

The goal of this study is to understand how a particular mutation in obscurin proteins leads to congenital heart disease.
Keywords: obscurin, phospholamban, heart disease, arrhythmia, Ca cycling, pressure overload, transaortic constriction, hypertrophic cardiomyopahty, dilated cardiomyopahty
Abstract
Obscurins are cytoskeletal proteins with structural and regulatory roles encoded by OBSCN. Mutations in OBSCN are associated with the development of hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM). Specifically, the R4344Q mutation present in immunoglobulin domain 58 (Ig58) was the first to be linked with the development of HCM. To assess the effects of R4344Q in vivo, we generated the respective knock-in mouse model. Mutant obscurins are expressed and incorporated normally into sarcomeres. The expression patterns of sarcomeric and Ca2+-cycling proteins are unaltered in sedentary 1-year-old knock-in myocardia, with the exception of sarco/endoplasmic reticulum Ca2+ adenosine triphosphatase 2 (SERCA2) and pentameric phospholamban whose levels are significantly increased and decreased, respectively. Isolated cardiomyocytes from 1-year-old knock-in hearts exhibit increased Ca2+-transients and Ca2+-load in the sarcoplasmic reticulum and faster contractility kinetics. Moreover, sedentary 1-year-old knock-in animals develop tachycardia accompanied by premature ventricular contractions, whereas 2-month-old knock-in animals subjected to pressure overload develop a DCM-like phenotype. Structural analysis revealed that the R4344Q mutation alters the distribution of electrostatic charges over the Ig58 surface, thus interfering with its binding capabilities. Consistent with this, wild-type Ig58 interacts with phospholamban modestly, and this interaction is markedly enhanced in the presence of R4344Q. Together, our studies demonstrate that under sedentary conditions, the R4344Q mutation results in Ca2+ deregulation and spontaneous arrhythmia, whereas in the presence of chronic, pathological stress, it leads to cardiac remodeling and dilation. We postulate that enhanced binding between mutant obscurins and phospholamban leads to SERCA2 disinhibition, which may underlie the observed pathological alterations.
INTRODUCTION
Cardiovascular disease (CVD) is a highly prevalent, multifactorial, and multifaceted disease accounting for one of every three deaths worldwide (1). CVD is subclassified into cardiomyopathies (CMs) and blood vessel diseases. Lifestyle preferences and complex genetics contribute to the predisposition and development of CM (1). Accordingly, congenital mutations in genes encoding sarcomeric proteins have been shown to causatively associate with the development of CM, with mutations in β-myosin heavy chain and cardiac myosin-binding protein C accounting for 30 to 50% of hereditary CM (2–6). Recently, mutations in genes encoding Ca2+-sensitive and Ca2+-handling proteins have also been linked to the pathogenesis of CM, including troponin C, phospholamban (PLN), junctophilin-2, ryanodine receptor 2 (RyR2), calsequestrin 2, and calreticulin 3 (7). These findings led to the realization that the molecular etiologies underlying CM are diverse and further implied an intimate interplay between sarcomeric disorganization and Ca2+ deregulation in CM development.
Sarcomeres contain structural, signaling, and accessory proteins that mediate the assembly, maintenance, and contraction of myofilaments, allowing them to sustain the physical distortion that occurs during repeating cycles of shortening and relaxation (5). Of the multitude of structural and signaling proteins, giant obscurins consisting of tandem adhesion and signaling motifs have a unique topography by wrapping around sarcomeric M-bands and Z-disks, where they provide binding sites for proteins present in different subcellular compartments (8, 9). Consistent with this, obscurins directly interact with sarcomeric proteins, the microtubule network, Ca2+-handling proteins residing in the sarcoplasmic reticulum (SR) membranes, as well as adhesion receptors and sarcolemmal proteins (10–12). Through those interactions, obscurins mediate sarcomeric assembly and stability (10, 13, 14), participate in intercellular adhesion (12), support sarcolemmal integrity (11), and serve as adaptors between the sarcomeric cytoskeleton and the SR membranes, potentially contributing to Ca2+ homeostasis (9).
Although the key role of obscurins as structural scaffolds is well established in different cell systems and organisms (10, 13), their importance in the regulation of Ca2+ homeostasis is less understood. Earlier studies have indicated an essential role for obscurins in the organization and sarcomeric alignment of the SR membranes, because primary cultures of skeletal myotubes depleted of obscurins and obscurin-null hearts exhibit poorly developed and misaligned SR membranes (13, 15). Similarly, body and pharyngeal wall muscles from Caenorhabditis elegans carrying mutant UNC89, the C. elegans obscurin homolog, exhibit dramatic disarray of SR membranes, as evidenced by mislocalization of the sarco/endoplasmic reticulum Ca2+ adenosine triphosphatase (ATPase) (SERCA) and RyR and the presence of delayed Ca2+ transients with reduced peak amplitudes (10). Thus, evidence has been building, pointing toward a key role for obscurins not only in the organization and sarcomeric alignment of the SR membranes but also in the regulation of Ca2+ cycling.
In line with the important roles of obscurins in diverse cellular processes, sequencing analysis has indicated the presence of missense, frameshift, and deletion mutations in the OBSCN gene that are directly associated with the development of hypertrophic CM (HCM) and dilated CM (DCM) (16–18). In particular, a missense mutation, R4344Q, in obscurin immunoglobulin 58 (Ig58) has been linked to the development of HCM in humans; however, the underlying molecular etiologies leading to disease pathogenesis remain elusive. To assess the functional ramifications of the obscurin R4344Q mutation in vivo, we generated a constitutive knock-in mouse model expressing full-length mutant obscurins that contain the R4344Q substitution. Our studies show that under sedentary conditions, knock-in myocardia do not develop hypertrophy but exhibit Ca2+ deregulation and develop arrhythmia by 1 year of age. Moreover, following exposure to pressure overload, knock-in myocardia undergo remodeling and develop a DCM-like phenotype. Remarkably, structural and biochemical evidence suggests that these functional alterations may result from SERCA2 disinhibition due to increased binding of mutant obscurins to monomeric PLN. Our study is the first to generate an in vivo model to assess the involvement of mutant obscurins in cardiac pathophysiology and to pinpoint a role for obscurins in Ca2+ homeostasis.
RESULTS
Mutations in the OBSCN gene have been recently associated with the development of heart disease (16–18). In particular, a single amino acid substitution, R4344Q, present in Ig58 was the first mutation that was causatively linked to the development of HCM in a 19-year-old male patient, who most likely inherited the mutation from his mother who also suffered from HCM (16).
To examine the pathological effects of the R4344Q mutation, we generated a knock-in mouse model expressing full-length obscurins carrying the R4344Q substitution (Fig. 1, A to B′, and fig. S1, A to C). Homozygous obscurin knock-in mice exhibit normal Mendelian genetics and are viable and fertile during adulthood (fig. S1, D and E).
Fig. 1. Examination of the hearts of 1-year-old homozygous female knock-in mice under sedentary conditions.
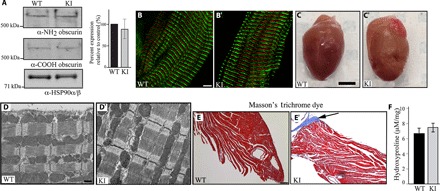
(A) Immunoblotting using antibodies to the NH2 and COOH termini of obscurins and protein lysates prepared from left ventricles of wild-type (WT) and knock-in (KI) hearts indicated similar expression levels of wild-type and mutant obscurins. Please note that obscurin A is the major giant isoform expressed in the heart, which is shown in our blots, whereas obscurin B is present in low amounts and is only detectable after longer exposure. The expression levels of obscurin A were quantified and found to be unaltered; n = 3 animals per group, two to three replica blots per heart; t test, P = 0.629; error bar represents SEM; heat shock protein 90 (HSP90)α/β served as loading and normalization control. (B and B′) Immunofluorescence combined with confocal optics and antibodies to the COOH terminus demonstrated that mutant obscurins containing the R4344Q substitution are incorporated normally into sarcomeric M-bands in knock-in left ventricles; obscurins are shown in red, and α-actinin, which is a Z-disk marker, is shown in green; scale bar, 5 μm. (C and C′) Gross morphology of wild-type and knock-in hearts showed that they have comparable sizes; scale bar, 3 mm. (D and D′) Transmission electron micrographs of wild-type and knock-in left ventricles revealed no major structural alterations; scale bar, 500 nm. (E and E′) Masson’s trichrome staining of wild-type and knock-in left ventricle sections illustrating the presence of peripheral fibrosis (arrow) in the latter; scale bar, 100 μm. (F) A trend of increased hydroxyproline content was found in the apices of knock-in left ventricles compared with wild-type tissue, which, however, did not reach statistical significance (t test, P = 0.202; n = 4 animals per group, three replica assays per heart).
Obscurin knock-in hearts exhibited modest morphological alterations at 1 year of age under sedentary conditions
Mutant obscurins were expressed in similar amounts to wild-type proteins in homozygous knock-in myocardia and incorporated normally into sarcomeres (Fig. 1 , A to B′). Morphometric and echocardiographic evaluation of female and male knock-in animals did not reveal any significant morphological or functional alterations compared to their wild-type littermates that would be indicative of the development of HCM (table S1). Consistent with this, knock-in hearts displayed comparable gross morphology, left ventricle mass–to–body weight or left ventricle mass–to–tibia length ratios, left ventricle wall thickness, ejection fraction (EF), and fractional shortening (FS) to age-matched wild-type hearts (Fig. 1, C and C′, and table S1).
Although neither gender exhibited an obvious cardiomyopathic phenotype, given the likelihood of gender-based differences in disease development (19, 20), we reasoned that combined evaluation of data from females and males might skew our conclusions. We therefore focused on the female population for the remainder of our studies, which has been greatly understudied in similar cardiac disease models (21).
