

Abstract
Methods for rapid and reliable microbial identification are essential in modern healthcare. The ability to detect and correctly identify pathogenic species and their resistance phenotype is necessary for accurate diagnosis and efficient treatment of infectious diseases. Bottom-up tandem mass spectrometry (MS) proteomics enables rapid characterization of large parts of the expressed genes of microorganisms. However, the generated data are highly fragmented, making downstream analyses complex. Here we present TCUP, a new computational method for typing and characterizing bacteria using proteomics data from bottom-up tandem MS. TCUP compares the generated protein sequence data to reference databases and automatically finds peptides suitable for characterization of taxonomic composition and identification of expressed antimicrobial resistance genes. TCUP was evaluated using several clinically relevant bacterial species (Escherichia coli, Pseudomonas aeruginosa, Staphylococcus aureus, Streptococcus pneumoniae, Moraxella catarrhalis, and Haemophilus influenzae), using both simulated data generated by in silico peptide digestion and experimental proteomics data generated by liquid chromatography-tandem mass spectrometry (MS/MS). The results showed that TCUP performs correct peptide classifications at rates between 90.3 and 98.5% at the species level. The method was also able to estimate the relative abundances of individual species in mixed cultures. Furthermore, TCUP could identify expressed β-lactamases in an extended spectrum β-lactamase-producing (ESBL) E. coli strain, even when the strain was cultivated in the absence of antibiotics. Finally, TCUP is computationally efficient, easy to integrate in existing bioinformatics workflows, and freely available under an open source license for both Windows and Linux environments.
Accurate and rapid typing and characterization of infectious bacteria are of great importance in modern healthcare and essential for correct diagnoses and effective treatments of patients. Particularly, with the rapid circulation of virulent strains of bacteria expressing multiresistance to antibiotics, timely and effective detection and identification are increasingly paramount for responding to infectious diseases. An extensive variety of methods to identify the bacterial content in clinical samples has been developed (1, 2). These methods range from traditional cultivation-based methods, profiling of resulting isolates into phenotypes, to more recently developed molecularly based techniques, including polymerase chain reaction (PCR) assays and DNA sequencing for detection of biomarker genes and for classification of genotypes and genetic lineages. Despite being well established, many of the existing methods for microbial characterization have significant drawbacks: cultivation-based methods are labor-intensive and inherently slow (2), and DNA-based methods, i.e. PCR-profiling and gene sequencing, are typically limited to applications targeting known features of bacteria. The last decade has seen rapid developments in next-generation sequencing (NGS) 1 technologies, which have enabled routine screening of bacteria by whole-genome sequence (WGS) determinations at decreasing costs (3). Microbial genome sequencing offers comprehensive analyses of pathogens with high throughput and is becoming an important tool for real-time diagnostics of bacterial infections (4). However, WGS does not provide any information about gene expression and is thus limited to the information present in the genotype. In contrast, the recent developments and improvements in the performance of tandem MS instrumentation enables characterization of significant parts of the expressed proteomes of bacteria. In relation to MALDI-TOF, which has recently been established as a versatile molecular diagnostics tool for infectious diseases (5, 6), MS/MS-based techniques provide information of the gene expression at the amino acid level of resolution. This enables fast and sensitive detection of both taxonomic delineators and expressed genotypic features, such as proteins associated with antibiotic resistance and virulence (7–9); thus, tandem MS has the potential to further improve the diagnostic analysis of bacterial infections.
Bottom-up (i.e. proteolytically generated peptide sequence-based) tandem MS identifies the sequences of peptides from digested proteins by measuring the mass-to-charge ratios of the molecular ions, followed by fragmentation of the peptides and matching of the fragment spectra to a database (10). However, the generated data are highly fragmented and noisy, which makes the downstream data analysis challenging, requiring dedicated methods for proper interpretation. Dworzanski et al. (11) developed a method, based on the matching of peptides against a reference database, that detects and identifies bacterial species (11–13). This method was later further developed into BACid (14, 15), which was shown to have correct peptide classification rates up to 89% (14, 15). Another approach, developed by Tracz et al. (16), instead uses the total number of matching spectra to identify the species present in a sample. This approach can be used also to estimate the relative abundance of species in mixed cultures. Furthermore, Pipasic (17) was developed specifically for analyzing proteomes from microbial communities. Pipasic uses peptide similarity estimation and expression level weighting to estimate the relative abundances of species in a sample. However, these existing methods are either 1) limited to pure cultures and not directly applicable to mixed populations that commonly occur in clinical applications, 2) require manual analysis steps, which are difficult to automate in computational workflows, or 3) are computationally inefficient and not applicable to the rapidly growing number of reference proteomes. Furthermore, none of these methods have the ability to combine the determination of taxonomic composition with the characterization of expressed antibiotic resistance markers or other genes of interest.
To address the lack of a suitable bioinformatics methodology, we developed TCUP, a new computational method for determining bacterial taxonomic compositions and detecting expressed antibiotic resistance proteins using bottom-up tandem MS data. TCUP operates by comparing the generated peptides to comprehensive reference databases, automatically identifying peptides that can be used for accurate discrimination between organisms and their expressed antimicrobial resistance genes. Evaluation of the method on simulated and experimental data from multiple clinically relevant bacteria shows highly correct classification rates for all tested microorganisms. The method is also computationally efficient making it well suited for integration into existing bioinformatics proteomics workflows. TCUP is freely available and open-source and runs on both Windows and Linux systems.
Materials and Methods
Implementation
The input to TCUP is a set of peptides predicted from spectra generated by bottom-up tandem MS specified as a file in FASTA format. TCUP is general and can be used with peptide data from any spectral matching software, including de novo methods (e.g. SEQUEST (18), X!Tandem (19, 20), TIDE (21), Mascot (22), PEAKS (23), PepNovo (24), and Lutefisk (25)). The output from TCUP is in Excel format and includes the following: 1) the relative abundances of all organisms identified in a sample at and below a user-specified taxonomic level; 2) specific genes in the reference genomes that are matched by peptides in the analysis; and 3) the relative abundances of identified antimicrobial resistance genes. TCUP is implemented in Python 3.5, and the code and usage documentation are freely available under the ISC license from the project's repository (https://bitbucket.org/chalmersmathbioinformatics/tcup).
The taxonomic composition of a sample is estimated as follows. The tandem MS-determined peptides are first aligned to a comprehensive high-quality reference database containing bacterial genome sequences. The comparison is done by aligning the peptides to the complete genomes translated into all six reading frames. Alignment of peptides to reference genome sequences is done using BLAT (26) in protein-to-DNA mode (command line arguments: “-out = blast8 -t = dnax -q = prot -tileSize = 5 -stepSize = 5 -minScore = 10 -minIdentity = 90”). Because BLAT is not available for the Windows platform, BLAST is used when running on Windows (running tblastn with command line arguments: “outfmt 6”). The reference database was assembled from NCBI RefSeq (27, 28) bacterial genomes (2785 genomes; downloaded Nov. 17, 2015). To remove sequences that move horizontally between organisms (i.e. mobile genetic elements), potentially appearing in multiple distantly related genomes, sequences shorter than 400,000 nucleotides or sequences annotated with any of the keywords “plasmid,” “phage,” “extrachromosomal,” “incision element,” or “transposon” were excluded. Sequences belonging to Shigella species were also excluded due to their similarity and systematic uncertainty to Escherichia coli (29). See supplemental file 1, reference genome database, for a complete listing of the sequences included in the database.
