

Abstract
Dynamic microtubules are fundamental to many cellular processes, and accurate measurements of microtubule dynamics can provide insight into how cells regulate these processes and how genetic mutations impact regulation. The quantification of microtubule dynamics in metazoan models has a number of associated challenges, including a high microtubule density and limitations on genetic manipulations. In contrast, the budding yeast model offers advantages that overcome these challenges. This protocol describes a method to measure the dynamics of single microtubules in living yeast cells. Cells expressing fluorescently tagged tubulin are adhered to assembled slide chambers, allowing for stable time-lapse image acquisition. A detailed guide for high-speed, four-dimensional image acquisition is also provided, as well as a protocol for quantifying the properties of dynamic microtubules in confocal image stacks. This method, combined with conventional yeast genetics, provides an approach that is uniquely suited for quantitatively assessing the effects of microtubule regulators or mutations that alter the activity of tubulin subunits.
Keywords: Cellular Biology, Issue 122, Cytoskeleton, Microtubule dynamics, Budding yeast, Microscopy, Live-cell imaging
Introduction
Microtubules are cytoskeletal polymers made of αβ-tubulin protein subunits and are used in a wide variety of cellular contexts, including intracellular transport, cell division, morphogenesis, and motility. To build microtubule networks for these diverse roles, cells must carefully regulate where and when microtubules form. This regulation is accomplished by controlling the reactions that either assemble αβ-tubulin subunits into microtubule polymers or disassemble microtubules into free subunits; this is known as microtubule dynamics.
A major goal of the microtubule field is to elucidate the molecular mechanisms that regulate microtubule dynamics, including studies of the αβ-tubulin subunits and extrinsic regulators that bind to tubulin and/or to microtubules. A well-established experimental approach is to reconstitute this system in vitro using purified αβ-tubulin protein, often in combination with purified extrinsic regulators. Although this is a useful approach, it is clear that microtubule dynamics in reconstituted systems differs strongly from that observed in living cells. For example, microtubules grow faster and shrink slower in vivo than in vitro. These differences may be attributed to the availability of known extrinsic regulators1, as well as to yet-undefined factors in cells. Therefore, it is critical to determine the activities of microtubule regulators and mutants that disrupt dynamics in a native cellular context.
Although metazoan models have proven to be the prevailing systems for investigating microtubule function and higher-order organization, several practical concerns severely limit the utility of these models for the precise measurement of microtubule dynamics. First, the high number of microtubules, ranging from dozens to thousands per cell, makes it difficult to confidently track individual microtubules over time. Many studies address this challenge by imaging proteins that selectively localize to microtubule ends, such as proteins in the end-binding (EB) family. However, these proteins are known to only localize to the ends of growing microtubules in metazoans2. Therefore, the utility of these proteins is limited to directly measuring growth rates, while only indirectly measuring other aspects of dynamics, such as frequency of catastrophe. Second, despite advances in genome editing technology, creating cells that stably express fluorescently labeled tubulin or introducing mutations to selectively manipulate tubulin or microtubule regulators remains a significant challenge. Moreover, the presence of many tubulin isotypes in metazoans confounds the study of how mutations impact individual tubulin genes.
The budding yeast system provides several important advantages for measuring in vivo microtubule dynamics. Yeast has a simplified microtubule network that permits the visualization of individual microtubules. In yeast, microtubules emanate from organizing centers known as spindle pole bodies (SPBs), which are embedded in the nuclear envelope3. The SPBs serve as scaffolds for γ-tubulin small complexes that nucleate microtubules4,5. SPBs nucleate two classes of microtubules, spindle microtubules and astral microtubules. Spindle microtubules project into the nucleoplasm and are important for attaching to chromosomes via kinetochore microtubules and for stabilizing the spindle via overlapping interpolar microtubules6. In contrast, astral microtubules project outwards into the cytosol and are relatively rare compared to the dense network of spindle microtubules. During mitosis, pre-anaphase cells have only 1-2 astral microtubules emanating from either SPB; these exist as individual microtubules rather than as bundles7. The role of astral microtubules during mitosis is to move the nucleus and spindle into the junction between the mother and bud compartments, known as the bud neck. This movement involves well-defined pathways that generate force on astral microtubules, pulling the nucleus and spindle toward and eventually into the bud neck8.
Another advantage of the yeast system is the utility of its genetics, which can be used to investigate microtubule regulators and tubulin subunits with unparalleled precision. Yeast also possess a simplified repertoire of tubulin isotypes: a single β-tubulin gene (TUB2) and two α-tubulin genes (TUB1 and TUB3). Mutations can be readily introduced into these genes and thereby studied in a homogenous tubulin population9,10. There are a number of widely available constructs for labeling microtubules, and these can be targeted for integration at chromosomal loci for stable expression (see the Discussion).
