

Abstract
There is a need for intraoperative imaging technologies to guide breast-conserving surgeries and to reduce the high rates of re-excision for patients in which residual tumor is found at the surgical margins during post-operative pathology analyses. Feasibility studies have shown that utilizing topically applied surface-enhanced Raman scattering (SERS) nanoparticles (NPs), in conjunction with the ratiometric imaging of targeted vs. untargeted NPs, enables the rapid visualization of multiple cell-surface biomarkers of cancer that are over-expressed at the surfaces of freshly excised breast tissues. In order to reliably and rapidly perform multiplexed Raman-encoded molecular imaging (REMI) of large numbers of biomarkers (with 5 or more NP flavors), an enhanced staining method has been developed in which tissue surfaces are cyclically dipped into a NP-staining solution and subjected to high-frequency mechanical vibration. This dipping and mechanical vibration (DMV) method promotes the convection of the SERS NPs at fresh tissues surfaces, which accelerates their binding to their respective biomarker targets. By utilizing a custom-developed device for automated DMV staining, we demonstrate the ability to simultaneously image 4 cell-surface biomarkers of cancer at the surfaces of fresh human breast tissues with a mixture of 5 flavors of SERS NPs (4 targeted and 1 untargeted control) topically applied for 5 min and imaged at a spatial resolution of 0.5 mm and a raster-scanned imaging rate of >5 cm2/min.
Keywords: nanomedicine, molecular imaging, biomedical optical imaging, cancer detection, Raman spectroscopy
1. Introduction
Breast-conserving surgery (a.k.a. partial mastectomy or lumpectomy) is carried out on a majority of the ~200,000 patients who are diagnosed with early-stage breast cancer each year in the United States.[1] Unfortunately, studies have shown that 20%−60% of lumpectomy patients require additional re-excision surgeries if post-operative pathology reveals that the resection margins are positive for tumor.[2] In the past, the existence of a close margin, in which post-operative pathology revealed the presence of tumor cells within 2 or 3 millimeters of the surgical margin surface (the “inked” surface), was often used as a criterion to warrant re-excision. However, recent consensus publications advocate defining a negative margin as “no tumor at the inked surface” (i.e. no tumor at the most superficial surface of the surgical excision), especially for invasive breast cancer.[3] Regardless of an institution’s criterion for re-excision, there is no debate that the presence of any subtype of breast tumor at the surgical margin surface (“tumor on ink”) is unacceptable and that there would be value in an intraoperative technology to comprehensively assess these surfaces for the presence of residual tumor at the final stages of lumpectomy. High specificity of tumor detection for such an intraoperative method would be necessary to minimize over-excision and to optimize patient cosmesis (a major goal of breast-conserving surgeries). However, if post-operative pathology could continue to be relied upon as a gold standard to assess the extent of resection, then even moderate tumor-detection sensitivity for an intraoperative method would greatly reduce the rate of re-excision surgeries and result in significant cost savings and benefits to patients (e.g. less risk of iatrogenic injury, less psychological and emotional distress from multiple surgeries, etc.).
Frozen-section pathology is utilized to guide the resection of certain tumor types. However, frozen sectioning is difficult to perform for breast tissues due to their high lipid content and suffers from severe sampling errors since only small numbers of tissue sections (typically 5-μm thick) can be rapidly prepared on slides and visualized under a microscope. As a result, frozen sectioning can yield significant false-negative rates.[4] In addition, frozen sections require the destruction of valuable tissues that should otherwise be assessed via archival formalin-fixed paraffin-embedded (FFPE) histopathology. There are other optical strategies in various stages of preclinical and/or clinical development, such as confocal mosaicing microscopy (CMM),[5] multiphoton microscopy,[6] light reflectance spectroscopy (LRS),[7] autofluorescence lifetime measurement (AFLM),[7b] intrinsic Raman spectroscopy,[8] and touch-prep cytology.[9] These techniques all have the potential to improve lumpectomy procedures, but with certain limitations,[10] and there are currently no intraoperative tools to enable surgeons to detect positive margins with high sensitivity and specificity, and to do so comprehensively for the entire surgical-margin surface.
The molecular imaging of overexpressed cell-surface biomarkers that are uniquely overexpressed by tumors is an approach with the potential to provide extremely high tumor-detection specificity.[11] However, in order to achieve high tumor-detection sensitivity, multiplexed biomarker detection is necessary since the molecular phenotypes of most tumors, including breast tumors, vary greatly between individuals as well as within a single tumor mass.[12] Recently, surface-enhanced Raman-scattering (SERS) nanoparticles (NPs), hereafter referred to as “SERS NPs” or “NPs”, have attracted interest due to their multiplexing capability.[13] These SERS NPs exist as various “flavors,” each of which generates a characteristic spectral “fingerprint” when illuminated by a single laser at one wavelength.[14] By conjugating various flavors of SERS NPs to different antibodies and applying them simultaneously on tissues, the NPs can target a panel of protein biomarkers of cancer. In order to perform quantitative imaging of multiple biomarkers at the surfaces of fresh tissues, we have been developing a Raman-encoded molecular imaging (REMI) technology that is enabled by a rapid topical-staining protocol with targeted SERS NPs.[10] A critical component of the REMI technique is the use of a ratiometric-imaging method, which allows for the accurate quantification of biomarker expression levels by utilizing one untargeted NP flavor to normalize for any nonspecific accumulation of the NPs, as for example due to off-target binding, uneven topical delivery and washout, and variations in tissue permeability and retention.[15]
We have recently demonstrated that the topical application and ratiometric imaging of three SERS NPs (two targeted and one isotype control) can enable the simultaneous quantification of two biomarkers in fresh tissues within 15 min.[10,15a,16] While these results were valuable for demonstrating the feasibility of the REMI approach for identifying residual tumors at the surfaces of fresh excised tissues, improving the degree of multiplexing would further improve the sensitivity of REMI for tumor detection. Previous studies have used untargeted SERS NPs to show that the multiplexed imaging of up to ten flavors of SERS NP is feasible.[14,17] However, the multiplexed imaging of more than two biomarkers with targeted SERS NPs has never been achieved in practice in real tissues.
In order to extend REMI for the imaging of four biomarkers (with a mixture of five NPs) or more, a major challenge to overcome is the fact that the detection limits and accuracy of REMI gradually deteriorates as increasing numbers of NP flavors are multiplexed. This is primarily due to spectral crosstalk and insufficient signal-to-noise ratios (SNR) of the detected Raman spectra (Figure S2, Supporting Information).[16b] To improve the degree of multiplexing for REMI, it is necessary to improve the SERS signal intensity (NP concentrations) in NP-stained tissues, while maximizing the ratio of targeted vs. untargeted NPs (molecular contrast) in biomarker-positive tissues. An additional goal is to improve the speed of staining (previously ~10 min) to improve the clinical utility of REMI.