Histological analysis using hematoxylin and eosin (H&E) staining (fig. S2, A and A′) and ultrastructural evaluation (Fig. 1, D and D′) revealed no apparent differences in the structural organization of knock-in myocardia compared to wild type. Moreover, the expression levels and localization of major sarcomeric proteins were indistinguishable between 1-year-old homozygous knock-in and wild-type hearts (fig. S2, B to E′). This was also the case for titin, which binds directly to the Ig58/Ig59 region of obscurin via its ZIg9/ZIg10 domains (fig. S2, B to B″) (16). Although staining of 1-year-old hearts with Masson’s trichrome revealed areas of peripheral fibrosis in knock-in but not wild-type myocardia (Fig. 1, E and E′). Quantification of collagen content via the hydroxyproline assay, however, did not reach statistical significance (Fig. 1F), indicating the presence of early stages of remodeling in the R4344Q myocardia.
Knock-in hearts carrying the obscurin R4344Q mutation exhibit altered Ca2+ cycling and arrhythmia
Previous studies have implicated obscurins directly in the organization and sarcomeric alignment of the SR membranes and indirectly in the modulation of Ca2+ transients (9, 10, 15). We therefore examined whether the expression levels and distribution of major Ca2+-handling proteins were affected in the R4344Q homozygous myocardia. We found that the expression levels of SERCA2 were significantly increased (~10%) in the left ventricles of 1-year-old knock-in hearts compared to wild-type hearts (Fig. 2, A, E, and E′), whereas the levels of pentameric but not monomeric PLN were reduced by ~15% (Fig. 2A). Notably, the amounts and localization of other major Ca2+-handling proteins remained unaltered (Fig. 2, A, B to D′, and F to G′). Because phosphorylation is a potent mechanism that regulates the activity of Ca2+-cycling proteins, we also examined the phosphorylation levels of SERCA2 and its regulator PLN as well as of RyR2 in knock-in hearts but did not observe any changes (Fig. 2H).
Fig. 2. Evaluation of the expression levels, phosphorylation status, and subcellular distribution of Ca2+-cycling proteins in 1-year-old homozygous female knock-in hearts.
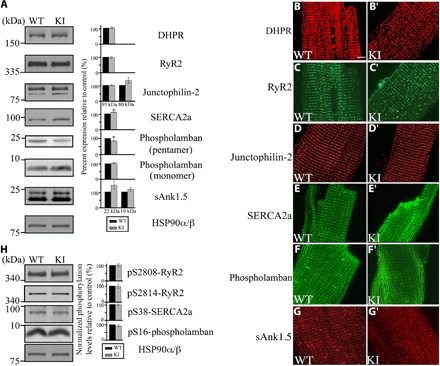
(A) Representative immunoblots and relative quantification of the expression levels of major Ca2+-handling proteins showed that the amounts of SERCA2 are significantly increased (~10%), whereas the amounts of pentameric (but not monomeric) PLN are significantly reduced (~15%) in knock-in compared to wild-type hearts; t test, *P < 0.05; error bars represent SEM; n = 3 animals per group, two replica blots per heart. HSP90α/β served as loading and normalization control. (B to G′) Immunolabeling of freshly isolated cardiomyocytes from wild-type and knock-in hearts with antibodies to Ca2+-cycling proteins did not show any apparent changes in their localization; scale bar, 5 μm; note that all images from wild-type and knock-in animals were obtained using the same laser power and microscope settings. (H) Representative immunoblots and relative quantification of the phosphorylation levels of select Ca2+-cycling proteins using antibodies to phospho-Ser2808 and phospho-Ser2814 of RyR2, phospho-Ser38 of SERCA2, and phospho-Ser16 of PLN did not reveal any differences between wild-type and knock-in hearts. Phosphorylation levels were normalized to total levels for each protein; t test, P > 0.05; error bars represent SEM; n = 3 animals per group, two replica blots per heart. As in (A), HSP90α/β served as loading and normalization control.
Given the increased amounts of SERCA2 in knock-in hearts, we then performed [Ca2+]i transient (Fig. 3, A to D″) and cell shortening (Fig. 3, E to F′) studies using isolated cardiomyocytes from 1-year-old wild-type and knock-in left ventricles. Following electrical stimulation to steady state, knock-in cardiomyocytes displayed a significantly higher peak amplitude of electrically stimulated [Ca2+]i transients and faster decay kinetics compared with wild-type cells (Fig. 3, B to C′). Knock-in cardiomyocytes also demonstrated an increase in SR Ca2+ content, as indicated by an increase in the amount of Ca2+ released from the SR upon application of caffeine (Fig. 3C″). However, fractional Ca2+ release from the SR, which is indicative of RyR2 activity, was unchanged between 1-year-old wild-type and knock-in cardiomyocytes, as measured from the ratio of Ca2+ released during electrical stimulation over Ca2+ released during caffeine application (Fig. 3D). The faster decay rate of the [Ca2+]i transients exhibited by the knock-in cardiomyocytes can be explained by either enhanced SERCA2 pumping of Ca2+ into the SR or increased extrusion of Ca2+ across the sarcolemma, which occurs predominantly via the Na+/Ca2+ exchanger (NCX) (22). To discriminate between these two possibilities, we compared the decay rate of the systolic [Ca2+]i transient, which depends on NCX and SERCA2 activity, to the decay rate of the caffeine-induced [Ca2+]i transient, which depends almost exclusively on NCX. Although the systolic decay rate was increased in knock-in cardiomyocytes compared with wild-type cells (Fig. 3C′), the caffeine-induced decay rate was not different between the two groups (Fig. 3D′), arguing against an alteration in NCX activity. By subtracting the two decay rates, we were able to isolate the contribution of SERCA2 activity, which was significantly increased (~30%) in knock-in cardiomyocytes (Fig. 3D″). Enhanced SERCA2 activity and up-regulation of SERCA2 can lead to elevated SR Ca2+ content and thus increased SR Ca2+ release upon stimulation (23), as seen in knock-in versus wild-type cardiomyocytes (Fig. 3, B to C″). Consistent with the enhanced Ca2+ release and more rapid Ca2+ reuptake via SERCA2, contractile magnitude (measured as a change in cell shortening, ΔSL) as well as contraction and relaxation velocities were significantly increased in 1-year-old knock-in cardiomyocytes compared to wild-type cells (Fig. 3, E to F″).
Fig. 3. Enhanced SERCA2 activity and increased contractility were observed in 1-year-old homozygous knock-in female hearts.
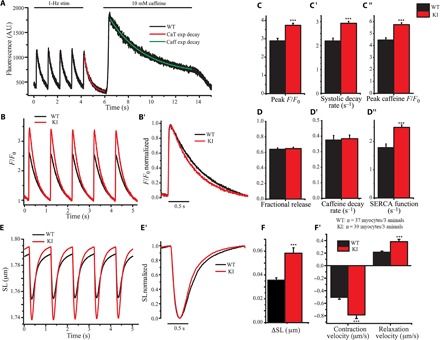
(A) Representative tracing of electrically stimulated and caffeine-induced Ca2+ transients in isolated wild-type ventricular myocytes. The final electrically stimulated and caffeine-induced Ca2+ transients were fit to a single exponential decay to calculate systolic (CaT) and caffeine (Caff) decay rates, respectively. A.U., arbitrary units. (B and C) Average traces of Ca2+ transients (B) revealed increased Ca2+ release during systolic contractions (C) in isolated ventricular myocytes from 1-year-old knock-in mice. (B′ and C′) Normalization of Ca2+ transients (B′) showed a faster decay rate (C′) in knock-in cardiomyocytes. (C″ to D″) Knock-in cardiomyocytes exhibited increased Ca2+ release after caffeine-induced contractions compared to wild-type cells (C″), indicating that more Ca2+ is stored in the SR. However, fractional release of Ca2+ during systolic contraction (D) and Ca2+ decay rates in the presence of caffeine (D′) remained unaltered, suggesting no clear change in RyR2 sensitivity or NCX activity. Consistent with this, calculations showed that SERCA2 activity is significantly increased (~30%) in knock-in hearts (D″). (E to F′) Isolated knock-in cardiomyocytes exhibit enhanced contractility (E and F) and accelerated contractility kinetics compared to wild-type cells, as indicated by their averaged (E) and normalized (E′) tracings of contractions that showed faster peak contraction and relaxation velocities (F′); t test, ***P < 0.001; error bars represent SEM; n = 3 animals per group, 40 cardiomyocytes per group. SL, sarcomeric length.
Given the increased amounts and activity of SERCA2 in isolated cardiomyocytes, we next investigated the global electrical activity of the whole heart by performing electrocardiography (ECG) in 1-year-old knock-in and wild-type animals. Knock-in animals consistently showed significantly faster heart rates than wild-type animals, indicating that they develop tachycardia (Fig. 4, A to A″). Knock-in animals were more susceptible to developing premature ventricular contractions (PVCs) compared to their wild-type littermates, given that four of five knock-in (n = 5) but not wild-type (n = 4) animals exhibited one or two episodes of PVC (P = 0.0397, Fisher’s exact test; Fig. 4B). The average lasting time of a PVC episode was ~290 ms and was occasionally accompanied by incidents of bigeminy and trigeminy (Fig. 4B′). Notably, an extreme case of a knock-in animal developed spontaneous ventricular tachycardia, which was evidenced throughout the 50-min-long monitoring period (Fig. 4C).
Fig. 4. One-year-old knock-in animals develop ventricular arrhythmia under sedentary conditions.
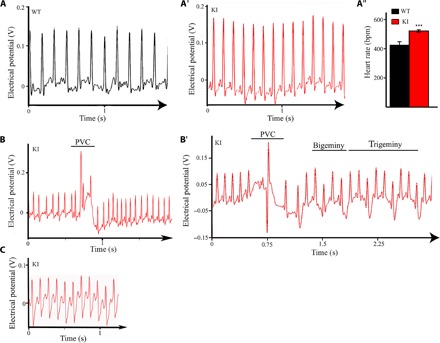
(A to A″) ECG tracings of 1-year-old wild-type (A) and homozygous obscurin knock-in (A′) female mice indicated that obscurin knock-in mice exhibit faster heart rate compared to wild-type mice (A″); t test, ***P < 0.001 (P = 0.00047); error bars represent SEM; wild-type, n = 4 animals; knock-in, n = 5 animals. bpm, beats per minute. (B) Knock-in but not wild-type mice exhibit frequent episodes of PVC during the 50-min monitoring period. (B′) Notably, one knock-in mouse also displayed incidents of bigeminy and trigeminy following a PVC episode. (C) An extreme case of a knock-in mouse developed spontaneous ventricular tachycardia that lasted throughout the 50-min monitoring period.