The overall goal of this method is to image single microtubules in living yeast cells in four dimensions (X, Y, Z, and T) for high-resolution measurements of microtubule dynamics. Methods for integrating constructs for the constitutive, low-level expression of fluorescently labeled tubulin in yeast cells are described. Prior to imaging, living cells are mounted into slide chambers coated with the lectin Concanavalin A to stabilize the cells for long-term imaging. The optimal parameters for image acquisition, as well as a workflow for data analysis, are also described.
Protocol
1. Preparing -LEU Dropout Plates
Add 800 mL of sterile, deionized water (DiH2O) to a 2 L flask. Add 26.71 g of dropout base (DOB) powder, 0.69 g of CSM-Leu, and 20 g of agar. Mix with a magnetic stir bar. Bring to 1 L with DiH2O.
Autoclave at 121 °C and 19 psi for 30 min.
Remove the flask from the autoclave and allow it to cool to ~65 °C with gentle stirring.
Pour 30 mL/plate into 100 mm diameter plates. Let these sit at room temperature for 36-48 h before storing at 4 °C.
2. Integrating GFP-Tub1 for the Constitutive Expression of GFP-labeled Tubulin
Boil a stock concentration of 10 mg/mL single-stranded DNA (ssDNA) for 3 min.
Combine 2 µL of boiled ssDNA with 400 ng of purified GFP-Tub1 plasmid DNA11. NOTE: There are a variety of plasmids that can be used for the stable, fluorescent labeling of microtubules in yeast that utilize alternative fluorophores and selectable markers. Some of these alternatives are described in the Discussion section.
Add the DNA mixture to a 50 µL aliquot of competent yeast cells. NOTE: Prepare competent yeast cells with sorbitol solution (SORB; 100 mM LiOAc, 10 mM Tris-HCl pH 8, 1 mM EDTA pH 8, and 1 M sorbitol, adjusted with dilute acetic acid to pH 8). For a detailed description of making competent yeast cells, see reference12.
Vortex thoroughly.
Add 6x the total volume of polyethylene glycol (PEG) solution (100 mM LiOAc, 10 mM Tris-HCl pH 8, 1 mM EDTA/NaOH pH 8, and 40% PEG3350) to the DNA-cell mixture.
Vortex thoroughly.
Incubate for at least 1 h at 30 °C with gentle agitation.
Add dimethyl sulfoxide (DMSO) to 10% of the total volume and vortex.
Incubate at 42 °C for 15 min.
Centrifuge at 2,500 x g for 5 min.
Remove the supernatant and suspend the cells in 100 µL of DiH2O.
Plate cells on an -LEU dropout plate. NOTE: The GFP-Tub1 construct contains a leucine auxotrophic marker, while other constructs could use an alternative marker. The dropout medium should be specific to the auxotrophic marker of the construct.
Allow the transformed cells to grow for 3-5 days at 30 °C.
Re-streak single transformation colonies on a fresh -LEU dropout plate by transferring colonies using an autoclaved toothpick. Incubate the plate overnight at 30 °C.
- Visually screen each transformant for a GFP signal by suspending approximately 1 µL of cells from a single colony from the plate in step 2.14 in 2 µL of water on a clean slide. Cover the suspended cells with a coverslip and observe using fluorescence microscopy.
- Excite the transformants with 488 nm light and look for the presence of a GFP signal. NOTE: This step can use a spinning disk confocal microscope, but a widefield microscope equipped with a 100X objective and appropriate optics for imaging GFP will suffice. Cells with no visible GFP-labeled microtubules, or those with abnormally high fluorescence, should be rejected. After this, the yeast strains are ready to be cultured for imaging.
3. Growing Liquid Yeast Culture
Inoculate ~1 µL of yeast cells from a single colony on a plate in 3 mL of synthetic complete medium. Allow the cells to grow overnight at 30 °C with shaking at 250 rpm.
Dilute the saturated culture the following day to an OD600 of <0.1 in the medium. Return the diluted culture to the 30 °C shaker for 3-4 h or until the OD600 is between 0.4 and 0.8, when the cells are ready to be loaded into chambers for imaging.
4. Preparing Flow Chamber Slides
Cut a sheet of paraffin film into a piece 4 in wide and 7 in long.
Place a standard microscope slide lengthwise on the 4 inch width of paraffin film and wrap the film around the slide 3-4 times such that the slide is covered in film that is 3-4 layers thick. Press the layers together to ensure there is a reasonable seal; this will make it easier to cut. Avoid pressing too hard or causing the film to wrinkle.
Cut the paraffin film along the outside edges of the slide using a clean box cutter razor. Use another slide to provide a straight edge for cutting.