Through extensive optimization studies, we have developed a method to significantly enhance the speed and the degree of multiplexing of REMI by periodically dipping tissue specimens into and out of a NP-staining solution in conjunction with high-frequency mechanical vibration (hereafter referred to as “DMV staining” – “dipping and mechanical vibration”). To validate our technique, we demonstrate that this method enables multiplexed imaging of 5 or more NP flavors after a 2- to 5-min topical application procedure. In addition, to perform DMV staining optimally and reproducibly, an automated staining device has been developed to simultaneously stain multiple tissues.
2. Results and discussion
2.1. Dipping and mechanical vibration (DMV) to enhance the efficiency of topical staining
Optimization experiments have been performed to improve the speed and effectiveness of the topical staining of fresh tissues surfaces with SERS NPs. A staining method was designed to enable the surface of a fresh tissue shaving (typically 4- to 8-mm thick) to be stained while preventing NPs from staining and generating interfering signal from the other side of the tissue shaving. A variety of staining methods were tested and are depicted in Figure 1a. The method labeled as “Static I” involves staining tissue shavings with NPs applied on the top surface of the tissue. However, this leads to NPs migrating to the bottom surface due to gravity, which is not ideal. With the “Static II” method, tissue shavings are placed on a glass slide, where the bottom surface of the tissue is stained in a pool of NPs. This method requires a low volume of NPs (~20 μL NPs at 150 pM per 1 cm2 of tissue surface) but has a low staining efficiency that often yields NP concentrations that are below the detection limits of our imaging system when 4 or more NP flavors are multiplexed (Figure 1b,c). To improve the staining efficiency, various mechanical methods were investigated, including shaking, ultrasonic vibration and high-frequency vibration (Figure 1a). However, improvements in staining efficiency were often confined to the tissue edges (Figure 1b). The most effective method to improve the efficiency of tissue staining was to cyclically dip the tissue into and out of a NP-staining solution, which increased the average NP concentration on the tissue surfaces by 3× and the minimum NP concentration by 4× compared with the static staining methods (Figure 1b,c). The minimum NP concentration, measured from the most poorly stained region of the tissue, is an important metric that should remain above the detection limit of the imaging system to ensure accurate REMI measurements across the entire tissue surface. In summary, the dipping method was found to be far superior to static or vibration-aided staining methods, and yielded NP concentrations that, according to prior limit-of-detection studies, should theoretically allow for the accurate multiplexed imaging of 5 or more NP flavors (Figure 1c, Figure S2, Supporting Information).
Figure 1.
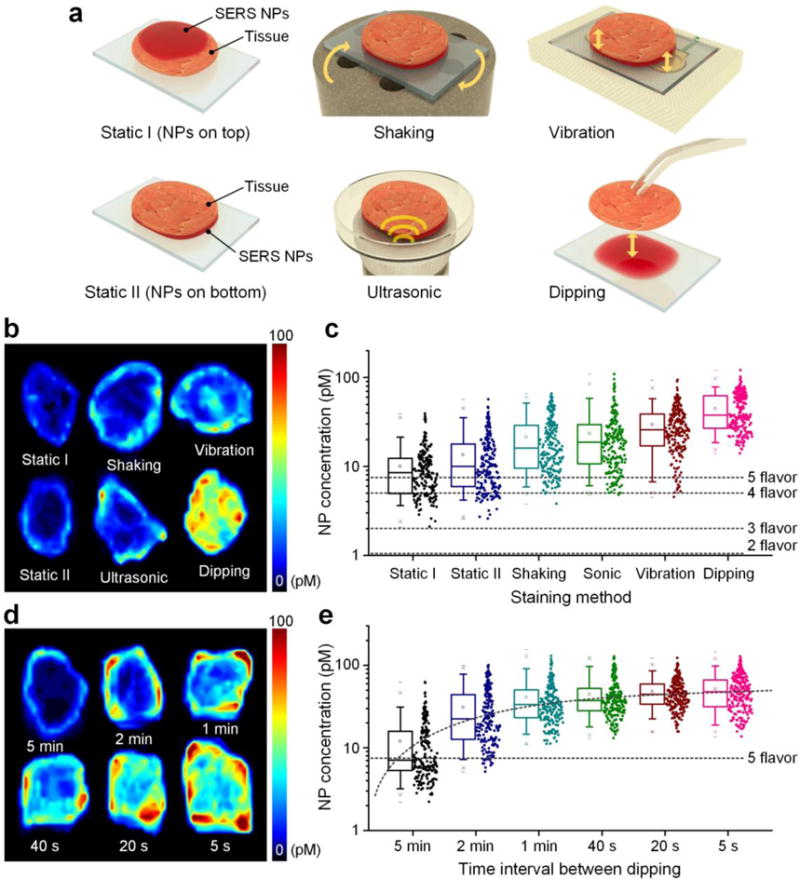
Comparison and optimization of methods to enhance the effectiveness of topical staining of fresh tissues with SERS NPs. Rat muscle tissues were topically stained for 10 min using a variety of methods, followed by a 10-s rinse in PBS and raster-scanned imaging of the NPs that remain at the tissue surface. a) Illustration of six staining methods, including staining the tissue surfaces from the top (Static I), staining from the bottom (Static II), low-frequency shaking with a vortex mixer (Shaking), ultrasonic vibration (Sonic), high-frequency (267 Hz) vibration, and low-frequency (once per 20 s) dipping of the tissue into and out of a NP-staining solution. (b, c) Comparison of NP concentrations from tissues that were stained using the six different methods: b) representative images of NP concentrations, and c) box plots of NP concentrations (n = 3 tissues, log scale) from the experiments shown in (a). The horizontal dashed lines in (c) indicate the detection limits of our spectral imaging system (at a pixel rate of 10 Hz), when 2 to 5 NP flavors are multiplexed (Figure S2, Supporting Information). d) Representative images and e) box plots (n = 3 tissues, log scale) of measured NP concentrations from tissues stained via dipping into and out of a staining solution at different frequencies.