Together, our findings indicate that the presence of the R4344Q obscurin mutation in the heart leads to up-regulation and overactivation of SERCA2, increased SR Ca2+ content, faster contraction and relaxation kinetics, and development of spontaneous ventricular arrhythmia by 1 year of age under sedentary conditions.
Knock-in animals carrying the obscurin R4344Q mutation develop a DCM-like phenotype following exposure to pressure overload
We next examined whether the knock-in animals are more susceptible to chronic, pathological stress compared to their wild-type littermates. To this end, 2-month-old wild-type and knock-in female mice were subjected to transverse aortic constriction (TAC) to simulate pressure overload; control groups were subjected to sham surgery (that is, surgery without aortic banding). Examination of the expression levels of mutant obscurins 8 weeks after TAC revealed a consistent up-regulation of obscurin B in knock-in–TAC hearts compared to controls; obscurin B is the giant kinase-bearing isoform that is present in relatively low amounts in healthy myocardia (Fig. 5A, asterisk) (12). This finding is consistent with a previous report documenting up-regulation of the transcript levels of obscurin-kinase isoforms in response to aortic stenosis (24) and suggests signaling alterations in the stressed knock-in myocardia possibly mediated by the kinase domains of obscurin B. The sarcomeric distribution of obscurins overlying M-bands, however, remained unaltered (Fig. 5, B to B‴).
Fig. 5. Homozygous female knock-in animals exhibit enlarged hearts and extensive fibrosis and calcification 8 weeks after TAC surgery.
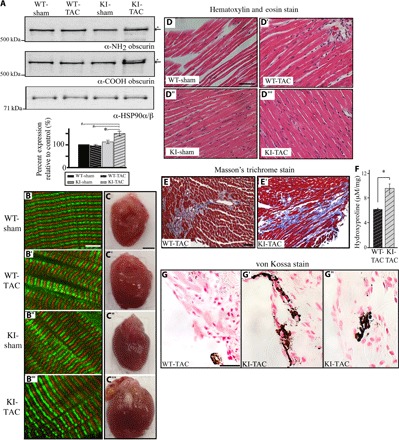
Two-month-old wild-type and knock-in female mice were subjected to either sham or TAC surgery and evaluated 8 weeks later. (A) Representative immunoblots of protein lysates prepared from left ventricles of sham- or TAC-subjected wild-type and knock-in animals using antibodies to the NH2 and COOH termini of obscurins. Relative quantification revealed increased levels of obscurins in the knock-in–TAC hearts due to up-regulation of obscurin B (asterisk). HSP90α/β served as loading control; n = 3 animals per group, two to three replica blots per heart; t test, *P < 0.05, #P < 0.005; error bars represent SEM. (B to B‴) Immunofluorescence combined with confocal optics and antibodies to the COOH terminus of obscurins demonstrated that their localization is unaltered in knock-in left ventricles subjected to sham or TAC surgery; obscurins occupying M-bands are shown in red, and α-actinin, which is a Z-disk marker, is shown in green; scale bar, 5 μm. (C to C‴) Gross evaluation of wild-type and knock-in hearts 8 weeks after sham or TAC surgery indicated that the KI-TAC hearts are notably enlarged; scale bar, 2 mm. (D to D‴) H&E staining of cardiac sections from left ventricles revealed no obvious structural abnormalities in any group; scale bar, 50 μm. (E and E′) Masson’s trichrome staining of sections from left ventricles of wild-type–TAC and knock-in–TAC hearts revealed the presence of excessive interstitial fibrosis in the latter; scale bar, 50 μm. (F) Left ventricles from knock-in–TAC animals contained significantly increased amounts of hydroxyproline compared to left ventricles from wild-type–TAC animals; t test, *P < 0.01; error bars represent SEM; n = 3 animals per group, three repeats per heart. (G to G″) von Kossa staining of left ventricles demonstrated the presence of more prominent peripheral (G′) and interstitial (G″) dystrophic calcification in knock-in–TAC versus wild-type–TAC (G) hearts; scale bar, 25 μm; 40× objective.
Knock-in–TAC mice exhibited significantly enlarged hearts compared to controls (Fig. 5, C to C‴, and table S2). Histological evaluation of the knock-in–TAC hearts with H&E staining did not show any apparent structural alterations (Fig. 5, D to D′); however, Masson’s trichrome staining revealed the presence of markedly increased interstitial fibrosis compared to wild-type–TAC hearts (Fig. 5, E and E′). In line with this, the knock-in–TAC group contained significantly higher amounts of hydroxyproline compared to the wild-type–TAC group (Fig. 5F). Moreover, von Kossa staining indicated the presence of a more prominent calcification in knock-in–TAC myocardia compared to controls (Fig. 5, G to G″). Consistent with these morphological alterations, evaluation of the knock-in–TAC animals using transthoracic echocardiography demonstrated that they developed a DCM-like phenotype manifested as reduced relative left ventricle wall thickness and increased left ventricle internal diameter during diastole, end diastolic volume, and isovolumetric relaxation time compared to wild-type–TAC animals (table S2). Assessment of the knock-in–sham animals showed that they developed an HCM-like phenotype, potentially as a compensatory response to the surgical operation stress, evidenced by the increased left ventricle anterior and posterior wall thickness in diastole and systole, reduced internal diameter during diastole and systole and end diastolic volume, and increased EF and FS compared to wild-type–sham animals (table S2). Collectively, these findings indicate that the presence of the R4344Q mutation renders the heart susceptible to stress, resulting either in maladaptive hypertrophy in the case of mild stress induced by sham surgery or in dilation and remodeling presented as fibrotic infiltration and Ca2+ deposition in the case of pathological mechanical stress induced by TAC surgery.
The R4344Q mutation causes an altered distribution of electrostatic charges at the surface of the obscurin Ig58 domain
To elucidate the molecular mechanisms through which the R4344Q mutation affects Ca2+ cycling and thus cardiac function, we solved the high-resolution structure of the human wild-type Ig58 domain using both nuclear magnetic resonance (NMR) and x-ray crystallography (Fig. 6, A and B). The Ig58 NMR ensemble includes greater than 14 restraints per residue, with no distance constraint violations greater than 0.4 Å and a Q factor of 0.27 (table S3). The NMR-based structures agree well with each other, with a backbone root mean square deviation (RMSD) of 0.471 Å for the 20 best structures. The x-ray structure of Ig58, at 2.41 Å resolution, has an Rfree value of 0.28 (table S4). Overall, the Ig58 structure folded in a canonical Ig-like configuration, forming a tightly packed β-sandwich (Fig. 6, A and B), with a high degree of similarity between the NMR and x-ray structures (heavy atom RMSD of 1.4 Å). Regions of the greatest difference lie in the B-C loop and the D-E loop (Fig. 6, A and B). These are long loops and likely dynamic, displaying slightly elevated B factors (a measure of atomic position certainty) in the x-ray structure and fewer than normal distance restraints in the solution ensemble. Mutant Ig58 containing the R4344Q substitution remains similarly folded to the wild-type protein, as demonstrated by only minor heteronuclear single-quantum coherence (HSQC) perturbations (Fig. 6C). Projecting these chemical shift changes onto the high-resolution wild-type Ig58 structure illustrates that residues in close physical proximity to the R4344Q mutation are mostly affected. These include residues E4360, Q4362, L4363, S4364, Q4365, E4392, and G4417, with the majority residing on neighboring β strands (Fig. 6C). Close inspection of the wild-type Ig58 electron density map revealed a series of side-chain charge-charge interactions between residues R4344 and Q4362, between residues R4344 and E4360, between residues E4360 and R4396, and, conceivably, between residues R4396 and E4392 (Fig. 6D). Such charge-charge pairs likely explain the cross-strand influence of the R4344Q mutation on the chemical shifts. Molecular dynamics simulations of the Ig58 structure further confirmed that the R4344-E4360, R4344-Q4362, and E4360-R4396 pairs can exist in stable (>10 ns) interactions (Fig. 6E) and that these electrostatic bonds stabilize and shape a significant part of the surface of the wild-type Ig58 domain. Thus, although the R4344Q mutation may affect target binding through direct ablation of coulombic interactions, it is equally feasible that it may disrupt or enhance target binding by globally altering the overall topology or dynamics of the Ig58 target binding site.
Fig. 6. Mutant obscurin Ig58 exhibits distorted electrostatic topology and target binding characteristics.
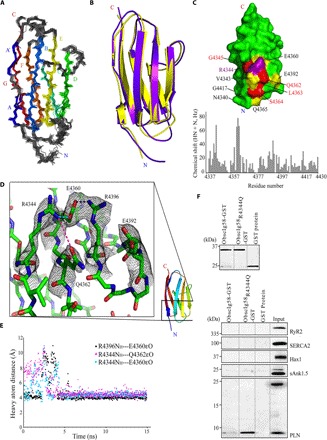
(A) An ensemble of the 20 best NMR structures of wild-type Ig58, colored by β strands A to G. (B) Overlay of the NMR and x-ray structure of wild-type Ig58. (C) Top: Chemical shift differences between mutant and wild-type Ig58 are mapped onto the wild-type Ig58 structure. Residues that exhibit greater than twofold and threefold the average NMR chemical shift change are colored yellow and red, respectively. The location of the R4344Q mutation is colored purple. Bottom: The residue-by-residue chemical shift differences between mutant and wild-type Ig58 HSQC spectra. (D) Electron density of the residues surrounding R4344 (in wild-type Ig58) involved in extensive charge-charge interactions. (E) Molecular dynamics plot of the side-chain N–O distances between surface residues of Ig58, showing long-lived electrostatic interactions. (F) The presence of the R4344Q mutation enhances the ability of Ig58 to interact with PLN. Equivalent amounts of wild-type and mutant glutathione S-transferase (GST)–tagged Ig58 as well as control GST-protein were bound to glutathione matrices (top) and incubated with protein homogenates prepared from adult cardiac muscle. Both wild-type and mutant GST-Ig58 but not control GST-protein efficiently adsorbed the monomeric form of native PLN. Remarkably, mutant GST-Ig58 carrying the R4344Q mutation did so more efficiently than wild-type protein (~10-fold higher). Moreover, neither wild-type nor mutant GST-Ig58 was able to retain other Ca2+-cycling proteins examined, including RyR2, SERCA2, Hax1, and sAnk1.5, further demonstrating the specific interaction between the wild-type and mutant Ig58 domain with PLN.