Peel the excess film off the working slide and discard. The remaining stacks of film will cover one face of the slide and will be used to make the walls between imaging chambers.
Using the second slide as a straight edge, cut the stacked film layers along the long axis of the slide into approximately ten strips, each 2-4 mm in width.
Cut the stacked paraffin film layers perpendicularly to the cuts made in step 4.5, along the short axis of the slide, to create strips 2-4 mm in width, 23-24 mm in length, and 3-4 layers thick.
Peel the cut strips using a razor at a 45° angle. Using forceps, evenly distribute the cut film strips on a clean microscope slide to create the desired numbers of chambers. Up to 3 chambers (made with 4 paraffin film strips) can be created on one slide.
Place a single coverslip on top of the strips of film and move it into position with forceps.
Place the prepared slide, coverslip up, on a hotplate set at ~95 °C (this is approximately the low-medium setting on most hotplates). Heat the slide until the film strips are translucent (approximately 3 min) and then seal the slide and coverslip together by gently pressing on the coverslip with the tweezers. NOTE: It is important to only press on the regions of the coverslip that are directly above the paraffin film strips, as scratching the regions above the chambers can diminish image quality. Take care not to overheat the slide; vaporized film will coat the inside of the chamber with a hydrophobic film that prevents the cells from adhering.
Use forceps to remove the slide from the hotplate (the slide will be hot) and set it aside to cool with the coverslip side facing up; as the slide cools, the film will return to a white, opaque coloration. NOTE: At this point, the slides are ready to be loaded with cultured yeast cells. Extra slides can be made to this point and stored in a clean and dry location for an indefinite amount of time.
5. Loading Yeast Cells into Prepared Flow Chamber Slides
Thaw a frozen, 200 µL aliquot of Concanavalin A (2 mg/mL in sterile DiH2O).
Add approximately 100 µL of thawed Concanavalin A to each chamber of the prepared slide by pipetting into one of the open ends. Add enough Concanavalin A to fill the chamber. For this step, the Concanavalin A will be pulled through the chamber via capillary action.
Hang the slide, coverslip-down, between two microfuge tube racks or pens for 5 min to allow the Concanavalin A to adhere to the coverslip (i.e., the chamber).
- Return the slide to a coverslip-up position. Wash the chamber with 2x the chamber volume (determined in step 5.2, above; approximately 200 µL) of synthetic complete medium to remove the Concanavalin A.
- Flow the medium through the chamber, using either the corner of a paper towel or a filter paper folded in half to draw fluid out of one end of the chamber while simultaneously pipetting fresh fluid into the other. Alternatively, use a vacuum aspirator to carefully draw fluid out of one end while pipetting fresh fluid into the other.
Load cells by flowing 2x the chamber volume (approximately 200 µL) of cell suspension (from step 3.4) using the directed flow method described in step 5.4.1. NOTE: The goal is to image a single layer of cells at the coverslip. Therefore, it is important to load a concentration of cells small enough that they do not pile on top of one another, but large enough that sufficient cells are present in the field to collect the maximum amount of data per image acquisition. The optimal concentration of cells within the chamber can vary and should be empirically determined. To increase the concentration of cells in the flow chamber, gently pellet the cell culture at 1,000 x g for 2 min and remove some of the supernatant. To decrease the concentration of cells, add fresh synthetic complete medium to the cell suspension. The concentration of cells can be evaluated after step 5.7 (below) by inspecting the chamber on a phase contrast microscope. At this point, additional cells can be added to the chamber by flowing in more cell suspension. However, reducing cell density at this point is not feasible and is best achieved by loading a new chamber with a diluted cell suspension.
Hang the slide coverslip-down, as described in step 5.3, for 10 min to allow the cells to adhere to the coverslip.
Flush out any unadhered cells by flowing through 4x the chamber volume (approximately 400 µL) of synthetic complete medium. This will eliminate free-floating cells in the chamber, which can contaminate image acquisition.
Carefully dry each end of the chamber with a clean corner of a paper towel, bringing the meniscus to the edge of the coverslip.
Seal each end of the chamber with warm (75 °C) sealant (e.g., VALAP; equal parts petroleum jelly, wool wax, and paraffin wax). NOTE: At this point, the slide is ready to image. The chambers can be imaged for up to 9 h, depending on the density of cells in each chamber. The cells will produce carbon dioxide (CO2) over time, which will gradually create bubbles that displace the cells.
6. Image Acquisition
Bring the slide to an inverted microscope equipped with a 1.45 NA 100X CFI Plan Apo objective, a piezoelectric stage, a spinning disk confocal scanner unit, an illumination laser, and an EMCCD camera. Maintain the temperature of the stage at room temperature (25 °C) during acquisition. NOTE: The high numerical aperture allows for a greater image resolution, and the piezoelectric stage enables high-speed z-stack acquisition. The minimum microscope requirements are the 100X objective, the illumination laser, and the EMCCD camera.
