

Cryo–electron microscopy shows how the ribosome prepares for take-off to bypass a noncoding mRNA stretch.
Keywords: translation, ribosome, bypassing, recoding, structure
Abstract
Bypassing is a recoding event that leads to the translation of two distal open reading frames into a single polypeptide chain. We present the structure of a translating ribosome stalled at the bypassing take-off site of gene 60 of bacteriophage T4. The nascent peptide in the exit tunnel anchors the P-site peptidyl-tRNAGly to the ribosome and locks an inactive conformation of the peptidyl transferase center (PTC). The mRNA forms a short dynamic hairpin in the decoding site. The ribosomal subunits adopt a rolling conformation in which the rotation of the small subunit around its long axis causes the opening of the A-site region. Together, PTC conformation and mRNA structure safeguard against premature termination and read-through of the stop codon and reconfigure the ribosome to a state poised for take-off and sliding along the noncoding mRNA gap.
INTRODUCTION
Bypassing is a recoding event that overrules the collinearity of mRNA and protein sequence in gene expression (1). Although these events were thought to be extremely rare for a long time, recent work has identified multiple ribosome bypassing elements in yeast mitochondria (2). The best-studied case of bypassing occurs upon translation of bacteriophage T4 gene 60, which encodes a bacteriophage-specific topoisomerase subunit (3, 4). During translation of gene 60, the ribosome translates the first 46 codons up to the take-off Gly codon (GGA) followed by a UAG stop codon, but instead of terminating translation, the ribosome bypasses the next 50 nucleotides of the mRNA and resumes translation after the landing Gly codon (Fig. 1A) (3, 4). The bypassing efficiency depends on a particular sequence in the nascent peptide and several mRNA secondary structure elements, including an mRNA hairpin that forms around the take-off Gly codon, as well as stem-loop (SL) structures 5′ and 3′ of the take-off helix (3–11). Both take-off and landing are efficient, but a fraction of the ribosomes is lost from the mRNA track during sliding (10). Despite the presence of the stop codon adjacent to the take-off site, bypassing is not inhibited by the overexpression of release factor 1 (RF1) (5, 7); likewise, read-through of the coding gap is negligible (10). The forward ribosome movement is coupled to mRNA secondary structure rearrangements (10–12). When approaching the landing site, the ribosome starts to scan the mRNA in search of optimal base-pairing interactions (11). How the stimulatory elements in the nascent peptide and mRNA guide the ribosome toward bypassing and avoid premature termination and erroneous read-through remains unclear.
Fig. 1. Ribosome stalling at a take-off codon upon translational bypassing on bacteriophage T4 gene 60.
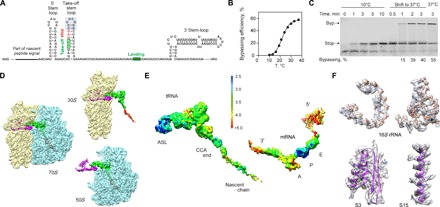
(A) Key regulatory elements in gene 60 mRNA, including the nascent peptide and the mRNA SL elements. The apical part of the take-off SL is highlighted in gray. (B) Temperature dependence of bypassing. (C) Reversibility of the temperature-induced ribosome stalling at the take-off codon. When translation is carried out at 10°C, only the stop peptide corresponding to codons 1 to 46 is synthesized. Switching the temperature to 37°C activates bypassing (Byp product). The last lane shows the maximum bypassing efficiency when translation is performed at 37°C. (D) Cryo-EM densities showing 30S (yellow) and 50S (blue) subunits, the P-site tRNAGly (green) carrying the nascent peptide (red), and the mRNA (magenta) (see also fig. S1). (E) Local resolution. The scale bar shows a color scale with resolution in angstroms (see also figs. S2 and S3). ASL, anticodon stem-loop. (F) Examples of the experimental cryo-EM density (transparent gray) and atomic model (ribbons), including 16S rRNA (nucleotides 766 to 779 and 586 to 639) and proteins S3 and S15 (residues 110 to 170 and 48 to 72, respectively).
RESULTS AND DISCUSSION
To elucidate the structural basis of bypassing, we assembled translating ribosome complexes from Escherichia coli on gene 60 mRNA in vitro. The efficiency of bypassing strongly depends on the incubation temperature (Fig. 1B). At low temperatures, translation of the first 46 codons is completed, but the ribosomes are stalled at the take-off site (Fig. 1C). The stalled complexes retain their ability to resume bypassing as soon as the temperature is raised, suggesting a functionally relevant state. The structure was solved to an overall resolution of 3.6 Å using cryo–electron microscopy (cryo-EM) and in silico sorting strategies (Fig. 1D and figs. S1 to S3). Local resolution measurement (Fig. 1E and fig. S3) suggests high flexibility for the mRNA, except for the P-site codon, which is well defined (Fig. 1E). Overall, the quality of the cryo-EM map is sufficient to model an atomic structure for the complex (Fig. 1F).