The R4344Q mutation alters binding of obscurin Ig58 to other proteins
Given the major alterations that we observed in Ca2+ regulation in the knock-in hearts (Fig. 3) and the effects of the R4344Q mutation on the topography of the electrostatic charges on the surface of Ig58 (Fig. 6C), we then tested the ability of wild-type and mutant Ig58 to interact with major Ca2+-cycling proteins using in vitro binding assays. We found that wild-type Ig58 interacts modestly with monomeric but not pentameric PLN or other key Ca2+-handling proteins, including RyR2, SERCA2, and its regulators Hax1 and sAnk1.5 (Fig. 6F). Remarkably, the direct interaction between obscurin Ig58 and PLN was significantly enhanced (~10-fold) in the presence of the R4344Q mutation (Fig. 6F). This finding is consistent with our structural data (Fig. 6, A to E), indicating that the presence of the R4344Q mutation disrupts electrostatic charge-charge pairs on the surface of Ig58, leading to altered (and, in the case of PLN, enhanced) binding interactions.
DISCUSSION
Recent studies have implicated mutations in the OBSCN gene in the development of hereditary HCM and DCM (16–18). The molecular etiologies that underlie the pathogenesis of either disease due to mutant obscurins, however, have remained largely elusive. In the current study, we generated an in vivo model to examine how the R4344Q mutation present in Ig58 of obscurins affects cardiac function. Sedentary homozygous knock-in mice develop normally, and their hearts do not exhibit any obvious morphological or structural alterations, with the exception of mild peripheral fibrosis. Accordingly, the amounts and localization of mutant obscurins and other major sarcomeric proteins remain unaltered in sedentary 1-year-old knock-in hearts. This is also the case for titin, which interacts directly with the obscurin Ig58-Ig59 region via its Z9-Z10 domains. Earlier work by Arimura and colleagues (16) reported that mutant obscurin Ig58-Ig59 protein incorporates less efficiently into sarcomeres than wild-type protein, potentially because of its (modestly) diminished binding to the titin Z9-Z10 region. The discrepancy between the study by Arimura et al. (16) and our study may arise from the use of different experimental systems (that is, in vitro versus in vivo) and exogenous short fragments versus endogenous full-length proteins. Remarkably, although the expression levels and activity of the SERCA2 pump are increased, the levels of pentameric PLN are decreased in 1-year-old knock-in cardiomyocytes, resulting in SR Ca2+ overload, increased amplitude of Ca2+-transients, and faster contractility kinetics. Consequently, sedentary 1-year-old female knock-in animals develop tachycardia and suffer from frequent episodes of PVC as a maladaptive response to these molecular alterations.
It is well established that the activity of SERCA2 is primarily modulated by the small phosphoprotein PLN (25), which exists in a monomeric and a pentameric form. Nonphosphorylated, monomeric PLN binds SERCA2 tightly and inhibits its Ca2+ pumping activity; this inhibitory effect of PLN on SERCA2 is relieved upon PLN phosphorylation and pentameric formation (26). Our biochemical studies revealed that monomeric but not pentameric PLN interacts directly with the obscurin Ig58 domain and that their binding is markedly enhanced (~10-fold) in the presence of the R4344Q mutation. It is therefore likely that mutant obscurins (carrying the R4344Q substitution) sequester monomeric PLN, which is consistent with the reduced levels of pentameric PLN that we observed (Fig. 2), rendering it unable to bind and inhibit the activity of SERCA2, thus leading to overactivation of the latter. Both our structural studies (Fig. 6) and molecular modeling simulations (Fig. 7) support such a scenario. Whereas the bulky and charged side chain of arginine (R)-4344 prohibits a tight contact between wild-type Ig58 and PLN, the shorter side chain of glutamine (Q) allows the α-helical cytoplasmic tail of PLN to be closely docked onto the surface of mutant Ig58. Moreover, as our molecular simulations indicate that the interaction between mutant Ig58 and PLN is stabilized through extensive electrostatic interactions between Q4344, E4356, E4360, Q4362, and E4392 on the surface of Ig58 and R9, R13, and R14 in the cytoplasmic tail of PLN, roughly half of these interactions are not possible in a model using wild-type Ig58 (Fig. 7). Consistent with this, comparison of the wild-type and mutant Ig58 HSQC spectra shows that four of these five obscurin residues exhibit significant chemical shift perturbations, demonstrating that our wet-lab work (Fig. 6) and computational modeling simulations (Fig. 7) point to similar molecular mechanisms. We therefore postulate that the presence of the R4344Q mutation increases the strength of the Ig58/PLN interaction by altering the topography of the electrostatic charges on the surface of Ig58, thus precluding PLN from binding to and inhibiting SERCA2, which, in turn, results in increased SR Ca2+ content, accelerated Ca2+ kinetics, and, ultimately, ventricular arrhythmia.
Fig. 7. The R4344Q substitution allows tighter binding of PLN to Ig58 of obscurins.
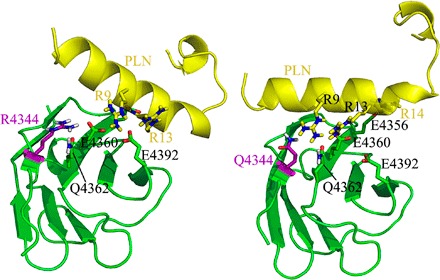
YASARA modeling of the interaction between Ig58 and PLN suggests that the bulky side chain of R4344 hinders PLN docking onto the surface of Ig58 (left), whereas the smaller side chain of the R4344Q substitution allows more significant PLN-Ig58 electrostatic interactions to occur (right). The obscurin Ig58 domain is colored green, and the cytoplasmic domain of PLN is colored yellow, with the R4344Q mutation on Ig58 colored purple.
Similar to the R4344Q obscurin knock-in animals, transgenic models carrying HCM-linked troponin T mutations (for example, I79N, F110I, and R278C) do not exhibit cardiac hypertrophy under sedentary conditions but exhibit altered Ca2+ kinetics and are susceptible to developing arrhythmia post-pause following electrical pacing (27–29). However, unlike the obscurin R4344Q knock-in model, the troponin T transgenic mice exhibit altered Ca2+ binding affinity at the level of the sarcomere instead of altered Ca2+ content and kinetics at the level of the SR (29). Thus, these findings underscore the key roles of sarcomeric proteins in regulating different aspects of Ca2+ homeostasis during contraction and relaxation. They also point out that congenital heart disease due to mutant sarcomeric proteins may not necessarily encompass structural perturbations (at least in a laboratory animal model that is not subjected to physiological or pathological stress) but may be characterized by the development of maladaptive responses, such as spontaneous or sustained ventricular arrhythmias, which may ultimately lead to myofibrillar disarray and heart failure (30).
The correlation between enhanced SERCA2 activity and the development of arrhythmia has been controversial (31–34). Our studies demonstrate that homozygous obscurin knock-in hearts exhibit increased SERCA2 activity and develop spontaneous tachycardia accompanied by incidents of PVC. Consistent with our findings, rats overexpressing SERCA2 suffer from more frequent episodes of ventricular tachycardia compared to wild-type animals following myocardial infarction (31). Conversely, mice with conditional deletion of SERCA2 subjected to myocardial infarction experience less frequent incidences of spontaneous ventricular extrasystoles than wild-type animals (32). Similarly, overloaded Ca2+ stores have been linked to the generation of Ca2+ waves and the development of arrhythmia (35, 36). By contrast, ectopic expression of SERCA2a in rat and porcine ischemia reperfusion models prevents the development of arrhythmia (33, 34). It therefore appears that whether enhanced SERCA2 expression and activity correlates with increased or decreased propensity of developing arrhythmia may depend on multiple factors, including the type of the stress stimulus (for example, mechanical versus oxidative stress).
Exertion of pressure overload demonstrated that the presence of the R4344Q mutation renders the heart susceptible to chronic, pathological stress. Accordingly, knock-in animals subjected to TAC surgery develop a DCM-like phenotype accompanied by extensive interstitial fibrosis and calcification, all of which are hallmarks of progressive heart disease [reviewed in previous studies (37, 38)]. Remarkably, knock-in animals subjected to sham surgery develop an HCM-like phenotype, potentially as an adaptive response to the mild surgical operation stress. Together, our findings indicate that the R4344Q mutation renders the heart susceptible to pathophysiological stressors, resulting in spontaneous arrhythmia as a function of aging, compensatory hypertrophy following acute mild stress, and modest dilation accompanied by remodeling due to chronic mechanical stress. Thus, our knock-in mouse model is suitable for study of the molecular etiologies underlying the development of heart disease owing to the R4344Q mutation under both sedentary and stress conditions.
A recent study postulated that the HCM-linked R4344Q obscurin mutation is a minor allele more commonly observed in non-Hispanic black Americans (15.3% frequency) compared to non-Hispanic white Americans (0.3% frequency) (39). Given the fact that non-Hispanic black Americans develop CVD at a considerably higher rate compared to non-Hispanic white Americans (44.4% versus 36.6% for males and 48.9% versus 32.4% for females, respectively) (40), it is highly likely that the presence of the R4344Q mutation may render the carriers more susceptible to develop CM. Obviously, additional factors, including genetic modifiers, individual lifestyle regimens, and environmental factors, may contribute to the penetrance and expressivity of the mutant allele and therefore to disease onset, progression, and severity among carriers.
Given the recent identification of missense, deletion, and frameshift mutations in the OBSCN gene that are causally associated with the development of HCM and DCM (16–18), it becomes apparent that obscurins are intimately linked to the maintenance of cardiac structure and function and the pathogenesis of heart disease by playing both scaffolding (14, 16) and regulatory (current study) roles, potentially via their multiple interactions with sarcomeric and membrane-associated proteins. Our studies demonstrate for the first time that obscurins contribute to the regulation of Ca2+ cycling, which is particularly compromised in heart disease. Future studies should therefore aim to decipher the potentially diverse molecular etiologies underlying the pathogenicity of congenital heart disease due to mutant obscurins.