After alignment to the translated reference genome sequences, each peptide is matched to zero, one, or multiple reference genomes. To remove matches that are too dissimilar and unlikely to contain any relevant information about the taxonomic affiliation, two filtering steps were applied. The first step requires matches to have an identity of at least 90% and a coverage of 100% (only complete peptide matches are considered). Also, peptides shorter than six amino acids are removed. In the second filtering step, all matches with sequence identity of at least 5% below the best match for that peptide are discarded.
After filtering, the remaining peptides are assigned to nodes in a taxonomic tree, using the lowest common ancestor algorithm (30). The taxonomic affiliation of a sample is then assigned based on the set of discriminative peptides, i.e. the peptides with an lowest common ancestor at a node that is at or below the user-specified taxonomic level. The taxonomic tree used in TCUP is based on the full NCBI Taxonomy (31) (taxdump downloaded Nov. 17, 2015), in which each reference genome is associated with a unique node. Our implementation extends the SQLite3 database used in the ETE3 package (32) with a table of taxonomic affiliations for all reference genome sequences included in the reference database. TCUP also has support for “blacklisting” reference sequences, disregarding them in the analysis without having to remove them from the database.
The number of discriminative peptides varies between organisms depending on the evolutionary distance to their closest relative. To provide more accurate estimates for samples containing multiple species, TCUP can adjust and normalize the results by dividing the estimated relative abundances with factors reflecting the expected proportion of discriminative fragments for the specific organisms and taxonomic level. These correction factors can be estimated in silico or from single species culture, and are provided to TCUP through a user-specified tab-separated text file. For the analyses in this paper, the normalization factors were computed at the species level in samples from pure cultures.
The antibiotic resistance gene content in a sample is estimated similarly to how the taxonomic composition is determined. In the first step, peptides are aligned to a database of reference antibiotic resistance gene sequences using BLAT or BLAST, depending on the operating system. BLAT is run with the following command line parameters: “-q = prot -t = prot -minIdentity = 90 -out = blast8.” BLAST is run with the following command line parameters: “-outfmt 6.” TCUP uses the freely available ResFinder database (33) as a source of antibiotic resistance gene sequences, which contains 2129 proteins in total (downloaded Jan. 3, 2016). The database was manually extended by adding antibiotic family assignments to all the included genes and then assembled into an SQLite3 database. Matches for each peptide are subject to the same two-step filtering method as described previously for taxonomic composition estimation. The first step required matches to have an identity threshold of at least 90% and a coverage of 100% (only complete peptide matches are considered). Also, peptides shorter than six amino acids are removed. The second filtering step discards all matches with lower percentage identities than the best match. For antibiotic resistance genes, discriminative peptides are defined as those peptides that match only a single antibiotic resistance gene family.
Generation of in Silico Digested Peptides
The performance of the computational method for prototyping was first evaluated on peptides generated by in silico digestion. The reference proteomes of four species of bacteria were downloaded from UniProt (34) as follows: E. coli (O138:H28/CCUG 49263); Pseudomonas aeruginosa (ATCC15692/CCUG 29297); Staphylococcus aureus (6850/CCUG 41582); and Streptococcus pneumoniae (R6). The proteomes were digested in silico using the EMBOSS tool digest (35), simulating trypsin cleavage, using command line arguments “-menu 1 -mono N -rformat2 srs.” From each digested proteome, six samples consisting of 10,000 randomly selected peptides were created. To mimic the sizes of peptide sequences observed in experimental MS data, only peptides from 6 to 45 amino acids long were considered. These peptide length thresholds were based on empirical observations of the typical lengths of peptides in a large number of datasets derived from bottom-up tandem MS. To estimate the robustness of the computational method under random sequencing errors, amino acid positions in peptides were substituted at six different rates (1–3, 5, and 10%) according to the probabilities specified by the PAM30 matrix supplied with NCBI BLAST (36). The true positive rate (TPR) was calculated as the ratio of correctly assigned discriminative peptides to the total number of discriminative peptides.
The effects of missing or incorrect genomes (i.e. either inaccuracies in genome sequence or incorrectly placed in the taxonomy) in the reference database on the computational method were evaluated by a leave-one-out simulation study, using the 62 E. coli genomes present in the NCBI RefSeq bacterial database (downloaded Oct. 2, 2015) (28, 37). One genome at a time was selected, and the corresponding proteome was digested in silico and randomly sampled as described above. The reduction in TPR was estimated based on the difference between 1) the TPR with the selected genome included in the reference database, and 2) the TPR with the selected genome excluded in the reference database.
Cultivation of Bacteria
The performance of the computational method was further evaluated using experimental shotgun LC-MS/MS proteomics data in different experiments. These included four single-species experiments with two Gram-negative and two Gram-positive bacteria: E. coli (K12/CCUG 49263); P. aeruginosa (PA01/CCUG 29297); S. aureus (NCTC_8325/CCUG 41582); and S. pneumoniae (CCUG 28588T). First, a mixture of the same four species were combined at ratios of 1:1:1:1, 4:2:2:1, and 1:2:2:4, respectively. Another experiment, simulating a respiratory tract sample with multiple species in a co-infection situation, consisted of mixtures of S. pneumoniae (CCUG 25588T), Haemophilus influenzae (CCUG 23945T), Moraxella catarrhalis (CCUG 353T), and S. aureus (NCTC_8325/CCUG 41582) at ratios of 1:1:1:1, 4:2:2:1, and 1:2:2:4, respectively. Finally, an experiment with an ESBL-positive E. coli (CCUG 62462) strain with and without antibiotic pressure was also evaluated. All experiments were replicated three times.
Bacterial strains were grown on Blood Agar medium. S. pneumoniae and M. catarrhalis were grown at 36 °C with 5% CO2 overnight, S. aureus at 37 °C overnight, and P. aeruginosa and E. coli at 30 °C overnight. H. influenzae was grown on chocolate agar medium at the same conditions as S. pneumoniae and M. catarrhalis. Bacterial biomass was collected and resuspended in phosphate-buffered saline (PBS). Bacterial densities were measured at A600 (A600 0.8  1 × 109 bacteria). For each experiment, the same amounts of bacterial biomass were established, by adjusting the A to 1.0 in 1.0 ml of PBS. The bacterial biomass was washed with PBS three times by centrifuging the sample for 5 min at 12,000 × g, discarding the supernatant, and resuspending the pellet in 1.0 ml of PBS. The bacteria were finally resuspended in 150 μl of PBS. The bacterial cell suspensions were transferred to 200-μl vials containing glass beads (Sigma-Aldrich, G1145). The bacterial cells were lysed by bead-beating, using a TissueLyser (Qiagen, 85220), with the following settings: frequency 1/25 s and 5 min. The bacterial lysates were frozen at −20 °C until analysis.
1 × 109 bacteria). For each experiment, the same amounts of bacterial biomass were established, by adjusting the A to 1.0 in 1.0 ml of PBS. The bacterial biomass was washed with PBS three times by centrifuging the sample for 5 min at 12,000 × g, discarding the supernatant, and resuspending the pellet in 1.0 ml of PBS. The bacteria were finally resuspended in 150 μl of PBS. The bacterial cell suspensions were transferred to 200-μl vials containing glass beads (Sigma-Aldrich, G1145). The bacterial cells were lysed by bead-beating, using a TissueLyser (Qiagen, 85220), with the following settings: frequency 1/25 s and 5 min. The bacterial lysates were frozen at −20 °C until analysis.