Independently of our work, a recent publication reported that periodically removing and replenishing a nanoparticle-staining solution significantly enhanced in vitro staining efficiencies compared with static staining or agitated staining.[18] In that work, a mechanistic hypothesis that was offered to explain their results is that the rapid binding of large NPs to surfaces (in vitro), along with their slow diffusion rates, lowers the nanoparticle concentrations near the bioassay surfaces. This so-called “NP depletion layer” was presumed to inhibit the targeted binding of the nanoparticles.[18] An alternative hypothesis that we have formulated is that a low-velocity boundary layer exists at the tissue surface (a classical fluid mechanics assumption) in which the velocity of the fluid (and hence the NPs) is extremely low (i.e. the “no-slip” condition in fluid mechanics). Since fluid motion (convection) is essential for our targeted NPs to encounter their protein targets, the existence of such a boundary layer would significantly impede the staining process. In addition, with tissues placed on a smooth staining surface, only a thin gap exists between the tissue and the surface upon which it sits, which further restricts fluid convection. The act of dipping tissues into and out of a staining mixture serves to disrupt any depletion layers or boundary layers that may be hampering the NP staining process. An observation that supports this view is that tissue staining is more efficient when the dipping occurs more frequently even if this results in the tissues spending less total time in the staining mixture (Figure 1d,e). Note that these mechanisms may be unique for large NPs with a size of > 100 nm, and that small molecules (e.g. peptides, fluorophores, and even antibodies with a size of < 10 nm) may be governed by a different regime of transport properties (e.g. Brownian motion, random-walk, etc.), making them potentially less affected by bulk fluid convection and depletion-layer/boundary-layer effects.
Based on optimization studies, a short dipping interval (5 s) was ultimately selected to maximize NP-staining efficiencies (Figure 1e). In addition, high-frequency (~270 Hz) mechanical vibration was incorporated, as it was shown to be the next-best method (after dipping) and provided further improvement in staining efficiency (Figure S3c, Supporting Information). Note that securing tissue shavings from their top surface minimizes interference with the bottom surface and improves the staining of that bottom surface (Figure S3a,b, Supporting Information).
2.2 Automated device for enhanced topical staining of tissues
In order to apply the DMV method to enhance the topical staining of various types/sizes of tissues with high reproducibility, an automated staining device was designed and developed (Figure 2, Figure S4, Supporting Information, and Video S1). This system utilizes a suction plate to secure tissue specimens from above (Figure 2b) while allowing their bottom surfaces (the surgical margin surfaces) to be stained with a multiplexed mixture of SERS NPs. A servo motor (RadioShack, 2730766) lowers the staining reservoir every 5 s to completely remove the NP mixture from the tissue surface, and then dips the tissue back into the mixture after 2 s (Figure 2d,e). Two vibration elements are positioned under the glass slide that holds the staining mixture (Figure 2c) in order to further enhance the staining efficiency (Figure S3c, Supporting Information). To allow the simultaneous staining of multiple tissue specimens, the automated device contains three separate suction plates. Each suction plate is adjustable in height in order to account for variable thicknesses amongst different tissue specimens. Figure 2f is a design schematic of the device with all circuitry enclosed in a sealed housing.
Figure 2.
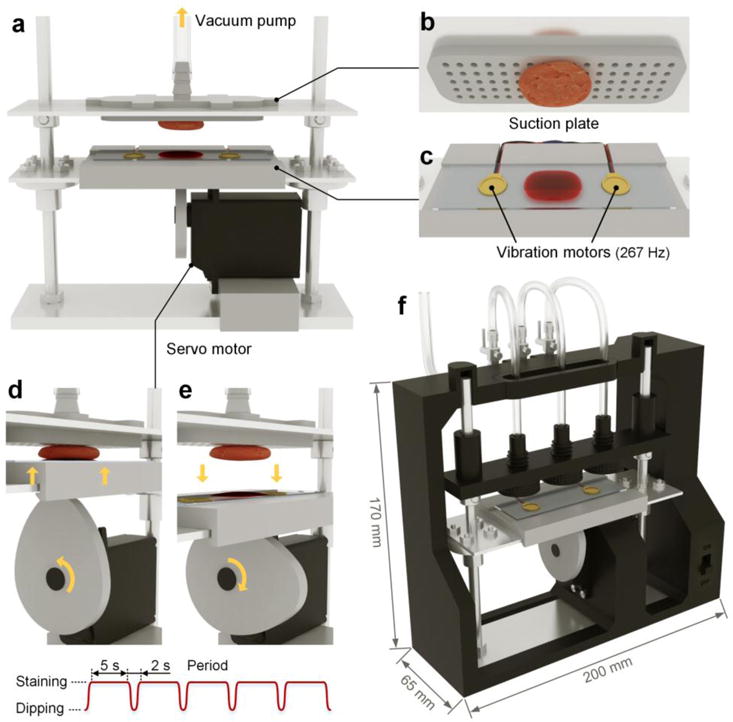
Automated staining device to enhance the speed and reproducibility of the REMI staining procedure, in which a multiplexed panel of SERS NPs is topically applied on the surfaces of fresh tissues. a) The staining device is equipped with a suction plate (b) to secure the tissue from above without interfering with the staining of the bottom surface of the tissue. The device incorporates a servo motor to dip the tissue into and out of a NP mixture (d,e), along with high-frequency vibration from two vibration motors (c) to further enhance the tissue staining. The fully assembled device (f) contains three adjustable suction plates to secure and stain multiple tissue specimens of varying thickness.
2.3 Improved ratiometric imaging through DMV staining
The ratiometric imaging of up to three SERS NPs has previously been demonstrated to be an effective strategy to quantify protein biomarkers in fresh tissues.[10,15a,15f,16a,17b,19] Prior to imaging fresh human tissues, the automated staining device was employed to study the effect of the DMV method for the ratiometric imaging of well-characterized tumor xenografts (Figure 3). Normal rat muscle and A431 tumor xenograft specimens were stained with an equimolar mixture of EGFR-NPs and isotype-NPs (150 pM/flavor) using either the “Static II” method in Figure 1a or the automated DMV staining device in Figure 2f, followed by a 10-s rinse in PBS and raster-scanned imaging.
Figure 3.
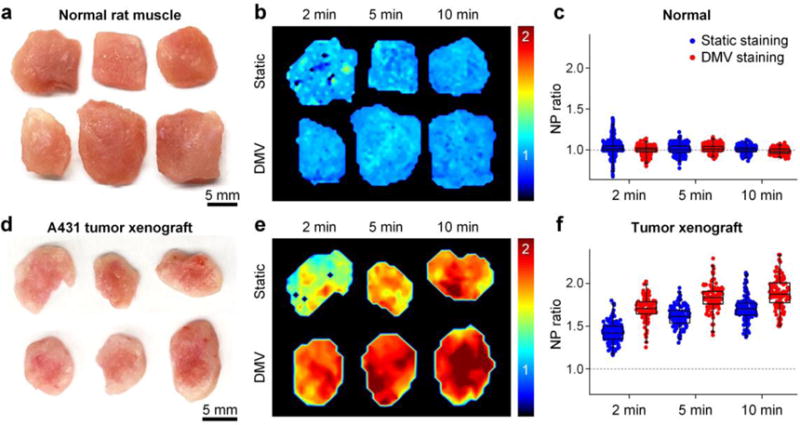
Comparison of static staining and DMV staining. Each tissue specimen was stained with a 2-flavor NP mixture (EGFR-NPs and isotype-NPs, 150 pM/flavor) for 2, 5 or 10 min through static staining or DMV staining, followed by a 10-s rinse in PBS and then raster-scanned imaging. a,d) Photographs of resected normal rat muscle (a) and A431 tumor xenograft specimens (d). b,e) Images showing the concentration ratio of EGFR-NPs vs. isotype-NPs on normal muscle (b) and tumor xenografts (e). c,f) Box plots of the NP ratio (targeted vs. untargeted) from three sets of experiments.