MATERIALS AND METHODS
Gene targeting and generation of obscurinR4344Q knock-in mice
ObscurinR4344Q knock-in mice were produced via a commercial source (GenOway). The gene-targeting construct was generated by subcloning isogenic genomic DNA spanning partial intron 59 to partial intron 65, with exon 60 carrying the nucleotides encoding the R4344Q mutation (fig. S1A). A Flippase recognition target (FRT) site-neomycin-FRT-loxP cassette was inserted in intron 59 along with a distal loxP site in intron 61. The linearized construct was electroporated into embryonic stem (ES) cells derived from the 129/Sv mouse strain, which has an agouti color. Clones carrying the targeting construct [including the neomycin (neo) cassette] were selected for resistance to G418. Genomic DNA from G418-resistant clones was screened for homologous recombination using primers complementary to sequences in exon 60 (table S5). Recombinant ES clones were injected into C57BL/6J blastocysts, and their offspring were identified by their agouti coat. The neo selection cassette was removed by breeding the animals with Flp recombinase–expressing deleter mice (GenOway). The genotypes of the animals were confirmed with polymerase chain reaction (PCR) and a primer set complementary to sequences in intron 59 and exon 60. F1 mice were backcrossed seven generations into the C57BL/6 mouse line before experimentation.
All animal care and experimental procedures were approved and performed in accordance with the ethical standards set by the Institutional Animal Care and Use Committees of the University of Maryland School of Medicine (UMSOM), the University of Pennsylvania, and the National Institutes of Health (NIH).
Transthoracic echocardiography
Cardiac function was assessed by echocardiography. Animals were shaved in the chest area and anesthetized with 4 to 5% isoflurane in oxygen in an induction chamber. Echocardiography measurements were obtained with a 40-MHz probe connected to the Vevo 2100 System (VisualSonics). During experimentation, the animals were placed on a warming pad and continually maintained with 2% isoflurane in oxygen. Two-dimensional (2D) cineloops and guided M-mode frames were recorded from the parasternal short axis. Absolute wall thickness and relative wall thickness were calculated during diastole (d) as PWTd + AWTd and (PWTd + AWTd)/EDD, respectively, where PWTd is the diastolic posterior wall thickness, AWTd is the diastolic anterior wall thickness, and EDD is the end-diastolic diameter. EF was calculated as (EDV − ESV)/EDV × 100%, where EDV is the end-diastolic volume and ESV is the end-systolic volume. EDV and ESV were calculated as 1.047 × EDD3 and 1.047 × ESD3, respectively, where ESD is the end-systolic diameter (41). Transthoracic echocardiography was performed in the Physiological Phenotyping Core of UMSOM.
Surface ECG
Three-lead ECG probes were inserted subcutaneously into 1-year-old wild-type and knock-in mice, which were placed supinely and under 1% isoflurane in pure oxygen via a face mask throughout data collection. The positive and negative electrodes were placed in the left hindlimb and right forelimb, respectively, whereas the ground electrode was placed in the left forelimb. The electrodes were linked to a biopotential amplifier (BIOPAC MEC100C) connected to a computer-interfaced BIOPAC MP150 system (BIOPAC Systems Inc.), which measures and records electrical signals. The animals were subjected to continuous monitoring via ECG for 50 min. Surface ECG was performed in the Physiological Phenotyping Core of UMSOM; n = 4 for wild-type and n = 5 for knock-in animals.
Cardiomyocyte isolation
Mice were terminally anesthetized by injection of pentobarbital, followed by excision of the heart and enzymatic isolation of ventricular myocytes using the cell isolation buffer [25 mM Hepes, 130 mM NaCl, 22 mM glucose, 5.4 mM KCl, 1 mM lactic acid, 0.5 mM MgCl2, 0.33 mM NaH2PO4, and 0.3 mM CaCl2, along with insulin (100 μU/ml), collagenase IIB (0.66 mg/ml; Worthington, LS004176), trypsin (0.033 mg/ml, Sigma-Aldrich T0303), and proteinase (0.033 mg/ml, Sigma-Aldrich P8038)], and perfused on a Langendorff perfusion column via cannulation of the aorta (42). Tissues were minced and subjected to additional digestion with 0.7 mM CaCl2 and bovine serum albumin (BSA; 2 mg/ml) at 37°C, until cardiomyocytes were easily dissociated by trituration. Cardiomyocytes were stored in normal Tyrode’s solution containing 140 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 0.5 mM MgCl2, 5 mM Hepes, 5 mM glucose, and 0.33 mM Na2HPO4. Experiments were performed the same day at room temperature (22°C).
[Ca2+]i transient and contractility measurements
Measurements of Ca2+ transients and contractility were performed in custom-fabricated cell chambers (Four Hour Day Foundation) mounted on a Zeiss LSM 510 inverted confocal microscope with a 40× oil objective (numerical aperture of 1.4). [Ca2+]i transients were evaluated with Fluo-4 by 15-min incubation with 2 μM Fluo-4-acetoxymethyl ester and 0.01% Pluronic F127. For measurements of contractility, sarcomere length was monitored with a high-speed video camera and fast Fourier transform analysis. Cells were allowed an additional 10 min for de-esterification and then scanned using a 488-nm argon ion laser in confocal line scan mode at 1.92 ms per line. Cells were paced to steady state using 1-Hz field stimulation through platinum electrodes. After 20 s of 1-Hz stimulation, all cells showed steady-state transients and contractions. At this point, five Ca2+ transients and five contractions were assayed and pooled for analysis. The number of cells analyzed was as follows: n = 40 myocytes/3 animals per group.
SERCA2a function was assessed in 1-year-old wild-type and knock-in cardiomyocytes. To evaluate SERCA2a function, we took advantage of the fact that the decay rate of the systolic Ca2+ transient is dependent on the activity of the NCX and SERCA2a (with a minor contribution of the plasma membrane Ca2+ ATPase), whereas decay of the caffeine-induced Ca2+ transient depends mainly on NCX. The difference in the two rates can thus be ascribed to SERCA2a activity (22). Following steady-state electrical field stimulation to produce systolic transients as described above, Ca2+ release from the SR was elicited by 10 mM caffeine by local superfusion. Systolic and caffeine Ca2+ transients were fitted with a single exponential Y = a + eкt, and the rate constant was defined as 1/к. SERCA2a activity was assessed by subtracting the caffeine-induced Ca2+ transient decay rate constant (кcaff) from the systolic Ca2+ transient decay rate constant (кsys): кsys − кcaff.
Lysate preparation and Western blotting
Cardiac lysates were prepared in urea-thiourea buffer [8 M urea, 2 M thiourea, 0.05 M tris-HCl (pH 6.8), 0.075 M dithiothreitol, 3% SDS, and 10% glycerol] or in Nonidet P-40 (NP-40) buffer (10 mM sodium phosphate, 2 mM EDTA, 10 mM NaN3, 0.9% NaCl, and 2% NP-40) supplemented with a cOmplete Mini Protease Inhibitor Cocktail Tablet (Roche). For lysates examined for the phosphorylation status of proteins, we included the following phosphatase inhibitors in the lysis buffer: 4 mM imidazole, 2 mM sodium fluoride, 2.3 mM sodium molybdate, 8 mM sodium tartrate dehydrate, 2 mM β-glycerophosphate, 2 mM sodium pyrophosphate, 0.2 mM EGTA, 2 mM sodium orthovanadate, and 1 mM okadaic acid. Protein lysates were separated by SDS–polyacrylamide gel electrophoresis (PAGE) and transferred to a nitrocellulose membrane. Membranes were probed with the appropriate primary antibodies and subsequently incubated with mouse or rabbit secondary antibodies conjugated to alkaline phosphatase. Immunoreactive bands were detected with chemiluminescence reagents (Tropix, Thermo Fisher Scientific). All immunoblots were performed with lysates prepared from three different hearts per group and were repeated at least twice. Relative quantification of the expression levels of the indicated proteins was performed with ImageJ software, and the reported values are averages obtained from all immunoblots normalized to HSP90α/β levels.
Electrophoresis and SYPRO Ruby staining of titin
Cardiac lysates prepared as described above were heated at 60°C for 20 min. Ten micrograms of lysates was loaded to a gel containing 1% (w/v) agarose and 3% (w/v) polyacrylamide in 50 mM tris, 0.384 M glycine, 2.5% (v/v) glycerol, 0.1% (w/v) SDS, 0.5 mM dithiothreitol, and 0.1% (w/v) ammonium persulfate. Electrophoresis was performed with the Mini-PROTEAN electrophoresis cell (Bio-Rad) at 110 V for 3 hours. SYPRO Ruby staining (Thermo Fisher Scientific) was performed according to the manufacturer’s protocol. The SYPRO Ruby–stained gel was imaged on a Typhoon FLA 9500 imager (GE Healthcare Bio-Sciences).
Preparation of frozen and paraffin-embedded cardiac sections
Frozen and paraffin-embedded cardiac sections were prepared, as previously described (12, 43). Mice (sedentary, 1-year-old animals) were sacrificed by perfusion with 2% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) under anesthesia. Hearts were excised and embedded in 7.5% gelatin and 15% sucrose in PBS and gradually frozen with 2-methylbutane. Frozen cardiac tissues were sectioned at a thickness of 10 to 14 μm. For paraffin sections, the tissue was fixed in formalin overnight at 4°C and sequentially dehydrated in 70, 80, and 95% absolute ethanol and two washes of xylene. The samples were subsequently embedded in paraffin wax at 58°C for 4 hours with one change in between and sectioned at a thickness of 5 μm.
Immunostaining
Immunostaining was performed, as previously described (12). Frozen cardiac sections and 2% PFA-fixed cardiomyocytes permeabilized with 0.1% Triton X-100 were incubated with blocking buffer [PBS, BSA (1 mg/ml), and 1 mM sodium azide] for 1 hour and then with the appropriate primary antibodies in blocking buffer overnight at 4°C. Sections and cardiomyocyte preparations were washed with blocking buffer three times for 5 min each and counterstained with Alexa Fluor 488 goat anti-mouse or Alexa Fluor 568 goat anti-rabbit secondary antibodies (1:200; Thermo Fisher Scientific). Following additional washing with blocking buffer, slides were mounted with VECTASHIELD (Vector Laboratories) and analyzed under a Zeiss LSM 510 confocal microscope (UMSOM Confocal Microscopy Facility). At least two animals per group were analyzed.