The ESBL-positive E. coli strain (CCUG 62462) encodes the β-lactamase CTX-M-15 and was isolated from a urine sample of an infected 2-year-old boy during an outbreak (previously described by Karami et al. (38) and Johnning et al. (56)). This strain together with the E. coli K12/CCUG 49263 strain (control) were cultured on Blood Agar medium, repeated twice from −70 °C freezer strain stocks, to obtain fresh bacterial cultures. A 0.5 McFarland bacterial suspension (corresponding to 1 × 108 CFU/ml) was prepared with PBS for both isolates and diluted with PBS 1:20 again to yield 5 × 106 CFU/ml. The final test concentration of bacteria was obtained by diluting 1:10 with Mueller-Hinton broth to yield 5 × 105 CFU/ml in a total volume of 5 ml, in a glass tube, and thereafter incubated at 37 °C for 16–20 h with shaking at 200 rpm. One milliliter of bacterial growth was centrifuged for 5 min at 12,000 × g, discarding the supernatant and washing the pellet three times with 1 ml of PBS. The bacteria were finally resuspended in 150 μl of PBS and transferred to 200-μl vials containing Sigma-Aldrich G1145 glass beads for LC MS/MS analysis. Two separate cultures of E. coli strain CCUG 62462 were prepared: one with cefotaxime (concentration 1000 μg/ml) and one without. The control sample of strain K12/CCUG 49263 was cultured without antibiotic.
Proteomics Analyses and Peptide Prediction
The bacterial lysates were injected into the LPI Hexalane FlowCell (Nanoxis Consulting AB, www.nanoxisconsulting.com; Patent Application No. WO2006068619), using a pipette to add 70 μl to fill the FlowCell channel. Bacterial proteins in cell lysates were immobilized to the FlowCell membrane, with a 1-h incubation at room temperature, to allow attachment. The FlowCell channels with bound proteins were washed with 400 μl of ammonium bicarbonate, using a syringe pump at a flow rate of 100 μl/min. Enzymatic digestions of the membrane-bound bacterial proteins were performed by injecting 80 μl of trypsin (2 μg/ml in 20 mm ammonium bicarbonate, pH ∼8) into the FlowCell channels and incubating for 1 h at room temperature. The generated peptides were eluted by injecting 200 μl of ammonium bicarbonate buffer (20 mm, pH ∼8) into the FlowCell channels. The eluted peptides were collected at the outlet ports, using a pipette, and transferred into Axygen tubes (2.0 ml). The peptide solutions were incubated at room temperature overnight and subsequently frozen at −20 °C until pending MS analyses. The peptides samples were not reduced or alkylated prior to analysis.
For analysis of mixtures, an in-solution digestion protocol was used to reduce the risk of binding efficiencies/kinetics biases to the LPI FlowCell surfaces of the bead-beaten membrane fractions from Gram-positive bacteria (Streptococcus and Staphylococcus) or Gram-negative bacteria (e.g. Moraxella and Haemophilus). The in-solution digestion was performed by adding trypsin to the suspension (2 μg/ml in 20 mm ammonium bicarbonate, pH 8, 80 μl) and the proteins were allowed to be digested for 1 h at 37 °C. The supernatant was removed from the glass beads and centrifuged at 13,000 rpm (18,000 × g) for 15 min to pellet biomass/debris. The pellet was discarded, and supernatants containing peptides were kept frozen until analysis.
For the detection of antibiotic resistance markers, an in-solution digestion protocol was employed. The suspension was transferred to 200-μl vials containing Sigma-Aldrich G1145 glass beads, and the bead beater used was a TissueLyser from Qiagen. Settings were as follows: frequency 1/25 s and continuous shaking for a total time of 5 min. The bead-beaten samples were frozen until analysis. The samples were thawed, and the bead-beating procedure was repeated. For the in-solution digestion, trypsin was added to the suspension (2 μg/ml in 20 mm ammonium bicarbonate, pH 8, 80 μl), and the proteins were allowed to be digested for 7 h at 37 °C. The supernatant was removed from the glass beads and centrifuged at 13,000 rpm (18,000 × g) for 15 min to pellet biomass/debris. The pellet was discarded, and supernatant containing peptides was kept frozen until analysis.
The tryptic peptides were desalted on Pep Clean C18 spin columns (Thermo Fisher Scientific, Inc., Waltham, MA), according to the manufacturer's guidelines, dried, and reconstituted with 15 μl of 0.1% formic acid (Sigma-Aldrich) in 3% gradient-grade acetonitrile (Merck KGaA, Darmstadt, Germany). A 2.0-μl sample was injected, with an Easy-nLC autosampler (Thermo Fisher Scientific), and analyzed, using an interfaced Q Exactive hybrid mass spectrometer (Thermo Fisher Scientific). The peptides were trapped on a pre-column (45 × 0.075-mm inner diameter) and separated on a reversed-phase column, 200 × 0.075 mm, packed in-house with 3-μm Reprosil-Pur C18-AQ particles (Dr. Maisch, Ammerbuch, Germany). The nanoLC (liquid chromatography) gradient was running at 200 nl/min, starting at 7% acetonitrile (ACN) in 0.2% formic acid, increased to 27% ACN for 25 min, then increased to 40% ACN for 5 min, and finally to 80% ACN for 5 min and held at 80% ACN for 10 min.
Electrospray ionization was applied under a voltage of 1.8 kV and a capillary temperature of 320 °C in data-dependent positive ion mode. Full scan (MS1) spectra were acquired in the Orbitrap over the m/z range 400–1600, with a charge range of 2–6, at a resolution of 70,000, until reaching an AGC target value of 1e6 at a maximum of 250 ms. MS/MS spectra were acquired, using higher energy collision dissociation, at 30% from m/z 110 for the 10 most abundant parent ions, at a resolution of 35,000, using a precursor isolation window of 2 Da until reaching an AGC target value of 1e5 during an injection time of 110 ms. Dynamic exclusion for 30 s after selection for MS/MS was enabled to allow for detection of as many precursors as possible.
The LC-MS/MS output was converted from the proprietary Thermo Xcalibur RAW format to the open source mzXML format (39), using ReAdW (40) (version 201411.xcalibur), with command line arguments: “–nocompress –gzip.” The X! Tandem spectrum search engine (version VENGEANCE Dec. 15, 2015) (20, 41) was used to identify peptides from the mass spectra with the following settings: fragment monoisotopic mass error = 20; parent monoisotopic mass error plus = 5; parent monoisotopic mass error minus = 5; fragment mass type monoisotopic, dynamic range = 100.0; total peaks = 50; maximum parent charge = 4; minimum parent m+h = 800.0; minimum fragment m/z = 100.0, minimum peaks = 15, potential modification mass = 16.0@M, maximum valid expectation value = 1.0. In addition, X! Tandem peptides were also filtered to only allow peptides with a hyperscore of >30 in downstream analyses. The hyperscore filtering resulted in a corresponding median E-value of 9.7 × 10−6 and a median false-positive rate (FPR) of 0.00407 (43) across all samples. E-values and FPRs for each individual sample are available in supplemental file 2, E-value and FPR per sample. Values for all X!Tandem settings are available in supplemental file 3, X! Tandem settings. The reference database used in this step was a customized database consisting of 56,967,781 non-redundant proteins from the NCBI GenBankTM NR (44) and 6,320,906 peptide sequences from the reference genomes archived within the Human Microbiome Project (45). All sequences containing unidentified peptides (“X”), as well as duplicates of sequences shared between the two databases, were removed. The resulting database used with X! Tandem contained a total of 59,349,300 distinct protein sequences. A listing of all the 279,986 identified peptides across all samples, along with their expectation values, hyperscores, charge, mass values, and observed modifications are provided in supplemental file 2, All identified peptides, E-values, and FPR per sample (raw files, X!Tandem XML files, and identified peptides in FASTA format also available via PRIDE, accession no. PXD004321).