Figures 3b,e show the concentration ratio of EGFR-NPs vs. isotype-NPs. For normal tissues, as the staining duration increases, the concentration ratio remains at unity while the variance becomes smaller (Figure 3c). The reason the variance decreases is that the NP concentrations increase with staining duration,[10] which reduces the error in the NP measurements. Figure 3c shows that 2 min of DMV staining yields ratiometric images with less noise compared with 2 min of static staining. For tumor xenografts that are statically stained (Figure 3e,f), 10 min of staining is necessary to yield optimal NP ratios (EGFR-NP vs. isotype-NPs), which is in agreement with previous studies.[10] In comparison, just 2 min of DMV staining yields similarly high NP ratios (Figure 3e,f). In other words, DMV staining shortens the time (2–5 min) that is required to maximize the specific vs. nonspecific ratio of the SERS NPs for the ratiometric quantification of biomarkers. Figure 4 shows that the large size of the SERS NPs (~120 nm) causes topically applied NPs to be confined to the tissue surface with a negligible penetration depth of 10–20 μm. Note that compared to static staining, DMV staining only enhances the NP binding and accumulation at the tissue surface, rather than increasing the NP penetration depth (Figure 4). This is important to ensure that unbound NPs are easily and rapidly washed out (rather than trapped in the tissue), which allows for maximum NP ratios to be achieved in biomarker-positive tumors, as well as maximum tumor-to-normal contrast.
Figure 4.
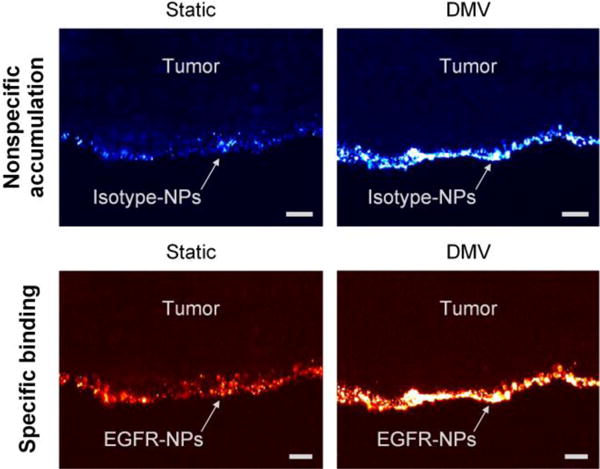
Fluorescence microscope image of a frozen section (10-μm tissue cross section) showing the penetration of SERS NPs near the surface of an A431 tumor. The specimen was topically stained with a mixture of EGFR-NPs and isotype-NPs (150 pM/flavor, 10 min) using either the static or DMV method. The images show that topically applied SERS NPs are localized to the tissue surface with a limited penetration depth of 10–20 μm. The scale bars represent 20 μm.
2.4 Highly multiplexed biomarker imaging through DMV staining
As discussed above, DMV staining greatly enhances the topical application efficiency of our SERS NPs and should enable the multiplexed imaging of 5 or more flavors of SERS NPs. Using the automated DMV-staining device, ex vivo studies with tumor xenografts and human breast tissues were performed to demonstrate the rapid imaging of 4 or 5 flavors of SERS NPs to quantify the expression of 3 or 4 protein biomarkers (respectively) at the surfaces of fresh tissue specimens.
A resected tumor xenograft specimen (A431) and a normal rat muscle specimen were simultaneously stained using the DMV staining device with an equimolar mixture of HER2-NPs, EGFR-NPs, CD44-NPs and isotype-NPs (150 pM/flavor, 5 min), followed by a quick rinse in PBS (10 s) and raster-scanned spectral imaging (< 2 min) (Figure 5). After spectral demultiplexing, three ratiometric images may be obtained from this single REMI experiment (Figure 5c). The ratiometric images provide a quantitative representation of EGFR, HER2 and CD44 expression that is consistent with flow cytometry and IHC results (Figure 5a,d,e).
Figure 5.
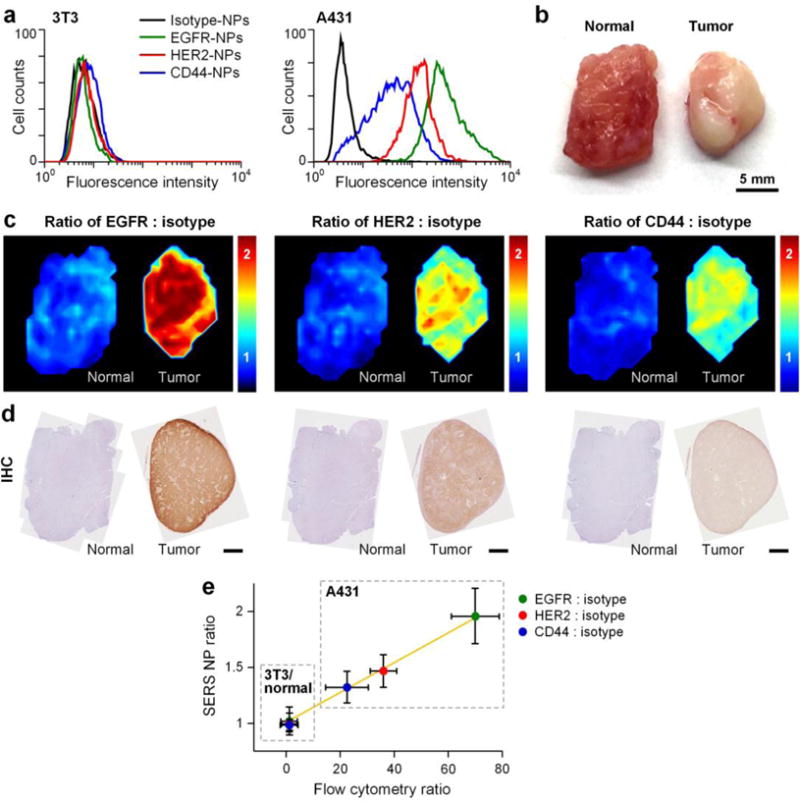
Multiplexed molecular imaging of a A431 tumor xenograft through automated DMV staining with a 4-flavor NP mixture (EGFR-NPs, HER2-NPs, CD44-NPs and isotype-NPs). a) Flow cytometry validation of conjugated NPs with cultured cells. EGFR-NPs, HER2-NPs, CD44-NPs and isotype-NPs were individually used to stain 3T3 (control) and A431 (cancer) cell lines. Fluorescence histograms from NP-stained cells are shown. b) Photograph of a resected normal rat muscle specimen and a A431 tumor xenograft. c) Ratiometric images of EGFR-NPs vs. isotype-NPs, HER2-NPs vs. isotype-NPs and CD44-NPs vs. isotype-NPs. d) Validation data: IHC for EGFR, HER2 and CD44. Unlabeled scale bars represent 2 mm. e) Plots showing the correlation between the NP ratios (targeted NP vs. untargeted NP) measured with REMI of xenograft tumors (in c) and the corresponding NP ratios measured via flow cytometry of NP-stained cells (in a). The NP ratios are the average and standard deviation from the REMI images of the tissues shown in (c). R > 0.98.