Antibodies
The following primary antibodies were used: mouse monoclonal antibodies to α-actinin [Western blotting (WB), 1:2500; immunofluorescence (IF), 1:400; A7811, Sigma-Aldrich], actin (WB, 300 ng/ml; A2171, Sigma-Aldrich), myomesin [WB/IF, 1:10; mMaC, Developmental Studies Hybridoma Bank (DSHB)], myosin (WB/IF, 1:10; MF20, DSHB), RyR2 (WB/IF, 1:1000; MA3-925, Thermo Fisher Scientific), SERCA2 (WB, 1:1000; IF, 1:100; MA3-919, Thermo Fisher Scientific), PLN [WB, 1:5000 (A010-14; Badrilla); WB, 2 μg/ml; IF, 1:200 (ab2865, Abcam)], titin (IF, 1:10; 9D10, DSHB), obscurin-NH2 (WB/IF, 1:10; tissue culture supernatant) (13), and Hax-1 (WB, 1:1000; clone 52; 610824, BD Biosciences); rabbit monoclonal antibody to HSP90 (WB, 1:1000; 4877, Cell Signaling); and rabbit polyclonal antibodies to dihydropyridine receptor (WB/IF, 1:200; ab58552, Abcam), junctophilin-2 (WB, 1 μg/ml; 40-5300, Invitrogen), titin-Z (IF, 2 μg/ml) (44), SERCA2 (IF, 1:100; Ab91032, Abcam), sAnk1.5 (WB, 300 ng/ml; IF, 3 μg/ml) (9), SERCA2-pSer38 (WB, 1:1000; IF, 100; A010-25AP, Badrilla), RyR2-pSer2808 (WB, 1:2000; IF, 1:100; ab59225, Abcam), RyR2-pSer2814 (WB, 1:1000; A010-31AP, Badrilla), PLN-pSer16 (WB, 1:5000; A010-12, Badrilla), PLN-pSer16 (IF, 1:200; 07-052, Millipore), obscurin-COOH (WB, 300 ng/ml) (9), and obscurin-Ig58/59 (IF, 3 μg/ml) (45).
Masson’s trichrome staining
Paraffin-embedded sections were deparaffinized in xylenes for 5 min with two changes and rehydrated by sequential incubation in 95 and 70% absolute alcohol, with two changes of 2 min for each. Sections were subsequently incubated with Bouin’s solution (4.8% acetic acid, 9.5% formaldehyde, and 0.93% picric acid) at 56°C for 1 hour. Samples were washed thoroughly in running water, rinsed in distilled water, and stained with Weigert’s iron hematoxylin solution (0.5% hematoxylin, 0.06 M HCl, 0.58% FeCl3, and 47.5% ethanol) for 10 min and then with Biebrich scarlet-acid fuchsin solution (0.89% Biebrich scarlet, 0.099% acid fuchsin, and 0.17 M acetic acid) for 2 min. Subsequently, the samples were incubated with phosphomolybdic-phosphotungstic acid solution (5% phosphomolybdic acid and 5% phosphotungstic acid) for 10 min and stained with aniline blue solution [2.45% (w/v) aniline blue and 0.33 M acetic acid] for 5 min, followed by incubation with 1% acetic acid for 3 min. Distilled water was applied between solutions. At the end of the staining process, samples were sequentially dehydrated in 95 and 100% ethanol and xylene (two times each) and then mounted with Permount (Thermo Fisher Scientific).
von Kossa staining
Cardiac sections were deparaffinized and rehydrated similar to the preparation for Masson’s trichrome staining. The deparaffinized and rehydrated sections were incubated in 5% silver nitrate solution for 1 hour and washed with distilled water four times. Sections were then incubated with 50% photographic developer solution for 2 min and 5% sodium thiosulfate for 5 min, followed by counterstaining with Nuclear Fast Red solution (Sigma-Aldrich) for 5 min. Between solutions, samples were rinsed with running tap water and then with distilled water. At the end of the staining, samples were dehydrated and rinsed with 95 and 100% ethanol, followed by xylenes for 2 min (two times each).
Hydroxyproline assay
The hydroxyproline assay was conducted as follows: 15 to 23 mg of flash-frozen left ventricles at the level of the apex was boiled in 0.2 ml of 6 M HCl at 110°C overnight. The hydrolyzed tissue was added to isopropanol in a 1:16 ratio and allowed to react with chloramine-T [final concentration: 0.448% (w/v) chloramine-T, 0.178 M sodium acetate, 44.5 μM citric acid, 11.14 μM NaOH, and 9.856% (v/v) isopropanol] for 5 min. Subsequently, isopropanol-Ehrlich solution [1.531 M p-dimethylaminobenzaldehyde; 76.1% (v/v) ethanol, 0.945 M sulfuric acid, and 23.1% (v/v) isopropanol] was added to the reaction in a 50:12.5 ratio and incubated in a water bath (55°C) for 1 hour. The absorbance of the final reaction was measured at 558 nm. A standard curve with 0 to 1 M trans-4-hydroxy-l-proline in 6 M HCl was generated, and the fibrotic content of the tissue sample was normalized to the sample mass; n = 4 animals per group, and each assay was repeated three times per heart.
Transverse aortic constriction
Pressure overload was induced on ~2-month-old wild-type and knock-in animals via TAC surgery, as previously described (46). Briefly, animals were anesthetized with 2% isoflurane in oxygen and placed in a supine position on a warming pad. The chest was cut open from the suprasternal notch to the third rib. A 28-gauge needle was placed next to the transverse aorta, and a 6-0 silk suture was used to tie the aorta with the tightness determined by the needle diameter. After the lungs were re-inflated, the chest wall was closed with 6-0 silk thread, and the skin was closed with 6-0 prolene thread. Mice were treated with 5 mg of carprofen (Pfizer) per kilogram of body weight and delivered subcutaneously immediately and 24 hours after surgery. Sham animals were subjected to the same surgical procedure without the aortic constriction. Number of animals used: wild-type–sham, n =11; knock-in–sham, n = 10; wild-type–TAC, n = 11; knock-in–TAC, n = 12.
Transmission electron microscopy
Mice were euthanized and perfusion-fixed with 4% PFA in PBS under anesthesia. Left ventricles were collected and stored in 4% PFA overnight at 4°C. Tissue was trimmed and further fixed in 2% PFA, 2.5% glutaraldehyde, and 2 mM CaCl2 in 0.1 M cacodylate buffer (pH 7.2) at room temperature for 60 min; quenched in 50 mM glycine in 0.1 M cacodylate buffer; and washed and postfixed in 1% (w/v) osmium tetroxide, 0.1 M cacodylate buffer for 30 min, and 0.5% (w/v) osmium tetroxide and 1.5% (w/v) potassium ferrocyanide for 30 min. Specimens were then treated with 1% (w/v) tannic acid for 60 min, washed, en bloc stained with 1% (w/v) uranyl acetate, and dehydrated using 30, 50, 70, 90, and 100% ethanol and 100% acetone in series. After dehydration, specimens were infiltrated and embedded in Araldite resin (Electron Microscopy Sciences) following the manufacturer’s recommendations. Ultrathin sections were cut on a Leica UC6 ultramicrotome (Leica Microsystems Inc.), poststained with uranyl acetate and lead citrate, and examined in an FEI Tecnai T12 transmission electron microscope operated at 80 kV. Digital images were acquired by an AMT bottom-mount charge-coupled device camera and AMT600 software. Tissue samples were prepared and examined in the Electron Microscopy Core Imaging Facility of the University of Maryland School of Dentistry. Two animals were analyzed per group.
Site-directed mutagenesis and GST pull-down experiments
The R4344Q mutation was introduced into the wild-type obscurin Ig58 using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s instructions using the mutagenesis primers listed in table S5. Introduction of the mutation was confirmed via sequencing. Wild-type and mutant obscurin Ig58 were produced in bacteria as GST-fusion proteins using the pGEX-4T1 vector system. Equivalent amounts of recombinant proteins and control GST were bound to glutathione beads and incubated at 4°C overnight with 0.5 mg of protein lysates prepared from the hearts of 2-month-old wild-type animals. Following incubation and extensive washing in wash buffer [PBS, 10 mM sodium azide, and 0.1% Tween 20 (pH 7.2)], beads were heated at 90°C for 5 min in 2× Laemmli sample buffer, and the supernatant was analyzed by SDS-PAGE and Western blotting using the indicated primary antibodies.
NMR spectroscopy and x-ray crystallography
Recombinant 15N-labeled, 15N-13C-labeled, and unlabeled wild-type obscurin Ig58 proteins were purified after overexpression in BL21(DE3) Escherichia coli cells using a pET24a vector system (Novagen), as previously described (47). The NMR samples were prepared in 20 mM tris (pH 7.5), 20 mM NaCl, 0.35 mM NaN3, and 10% D2O, with 0.5 to 2.5 mM recombinant Ig58 or the mutant Ig58 carrying the R4344Q mutation. All NMR spectra were collected on a 600-MHz Bruker Avance II spectrometer equipped with a 5-mm TXI room temperature probe and z-axis pulse field gradient coils. We collected 2D HSQC and standard triple-resonance spectra, including HNCACB, CBCA(CO)NH, HNCO, HN(CA)CO, C(CO)NH, H(CCCO)NH, 15N-edited TOCSY (total correlation spectroscopy), 15N-edited nuclear Overhauser effect spectroscopy (NOESY), 13C-edited NOESY, and a pseudo-3D IPAP (in-phase anti-phase) experiment, as previously described (47). Most experiments were collected with 128, 64, and 1024 points in the T1, T2, and T3 dimensions, respectively. NMR data were processed with NMRPipe (48) and extended in the indirect dimension via linear prediction, and the resulting spectra were analyzed via Sparky.