Bring the cells into focus. Ensure that the acquisition field has at least 5-6 cells clustered together by searching the slide for cells grouped together. NOTE: Although it is not necessary to image more than one cell at a time, imaging multiple cells in the same field improves the efficiency of data acquisition without impairing the quality of data. However, it is critical that the cells are on the same focal plane and not piled on top of one another.
- Program a multi-dimensional acquisition to collect multiple z-series over time.
- Adjust the settings within the active acquisition settings to the following: total Z-depth = 6 µm, to encompass the entire yeast cell; Z-step size = 300 nm, to maximize light collection without oversampling; total time duration = 10 min; time intervals = Z-series every 4 s; and exposure time = 90 ms for each z-plane. NOTE: The optimal exposure time will vary depending on the camera, the excitation light source, and other factors. In general, the optimal exposure time will be the minimum time necessary to achieve a 3:1 ratio of signal from GFP-labeled astral microtubules to signal from the cytoplasm based on pixel values measured by the EMCCD.
Begin the acquisition by clicking 'Run'. Allow it to run for the full 10 min duration. Save the data as an image series (.tiff or .nd2).
Select a new field to image within the chamber and repeat steps 6.2-6.4. NOTE: A single chamber should yield between 15 and 20 fields of cells, depending on the cell density within the chamber. Higher cell densities will yield lower total fields, as the CO2 in the chamber will build up faster.
7. Image Analysis
- Open the image file in ImageJ (https://imagej.nih.gov) as a hyperstack by using the bio-formats importer plugin.
- Open ImageJ and drag the acquired image stack from section 6 directly onto the control panel. The bio-formats importer will start automatically. Select "Ok" to load the image stack as a hyperstack. NOTE: The "Bio-formats Import Options" prompt will open and ensure that under the "Stack Viewing" section, "View stack with:" is set to "Hyperstack" and "Stack Order:" is set to "XYCZT."
- Collapse each Z-series into a maximum-intensity, 2-dimensional projection using the Z-project function.
- Select the "Image" menu, the "Stack" submenu, and the "Z-project" feature. Select "Maximum projection" from the drop-down list and click "Ok."
Apply a median filter to the image stack to reduce speckling noise by selecting the "Process" menu, the "Filters" submenu, and the "Median" feature. Enter 2.0 pixels into the radius box and click "Ok."
Identify pre-anaphase cells in the field. NOTE: These are characterized by medium-sized buds and a short mitotic spindle, 1-1.5 µm in length (Figure 1; the arrowhead points to a short mitotic spindle). Cells at this stage are ideal for analyzing microtubule dynamics because they typically exhibit only a few astral microtubules emanating the spindle poles7. Astral microtubules can be identified as linear filaments that exhibit a uniform GFP signal intensity from the minus end, at the SPB, to the plus end, in the cytoplasm.
Starting at the first timepoint of the image series, use the segmented line tool to draw a line along a single astral microtubule, from the minus end, located at the junction between the astral microtubule and the more intensely labeled spindle microtubules, to the plus end, which is in the cytoplasm (see the red lines in Figure 1). Double-click on the end of the microtubule to complete the segmented line.
Record the measurement in the results table using the "measure" feature by pressing the "M" button on the keyboard. NOTE: Additional features, such as gray intensity, can be recorded by setting the measurements in the "Analyze" menu and "Set Measurements" submenu.
Advance to the next timepoint by either clicking the right arrow in the Z-slider bar or pressing the "./>" key on the keyboard. Repeat steps 7.5 and 7.6 for all timepoints in the image sequence.
Copy the data from the results table and paste them into a spreadsheet. Save the data by selecting "Save As." NOTE: These measurements will include several values, but the most important values are the slice, which indicates the timepoint in the series, and the length, which indicates the length of the line (i.e., the astral microtubule). Columns containing other measurements (e.g., the area, mean pixel value, angle, etc.) can either be kept or discarded.
Create a new column in the spreadsheet by right-clicking and using the "Insert" function. Input the time (s) of each timepoint.
- Create an XY scatter plot of microtubule length (µm) as a function of time (s) using the "Insert Line Chart" feature; select the length measurements as "Y" and the time column as "X."
- Identify regions of polymerization, depolymerization, and pause by looking for positive and negative changes in length over time on the scatter plot from step 7.10. NOTE: Polymerization events increase microtubule length by at least 0.5 µm across a minimum of 3 timepoints and fit a linear regression with an R2 ≥ 0.9 and an R-value > 0 (Figure 1B and D, green points). Depolymerization events decrease microtubule length by at least 0.5 µm across a minimum of 3 timepoints and fit a linear regression with an R2 ≥ 0.9 and an R-value < 0 (Figure 1B and D, pink points). Pause events are separate from polymerization and depolymerization. Pauses are defined as net changes in microtubule length less than 0.5 µm across a minimum of 3 timepoints; fit a linear regression with an R-value between -0.4 and 0.4; and have a slope that is not statistically different from zero, as determined by an F-test.