Genome Sequencing of ESBL-positive E. coli
Total DNA from E. coli strain CCUG 62462 was extracted using a PureLink Genomic DNA mini kit (Invitrogen), and the DNA purity and concentration were estimated by NanoDrop ND-1000 (Thermo Fisher Scientific), Qubit 2.0 fluorometer (Invitrogen), and agarose gel. The DNA was sequenced on the Illumina MiSeq system, generating 250-bp paired-end reads. Residual adapter sequences and low quality sequences were trimmed using Trim Galore! version 0.3.7 (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), with command line arguments: “–stringency 3 -retain_unpaired”). Remaining high quality reads were assembled de novo with SPAdes version 3.7.0 (46), with command line arguments: “–careful -cov_cutoff 5.” The resulting 5273-kb assembly consisted of 199 contigs with an N50 value of 328,690 bp and an L50 value of 7. The assembly was annotated for mobile antibiotic resistance genes, using ResFinder (33) with default parameters.
RESULTS
Overview of the Method
In this paper we present TCUP, a computational method for typing and characterization of microorganisms using proteomics data generated by bottom-up tandem MS. An overview of the TCUP workflow is presented in Fig. 1, which starts with peptides derived from sets of MS spectra and returns the taxonomic composition and the expressed antibiotic resistance genes present in the sample. The taxonomic composition is provided in relative abundances at each taxonomic level (down to strain level), and the method handles single species samples as well as species mixtures. Determination of taxonomic composition is based on alignment of the peptides against a translated reference genome sequence database, and the method identifies the taxonomic level at which each peptide is discriminative (i.e. at what taxonomic level each peptide can be used to provide information on the taxonomic content of the sample). The relative abundances of microorganisms are then estimated based on the proportions of discriminative peptides from the taxonomic entities in the sample. The detection of expressed antibiotic resistance proteins is based on comparisons of the peptides against a reference database of antibiotic resistance genes. The abundances of the particular antibiotic resistance genes expressed in the sample are then estimated from those discriminatively matched peptides. TCUP is open source and is freely available for both Windows and Linux platforms (https://bitbucket.org/chalmersmathbioinformatics/tcup). See under “Materials and Methods” for full details about the implementation of TCUP.
Fig. 1.
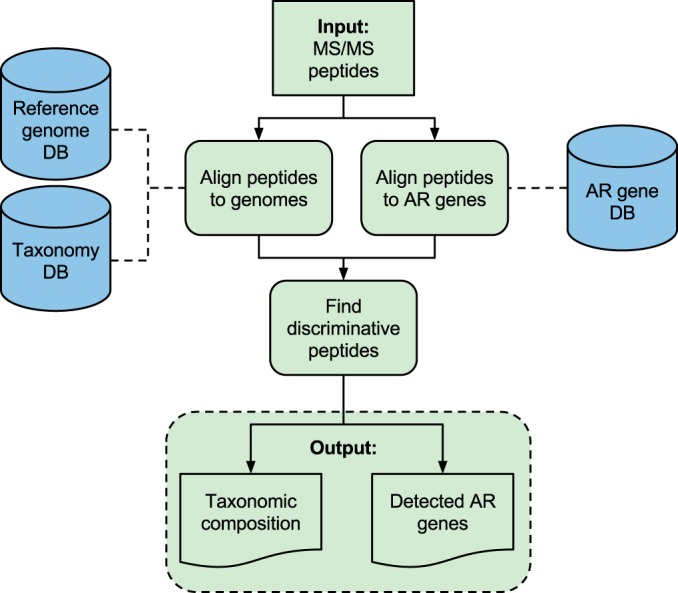
TCUP overview. Left track, taxonomic composition estimation. Peptides are aligned to reference genome sequences. The lowest common ancestor algorithm is used to find discriminative peptides that uniquely identify organisms in the sample. Right track, antibiotic resistance (AR) protein detection. Peptides are aligned to a database of reference antibiotic resistance gene sequences. The method outputs the estimated relative abundances of all taxonomic entities detected in the sample and a list of detected expressed antibiotic resistance proteins.
Evaluation of Taxonomic Composition Estimation
The performance of TCUP was first evaluated using simulated data from the four bacterial species E. coli, P. aeruginosa, S. aureus, and S. pneumoniae. For each species, 10,000 peptides were sampled from their in silico digested proteomes and analyzed by TCUP. The number of peptides that were classified as discriminative at the species level and thus exhibited unique taxonomic affiliations was highest for P. aeruginosa (55.8%) followed by S. aureus (30.5%), E. coli (27.1%), and S. pneumoniae (17.9%). The proportion of correctly assigned discriminative peptides was high for all four species with estimated true positive rates of 99.9, 99.8, 99.6, and 99.8% for P. aeruginosa, S. aureus, E. coli, and S. pneumoniae, respectively (Fig. 2). When substitutions were randomly introduced into the peptides, the number of discriminative peptides decreased (supplemental file 4, in silico results). The true positive rates were also reduced, although the effect was much smaller (Fig. 2). At the highest substitution rate of 10%, the true positive rate decreased with 4.59 percentage points (pp), 7.02, 9.08, and 9.43 pp for P. aeruginosa, S. aureus, E. coli, and S. pneumoniae, respectively. These results show that the assignment of discriminative fragments performed by TCUP is robust and has high performance, even in noisy data.
Fig. 2.

True positive rates for E. coli, P. aeruginosa, S. aureus, and S. pneumoniae at mutation rate of 0–3, 5, and 10%. The decrease in true positive rates is most prominent for E. coli and S. pneumoniae, whereas the performance is more robust for P. aeruginosa and S. aureus.
Furthermore, the robustness of the taxonomic assignments to an incomplete reference database was evaluated by a leave-one-out experiment using the 62 E. coli genomes available in NCBI RefSeq. When the E. coli strain from which the simulated peptides were generated was excluded, the number of discriminative peptides decreased an average of 2.3 pp (S.E. = 0.41 pp), and the true positive rates decreased an average of 0.52 pp (S.E. = 0.10 pp). Only three of the 62 E. coli strains showed a decrease in true positive rates greater than 2.0 pp (O127:H6 E2348/69, SMS-3–5, UMNK88, with decreases of 4.5, 2.3, and 2.2 pp, respectively) (supplemental file 5, results from leave-one-out evaluation).
Next, TCUP was evaluated using data from bacterial cultures generated by bottom-up tandem MS (Table 1). The average proportions of discriminative peptides were lower than for the in silico generated data, 11.13, 44.51, 22.48, and 11.04% for E. coli, P. aeruginosa, S. aureus, and S. pneumoniae, respectively. All of the four different species were correctly identified with high correct peptide classification rates 90.30, 98.52, 97.58, and 90.30%, respectively (Fig. 3). The results were highly reproducible between technical replicates, with S.E. <1.1% for all species. If only species detected in the sample with at least five discriminative fragments were considered, the correct classification rates increased to 99.43, 100.00, 100.00, and 94.77%. When mixed species samples containing an equal ratio of S. pneumoniae, E. coli, P. aeruginosa, and S. aureus were analyzed, TCUP estimated the average normalized relative abundances (see “Materials and Methods”) to 31.56, 23.62, 22.52, and 22.30%, respectively (Fig. 4A). For mixed samples with an equal ratio of H. influenzae, M. catarrhalis, S. aureus, and S. pneumoniae, the average relative abundances were estimated to be 32.63, 25.40, 20.00, and 21.93%, respectively (Fig. 4B). Samples containing S. pneumoniae, P. aeruginosa, E. coli, and S. aureus in 4:2:2:1 ratios were estimated to average relative abundances of 45.75, 26.34, 17.93, and 9.96%, respectively (Fig. 4C). Samples containing S. aureus, H. influenzae, M. catarrhalis, and S. pneumoniae in 4:2:2:1 ratios were estimated to average relative abundances of 36.11, 31.94, 23,58, and 8.35%, respectively (Fig. 4D). All results exhibited low variability, with S.E. <1.38% for all species across all mixtures (more detailed figures available in supplemental file 6, mixed-species samples). Expected proportions for equal ratio mixes were 25% per species and 44, 22, 22, and 11% for 4:2:2:1 ratio mixes.