Interproton distance constraints were derived from 3D NOESY experiments, as previously reported (49). Dihedral constraints derived from TALOS+ were included on the basis of the chemical shift index of 1Hα and 13Cα atoms (50). Hydrogen bond constraints of rHN–O = 1.5 to 2.8 Å and rN–O = 2.4 to 2.5 Å were included in the final stage of structure calculations. Pseudopotentials for secondary 13Cα and 13Cβ chemical shifts and a conformational database potential were included in the final simulated annealing refinements using the computer program XPLOR-NIH (51). The internuclear dipolar coupling (in hertz) was incorporated into the final structure calculation, as previously described (49). Q factors were calculated by randomly removing ~10% of both the N-HN residual dipolar coupling data and then comparing these values to those back-calculated from the structure. The final 20 structures were selected (from 200) on the basis of lowest energy and were of high quality based on the statistical criteria listed in table S3. Chemical shifts for Ig58 have been deposited in the Biological Magnetic Resonance Bank (BMRB) under accession number 25308, and the 20 best structures have been deposited in the Protein Data Bank (PDB) under accession number 2MWC.
Crystals of wild-type Ig58 were grown in 0.3 to 0.5 M NaCl, 19 to 23% polyethylene glycol 3350, and Ig58 (10.5 to 11.4 mg/ml) at 25°C for 8 days via hanging drop vapor diffusion. Crystals were harvested using MicroMeshes (MiTeGen) and cryoprotected in 50% glycerol (v/v) before freezing in liquid nitrogen. Crystallographic reflections data were collected at the Structural Biology Center beamline 19-ID-D at the Advanced Photon Source (Argonne National Laboratory) to 2.411 Å. The final data set used to solve the structure was collected from two different crystals.
Indexing and processing were performed with HKL2000 by integration of single-crystal diffraction data from an area detector with a sigma cutoff of 2. Data were subsequently scaled and merged with the programs Pointless, Aimless, and Scala-truncate within the CCP4 program suite (52). Phasing of the Ig58 reflections data was solved in PHENIX by molecular replacement using PHASER-MR crystallographic software with PDB #2YZ8 as the search model (53). Autobuild was used to generate a starting structure that then underwent 160 iterative rounds of manual model building and refinement and performed with COOT and PHENIX Refine (53). Atomic coordinates and reflections have been deposited in the Research Collaboratory for Structural Bioinformatics (RCSB) Data Bank under accession number 4RSV.
Computational modeling of PLN and Ig58 interactions
Computational modeling was carried out using the Ig58 x-ray structure or the Ig58 R4344Q mutation model, which was generated with the “mutate” function in the computer program YASARA (54). The solvent-accessible region of human PLN (PDB 2HYN; residues 1 to 19) was used as a target. The structures were loaded into the HADDOCK server, and the best cohort of structures was then loaded into YASARA (54). At least 10 ns of molecular dynamics was conducted on each complex in explicit solvent at 310 K and 150 mM NaCl. At the end of these simulations, models were assessed for whether the two proteins remained bound to each other in roughly the same orientations as the input models.
Statistical analysis
Statistical significance between control (wild-type) and experimental (homozygous knock-in) groups was determined by two-sided Student’s t test or Fisher’s exact test, as noted. Sample sizes are described in Materials and Methods and in the corresponding figure legends.
Supplementary Material
Acknowledgments
We thank M. Shriver, N. Lieberman, M. Contreras, and J. Valenti for their technical support in animal maintenance; C. Chang for the collection of diffraction data; and M. Russell for valuable discussions on the phenotypic manifestations of HCM and DCM. The structural results shown in this report are derived from work performed at the Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. In addition, we wish to dedicate this study to the memory of the late W. Stanley. Funding: This work was supported by NIH [grants 2T32AR7592-16 (L.-Y.R.H.), HL116778 (M.A.A.), HL114879 (B.L.P.), HL106059 (W.J.L.), and U01 HL116321 (W.J.L.)], the Leducq North American-European Atrial Fibrillation Research Alliance (W.J.L.), the European Community’s Seventh Framework Programme FP7/2007-2013 under grant agreement no. HEALTH-F2-2009-241526, EUTrigTreat (W.J.L.), the Jeffress Memorial Trust Award J-1041 (N.T.W.), the Research Corporation Cottrell College Award 22450 (N.T.W.), the NSF Research Experiences for Undergraduates award (CHE-1062629) (to N.T.W.), the 4-VA Organization (C.E.B.), and the American Heart Association (14GRNT20380360 and 16GRNT31290010 to A.K.-K.). Author contributions: A.K.-K. conceived the study; L.-Y.R.H., M.A.A., B.L.P., N.T.W., W.J.L., and A.K.-K. designed the study; L.-Y.R.H., M.A.A., P.A.H., B.L.P., B.K., K.A.O., A.G., L.C.M., and C.E.B. performed the experiments of the study; and L.-Y.R.H., M.A.A., P.A.H., B.L.P., N.T.W., and A.K.-K. analyzed the data and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The structures of Ig58 domains are deposited in the RCSB Data Bank under accession numbers 4RSV and 2MWC. Chemical shifts for Ig58 have been deposited in BMRB under accession number 25308.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1603081/DC1
fig. S1. Generation of obscurin knock-in mice carrying the R4344Q mutation in Ig58.
fig. S2. Sarcomeric organization is unaltered in 1-year-old homozygous knock-in female mice.
table S1. Morphometric and echocardiographic analyses of sedentary wild-type and homozygous knock-in hearts.
table S2. Morphometric and echocardiographic analyses of sham- and TAC-subjected wild-type and knock-in hearts.
table S3. NMR-derived restraints and statistics of 20 NMR structures of wild-type Ig58.
table S4. Statistics of wild-type Ig58 crystal diffraction.
table S5. List of primers used for confirmation of gene targeting, animal genotyping, and site-directed mutagenesis.
REFERENCES AND NOTES
- 1.Mozaffarian D., Benjamin E. J., Go A. S., Arnett D. K., Blaha M. J., Cushman M., Das S. R., de Ferranti S., Després J.-P., Fullerton H. J., Howard V. J., Huffman M. D., Isasi C. R., Jiménez M. C., Judd S. E., Kissela B. M., Lichtman J. H., Lisabeth L. D., Liu S., Mackey R. H., Magid D. J., McGuire D. K., Mohler E. R., Moy C. S., Muntner P., Mussolino M. E., Nasir K., Neumar R. W., Nichol G., Palaniappan L., Pandey D. K., Reeves M. J., Rodriguez C. J., Rosamond W., Sorlie P. D., Stein J., Towfighi A., Turan T. N., Virani S. S., Woo D., Yeh R. W., Turner M. B. American Heart Association Statistics Committee; Stroke Statistics Subcommittee; Writing Group Members , Heart disease and stroke statistics—2016 update: A report from the American Heart Association. Circulation 133, e38–e360 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Bos J. M., Towbin J. A., Ackerman M. J., Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 54, 201–211 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Tardiff J. C., Thin filament mutations: Developing an integrative approach to a complex disorder. Circ. Res. 108, 765–782 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colegrave M., Peckham M., Structural implications of β-cardiac myosin heavy chain mutations in human disease. Anat. Rec. 297, 1670–1680 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Hu L.-Y., Ackermann M. A., Kontrogianni-Konstantopoulos A., The sarcomeric M-region: A molecular command center for diverse cellular processes. Biomed. Res. Int. 2015, 714197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neiva-Sousa M., Almeida-Coelho J., Falcão-Pires I., Leite-Moreira A. F., Titin mutations: The fall of Goliath. Heart Fail. Rev. 20, 579–588 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Landstrom A. P., Ackerman M. J., Beyond the cardiac myofilament: Hypertrophic cardiomyopathy-associated mutations in genes that encode calcium-handling proteins. Curr. Mol. Med. 12, 507–518 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukuzawa A., Lange S., Holt M., Vihola A., Carmignac V., Ferreiro A., Udd B., Gautel M., Interactions with titin and myomesin target obscurin and obscurin-like 1 to the M–band: Implications for hereditary myopathies. J. Cell Sci. 121, 1841–1851 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Kontrogianni-Konstantopoulos A., Jones E. M., Van Rossum D. B., Bloch R. J., Obscurin is a ligand for small ankyrin 1 in skeletal muscle. Mol. Biol. Cell 14, 1138–1148 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spooner P. M., Bonner J., Maricq A. V., Benian G. M., Norman K. R., Large isoforms of UNC-89 (obscurin) are required for muscle cell architecture and optimal calcium release in Caenorhabditis elegans. PLOS ONE 7, e40182 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Randazzo D., Giacomello E., Lorenzini S., Rossi D., Pierantozzi E., Blaauw B., Reggiani C., Lange S., Peter A. K., Chen J., Sorrentino V., Obscurin is required for ankyrinB-dependent dystrophin localization and sarcolemma integrity. J. Cell Biol. 200, 523–536 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu L.-Y., Kontrogianni-Konstantopoulos A., The kinase domains of obscurin interact with intercellular adhesion proteins. FASEB J. 27, 2001–2012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kontrogianni-Konstantopoulos A., Catino D. H., Strong J. C., Sutter S., Borisov A. B., Pumplin D. W., Russell M. W., Bloch R. J., Obscurin modulates the assembly and organization of sarcomeres and the sarcoplasmic reticulum. FASEB J. 20, 2102–2111 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Borisov A. B., Sutter S. B., Kontrogianni-Konstantopoulos A., Bloch R. J., Westfall M. V., Russell M. W., Essential role of obscurin in cardiac myofibrillogenesis and hypertrophic response: Evidence from small interfering RNA-mediated gene silencing. Histochem. Cell Biol. 125, 227–238 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Lange S., Ouyang K., Meyer G., Cui L., Cheng H., Lieber R. L., Chen J., Obscurin determines the architecture of the longitudinal sarcoplasmic reticulum. J. Cell Sci. 122, 2640–2650 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arimura T., Matsumoto Y., Okazaki O., Hayashi T., Takahashi M., Inagaki N., Hinohara K., Ashizawa N., Yano K., Kimura A., Structural analysis of obscurin gene in hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 362, 281–287 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Marston S., Montgiraud C., Munster A. B., Copeland O., Choi O., dos Remedios C., Messer A. E., Ehler E., Knöll R., OBSCN mutations associated with dilated cardiomyopathy and haploinsufficiency. PLOS ONE 10, e0138568 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu J., Li Z., Ren X., Dong M., Li J., Shi X., Zhang Y., Xie W., Sun Z., Liu X., Dai Q., Investigation of pathogenic genes in chinese sporadic hypertrophic cardiomyopathy patients by whole exome sequencing. Sci. Rep. 5, 16609 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shioura K. M., Farjah M., Geenen D. L., Solaro R. J., Goldspink P. H., Myofilament calcium sensitization delays decompensated hypertrophy differently between the sexes following myocardial infarction. Am. J. Physiol. Regul. Integr. Comp. Physiol. 