as described in RESULTS to scale it to these melting profiles.
Acknowledgments
We are grateful to B. Kuhlman for pointing out the importance of cold denaturation, to A. Doig for discussion of the unusual CD spectrum, to D. Herschlag and S. Benkovic for useful discussion of the problems associated with transition state binding by a molten globule, and to L. Martinez for demonstrating that renaturation was a viable purification.
This work was supported, in whole or in part, by the National Institutes of Health grant GM078227 through the NIGMS (to C. W. C. Jr.) and GM 083059 through the NIGMS (to A.L.).
Abbreviations
- TrpRS
tryptophanyl-tRNA synthetase
- MBP
maltose-binding protein
- CD
circular dichroism
- HSQC
heteronuclear single quantum coherence
- TLC
thin-layer chromatography
- CP1
connecting peptide 1
Footnotes
Author Contributions: CWC Jr and AL designed the NMR experiment, LL prepared the isotopically labeled TrpRS Urzyme and purified the monomeric form by gel filtration, TW performed the enzymatic assay and Thermofluor analysis, CWC Jr performed the CD spectroscopy, CWC Jr, AL, and PS wrote the manuscript, all authors approved the manuscript.
References
- 1.Wolfenden R. Benchmark Reaction Rates, the Stability of Biological Molecules in Water, and the Evolution of Catalytic Power in Enzymes. Ann Rev Biochem. 2011;80:645–667. doi: 10.1146/annurev-biochem-060409-093051. [DOI] [PubMed] [Google Scholar]
- 2.Wolfenden R, Snider MJ. The Depth of Chemical Time and the Power of Enzymes as Catalysts. Acc Chem Res. 2001;34:938–945. doi: 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- 3.Martinez L, Jimenez-Rodriguez M, Gonzalez-Rivera K, Williams T, Li L, Weinreb V, Niranj Chandrasekaran S, Collier M, Ambroggio X, Kuhlman B, Erdogan O, Carter CWJ. Functional Class I and II Amino Acid Activating Enzymes Can Be Coded by Opposite Strands of the Same Gene. J Biol Chem. 2015;290:19710–19725. doi: 10.1074/jbc.M115.642876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carter CWJ. Urzymology: Experimental Access to a Key Transition in the Appearance of Enzymes. J Biol Chem. 2014;289:30213–30220. doi: 10.1074/jbc.R114.567495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li L, Francklyn C, Carter CW., Jr Aminoacylating Urzymes Challenge the RNA World Hypothesis. J Biol Chem. 2013;288:26856–26863. doi: 10.1074/jbc.M113.496125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li L, Weinreb V, Francklyn C, Carter CW., Jr Histidyl-tRNA Synthetase Urzymes: Class I and II Aminoacyl-tRNA Synthetase Urzymes have Comparable Catalytic Activities for Cognate Amino Acid Activation. J Biol Chem. 2011;286:10387–10395. doi: 10.1074/jbc.M110.198929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pham Y, Kuhlman B, Butterfoss GL, Hu H, Weinreb V, Carter CW., Jr Tryptophanyl-tRNA synthetase Urzyme: a model to recapitulate molecular evolution and investigate intramolecular complementation. J Biol Chem. 2010;285:38590–38601. doi: 10.1074/jbc.M110.136911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carter CW., Jr What RNA World? Why a Peptide/RNA Partnership Merits Renewed Experimental Attention. Life. 2015;5:294–320. doi: 10.3390/life5010294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carter CWJ, Li L, Weinreb V, Collier M, Gonzales-Rivera K, Jimenez-Rodriguez M, Erdogan O, Chandrasekharan SN. The Rodin-Ohno Hypothesis That Two Enzyme Superfamilies Descended from One Ancestral Gene: An Unlikely Scenario for the Origins of Translation That Will Not Be Dismissed. Biology Direct. 2014;9:11. doi: 10.1186/1745-6150-9-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dantas G, Kuhlman B, Callender D, Wong M, Baker D. A Large Scale Test of Computational Protein Design: Folding and Stability of Nine Completely Redesigned Globular Proteins. J Mol Biol. 2003;332:449–460. doi: 10.1016/s0022-2836(03)00888-x. [DOI] [PubMed] [Google Scholar]
- 11.Fersht AR, Ashford JS, Bruton CJ, Jakes R, Koch GLE, Hartley BS. Active Site Titration and Aminoacyl Adenylate Binding Stoichiometry of Amionacyl-tRNA Synthetases. Biochem. 1975;14:1–4. doi: 10.1021/bi00672a001. [DOI] [PubMed] [Google Scholar]