Table I. TCUP classification accuracy averages on cultures of four bacterial species (numbers are averages from three technical replicates).
| E. coli | P. aeruginosa | S. aureus | S. pneumoniae | |
|---|---|---|---|---|
| No. of peptides | 4132 | 3964 | 3707 | 5043 |
| Proportion of discriminative peptides (%) | 11.13 | 44.51 | 22.48 | 11.05 |
| Proportion of peptides discriminative to the correct species (%) | 10.00 | 43.85 | 21.98 | 9.99 |
| True positive rate (%) | 90.3 | 98.5 | 97.5 | 90.3 |
Fig. 3.

Bar plot showing the mean true positive rates and standard errors for pure cultures of E. coli, P. aeruginosa, S. aureus, and S. pneumoniae. E. coli and S. pneumoniae display lower true positive rates than P. aeruginosa and S. aureus, likely caused by the large number of closely related sequences in the reference database.
Fig. 4.

Abundance estimations and standard errors for mixed samples containing S. pneumoniae, E. coli, P. aeruginosa, and S. aureus in 1:1:1:1 ratio (A); H. influenzae, M. catarrhalis, S. pneumoniae, and S. aureus in 1:1:1:1 ratio (B); S. pneumoniae, P. aeruginosa, E. coli, and S. aureus in 4:2:2:1 ratio (C); and S. pneumoniae, H. influenzae, M. catarrhalis, and S. aureus in 4:2:2:1 ratio (D). The number of discriminative peptides for each species has been adjusted by the expected proportion of discriminative peptides estimated from pure culture samples, and then each sample has been normalized by the total number of discriminative peptides in the sample. The horizontal red lines indicate the expected abundance of each species based on their ratios in the mixture.
The impact in the performance of TCUP when using the large comprehensive X!Tandem database compared with a small database containing only the proteome of the target species was evaluated based on three samples of E. coli cultures (details are available in supplemental file 7, large versus small protein database). The comparison showed that the TPR was slightly higher for the targeted database (100%) compared with the comprehensive database (98.21%). There is thus a small advantage of using a targeted database, but this requires knowledge about the sample contents. Using a comprehensive database, however, results in a minor impact on the true positive rate but enables identification of a wide range of clinically relevant species without any prior information of the sample contents.
Evaluation of Antibiotic Resistance Detection
TCUP was evaluated on data generated from pure cultures of the CTX-M-15-positive E. coli strain CCUG 62462, cultured with and without cefotaxime (a third generation cephalosporin). TCUP correctly identified the isolate as E. coli (an average 86.78% of the discriminative peptides matched to E. coli at the species level when grown without cefotaxime). Furthermore, the expression of CTX-M was detected and identified under both conditions, with a mean relative abundance of 0.322% (S.E. = 1.39 × 10−3%; average 62.33 peptides detected) when grown with cefotaxime, and 0.0521% (S.E. = 1.42 × 10−2%; average 14.66 peptides detected) on standard media (Fig. 5). Among the seven resistance genes identified by WGS (“Materials and Methods” and supplemental file 8, antibiotic resistance profile analysis of E. coli strain CCUG 62462), TCUP identified the expression of three antibiotic resistance factors in addition to CTX-M: the β-lactamase blaTEM-1, the aminoglycoside acetyltransferase aac(3)-II (detected in five of six samples), and the macrolide phosphorylase mph(A). All of these genes were detectable in the cultures grown from both selective and standard media. The method did not detect any peptides from the remaining three resistance genes (dfrA17, sul1, and aac-(3”)-la). The negative control, E. coli strain K12/CCUG 49263, was grown under identical conditions but did not result in any matches to antibiotic resistance genes (Fig. 5).
Fig. 5.
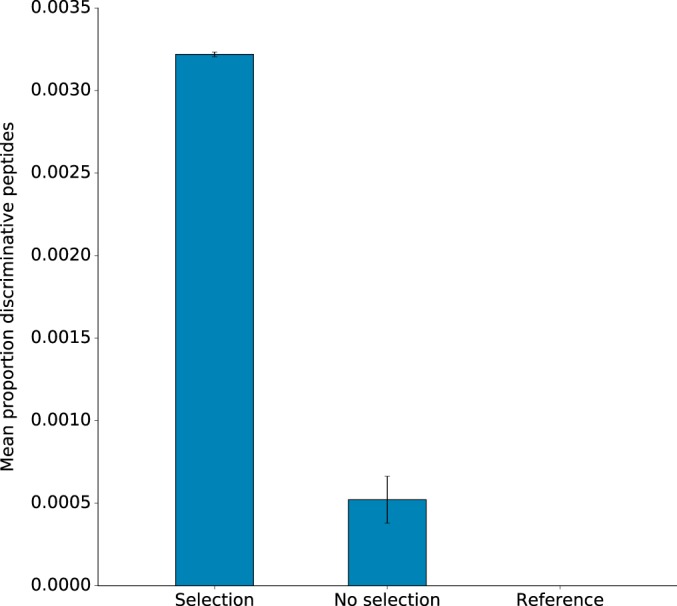
Bar plot showing the proportion and number of discriminative peptides assigned to CTX-M gene products. The left bar corresponds to E. coli strain CCUG 62462 grown under antibiotic selection pressure by cefotaxime; the center bar corresponds to E. coli strain CCUG 62462 grown without antibiotic selection pressure; and the right bar corresponds to E. coli reference strain K12/CCUG 49263 grown without antibiotic selection pressure.
DISCUSSION
Here we present TCUP, a new computational method for bacterial typing and characterization using proteomics, capable of estimating taxonomic composition and detecting antibiotic resistance proteins in bacterial samples. The method has been optimized for the fragmented data produced by shotgun proteomics techniques, in particular, bottom-up tandem MS. TCUP can automatically find peptides that uniquely identify organisms in samples, making the method capable of analyzing data generated from single-species cultures and mixed-species samples, without any prior information about their contents. The performance of TCUP was investigated using in silico digested proteomes from four microbial species, two Gram-negative (E. coli and P. aeruginosa) and two Gram-positive bacteria (S. aureus and S. pneumoniae). These results showed that the proportion of peptides that provide discrimination at the species level varied between species from 18% for S. pneumoniae to 55% for P. aeruginosa. Thus, the information present in the peptide sequence data differs between species and is dependent on the variability of their genomes and proteomes and the similarities to their closest relatives. In particular, S. pneumoniae (pneumococcus) is known to be difficult to distinguish based on known phenotypic and genotypic features due to the close phylogenetic relationships with related species, e.g. Streptococcus mitis and Streptococcus pseudopneumoniae (47). In contrast, P. aeruginosa has a more variable genome and fewer closely related species present in the reference database (48, 49). However, the proportion of discriminative peptides that were correctly classified was high, with true positive rates above 99% for all species. The true positive rates remained high both when mutations were introduced into the peptide fragments and when the genome of the correct strain was excluded from the reference data base. Taken together, the in silico analysis demonstrates that the method has high potential for correct and robust species identification using peptides generated by bottom-up tandem MS.