- Using the scatter plot as a guide, identify the sections of time when the length change is positive and highlight them in a green color. Highlight regions of time when the length change is negative in a pink color.
- Calculate the linear regression of each highlighted section and record the R- and R2-value for each section in the columns proximal to the regions. Classify each colored region as either a polymerization event or a depolymerization event.
Calculate the polymerization rates by dividing the net change in length by the change in time for each growth event. Record these results in the column adjacent to the linear regression values from step 7.10.3.
Calculate the depolymerization rates by dividing the net change in length by the change in time for each shrinking event. Record these results in the column adjacent to the polymerization rate values from step 7.11.
Calculate the catastrophe frequency by dividing the number of polymerization-to-depolymerization transitions by the total time of all growth events13. Record these results in the column adjacent to the depolymerization rate values from step 7.12.
Calculate the rescue frequency by dividing the number of depolymerization-to-polymerization transitions by the total time of all shrinkage events13. Record these results in the column adjacent to the catastrophe frequency values from step 7.13.
Calculate the dynamicity by dividing the sum of the absolute value of all length changes by the total duration of the image acquisition, calculated in step 7.10.1, and multiplying by 27.0833 to obtain tubulin subunits per second14. Record these results in the column adjacent to the rescue frequency values from step 7.14 and save the results table. NOTE: It is possible to automate the analysis process described in steps 7.10-7.15 using a custom-made program (Estrem, et al., manuscript submitted). The program is designed to identify periods of polymerization and depolymerization in the length measurements by following the parameters detailed above.
Representative Results
Measuring microtubule dynamics in living yeast cells provides a compelling tool to assess how mutations in genes encoding microtubule regulators or tubulin subunits impact polymerization and depolymerization rates, as well as the frequency of transition between these states. Figure 1 displays a time series of astral microtubule dynamics in a wild-type cell and a mutant cell with a mutation in β-tubulin (tub2-430Δ). Microtubules are labeled with GFP-tagged α-tubulin to visualize microtubule length. The red arrows trace the length of the astral microtubule in each image (Figure1A and 1C). Microtubule lengths were measured at each timepoint for 8 min, and these compiled lengths are depicted in life plots (Figure 1B and 1D). These data were used to determine the polymerization rate, depolymerization rate, polymerization duration, depolymerization duration, rescue frequency, catastrophe frequency, and dynamicity.
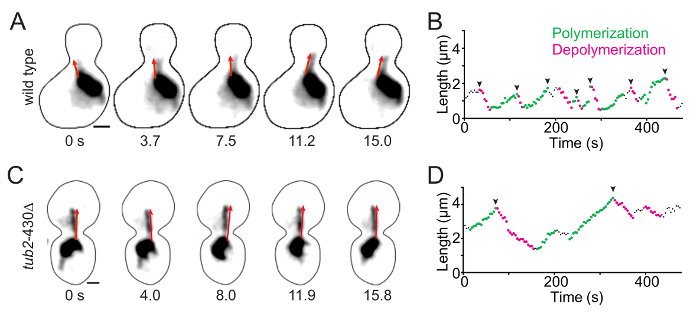 Figure 1. Time-lapse Imaging of GFP-labeled Microtubules and Measurements of Astral Microtubule Dynamics. (A and C) The first five sequential images of time-lapse imaging for the wild-type and tub2-430Δ mutant cells. Microtubules are labeled with GFP-Tub1 and imaged in pre-anaphase of the cell cycle. Each image is a maximum-intensity projection from a confocal Z-series. Scale bars: 1 µm. Timestamps shows the total time at each frame acquisition. The red arrows follow along the total length of an astral microtubule, with the arrowheads denoting the plus ends. (B and D) Microtubule life plots display the lengths of single astral microtubules over time. The green points denote polymerization and the pink points denote depolymerization. The arrowheads point to catastrophe events. Please click here to view a larger version of this figure.
Figure 1. Time-lapse Imaging of GFP-labeled Microtubules and Measurements of Astral Microtubule Dynamics. (A and C) The first five sequential images of time-lapse imaging for the wild-type and tub2-430Δ mutant cells. Microtubules are labeled with GFP-Tub1 and imaged in pre-anaphase of the cell cycle. Each image is a maximum-intensity projection from a confocal Z-series. Scale bars: 1 µm. Timestamps shows the total time at each frame acquisition. The red arrows follow along the total length of an astral microtubule, with the arrowheads denoting the plus ends. (B and D) Microtubule life plots display the lengths of single astral microtubules over time. The green points denote polymerization and the pink points denote depolymerization. The arrowheads point to catastrophe events. Please click here to view a larger version of this figure.