- 12.Francklyn CS, First EA, Perona JJ, Hou YM. Methods for kinetic and thermodynamic analysis of aminoacyl-tRNA synthetases. Methods. 2008;44:100–118. doi: 10.1016/j.ymeth.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinreb V, Li L, Chandrasekaran SN, Koehl P, Delarue M, Carter CW., Jr Enhanced Amino Acid Selection in Fully-Evolved Tryptophanyl-tRNA Synthetase, Relative to its Urzyme, Requires Domain Movement Sensed by the D1 Switch, a Remote, Dynamic Packing Motif. J Biol Chem. 2014;289:4367–4376. doi: 10.1074/jbc.M113.538660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandrasekaran SN, Yardimci G, Erdogan O, Roach JM, Carter CW., Jr Statistical Evaluation of the Rodin-Ohno Hypothesis: Sense/Antisense Coding of Ancestral Class I and II Aminoacyl-tRNA Synthetases. Mol Biol Evol. 2013;30:1588–1604. doi: 10.1093/molbev/mst070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li L, Carter CW., Jr Full Implementation of the Genetic Code by Tryptophanyl-tRNA Synthetase Requires Intermodular Coupling. J Biol Chem. 2013;288:34736–34745. doi: 10.1074/jbc.M113.510958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weinreb V, Li L, Carter CW., Jr A Master Switch Couples Mg2+-Assisted Catalysis to Domain Motion in B. stearothermophilus Tryptophanyl-tRNA Synthetase. Structure. 2012;20:128–138. doi: 10.1016/j.str.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinreb V, Li L, Kaguni LS, Campbell CL, Carter CW., Jr Mg2+-Assisted Catalysis by B. stearothermophilus TrpRS is Promoted by Allosteric Effects. Structure. 2009;17:952–964. doi: 10.1016/j.str.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinreb V, Carter CW., Jr Mg2+-free B. stearothermophilus Tryptophanyl-tRNA Synthetase Activates Tryptophan With a Major Fraction of the Overall Rate Enhancement. J Am Chem Soc. 2008;130:1488–1494. doi: 10.1021/ja076557x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pham Y, Li L, Kim A, Erdogan O, Weinreb V, Butterfoss G, Kuhlman B, Carter CW., Jr A Minimal TrpRS Catalytic Domain Supports Sense/Antisense Ancestry of Class I and II Aminoacyl-tRNA Synthetases. Mol Cell. 2007;25:851–862. doi: 10.1016/j.molcel.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 20.Pham Y, Li L, Kim A, Weinreb V, Butterfoss G, Kuhlman B, Carter CW., Jr A Minimal TrpRS Catalytic Domain Supports Sense/Antisense Ancestry of Class I and II Aminoacyl-tRNA Synthetases. Mol Cell. 2007;25:851–862. doi: 10.1016/j.molcel.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 21.Uversky VN. Size-Exclusion Chromatography in Structural Analysis of Intrinsically Disordered Proteins. In: Uversky VN, Dunker AK, editors. Intrinsically Disordered Protein Analysis Volume 2, Methods and Experimental Tools. Humana Press; New York: 2012. pp. 179–193. [DOI] [PubMed] [Google Scholar]
- 22.Tropsha A, Carter CWJ, Cammer S, Vaisman II. Simplicial Neighborhood Analysis of Protein Packing (SNAPP): A Computational Geometry Approach to Studying Proteins. Methods in Enzymology. 2003;374:509–544. doi: 10.1016/S0076-6879(03)74022-1. [DOI] [PubMed] [Google Scholar]
- 23.Carter CW, Jr, LeFebvre B, Cammer SA, Tropsha A, Edgell MH. Four-body potentials reveal protein-specific correlations to stability changes caused by hydrophobic core mutations. Journal of Molecular Biology. 2001;311:625–638. doi: 10.1006/jmbi.2001.4906. [DOI] [PubMed] [Google Scholar]
- 24.Formaggio F, Crisma M, Rossi P, Scrimin P, Kaptein B, Broxterman QB, Kamphuis J, Toniolo C. The First Water-Soluble 310-Helical Peptides. Chemistry A European Journal. 2000;6:4498–4504. doi: 10.1002/1521-3765(20001215)6:24<4498::aid-chem4498>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 25.Micsonaia A, Wienb F, Kernyaa L, Leec YH, Gotoc Y, Réfrégiersb M, Kardos J. Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc Nat Acad Sci USA. 2015 Jun 2;:E3095–E3103. doi: 10.1073/pnas.1500851112. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishii I, Kataoka M, Tokunaga F, Goto Y. Cold Denaturation of the Molten Globule States of Apomyoglobin and a Profile for Protein Folding. Biochem. 1994;33:4903–4909. doi: 10.1021/bi00182a019. [DOI] [PubMed] [Google Scholar]