To demonstrate the feasibility of REMI, with convection-enhanced DMV staining, to rapidly assess a large panel of biomarkers in human surgical specimens, we imaged 8 fresh human breast tissue specimens excised from five patients (Figure 6). Using the automated DMV staining device, each tissue specimen was stained with a 5-flavor NP mixture (EGFR-NPs, HER2-NPs, CD44-NPs, CD24-NPs and isotype-NPs, 150 pM/flavor) for 5 min, followed by a 10-s rinse step in PBS. An area of up to 4×4 cm2 was raster-scanned within 3 min (i.e. >5 cm2/min). After spectral demultiplexing, four ratiometric images may be obtained from each single REMI experiment (Figure 6b). The entire REMI procedure (staining, rinsing, and imaging) was performed in less than 10 min, a time frame that is consistent with current intraoperative guidance techniques such as specimen X-ray and frozen-section pathology (which typically requires > 20 min and suffers from sampling errors). Ratiometric REMI accurately quantifies HER2, EGFR, CD44 and CD24 expression levels in agreement with IHC validation data (Figure 6b,c). The concentration ratios of biomarker-targeted NPs vs. isotype-NPs on IHC-validated biomarker-positive regions are significantly elevated (Figure 6d), indicating preferential binding of targeted NPs to their biomarker targets with high tumor-to-normal contrast. In summary, the results show that DMV staining improves the capability of REMI to visualize a large panel of overexpressed tumor biomarkers in 10 min (~5 min for staining and ~5 min for imaging), with the potential to assess surgical margins intraoperatively with high specificity and sensitivity and to reduce the need for re-excision surgeries.
Figure 6.
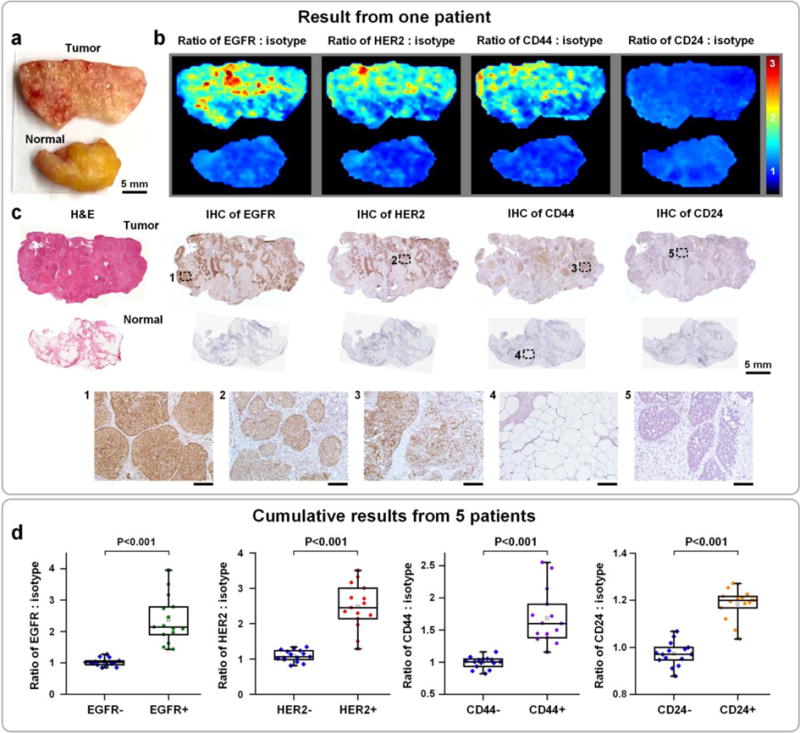
Multiplexed molecular imaging of freshly excised breast tissues with REMI, in which automated DMV staining of a 5-flavor mixture of NPs is employed. Each tissue specimen is stained with an equimolar mixture of HER2-NPs, EGFR-NPs, CD44-NPs, CD24-NPs and isotype-NPs (5 min), followed by a quick rinse in PBS (10 s) and raster-scanned imaging (< 3 min) to simultaneously quantify the expression of four biomarkers: EGFR, HER2, CD44 and CD24. a) Photograph of a human breast tumor and a normal tissue specimen from one patient. b) Ratiometric images of EGFR-NPs vs. isotype-NPs, HER2-NPs vs. isotype-NPs, CD44-NPs vs. isotype-NPs and CD24-NPs. c) Validation data: H&E and IHC for EGFR, HER2, CD44 and CD24. Unlabeled scale bars represent 200 μm. d) Cumulative results from multiple regions of interest from a total of 5 patient specimens: measured NP ratios on IHC-validated biomarker-negative and biomarker-positive tissue regions. Each data point in the plots is the average ratio from one region of interest.
3. Conclusion
Ratiometric imaging of topically applied SERS NPs (i.e. Raman-encoded molecular imaging, REMI) has been demonstrated as a quantitative approach to simultaneously visualize the expression of multiple protein biomarkers at fresh tissue surfaces, and to potentially guide tumor-resection procedures such as breast conserving surgeries.[10] However, previous topical application methods suffered from poor staining efficiency, which led to inaccurate NP measurements and limited the capability of REMI to image large multiplexed panels of biomarkers. Through a series of optimization experiments (Figure 1), we have developed an enhanced topical application method, the DMV method, to greatly improve the NP binding efficiency for REMI, and thereby increase the degree of multiplexing as well. Compared to conventional staining techniques, DMV staining significantly improves staining rates, and most importantly, the rate of specific vs. nonspecific NP accumulation. As a result, the overall staining time for REMI has been reduced to 5 min or less, as compared to previous studies that utilized staining times of 10 – 15 min,[10,15a] along with the added benefit of a greater degree of multiplexing (5 NPs) with no loss in accuracy.