Figure 1 demonstrates altered microtubule dynamics between a wild-type strain and a mutant strain lacking the 27 amino acids of β-tubulin on the C-terminal, a mutant that was previously shown to have more stable microtubules15. As displayed in the microtubule life plots, the microtubule in the tub2-430Δ mutant is longer and exhibits fewer catastrophes than the wild-type microtubule (Figure 1B and D arrowheads; Table 1). In addition, the polymerization and depolymerization rates, determined from the slopes of the ascending and descending microtubule lengths, are decreased in the mutant compared to the wild-type (Figure 1B and 1D; Table 1). Finally, the microtubule in the tub2-430Δ mutant cell spends more time in states of polymerization and depolymerization and has decreased dynamicity compared to the wild-type (Table 1).
| Strain | Dynamicity (subunits/s) | Polymerization rate (µm/min) | % Time in Polymerization | Depolym rate (µm/min) | % Time in Depoly- merization | % Time in Pause | Rescue frequency (events/min) | Catastrophe frequency (events/min) |
| Wild-type n=1 | 54 | 1.8 ± 0.5 | 48 | 3.0 ± 0.3 | 29 | 3 | 1.8 | 1.4 |
| tub2-430∆ n=1 | 40 | 1.4 ± 0.1 | 41 | 1.5 ± 0.2 | 32 | 0 | 0.8 | 0.7 |
| Abbreviations are as follows: µm, micron; min, minute; s, second; n, single microtubule analyzed from microtubule life plot (Fig1B,D). Values are mean ± standard error of the mean. |
Table 1: Microtubule Dynamics Measurements for Cells in Figure 1. The values are the mean ± standard error of the mean from measurements of the single microtubules shown in Figure 1.
Data such as this can be acquired from individual life plots of multiple microtubules (on average, at least 50 microtubules are analyzed) and used in statistical analysis to determine significant changes in microtubule dynamics across a population.
Discussion
The budding yeast model offers major advantages for gathering high-resolution measurements of microtubule dynamics in an in vivo setting, including the ability to image single microtubules over time and the ability to manipulate tubulins and microtubule regulators using the tools of yeast genetics.
The Concanavalin A-coated chambers provide a number of advantages over previously described apparatuses, including molten agar pads. Slides with chambers can be pre-made and stored long term or can be used immediately. Additionally, the Concanavalin A-coated chambers are capable of culturing yeast cells for >6 h in the same medium in which the cells were originally cultured. It is important to note that the number of cells within the chamber will dictate how long the chamber can be imaged. The coated chambers ensure that only a single layer of yeast cells adheres to the coverslip. Finally, creating multiple chambers allows for several different yeast strains to be imaged on the same slide.
There are major challenges to imaging microtubule dynamics in budding yeast, including relatively low signal levels and the highly dynamic nature of the process. Widefield microscopes are adequate for capturing single-timepoint images of labeled microtubules. However, widefield microscopy causes faster photobleaching due to longer exposure times and the bombardment of the sample with out-of-focus light. Likewise, laser scanning confocal microscopes are inadequate due to faster photobleaching. Spinning disk confocal microscopy is uniquely suited for imaging microtubule dynamics in budding yeast. The spinning disk confocal microscope system used here was designed with these requirements in mind.
The microscope system must be designed for high speed and high sensitivity. Although a variety of microscope systems may prove suitable, several components are essential. A 100X objective with a high numerical aperture (1.45NA) is necessary to resolve individual microtubules and length changes on the order of 100 nm. A spinning disk confocal scanner is necessary to minimize photobleaching and toxicity, by blocking out-of-focus light, and to maximize resolution and sensitivity, by gathering only in-focus light. The EMCCD camera must be fast and sensitive to collect enough GFP-Tub1 signal during each 90-ms of the time-lapse acquisition. Currently, EMCCD cameras are the best choice for this application. Current CMOS sensor cameras lack the sensitivity of high-end EMCCDs; however, this technology is improving significantly and may soon be suitable. Finally, a piezoelectric stage is needed for fast and precise movement in Z, which can be the rate-limiting step in four-dimensional image acquisition.