The nascent peptide exits the ribosome through the ~100 Å–long tunnel that traverses the 50S subunit of the ribosome (Fig. 2). Single amino acid substitutions in the middle part of the nascent chain can abolish bypassing (4, 5, 10). In the take-off complex, the nascent peptide forms a well-defined α helix in the central part of the tunnel (Fig. 2A). The main contacts (Fig. 2B) are between Tyr16 of the nascent peptide and Arg95 of L22, as well as between Arg24 and Ser28 of the nascent peptide and Arg67 and Arg61 of L4, respectively, in line with results from mutational analysis (10, 11). In addition, peptide residues 30 to 42 appear to adopt an α-helical conformation (albeit less well resolved; Fig. 1E) in the upper part of the tunnel between the peptidyl transferase center (PTC) and the constriction formed by ribosomal proteins L4 and L22 (Fig. 2A). Here, the nascent peptide makes contact with 23S ribosomal RNA (rRNA), Tyr33 with A2609 and A752, Met39 with A2062, Thr40 with U2586, and His44 with U2506. The density corresponding to the nascent peptide is poorly defined in the lower part of the tunnel toward the exit port. Interactions between Lys5 and Ile6 of the nascent chain with A1321 and Gln72 of L23, respectively, can only be visualized at low isosurface levels (Fig. 2, A and B). The high flexibility (Fig. 1E) may explain why mutations in the protein L23 signaling loop (13) had little effect on bypassing (10).
Fig. 2. The peptide exit tunnel and the PTC.
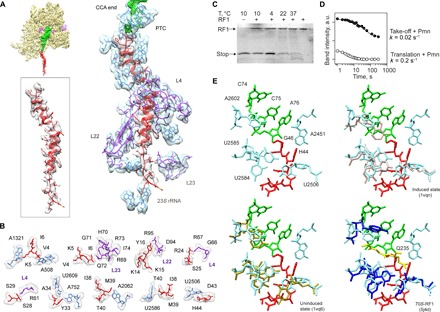
(A) Nascent chain interactions in the exit tunnel. The cryo-EM density is presented in transparent gray (peptide) and blue/purple (the 50S subunit 23S rRNA and proteins, respectively). The peptide density is shown at a lower contour level. (B) Closeup view of the interactions. The cryo-EM density is shown in transparent gray. (C) RF1 is inactive on the take-off complexes at low temperatures. Ribosome complexes formed in the presence of RF1 were isolated by gel filtration, which separates the complexes from free RF1 and released nascent chains. The amounts of RF1 and nascent chains copurifying in the isolated complex were analyzed by SDS–polyacrylamide gel electrophoresis (PAGE) and imaging of the fluorescence reporters on RF1 and the nascent chain. The last lane shows the control with RF1 alone. (D) Kinetics of the Pmn reaction of two peptidyl-tRNAs, one stalled before bypassing at the take-off codon and carrying the stop peptide (Take-off + Pmn; closed circles) and another carrying a shorter peptide, which appears as a transient intermediate of translation at 10°C (Translation + Pmn; open circles) (see also fig. S5). a.u., arbitrary units. (E) Conformation of the PTC in the take-off complex (upper left), compared to the induced (upper right) and uninduced (lower left) conformations, and in a complex with RF1 (lower left; the GGQ motif is shown in yellow) (53).