Evaluation of TCUP on peptides from experimental data generated from bacterial cultures showed high proportions of discriminative peptides and true positive rates between 90.3 and 98.5% for the investigated species. These values were further improved to between 94.7 and 100.0% when spurious species detected with less than five discriminative peptides were removed from the analysis. However, the use of such low abundance filters is reasonable only when the peptide sampling depth is high, or it is known beforehand that the sample contains a high abundance of peptides from a limited number of separate species, and thus is not suitable for uncultured clinical samples. TCUP was also able to identify and estimate the relative abundance of individual species in mixed samples. Furthermore, we noticed that the true positive rates from the experimental data were slightly lower than in silico generated data (average reduction in TPR between 1.41 and 9.53 pp). These discrepancies are likely caused by the many sources of noise affecting the experimental data, introduced by cell cultivation, sample preparation and digestion, the MS analysis, and the matching of spectrum-peptide matching process. Parts of the noise are potentially species-specific and may affect the number of discriminative peptides detected and the true positive rate negatively. The experimental data are also dependent on gene expression, e.g. the sampling occasion, and highly expressed genes, which will be represented with a larger number of peptides, are known to exhibit lower mutation rates (50, 51). This will likely result in lower ratios of discriminative peptides and thus lower rates of correct classifications. Nevertheless, the evaluation using experimental data showed that TCUP has an overall high performance to accurately identify organisms in single-species cultures and mixed-species samples.
The ability to detect highly expressed antibiotic resistance proteins was evaluated using a clinical E. coli strain known to encode the ESBL enzyme CTX-M-15 gene. Here, TCUP was able to correctly identify peptides from this gene even when the strain was cultivated on media without any antibiotics. The strain was known to contain six additional antibiotic resistance genes, of which expression from three were found in all experimental replicates. It is likely that several of the remaining three antibiotic resistance genes, which could not be consistently detected, were expressed only at low levels under the investigated growing conditions. The phenotypic screen showed, for example, resistance to tobramycin (supplemental file 8, antibiotic resistance profile analysis of E. coli strain CCUG 62462), and the gene providing resistance to that aminoglycoside (aac(3)-II) was not detected in one of the samples grown without selection pressure (supplemental file 9, detailed results from antibiotic resistance detection). In addition, in silico digestion of the detected resistance genes verified that all seven genes were expected to produce peptide fragments of lengths suitable for detection with the applied tandem MS protocol. Thus, our results suggest that the sequence depth provided by one run of the used tandem MS setup may not be enough to provide a full view of all expressed antibiotic resistance genes. It should, however, be emphasized that many resistance mechanisms are based on highly expressed genes. Overexpression of chromosomal genes can, for example, cause clinical resistance to important antibiotics and is commonly encountered in Enterobacteriaceae and P. aeruginosa, for example (52, 53). These forms of resistance are often caused by mutations in promoters and transcription factors, which are notoriously hard to identify using DNA-based diagnostics methods such as WGS (54). In these cases, proteomics, including bottom-up tandem MS approaches, provide a powerful alternative that can complement existing diagnostics methods. For such applications, TCUP provides the necessary means to accurately and robustly assign the generated peptides to the correct resistance genes.
TCUP relies heavily upon comprehensive reference genome and resistance gene databases. Incomplete databases may therefore result in reductions of overall performance. However, the leave-one-out experiments of the 62 E. coli strains showed that the impact was, in general, relatively small, with an average reduction in true positive rate of only 0.52 pp. A few strains showed a more dramatic reduction in performance with a reduced true positive rate of up to 4.5 pp. Careful examination of the incorrectly classified peptides revealed that many of the misclassified peptides originated from genes located on horizontally transferred genetic elements that were not present in any other E. coli strains in the reference database (phages, conjugative elements, etc.). These results demonstrate that, although the average reduction in true positive rate is low, strains of species with plastic genomes, carrying relatively large amounts of mobile genetic material, can be more sensitive to an incomplete reference database. Thus, we cannot rule out that the rates of correctly classified peptides will be lower for newly encountered strains of species for which sequenced genomes are lacking, especially if they carry large amounts of mobile genetic material. This effect is also likely to be larger for other taxonomic groups for which fewer representative genomes are available. However, the recent developments of NGS have enabled fast and cost-efficient characterization of bacterial genomes. The number of bacterial strains with fully sequenced genomes available in the public repositories is therefore rapidly growing, and this will, over time, further increase the completeness of the reference database and thereby further improve the performance and robustness of TCUP.
Clinical samples can contain proteins from multiple species, including both bacteria and eukaryotes (e.g. human and fungi). Therefore, a comprehensive protein database was used to ensure that as many of the MS spectra as possible had satisfactory peptide matches. Moreover, our analysis showed that the size of the database used in the spectral matching had little impact on the overall performance. In fact, when the spectra from pure cultures were instead matched to a specialized database containing only the single species present in the sample, true positive rates were not greatly affected. For the E. coli sample, we observed an average decrease in TPR of 1.79 pp when the comprehensive database was replaced with a database containing only the E. coli proteome, whereas the average number of discriminative peptides did not change (using the large database identified an average of 0.33 more discriminative peptides). It should, however, be pointed out that the case with a single reference proteome gives results that are too optimistic, because in most cases where TCUP will be applied the species and thus the correct proteome will be unknown. Consequently, using a comprehensive protein database with millions of reference proteins enables identification of a wide range of species that potentially can be present in the sample, and it does not have any substantial negative impact on the performance (complete details are available in supplemental file 7, large versus small protein database). One important reason for this is that TCUP, in contrast to many other previously suggested algorithms (11–16), has separated the identification of peptides from MS spectra and the taxonomic assignment into two independent steps. In the first step, an extensive protein database is preferable to maximize the likelihood that each spectrum can be matched to the correct peptide. In the second step, however, TCUP uses a smaller curated database that ensures a high accuracy of the taxonomic assignments. Keeping the processes separated is essential to classify samples without a priori information on their contents, as it is impossible to create a protein database specifically designed for each combination of species that will potentially occur in a sample. It also adds flexibility because it makes it possible to combine TCUP with virtually any type of spectrum-matching algorithm or even de novo peptide prediction from observed mass spectra.
TCUP estimates the taxonomic composition in a sample based on normalized relative abundances of discriminative peptides. An alternative approach, which is used by Pipasic, for example, is to weight the relative abundance of each peptide based on its similarity between the reference proteomes (17, 55). This enables the use of a larger set of peptides and can thus provide increased accuracy. Calculating the reference proteome similarities requires sequence comparison of all possible peptides, which is computationally complex. This makes Pipasic intractable in many clinical settings where time, from the collected sample to the final results, is often of essence and hundreds of different pathogens and commensals can potentially be present in a single sample. In contrast, TCUP bases its estimation on the proportion of discriminative peptides, determined by the lowest common ancestor algorithm. Normalization is done by the number of discriminative fragments observed in pure culture or, alternatively, based on in silico digestion (supplemental file 10,example comparison of experimental and in silico-basedcorrection). This approach is computationally much more efficient and enables the use of a reference database with thousands of species. Furthermore, it should be pointed out that TCUP matches identified peptides directly to complete reference genomes rather than to proteomes. This has the added benefit of making the method completely annotation agnostic and thus robust against errors related to the prediction of open reading frames (42). Many years of research have been put into the development of efficient and accurate sequence alignment algorithms, and TCUP leverages that information by using regular sequence programs (BLAT and BLAST) to infer the taxonomic affiliation of each peptide. Thanks to the use of an efficient SQLite3-based back-end for storing the taxonomic structure, sequence annotations, and sequence to taxa connections, the main algorithm driving TCUP is fast and requires relatively little computing resources.