300, R361–R368 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller L. R., Marks C., Becker J. B., Hurn P. D., Chen W.-J., Woodruff T., McCarthy M. M., Sohrabji F., Schiebinger L., Wetherington C. L., Makris S., Arnold A. P., Einstein G., Miller V. M., Sandberg K., Maier S., Cornelison T. L., Clayton J. A., Considering sex as a biological variable in preclinical research. FASEB J. 31, 29–34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsich E. M., Piña I. L., Heart failure in women: A need for prospective data. J. Am. Coll. Cardiol. 54, 491–498 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Diaz M. E., Graham H. K., Trafford A. W., Enhanced sarcolemmal Ca2+ efflux reduces sarcoplasmic reticulum Ca2+ content and systolic Ca2+ in cardiac hypertrophy. Cardiovasc. Res. 62, 538–547 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Periasamy M., Huke S., SERCA pump level is a critical determinant of Ca2+ homeostasis and cardiac contractility. J. Mol. Cell. Cardiol. 33, 1053–1063 (2001). [DOI] [PubMed] [Google Scholar]
- 24.Borisov A. B., Raeker M. O., Kontrogianni-Konstantopoulos A., Yang K., Kurnit D. M., Bloch R. J., Russell M. W., Rapid response of cardiac obscurin gene cluster to aortic stenosis: Differential activation of Rho-GEF and MLCK and involvement in hypertrophic growth. Biochem. Biophys. Res. Commun. 310, 910–918 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Stammers A. N., Susser S. E., Hamm N. C., Hlynsky M. W., Kimber D. E., Scott Kehler D., Duhamel T. A., The regulation of sarco(endo)plasmic reticulum calcium-ATPases (SERCA). Can. J. Physiol. Pharmacol. 93, 843–854 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Haghighi K., Bidwell P., Kranias E. G., Phospholamban interactome in cardiac contractility and survival: A new vision of an old friend. J. Mol. Cell. Cardiol. 77, 160–167 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller T., Szczesna D., Housmans P. R., Zhao J., de Freitas F., Gomes A. V., Culbreath L., McCue J., Wang Y., Xu Y., Kerrick W. G. L., Potter J. D., Abnormal contractile function in transgenic mice expressing a familial hypertrophic cardiomyopathy-linked troponin T (I79N) mutation. J. Biol. Chem. 276, 3743–3755 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Hernandez O. M., Szczesna-Cordary D., Knollmann B. C., Miller T., Bell M., Zhao J., Sirenko S. G., Diaz Z., Guzman G., Xu Y., Wang Y., Kerrick W. G. L., Potter J. D., F110I and R278C troponin T mutations that cause familial hypertrophic cardiomyopathy affect muscle contraction in transgenic mice and reconstituted human cardiac fibers. J. Biol. Chem. 280, 37183–37194 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Schober T., Huke S., Venkataraman R., Gryshchenko O., Kryshtal D., Hwang H. S., Baudenbacher F. J., Knollmann B. C., Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia. Circ. Res. 111, 170–179 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ellis E. R., Josephson M. E., What about tachycardia-induced cardiomyopathy? Arrhythm. Electrophysiol. Rev. 2, 82–90 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y., Escoubet B., Prunier F., Amour J., Simonides W. S., Vivien B., Lenoir C., Heimburger M., Choqueux C., Gellen B., Riou B., Michel J.-B., Franz W. M., Mercadier J.-J., Constitutive cardiac overexpression of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase delays myocardial failure after myocardial infarction in rats at a cost of increased acute arrhythmias. Circulation 109, 1898–1903 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Stokke M. K., Hougen K., Sjaastad I., Louch W. E., Briston S. J., Enger U. H., Andersson K. B., Christensen G., Eisner D. A., Sejersted O. M., Trafford A. W., Reduced SERCA2 abundance decreases the propensity for Ca2+ wave development in ventricular myocytes. Cardiovasc. Res. 86, 63–71 (2010). [DOI] [PubMed] [Google Scholar]
- 33.del Monte F., Lebeche D., Guerrero J. L., Tsuji T., Doye A. A., Gwathmey J. K., Hajjar R. J., Abrogation of ventricular arrhythmias in a model of ischemia and reperfusion by targeting myocardial calcium cycling. Proc. Natl. Acad. Sci. U.S.A. 101, 5622–5627 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prunier F., Kawase Y., Gianni D., Scapin C., Danik S. B., Ellinor P. T., Hajjar R. J., del Monte F., Prevention of ventricular arrhythmias with sarcoplasmic reticulum Ca2+ ATPase pump overexpression in a porcine model of ischemia reperfusion. Circulation 118, 614–624 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Jiang D., Xiao B., Yang D., Wang R., Choi P., Zhang L., Cheng H., Chen S. R. W., RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc. Natl. Acad. Sci. U.S.A. 101, 13062–13067 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Venetucci L. A., Trafford A. W., O’Neill S. C., Eisner D. A., The sarcoplasmic reticulum and arrhythmogenic calcium release. Cardiovasc. Res. 77, 285–292 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Biernacka A., Frangogiannis N. G., Aging and cardiac fibrosis. Aging Dis. 2, 158–173 (2011). [PMC free article] [PubMed] [Google Scholar]
- 38.Simchen M. J., Toi A., Silver M., Smith C. R., Hornberger L. K., Taylor G., Chitayat D., Fetal cardiac calcifications: Report of four prenatally diagnosed cases and review of the literature. Ultrasound Obstet. Gynecol. 27, 325–330 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Manrai A. K., Funke B. H., Rehm H. L., Olesen M. S., Maron B. A., Szolovits P., Margulies D. M., Loscalzo J., Kohane I. S., Genetic misdiagnoses and the potential for health disparities. N. Engl. J. Med. 375, 655–665 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Go A. S., Mozaffarian D., Roger V. L., Benjamin E. J., Berry J. D., Borden W. B., Bravata D. M., Dai S., Ford E. S., Fox C. S., Franco S., Fullerton H. J., Gillespie C., Hailpern S. M., Heit J. A., Howard V. J., Huffman M. D., Kissela B. M., Kittner S. J., Lackland D. T., Lichtman J. H., Lisabeth L. D., Magid D., Marcus G. M., Marelli A., Matchar D. B., McGuire D. K., Mohler E. R., Moy C. S., Mussolino M. E., Nichol G., Paynter N. P., Schreiner P. J., Sorlie P. D., Stein J., Turan T. N., Virani S. S., Wong N. D., Woo D., Turner M. B. American Heart Association Statistics Committee; Stroke Statistics Subcommittee , Heart disease and stroke statistics—2013 update: A report from the American Heart Association. Circulation 127, e6–e245 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hecker P. A., Galvao T. F., O’Shea K. M., Brown B. H., Henderson R. Jr, Riggle H., Gupte S. A., Stanley W. C., High-sugar intake does not exacerbate metabolic abnormalities or cardiac dysfunction in genetic cardiomyopathy. Nutrition 28, 520–526 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kerr J. P., Robison P., Shi G., Bogush A. I., Kempema A. M., Hexum J. K., Becerra N., Harki D. A., Martin S. S., Raiteri R., Prosser B. L., Ward C. W., Detyrosinated microtubules modulate mechanotransduction in heart and skeletal muscle. Nat. Commun. 6, 8526 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ackermann M. A., Shriver M., Perry N. A., Hu L.-Y., Kontrogianni-Konstantopoulos A., Obscurins: Goliaths and Davids take over non-muscle tissues. PLOS ONE 9, e88162 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kontrogianni-Konstantopoulos A., Catino D. H., Strong J. C., Bloch R. J., De novo myofibrillogenesis in C2C12 cells: Evidence for the independent assembly of M bands and Z disks. Am. J. Physiol. Cell Physiol. 290, C626–C637 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Shriver M., Stroka K. M., Vitolo M. I., Martin S., Huso D. L., Konstantopoulos K., Kontrogianni-Konstantopoulos A., Loss of giant obscurins from breast epithelium promotes epithelial-to-mesenchymal transition, tumorigenicity and metastasis. Oncogene 34, 4248–4259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hecker P. A., Lionetti V., Ribeiro R. F., Rastogi S., Brown B. H., O’Connell K. A., Cox J. W., Shekar K. C., Gamble D. M., Sabbah H. N., Leopold J. A., Gupte S. A., Recchia F. A., Stanley W. C., Glucose 6-phosphate dehydrogenase deficiency increases redox stress and moderately accelerates the development of heart failure. Circ. Heart Fail. 6, 118–126 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rudloff M. W., Woosley A. N., Wright N. T., Biophysical characterization of naturally occurring titin M10 mutations. Protein Sci. 24, 946–955 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A., NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 49.Wright N. T., Varney K. M., Ellis K. C., Markowitz J., Gitti R. K., Zimmer D. B., Weber D. J., The three-dimensional solution structure of Ca2+-bound S100A1 as determined by NMR spectroscopy. J. Mol. Biol. 353, 410–426 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Shen Y., Delaglio F., Cornilescu G., Bax A., TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schwieters C. D., Kuszewski J. J., Tjandra N., Clore G. M., The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 160, 65–73 (2003). [DOI] [PubMed] [Google Scholar]
- 52.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krieger E., Vriend G., YASARA View—Molecular graphics for all devices—From smartphones to workstations. Bioinformatics 30, 2981–2982 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1603081/DC1
fig. S1. Generation of obscurin knock-in mice carrying the R4344Q mutation in Ig58.
fig. S2. Sarcomeric organization is unaltered in 1-year-old homozygous knock-in female mice.
table S1. Morphometric and echocardiographic analyses of sedentary wild-type and homozygous knock-in hearts.
table S2. Morphometric and echocardiographic analyses of sham- and TAC-subjected wild-type and knock-in hearts.
table S3. NMR-derived restraints and statistics of 20 NMR structures of wild-type Ig58.
table S4. Statistics of wild-type Ig58 crystal diffraction.
table S5. List of primers used for confirmation of gene targeting, animal genotyping, and site-directed mutagenesis.