Through imaging experiments with tumor xenografts and human breast tissues, we have demonstrated that the DMV method enables the simultaneous quantification of at least 4 biomarkers on large tissue surfaces after a 2- to 5-min staining duration (Figure 3–6). This reduction in staining time is not only of great clinical benefit, but could also enable the spatial resolution of REMI to be improved through the use of a smaller illumination spot size coupled with a finer sampling pitch for this raster-scanned imaging method (e.g. a 100-μm resolution for the identification of low numbers of tumor cells). Future clinical studies will assess the ability of our optimized REMI technology to rapidly and comprehensively assess entire surgical margins for the presence of residual tumor without the sampling errors that are inevitable with histopathology.
4. Experimental Section
REMI system
A customized spectral-imaging system has been developed to measure the concentration and concentration ratio of SERS NPs applied on tissue specimens.[10] The raster-scanned imaging of a tissue surface was performed by using a fixed spectral-imaging probe (FiberTech Optica Inc.) to image a thin tissue specimen that was translated with a two-axis stage (Newmark systems Inc., ET-50-11). The imaging probe utilizes a multimode fiber (100-μm core, 0.10 NA) at the center of the probe for illumination, which is surrounded by 27 multimode fibers (200-μm core, 0.22 NA) for collection of Raman, autofluorescence, and back-scattered laser light. A 785-nm diode laser (15 mW at the tissue) is used to illuminate the tissue, creating a laser spot with a diameter of 0.5 mm (imaging resolution). Photons collected by the 27 multimode fibers are transmitted to a customized spectrometer (Andor Holospec), where they are filtered (to remove autofluorescence and back-scattered laser light) and then dispersed onto a cooled deep-depletion spectroscopic CCD (Andor, Newton DU920P-BR-DD). The imaging probe was angled at 45 deg with respect to the tissue surface to minimize the collection of specular reflections. The detector integration time (i.e. the spectral acquisition rate, which equals the pixel rate) utilized in this study was 0.1 s. A direct-classical-least-squares (DCLS) algorithm was employed to calculate the concentration and ratio of various SERS NP flavors as described previously.[10,16b] In short, the acquired raw spectra are demultiplexed using the DCLS algorithm to calculate the weight of spectral components based on their reference spectra (e.g. reference spectra of the SERS NPs, the principal components of tissue-background spectra, etc.). The NP weights are then converted to NP concentrations based on calibration measurements with stock NPs of known concentrations. The NP concentrations are used to calculate concentration ratios for ratiometric mapping (of specific vs. nonspecific NP accumulation). A high degree of measurement linearity for NP concentrations in the range of 1–400 pM has been demonstrated (Figure S2, Supporting Information and previous publications[15a,15e,16b,20]).
Tissue staining and imaging
Prior to staining, tissue specimens were raster scanned to acquire a set of background spectra for the calculation of their principal components (to account for background variations during least-squares demultiplexing). The tissue surfaces were then stained using the methods discussed in the Results section (Figure 1,2). NP mixtures were applied with approximately 20 μL per 1 cm2 of tissue area (150 pM per flavor) and contained 1% BSA to minimize nonspecific binding (an optimal staining condition found in our previous studies[10]). For the “shaking” method in Figure 1a, a microplate vortex mixer (Fisher, 02216101), set to a speed of 300 rpm, was used to shake the NP-staining solution and tissue on a glass slide. For the “ultrasonic” method, an ultrasonic cleaner (Etekcity, CD-2800) was used to introduce an ultrasonic vibration (42 kHz) to the NP-staining solution. The tissue was stained in a covered petri dish to prevent aerosolized NPs from escaping to the air. For the “vibration” method, two vibration motors (Yuesui, B1034.FL45-00-015) were positioned under the glass slide to introduce a high-frequency vibration (adjustable from 167 Hz to 267 Hz) to the staining solution and tissue. After each of the staining methods was performed, the tissue sample was rinsed in 50-mL PBS with gentle agitation for 10 s, followed by raster-scanned imaging of the entire stained tissue surface.
SERS NPs and functionalization
SERS NPs were purchased from BD (Becton, Dickinson and Company). These NPs consist of a 60-nm-diameter gold core, a unique layer of Raman reporters adsorbed onto the surface of the gold cores, surrounded by a 60-nm-thick silica coating, resulting in an overall diameter of ~120 nm (Figure S1a, Supporting Information). The silica shell makes the SERS NP signals insensitive to the environment and immune from signal changes induced by aggregation of the NPs (since the gold cores in different NPs will never touch each other). Previous studies have demonstrated excellent linearity of NP measurements in a variety of animal and human tissues over a wide range of concentrations.[14,17c] Five “flavors” of NPs were used in this study, identified as S420, S421, S440, S481 and S493, each of which emits a characteristic Raman spectrum due to chemical differences in the Raman reporter layer (Figure S1b, Supporting Information).
Using a previously described conjugation protocol,[15a] different SERS NP flavors were functionalized with different monoclonal antibodies (mAb) targeting either the epidermal growth factor receptor (EGFR), the human epidermal growth factor receptor 2 (HER2), CD44 or CD24. In addition, negative-control NPs were prepared by conjugating one NP flavor with an isotype control antibody (mouse IgG1). The NPs, which contain reactive thiols at their surface, were first reacted with a fluorophore, DyLight 650 Maleimide (Thermo Scientific, 62295), for the purposes of flow-cytometry characterization. The NPs were then conjugated with either an isotype control (Thermo Scientific, MA110407), an anti-EGFR (Thermo Scientific, MS378PABX), an anti-HER2 (Thermo Scientific, MS229PABX), an anti-CD44 (Abcam plc., ab6124) or an anti-CD24 (Abcam plc., ab31622) mAb at 500 molar equivalents per NP. The NP conjugates were stored at 4°C and protected from light. UV-VIS spectrophotometry was used to measure the concentration of the SERS NP conjugates (by comparing the absorbance of NP conjugates with the absorbance of stock NPs whose concentration is known).[15a]
Cell culture and flow cytometry
The two cell lines employed in this study were A431 (ATCC, CRL-1555) and 3T3 (ATCC, CRL-1658). The cells were cultured in DMEM medium (Lonza, 12-604F), which were supplemented with 10% fetal bovine serum (FBS, Thermo Scientific, SH3008803) and 1% penicillin-streptomycin (Lonza, 17-602E). All cells were cultured at 37 °C with 5% CO2. Trypsin EDTA 1X (Mediatech, MT25051CI) was used to detach cells.
Flow cytometry samples were prepared by mixing 25-uL cell suspensions (0.2 million cells) with 25 μL of NP conjugates (100 pM) for 15 min at 20 °C with gentle agitation at 300 rpm, followed by three rounds of purification via centrifugation (400 g for 5 min) and supernatant-replacement (500 μL per rinse) with FACS buffer (20% FBS in PBS). Each cell line was split into equally sized samples that were individually stained by EGFR-NPs, HER2-NPs, CD44-NPs or isotype-NPs.