In conclusion, we have presented TCUP, a new computational method for typing and characterizing bacteria, estimating bacterial compositions in samples, and detecting and identifying expressed antibiotic resistance proteins in bottom-up MS/MS data. The method has shown good performance on both pure cultures of single species and mixed-species culture model systems samples. The method is computationally efficient and publicly available under an open source license for both the Windows and Linux platforms. TCUP has already been used in high performance cluster environments and can, thanks to its easily parallelizable implementation, efficiently process hundreds of samples. Thus, TCUP has the potential to further enable the use of bottom-up tandem MS in clinical settings, thereby improving the detection, characterization, and typing of pathogenic bacteria for diagnostics of infectious diseases.
DATA AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD004321. Data from the evaluation of between using a large and a small reference protein database for X!Tandem are available via PRIDE, accession PXD005924.
Supplementary Material
Acknowledgments
We thank Shora Yazdanshenas for technical assistance and Diarmuid Kenny for suggestions and support in finding suitable MS search engines. The Culture Collection University of Gothenburg (CCUG; www.ccug.se) was supported by the Department of Clinical Microbiology of The Sahlgrenska University Hospital. The mass spectrometry analyses were performed by The Proteomics Core Facility at the Sahlgrenska Academy, Gothenburg University.
Footnotes
Author contributions: F.B., R.K., L.G., E.R.B.M., and E.K. designed research; F.B., R.K., L.G., A.J., N.K., O.A., C.A., E.R.B.M., and E.K. performed research; F.B., R.K., and L.G. contributed new reagents or analytic tools; F.B., R.K., A.J., and E.K. analyzed data; F.B., R.K., L.G., A.J., N.K., O.A., C.A., E.R.B.M., and E.K. wrote the paper.
* This work was supported in part by European Commission, Seventh Framework Programme FP7, under REA Grant HEALTH-F3-602860-2013 (TAILORED-Treatment); Life Science Area of Advance, Chalmers University of Technology, Swedish Research Council; Västra Götaland Region Project No. ALFGBG-437221; and the Wallenberg Foundation.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- NGS
- next-generation sequencing
- WGS
- whole-genome sequence
- ESBL
- extended spectrum β-lactamase
- TPR
- true positive rate
- ACN
- acetonitrile
- FPR
- false positive rate.
REFERENCES
- 1. Braga P. A., Tata A., Santos V. G., Barreiro J. R., Schwab N. V., Santos M. V., et al. (2012) Bacterial identification: from the agar plate to the mass spectrometer. RSC Adv. 2012, 994–1008 [Google Scholar]
- 2. Emerson D., Agulto L., Liu H., and Liu L. (2008) Identifying and characterizing bacteria in an era of genomics and proteomics. Bioscience 58, 925–936 [Google Scholar]
- 3. Loman N. J., Constantinidou C., Chan J. Z., Halachev M., Sergeant M., Penn C. W., Robinson E. R., and Pallen M. J. (2012) High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat. Rev. Microbiol. 10, 599–606 [DOI] [PubMed] [Google Scholar]
- 4. Gardy J., Loman N. J., and Rambaut A. (2015) Real-time digital pathogen surveillance–the time is now. Genome Biol. 16, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang W., Xi H., Huang M., Wang J., Fan M., Chen Y., Shao H., and Li X. (2014) Performance of mass spectrometric identification of bacteria and yeasts routinely isolated in a clinical microbiology laboratory using MALDI-TOF MS. J. Thorac. Dis. 6, 524–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lévesque S., Dufresne P. J., Soualhine H., Domingo M. C., Bekal S., Lefebvre B., and Tremblay C. (2015) A side by side comparison of Bruker Biotyper and VITEK MS: utility of MALDI-TOF MS technology for microorganism identification in a Public Health Reference Laboratory. PLoS ONE 10, e0144878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lima T. B., Pinto M. F., Ribeiro S. M., de Lima L. A., Viana J. C., Gomes Júnior N., Cândido Ede S., Dias S. C., and Franco O. L. (2013) Bacterial resistance mechanism: What proteomics can elucidate. FASEB J. 27, 1291–1303 [DOI] [PubMed] [Google Scholar]
- 8. Radhouani H., Pinto L., Poeta P., and Igrejas G. (2012) After genomics, what proteomics tools could help us understand the antimicrobial resistance of Escherichia coli? J. Proteomics 75, 2773–2789 [DOI] [PubMed] [Google Scholar]
- 9. Sauer S., and Kliem M. (2010) Mass spectrometry tools for the classification and identification of bacteria. Nat. Rev. Microbiol. 8, 74–82 [DOI] [PubMed] [Google Scholar]
- 10. Karlsson R., Gonzales-Siles L., Boulund F., Svensson-Stadler L., Skovbjerg S., Karlsson A., Davidson M., Hulth S., Kristiansson E., and Moore E. R. (2015) Prototyping: proteomic characterization, classification and identification of microorganisms–a prospectus. Syst. Appl. Microbiol. 38, 246–257 [DOI] [PubMed] [Google Scholar]
- 11. Dworzanski J. P., Snyder A. P., Chen R., Zhang H., Wishart D., and Li L. (2004) Identification of bacteria using tandem mass spectrometry combined with a proteome database and statistical scoring. Anal. Chem. 76, 2355–2366 [DOI] [PubMed] [Google Scholar]
- 12. Dworzanski J. P., and Snyder A. P. (2005) Classification and identification of bacteria using mass spectrometry-based proteomics. Expert Rev. Proteomics 2, 863–878 [DOI] [PubMed] [Google Scholar]
- 13. Dworzanski J. P., Deshpande S. V, Chen R., Jabbour R. E., Snyder A. P., Wick C. H., and Li L. (2006) Mass spectrometry-based proteomics combined with bioinformatic tools for bacterial classification. J. Proteome Res. 5, 76–87 [DOI] [PubMed] [Google Scholar]
- 14. Jabbour R. E., Deshpande S. V., Wade M. M., Stanford M. F., Wick C. H., Zulich A. W., Skowronski E. W., and Snyder A. P. (2010) Double-blind characterization of non-genome-sequenced bacteria by mass spectrometry-based proteomics. Appl. Environ. Microbiol. 76, 3637–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jabbour R. E., Deshpande S. V., Stanford M. F., Wick C. H., Zulich A. W., and Snyder A. P. (2011) A protein processing filter method for bacterial identification by mass spectrometry-based proteomics. J. Proteome Res. 10, 907–912 [DOI] [PubMed] [Google Scholar]
- 16. Tracz D. M., McCorrister S. J., Chong P. M., Lee D. M., Corbett C. R., and Westmacott G. R. (2013) A simple shotgun proteomics method for rapid bacterial identification. J. Microbiol. Methods 94, 54–57 [DOI] [PubMed] [Google Scholar]
- 17. Penzlin A., Lindner M. S., Doellinger J., Dabrowski P. W., Nitsche A., and Renard B. Y. (2014) Pipasic: similarity and expression correction for strain-level identification and quantification in metaproteomics. Bioinformatics 30, 149–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eng J. K., McCormack A. L., and Yates J. R. (1994) An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5, 976–989 [DOI] [PubMed] [Google Scholar]
- 19. Craig R., and Beavis R. C. (2003) A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid Commun. Mass Spectrom. 17, 2310–2316 [DOI] [PubMed] [Google Scholar]