The specific microscope system described in this protocol may present a barrier if similar equipment is not readily available. Alterations can be made to accommodate different imaging systems, but these modifications may come at the expense of temporal and/or spatial resolution. A spindle-driven stage motor can be used in place of the piezoelectric stage. However, these motors are slower, less accurate, and prone to drift. Post-acquisition image-stabilizing plug-ins can be used to minimize the drift in the XY dimensions associated with these motor-driven stages. In addition, alternate cameras with slower readout speeds can be used. To accommodate a slower acquisition rate, Z-series could be collected less often (i.e., more time between Z-series). However, increasing the time between Z-series risks missing transitions that may occur between timepoints. Alternatively, the distance between Z-planes can be increased as a way of decreasing the number of images collected per Z-series. However, increasing this spacing risks losing image information between planes, which will impair the experimenter's ability to accurately identify microtubule ends. Clearly, alterations can be made to accommodate alternative imaging systems, but these alterations must be weighed against the potential loss of image information.
Another limitation of this protocol is the loss of spatial information when three-dimensional image stacks are converted to two-dimensional image projections. An individual astral microtubule in a pre-anaphase yeast cell is typically between 0.5 and 2.0 µm in length and projects across the three-dimensional space of the cytoplasm. It is therefore critical to acquire images from a series of Z-positions to collect all the spatial information of an individual microtubule. In a time-lapse acquisition, this can produce a complex data set, with 20 Z-slices at each of the 150 timepoints, for a total of 3,000 images. To address this challenge and to simplify the image analysis, the spatial information of the Z-stacks is compressed during the analysis. While this benefits analysis, it comes at the cost of spatial information from the Z-dimension. How much information is lost when the Z-dimension is compressed? To estimate this loss, experiments were conducted to measure the distance between SPBs in pre-anaphase cells, which are relatively easy to measure because Spc110-GFP-labeled SPBs exhibit spherical foci with high signal-to-noise ratios. Although the spindle may exhibit different displacement in Z than astral microtubules, the average length is similar (~1.5 µm) and therefore provides a useful proxy. Spindle lengths are, on average, 15% longer when measured in full three-dimensional image stacks than when the same spindles are measured in two-dimensional projections (J.M., unpublished results). Thus, the benefit of the simplified data set and measurements should be weighed against the cost of discarded Z information.
The yeast cell biology community has developed a versatile toolkit for imaging microtubule networks. This includes a variety of fluorescent proteins fused to α-tubulin/Tub1 and marked with various auxotrophic markers. These constructs include LEU2::GFP-TUB1 (pSK1050)11, which integrates at the chromosomal LEU2 locus, and several fusions to GFP and other fluorophores, which integrate at the URA3 locus, including URA3::GFP-TUB1 (pAFS125)16, URA3::CFP-TUB1 (pAFS125-CFP), and URA3::pHIS3-mCherry-TUB1 (pAK011)17. In addition, a set of Tub1 fusions to diverse fluorescent proteins has recently been created that target integration to either the chromosomal URA3 locus or to a locus adjacent to the native TUB1 locus18. This extensive palette of fluorophores and markers is very useful for co-localization experiments with differentially tagged proteins. The GFP-Tub1 fusion remains the best tool for the time-lapse imaging of microtubule dynamics due to its brightness and photostability. The GFP-Tub1 fusion created by Song & Lee (pSK1050)11 contains a fusion of GFP to the amino-terminus of α-tubulin/Tub1 and integrates at the LEU2 locus to rescue leucine auxotrophy. Thus, the fusion protein is expressed ectopically in addition to native α-tubulin and comprises ~20% of the total α-tubulin in the cell9. Importantly, the GFP-Tub1 fusion does not rescue the function of native α-tubulin/Tub1 (J.M., unpublished results). This is also true of other TUB1 fusion constructs18.
With the available tools to visualize microtubules and their binding proteins, budding yeast is an ideal system to study mutations in tubulins and other microtubule regulators. Additionally, the limited number of tubulin genes allows for the direct study of a homogenous pool of mutant tubulin. In addition to mutations in tubulin genes, budding yeast has a number of microtubule-associated proteins (MAPs) that are homologous to metazoan counterparts. These MAPs can be studied directly, and the effects that mutations have on single microtubule filaments can also be examined. The protocol detailed here allows for the investigation of both the intrinsic and extrinsic processes that influence microtubule dynamics.
In summary, microtubules must be dynamic to function properly during many cellular processes. It is necessary to understand the factors intrinsic to tubulin and the extrinsic associated proteins that regulate microtubule dynamics. Budding yeast is an ideal system to study the effect of these factors by directly quantifying the dynamics of a single microtubule. The protocol detailed above describes how to stably integrate a fluorescent fusion protein into the yeast genome and how to acquire and analyze confocal image stacks to quantify microtubule dynamics in vivo. Finally, this versatile protocol can be adapted to study the dynamics of any protein in budding yeast, so long as the protein of interest is fused to a suitable fluorescent protein.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We thank Kerry Bloom (University of North Carolina), Kyung Lee (NCI), Steven Markus (Colorado State University), and Elmar Schiebel (Universität Heidelberg) for sharing various FP-TUB1 plasmids. We are grateful to Melissa Gardner (University of Minnesota) for training us in the slide chamber preparation method. This work was supported by the National Institutes of Health (NIH) grant R01GM112893-01A1 (to J.K.M.) and T32GM008730 (to C.E.).