- 27.Thompson KS, Vinson CR, Freire E. Thermodynamic Characterization of the Structural Stability of the Coiled-Coil Region of the bZIP Transcription Factor GCN4. Biochem. 1993;32:5491–5496. doi: 10.1021/bi00072a001. [DOI] [PubMed] [Google Scholar]
- 28.Kitakuni E, Kuroda Y, Oobatake M, Tanaka T, Nakamura H. Thermodynamic characterization of an artificially designed amphiphilic a-helical peptide containing periodic prolines: Observations of high thermal stability and cold denaturation. Prot Sci. 1994;3:831–837. doi: 10.1002/pro.5560030512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Richardson JM, Lopez MM, Makhatadze GI. Enthalpy of helix-coil transition: missing link in rationalizing the thermodynamics of helix-forming propensities of the amino acid residues. Proc Nat Acad Sci USA. 2005;102:1413–1418. doi: 10.1073/pnas.0408004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richardson JM, Makhatadze G. Temperature dependence of the thermodynamics of helix-coil transition. J Mol Biol. 2004;335:1029–1037. doi: 10.1016/j.jmb.2003.11.027. [DOI] [PubMed] [Google Scholar]
- 31.Màlnàsi-Csizmadia A, Hegyi G, Tölgyesi F, Szent-Györgyi AG, Nyitray L. Fluorescence measurements detect changes in scallop myosin regulatory domain. Eur J Biochem. 1999;261:452–458. doi: 10.1046/j.1432-1327.1999.00290.x. [DOI] [PubMed] [Google Scholar]
- 32.Weber PC, Wendoloski JJ, Pantoliano MW, Salemme FR. Crystallographic and Thermodynamic Comparison of Natural and Synthetic Ligands Bound to Streptavidin. J Am Chem Soc. 1992;114:3197–3200. [Google Scholar]
- 33.Lavinder JJ, Hari SB, Sullivan BJ, Magliery TJ. High-Throughput Thermal Scanning: A General, Rapid Dye-Binding Thermal Shift Screen for Protein Engineering. J Am Chem Soc. 2009;131:3794–3795. doi: 10.1021/ja8049063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matulis D, Kranz JK, Salemme FR, Todd MJ. Thermodynamic Stability of Carbonic Anhydrase: Measurements of Binding Affinity and Stoichiometry Using Thermofluor. Biochem. 2005;44:5258–5266. doi: 10.1021/bi048135v. [DOI] [PubMed] [Google Scholar]
- 35.Retailleau P, Huang X, Yin Y, Hu M, Weinreb V, Vachette P, Vonrhein C, Bricogne G, Roversi P, Ilyin V, Carter CW., Jr Interconversion of ATP binding and conformational free energies by Tryptophanyl-tRNA Synthetase: structures of ATP bound to open and closed, pre-transition conformations. Journal of Molecular Biology. 2003;325:39–63. doi: 10.1016/s0022-2836(02)01156-7. [DOI] [PubMed] [Google Scholar]
- 36.Chuang WJ, Abeygunawardana C, Pedersen PL, Mildvan AS. Two-Dimensional NMR, Circular Dichroism, and Fluorescence Studies of PP-50, a Synthetic ATP-Binding Peptide from the b-Subunit of Mitochondrial ATP Synthase. Biochem. 1992;31:7915–7921. doi: 10.1021/bi00149a024. [DOI] [PubMed] [Google Scholar]
- 37.Chuang WJ, Abeygunawardana C, Gittis AG, Pedersen PL, Mildvan AS. Solution Structure and Function in Trifluoroethanol of PP-50, an ATP-Binding Peptide from F1ATPase. Arch Biochem Biophys. 1992;319:110–122. doi: 10.1006/abbi.1995.1272. [DOI] [PubMed] [Google Scholar]
- 38.Abrahams JP, Leslie AGW, Lutter R, Walker JE. Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature. 1994;370:621–628. doi: 10.1038/370621a0. [DOI] [PubMed] [Google Scholar]
- 39.Pervushin K, Vamvaca K, Vogeli B, Hilvert D. Structure and dynamics of a molten globular enzyme. Nat Struct Mol Biol. 2007;14:1202–1206. doi: 10.1038/nsmb1325. [DOI] [PubMed] [Google Scholar]
- 40.Hu H. Wild-type and molten globular chorismate mutase achieve comparable catalytic rates using very different enthalpy/entropy compensations. Science China. 2014;57:156–164. [Google Scholar]
- 41.Saha R, Dasgupta S, Banerjee R, Mitra-Bhattacharyya A, Söll D, Basu G, Roy S. A Functional Loop Spanning Distant Domains of Glutaminyl-tRNA Synthetase Also Stabilizes a Molten Globule State. Biochem. 2012;51:4429–4437. doi: 10.1021/bi300221t. [DOI] [PubMed] [Google Scholar]