- 20. Craig R., and Beavis R. C. (2004) TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 21. Diament B. J., and Noble W. S. (2011) Faster SEQUEST searching for peptide identification from tandem mass-spectra. J. Proteome Res. 10, 3871–3879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perkins D. N., Pappin D. J., Creasy D. M., and Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 23. Ma B., Zhang K., Hendrie C., Liang C., Li M., Doherty-Kirby A., and Lajoie G. (2003) PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun. Mass Spectrom. 17, 2337–2342 [DOI] [PubMed] [Google Scholar]
- 24. Frank A., and Pevzner P. (2005) PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal. Chem. 77, 964–973 [DOI] [PubMed] [Google Scholar]
- 25. Johnson R. S., and Taylor J. A. (2002) Searching sequence databases via de novo peptide sequencing by tandem mass spectrometry. Mol. Biotechnol. 22, 301–315 [DOI] [PubMed] [Google Scholar]
- 26. Kent W. J. (2002) BLAT—The BLAST-like alignment tool. Genome Res. 12, 656–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tatusova T., Ciufo S., Federhen S., Fedorov B., McVeigh R., O'Neill K., Tolstoy I., and Zaslavsky L. (2015) Update on RefSeq microbial genomes resources. Nucleic Acids Res. 43, D599–D605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pruitt K. D., Tatusova T., and Maglott D. R. (2007) NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35, D61–D65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lan R., and Reeves P. R. (2002) Escherichia coli in disguise: molecular origins of Shigella. Microbes Infect. 4, 1125–1132 [DOI] [PubMed] [Google Scholar]
- 30. Hecht M. S., and Ullman J. D. (1973) Analysis of a simple algorithm for global data flow problems in Proceedings of the 1st annual ACM SIGACT-SIGPLAN symposium on Principles of programming languages, pp. 207–217, ACM [Google Scholar]
- 31. Federhen S. (2012) The NCBI Taxonomy database. Nucleic Acids Res. 40, D136–D143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huerta-Cepas J., Dopazo J., and Gabaldón T. (2010) ETE: a python environment for tree exploration. BMC Bioinformatics 11, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zankari E., Hasman H., Cosentino S., Vestergaard M., Rasmussen S., Lund O., Aarestrup F. M., and Larsen M. V. (2012) Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Apweiler R., Bairoch A., Wu C. H., Barker W. C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., Martin M. J., Natale D. A., O'Donovan C., Redaschi N., and Yeh L. S. (2004) UniProt: the universal protein knowledgebase. Nucleic Acids Res. 32, D115–D119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rice P., Longden I., and Bleasby A. (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends in Genetics 16, 276–277 [DOI] [PubMed] [Google Scholar]
- 36. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., and Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tatusova T., Ciufo S., Fedorov B., O'Neill K., and Tolstoy I. (2014) RefSeq microbial genomes database: new representation and annotation strategy. Nucleic Acids Res. 42, 553–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karami N., Helldal L., Welinder-Olsson C., Ahrén C., and Moore E. R. (2013) Sub-typing of extended-spectrum-β-lactamase-producing isolates from a nosocomial outbreak: application of a 10-loci generic Escherichia coli multi-locus variable number tandem repeat analysis. PLoS ONE 8, e83030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pedrioli P. G., Eng J. K., Hubley R., Vogelzang M., Deutsch E. W., Raught B., Pratt B., Nilsson E., Angeletti R. H., Apweiler R., Cheung K., Costello C. E., Hermjakob H., Huang S., Julian R. K., et al. (2004) A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 22, 1459–1466 [DOI] [PubMed] [Google Scholar]
- 40. Seattle Proteome Center. Software (2009) ReAdW (Internet) (cited July 10, 2015) Available from: http://tools.proteomecenter.org/wiki/index.php?title=Software:ReAdW
- 41. Bjornson R. D., Carriero N. J., Colangelo C., Shifman M., Cheung K. H., Miller P. L., and Williams K. (2008) X!!Tandem, an improved method for running X!Tandem in parallel on collections of commodity computers. J. Proteome Res. 7, 293–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Warren A. S., Archuleta J., Feng W.-C., and Setubal J. C. (2010) Missing genes in the annotation of prokaryotic genomes. BMC Bioinformatics 11, 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gupta N., Bandeira N., Keich U., and Pevzner P. A. (2011) Target-decoy approach and false discovery rate: when things may go wrong. J. Am. Soc. Mass Spectrom. 22, 1111–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Benson D. A., Boguski M. S., Lipman D. J., Ostell J., Ouellette B. F., Rapp B. A., and Wheeler D. L. (1999) GenBankTM. Nucleic Acids Res. 27, 12–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Human Microbiome Project Consortium (2012) Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., Lesin V. M., Nikolenko S. I., Pham S., Prjibelski A. D., Pyshkin A. V., Sirotkin A. V., Vyahhi N., Tesler G., Alekseyev M. A., et al. (2012) SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fraser C., Alm E. J., Polz M. F., Spratt B. G., and Hanage W. P. (2009) The bacterial species challenge: ecological diversity. Science 323, 741–746 [DOI] [PubMed] [Google Scholar]
- 48. Stover C. K., Pham X. Q., Erwin A. L., Mizoguchi S. D., Warrener P., Hickey M. J., Brinkman F. S., Hufnagle W. O., Kowalik D. J., Lagrou M., Garber R. L., Goltry L., Tolentino E., Westbrock-Wadman S., Yuan Y., et al. (2000) Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964 [DOI] [PubMed] [Google Scholar]
- 49. Nikbin V. S., Aslani M. M., Sharafi Z., Hashemipour M., Shahcheraghi F., and Ebrahimipour G. H. (2012) Molecular identification and detection of virulence genes among Pseudomonas aeruginosa isolated from different infectious origins. Iran J. Microbiol. 4, 118–123 [PMC free article] [PubMed] [Google Scholar]
- 50. Dötsch A., Klawonn F., Jarek M., Scharfe M., Blöcker H., and Häussler S. (2010) Evolutionary conservation of essential and highly expressed genes in Pseudomonas aeruginosa. BMC Genomics 11, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rocha E. P., and Danchin A. (2004) An analysis of determinants of amino acids substitution rates in bacterial proteins. Mol. Biol. Evol. 21, 108–116 [DOI] [PubMed] [Google Scholar]
- 52. Pfeifer Y., Cullik A., and Witte W. (2010) Resistance to cephalosporins and carbapenems in Gram-negative bacterial pathogens. Int. J. Med. Microbiol. 300, 371–379 [DOI] [PubMed] [Google Scholar]
- 53. Poole K. (2005) Efflux-mediated antimicrobial resistance. J. Antimicrob. Chemother. 56, 20–51 [DOI] [PubMed] [Google Scholar]
- 54. Kos V. N., Déraspe M., McLaughlin R. E., Whiteaker J. D., Roy P. H., Alm R. A., Corbeil J., and Gardner H. (2015) The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob Agents Chemother. 59, 427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lindner M. S., and Renard B. Y. (2013) Metagenomic abundance estimation and diagnostic testing on species level. Nucleic Acids Res. 41, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Johnning A., Jakobsson H. E., Boulund F., Salvà-Serra F., Moore E. R. B., Åhrèn C., Karami N., and Kristiansson E. (2016) Draft genome sequence of extended-spectrum-β-lactamase-producing Escherichia coli strain CCUG 62462, isolated from a urine sample. Genome Announc. 4, e01382–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD004321. Data from the evaluation of between using a large and a small reference protein database for X!Tandem are available via PRIDE, accession PXD005924.