Mouse xenograft model and human breast tissues
Nude mice (Taconic Farms Inc, model NCRNU-F) were used to develop A431 tumor xenografts. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Washington IACUC (#4345-01). A431 cancer cells (1 × 106) were suspended in matrigel (BD biosciences, 354234) at a 1:1 volume ratio to form a 100 μL mixture. Nude mice (5–8 weeks) were subcutaneously implanted in their flanks with the cell/matrigel mixture. The mice were euthanized by CO2 inhalation when tumors reached a size of 10 mm, followed by the surgical removal of the implanted tumors.
De-identified human breast tissue specimens were obtained from consenting patients and imaged within 1–2 hours after lumpectomy or mastectomy at the University of Washington Medical Center. Tissue collection was managed by the Northwest BioTrust (NWBT) under an IRB exemption for these de-identified tissues. After imaging, the tissues were fixed with 10% formalin and submitted for histopathology (IHC and H&E staining).
Statistical analysis
Statistical analysis was performed in Matlab or Origin. All values in the figures are presented as mean ± standard deviation unless otherwise noted in the text and figure captions. Statistical significance was calculated by a student’s t-test (two-sample, unpaired), and the level of significance was set at P < 0.001. For all the box plots, the bottom and top of the box represent the 1st and 3rd quartiles of the dataset, respectively, and the band inside the box represents the median (2nd quartile) of the data.
Supplementary Material
Acknowledgments
The authors acknowledge support from the NIH/NIBIB – R21 EB015016 (Liu), the Department of Mechanical Engineering at the University of Washington, and the department of education GAANN fellowship program (SK). The Northwest BioTrust (NWBT) is supported, in part, by the NIH (P30-CA015704).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Yu “Winston” Wang, Department of Mechanical Engineering, University of Washington, Seattle, WA 98195, USA.
Josh D. Doerksen, Department of Mechanical Engineering, University of Washington, Seattle, WA 98195, USA
Soyoung Kang, Department of Mechanical Engineering, University of Washington, Seattle, WA 98195, USA.
Daniel Walsh, Department of Mechanical Engineering, University of Washington, Seattle, WA 98195, USA.
Qian Yang, Department of Pharmacy, Chengdu Medical College, Chengdu, Sichuan 615000, China; Department of Mechanical Engineering, University of Washington, Seattle, WA 98195, USA.
Daniel Hong, Department of Mechanical Engineering, University of Washington, Seattle, WA 98195, USA.
Jonathan T.C. Liu, Department of Mechanical Engineering, University of Washington, Seattle, WA 98195, USA
References
- 1.a) DeSantis C, Ma J, Bryan L, Jemal A. CA-Cancer J Clin. 2014;64:52. doi: 10.3322/caac.21203. [DOI] [PubMed] [Google Scholar]; b) Hou N, Huo D. Breast Cancer Res Treat. 2013;138:633. doi: 10.1007/s10549-013-2434-0. [DOI] [PubMed] [Google Scholar]
- 2.a) Jacobs L. Ann Surg Oncol. 2008;15:1271. doi: 10.1245/s10434-007-9766-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Waljee JF, Hu ES, Newman LA, Alderman AK. Ann Surg Oncol. 2008;15:1297. doi: 10.1245/s10434-007-9777-x. [DOI] [PubMed] [Google Scholar]; c) Jeevan R, Cromwell DA, Trivella M, Lawrence G, Kearins O, Pereira J, Sheppard C, Caddy CM, van der Meulen JHP. BMJ. 2012;345 doi: 10.1136/bmj.e4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Moran MS, Schnitt SJ, Giuliano AE, Harris JR, Khan SA, Horton J, Klimberg S, Chavez-MacGregor M, Freedman G, Houssami N, Johnson PL, Morrow M. J Clin Oncol. 2014 doi: 10.1200/JCO.2013.53.3935. [DOI] [PubMed] [Google Scholar]; b Adams BJ, Zoon CK, Stevenson C, Chitnavis P, Wolfe L, Bear HD. Ann Surg Oncol. 2013;20:2250. doi: 10.1245/s10434-012-2831-3. [DOI] [PubMed] [Google Scholar]; c) Morrow M, Harris JR, Schnitt SJ. N Engl J Med. 2012;367:79. doi: 10.1056/NEJMsb1202521. [DOI] [PubMed] [Google Scholar]; d) Schnitt SJ, Morrow M. Am J Clin Pathol. 2012;138:635. doi: 10.1309/AJCPPQ1JGV0GJIAB. [DOI] [PubMed] [Google Scholar]; e) Singletary SE. Am J Surg. 2002;184:383. doi: 10.1016/s0002-9610(02)01012-7. [DOI] [PubMed] [Google Scholar]
- 4.a) Jorns JM, Visscher D, Sabel M, Breslin T, Healy P, Daignaut S, Myers JL, Wu AJ. Am J Clin Pathol. 2012;138:657. doi: 10.1309/AJCP4IEMXCJ1GDTS. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jorns JM, Daignault S, Sabel MS, Wu AJ. Am J Clin Pathol. 2014;142:601. doi: 10.1309/AJCPRSOA2G8RLEXY. [DOI] [PubMed] [Google Scholar]; c) Cendan JC, Coco D, Copeland EM., 3rd J Am Coll Surg. 2005;201:194. doi: 10.1016/j.jamcollsurg.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 5.a) Abeytunge S, Li Y, Larson B, Peterson G, Seltzer E, Toledo-Crow R, Rajadhyaksha M. J Biomed Opt. 2013;18:61227. doi: 10.1117/1.JBO.18.6.061227. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gareau DS, Patel YG, Li Y, Aranda I, Halpern AC, Nehal KS, Rajadhyaksha M. J Microsc. 2009;233:149. doi: 10.1111/j.1365-2818.2008.03105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tao YK, Shen D, Sheikine Y, Ahsen OO, Wang HH, Schmolze DB, Johnson NB, Brooker JS, Cable AE, Connolly JL, Fujimoto JG. Proc Natl Acad Sci U S A. 2014;111:15304. doi: 10.1073/pnas.1416955111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Bydlon TM, Barry WT, Kennedy SA, Brown JQ, Gallagher JE, Wilke LG, Geradts J, Ramanujam N. PloS One. 2012;7:e51418. doi: 10.1371/journal.pone.0051418. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sharma V, Shivalingaiah S, Peng Y, Euhus D, Gryczynski Z, Liu H. Biomed Opt Express. 2012;3:1825. doi: 10.1364/BOE.3.001825. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Nichols BS, Schindler CE, Brown JQ, Wilke LG, Mulvey CS, Krieger MS, Gallagher J, Geradts J, Greenup RA, Von Windheim JA, Ramanujam N. PloS One. 2015;10:e0127525. doi: 10.1371/journal.pone.0127525. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Brown JQ, Bydlon TM, Kennedy SA, Caldwell ML, Gallagher JE, Junker M, Wilke LG, Barry WT, Geradts J, Ramanujam N. PloS One. 2013;8:e69906. doi: 10.1371/journal.pone.0069906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Keller MD, Vargis E, de Matos Granja N, Wilson RH, Mycek MA, Kelley MC, Mahadevan-Jansen A. J Biomed Opt. 2011;16:077006. doi: 10.1117/1.3600708. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu CH, Zhou Y, Sun Y, Li JY, Zhou LX, Boydston-White S, Masilamani V, Zhu K, Pu Y, Alfano RR. Technol Cancer Res Treat. 2013;12:371. doi: 10.7785/tcrt.2012.500325. [DOI] [PubMed] [Google Scholar]; c) Kong K, Rowlands CJ, Varma S, Perkins W, Leach IH, Koloydenko AA, Williams HC, Notingher I. Proc Natl Acad Sci U S A. 2013;110:15189. doi: 10.1073/pnas.1311289110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Valdes EK, Boolbol SK, Cohen JM, Feldman SM. Ann Surg Oncol. 2007;14:1045. doi: 10.1245/s10434-006-9263-x. [DOI] [PubMed] [Google Scholar]; b) Saarela AO, Paloneva TK, Rissanen TJ, Kiviniemi HO. J Surg Oncol. 1997;66:248. doi: 10.1002/(sici)1096-9098(199712)66:4<248::aid-jso5>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Kang S, Khan A, Ruttner G, Leigh SY, Murray M, Abeytunge S, Peterson G, Rajadhyaksha M, Dintzis S, Javid S, Liu JTC. Sci Rep. 2016;6:21242. doi: 10.1038/srep21242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Freise AC, Wu AM. Mol Immunol. 2015;67:142. doi: 10.1016/j.molimm.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pysz MA, Gambhir SS, Willmann JK. Clin Radiol. 2010;65:500. doi: 10.1016/j.crad.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Weissleder R, Ross BD, Rehemtulla A, Gambhir SS, People’s Medical Publishing House 2010 [Google Scholar]
- 12.a) Samoylova TI, Morrison NE, Globa LP, Cox NR. Anti-Cancer Agents Med Chem. 2006;6:9. doi: 10.2174/187152006774755492. [DOI] [PubMed] [Google Scholar]; b) Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, Aas T, Alexandrov LB, Larsimont D, Davies H, Li Y, Ju YS, Ramakrishna M, Haugland HK, Lilleng PK, Nik-Zainal S, McLaren S, Butler A, Martin S, Glodzik D, Menzies A, Raine K, Hinton J, Jones D, Mudie LJ, Jiang B, Vincent D, Greene-Colozzi A, Adnet PY, Fatima A, Maetens M, Ignatiadis M, Stratton MR, Sotiriou C, Richardson AL, Lonning PE, Wedge DC, Campbell PJ. Nat Med. 2015;21:751. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zardavas D, Irrthum A, Swanton C, Piccart M. Nat Rev Clin Oncol. 2015;12:381. doi: 10.1038/nrclinonc.2015.73. [DOI] [PubMed] [Google Scholar]
- 13.a) Wang Y, Yan B, Chen L. Chem Rev. 2013;113:1391. doi: 10.1021/cr300120g. [DOI] [PubMed] [Google Scholar]; b) Samanta A, Jana S, Das RK, Chang YT. Nanomedicine (London, U K) 2014;9:523. doi: 10.2217/nnm.13.222. [DOI] [PubMed] [Google Scholar]
- 14.Zavaleta CL, Garai E, Liu JT, Sensarn S, Mandella MJ, Van de Sompel D, Friedland S, Van Dam J, Contag CH, Gambhir SS. Proc Natl Acad Sci U S A. 2013;110:E2288. doi: 10.1073/pnas.1211309110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Wang YW, Khan A, Som M, Wang D, Chen Y, Leigh SY, Meza D, McVeigh PZ, Wilson BC, Liu JT. Technology. 2014;2:118. doi: 10.1142/S2339547814500125. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu JTC, Helms MW, Mandella MJ, Crawford JM, Kino GS, Contag CH. Biophys J. 2009;96:2405. doi: 10.1016/j.bpj.2008.12.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tichauer KM, Samkoe KS, Gunn JR, Kanick SC, Hoopes PJ, Barth RJ, Kaufman PA, Hasan T, Pogue BW. Nat Med. 2014;20:1348. doi: 10.1038/nm.3732. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Tichauer KM, Wang Y, Pogue BW, Liu JT. Phys Med Biol. 2015;60:R239. doi: 10.1088/0031-9155/60/14/R239. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Wang YW, Khan A, Leigh SY, Wang D, Chen Y, Meza D, Liu JTC. Biomed Opt Express. 2014;5:2883. doi: 10.1364/BOE.5.002883. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Mallia RJ, McVeigh PZ, Fisher CJ, Veilleux I, Wilson BC. Nanomedicine (London, U K) 2015;10:89. doi: 10.2217/nnm.14.80. [DOI] [PubMed] [Google Scholar]
- 16.a) Wang YW, Kang S, Khan A, Bao PQ, Liu JTC. Biomed Opt Express. 2015;6:3714. doi: 10.1364/BOE.6.003714. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang Y, Kang S, Doerksen J, Glaser A, Liu J. IEEE J Sel Top Quantum Electron. 2016;22:1. doi: 10.1109/JSTQE.2015.2507358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Bohndiek SE, Wagadarikar A, Zavaleta CL, Van de Sompel D, Garai E, Jokerst JV, Yazdanfar S, Gambhir SS. Proc Natl Acad Sci U S A. 2013;110:12408. doi: 10.1073/pnas.1301379110. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Garai E, Sensarn S, Zavaleta CL, Van de Sompel D, Loewke NO, Mandella MJ, Gambhir SS, Contag CH. J Biomed Opt. 2013;18:096008. doi: 10.1117/1.JBO.18.9.096008. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zavaleta CL, Smith BR, Walton I, Doering W, Davis G, Shojaei B, Natan MJ, Gambhir SS. Proc Natl Acad Sci U S A. 2009;106:13511. doi: 10.1073/pnas.0813327106. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Garai E, Sensarn S, Zavaleta CL, Loewke NO, Rogalla S, Mandella MJ, Felt SA, Friedland S, Liu JT, Gambhir SS, Contag CH. PloS One. 2015;10:e0123185. doi: 10.1371/journal.pone.0123185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Zrazhevskiy P, Gao X. Small. 2016;12:1035. doi: 10.1002/smll.201503101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinha L, Wang Y, Yang C, Khan A, Brankov JG, Liu JTC, Tichauer KM. Sci Rep. 2015;5:8582. doi: 10.1038/srep08582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leigh SY, Som M, Liu JTC. PloS One. 2013;8:e62084. doi: 10.1371/journal.pone.0062084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.