References
- Zanic M, Widlund PO, Hyman AA, Howard J. Synergy between XMAP215 and EB1 increases microtubule growth rates to physiological levels. Nat Cell Biol. 2013;15(6):688–693. doi: 10.1038/ncb2744. [DOI] [PubMed] [Google Scholar]
- Mimori-Kiyosue Y, Shiina N, Tsukita S. The dynamic behavior of the APC-binding protein EB1 on the distal ends of microtubules. Curr Biol. 2000;10(14):865–868. doi: 10.1016/s0960-9822(00)00600-x. [DOI] [PubMed] [Google Scholar]
- Byers B, Goetsch L. Behavior of spindles and spindle plaques in the cell cycle and conjugation of Saccharomyces cerevisiae. J Bacteriol. 1975;124(1):511–523. doi: 10.1128/jb.124.1.511-523.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marschall LG, Jeng RL, Mulholland J, Stearns T. Analysis of Tub4p, a yeast gamma-tubulin-like protein: implications for microtubule-organizing center function. J Cell Biol. 1996;134(2):443–454. doi: 10.1083/jcb.134.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang A, Geissler S, Grein K, Schiebel E. gamma-Tubulin-like Tub4p of Saccharomyces cerevisiae is associated with the spindle pole body substructures that organize microtubules and is required for mitotic spindle formation. J Cell Biol. 1996;134(2):429–441. doi: 10.1083/jcb.134.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winey M, Bloom K. Mitotic spindle form and function. Genetics. 2012;190(4):1197–1224. doi: 10.1534/genetics.111.128710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta MLJ, et al. beta-Tubulin C354 mutations that severely decrease microtubule dynamics do not prevent nuclear migration in yeast. Mol Biol Cell. 2002;13(8):2919–2932. doi: 10.1091/mbc.E02-01-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK, Cooper JA. Coordinating mitosis with cell polarity: Molecular motors at the cell cortex. Semin Cell Dev Biol. 2010;21(3):283–289. doi: 10.1016/j.semcdb.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartz Hanson M, et al. Novel alpha-tubulin mutation disrupts neural development and tubulin proteostasis. Dev Biol. 2016;409(2):406–419. doi: 10.1016/j.ydbio.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fees CP, Aiken J, O'Toole ET, Giddings THJ, Moore JK. The negatively charged carboxy-terminal tail of beta-tubulin promotes proper chromosome segregation. Mol Biol Cell. 2016;27(11):1786–1796. doi: 10.1091/mbc.E15-05-0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Lee KS. A novel function of Saccharomyces cerevisiae CDC5 in cytokinesis. J Cell Biol. 2001;152(3):451–469. doi: 10.1083/jcb.152.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, et al. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast. 1999;15(10B):963–972. doi: 10.1002/(SICI)1097-0061(199907)15:10B<963::AID-YEA399>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Kosco KA, et al. Control of microtubule dynamics by Stu2p is essential for spindle orientation and metaphase chromosome alignment in yeast. Mol Biol Cell. 2001;12(9):2870–2880. doi: 10.1091/mbc.12.9.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toso RJ, Jordan MA, Farrell KW, Matsumoto B, Wilson L. Kinetic stabilization of microtubule dynamic instability in vitro by vinblastine. Biochemistry. 1993;32(5):1285–1293. doi: 10.1021/bi00056a013. [DOI] [PubMed] [Google Scholar]
- Aiken J, Sept D, Costanzo M, Boone C, Cooper JA, Moore JK. Genome-wide analysis reveals novel and discrete functions for tubulin carboxy-terminal tails. Curr Biol. 2014;24(12):1295–1303. doi: 10.1016/j.cub.2014.03.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight AF, Marshall WF, Sedat JW, Murray AW. Mitosis in living budding yeast: anaphase A but no metaphase plate. Science. 1997;277(5325):574–578. doi: 10.1126/science.277.5325.574. [DOI] [PubMed] [Google Scholar]
- Khmelinskii A, Lawrence C, Roostalu J, Schiebel E. Cdc14-regulated midzone assembly controls anaphase B. J Cell Biol. 2007;177(6):981–993. doi: 10.1083/jcb.200702145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus SM, Omer S, Baranowski K, Lee WL. Improved Plasmids for Fluorescent Protein Tagging of Microtubules in Saccharomyces cerevisiae. Traffic. 2015;16(7):773–786. doi: 10.1111/tra.12276. [DOI] [PMC free article] [PubMed] [Google Scholar]