One open question is why RF1, which could read the UAG stop codon adjacent to the take-off codon, does not abort bypassing (5). The gene 60 stop peptide is not released even when RF1 is bound to the ribosome (Fig. 2C and fig. S4). Furthermore, the rate of the reaction with puromycin (Pmn), a diagnostic assay for PTC activity, is 10-fold lower for the P-site peptidyl-tRNAGly stalled at the take-off codon than for peptidyl-tRNAs during ongoing translation, even if compared to the slowly reacting peptidyl-tRNAs that accumulate as a result of transient ribosome pausing (Fig. 2D and fig. S5). The reactions at the PTC require accurate placement of the substrates to allow nucleophilic attack of a water molecule (during peptide release) or of the α-amino group of the aminoacyl-tRNA (during peptide bond formation) on the carbonyl carbon of the P-site peptidyl-tRNA (14–21), which is facilitated by a network of interactions with the nucleotides at the PTC (22). When the A site is empty, the PTC adopts a conformation unfavorable for catalysis (uninduced state). Binding of RF1 or aminoacyl-tRNA induces a reorientation of U2584 and U2585 toward the catalytically active state (15–17). In our structure, these nucleotides are locked in a conformation that is closer to the uninduced state, which may be caused by the interaction of U2586 with the nascent peptide (Fig. 2, A and B). Formation of the induced PTC conformation also requires the rotation of U2506 toward the P site (15). However, in the complex, the side chain of the nascent peptide His44 limits this movement. The position of His44 might also obstruct productive binding of RF1 at the PTC because the conformation of His44 would clash with the conserved Gln from the GGQ motif of RF1 (Fig. 2E), the amino acid that stabilizes the water molecule for in-line attack on peptidyl-tRNA (23). A similar locked conformation of the PTC was seen in the ribosome complexes with nascent peptides that mediate ribosome stalling (fig. S6) (24–28), suggesting a similar mechanism to induce translational arrest (29).
Gene 60 mRNA is known to be highly structured in solution (12). Several secondary structure elements regulate bypassing (10), including a part of the take-off helix (Fig. 1A) (4, 6, 9). In the stalled take-off complex, mRNA forms a small (12 nucleotides) hairpin in the decoding A site (Fig. 3A). The hairpin is formed by three consecutive C-G base pairs that form the double helix, which is closed by an unpaired UUCG loop, and contains the stop codon next to the take-off site. The overall lower local resolution of the hairpin (Fig. 1E) suggests that the observed structure is an ensemble of secondary structures with similar configurations. Despite its limited stability, folding of the mRNA in the A site may explain why bypassing does not occur at low temperatures. When the temperature is raised, the unfolded state is favored, making the ribosome more prone to bypass. In addition, formation of the A-site mRNA structure may protect the complex from premature termination by hindering the access of release factors to the stop codon, as proposed earlier (30). RF1 is more prone to termination when the stability of the hairpin is reduced by removing one G-C base pair, particularly at high temperature (Fig. 3, C and D, and fig. S4). Moreover, hairpin stability appears crucial to prevent read-through because lowering the stability leads to the appearance of the read-through product (Fig. 3D). Thus, the A-site hairpin, together with the uninduced conformation of PTC, protects the bypassing complex from premature termination or erroneous read-through.
Fig. 3. The mRNA path.
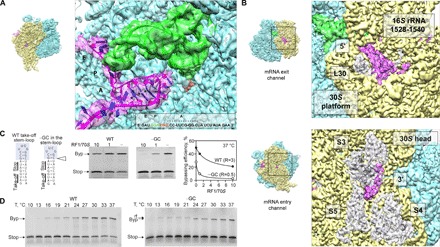
(A) mRNA hairpin in the A site. Closeup view of the mRNA (magenta), P-site peptidyl-tRNAGly (green), and the modeled sequence. Potential G-C pairs are marked by asterisks. The mRNA density is shown at a lower contour level. (B) Visualization of the mRNA densities of the 5′ and 3′ regions. Top: The 5′ region of the mRNA. The 3′ end of 16S rRNA (nucleotides 1528 to 1540) and protein L30 are highlighted. Bottom: The 3′ end of the mRNA as it emerges from the channel. Proteins S3, S4, and S5 are also highlighted. (C) Importance of the A-site hairpin for the discrimination against RF1. Left: mRNA constructs used, with the native sequence of gene 60 [wild type (WT)] or lacking one G-C base pair from the apical part of the take-off hairpin (−GC). Middle: Translation of WT and –GC mRNAs in the presence of different concentrations of RF1 at 37°C. Right: Dependence of the bypassing efficiency on the RF1 concentration. R, RF1/70S ratio under half-inhibition conditions (see also fig. S4). (D) A-site hairpin as a guard against UAG read-through (rt). Translation of the WT mRNA (left) and −GC mRNA (right) at different temperatures. Note the accumulation of the read-through product at low temperatures during translation of −GC mRNA.
The atomic model includes 29 nucleotides of the mRNA protected inside the channel that wraps around the 30S neck, from mRNA position −3 on the 5′ side to position +23 on the 3′ side, with respect to the take-off codon. The 3′ and 5′ extremes of the mRNA outside the channel are not well ordered; however, at lower isosurface thresholds, continuous densities corresponding to the ends at the entry and exit points can be visualized (Fig. 3B). The mRNA 5′ end arranges at the platform of the 30S subunit, apparently configured in a “loose” conformation. Because the ribosome slides over the coding gap and further mRNA portions emerge from the channel, the mRNA can form the 5′ SL regulatory element reported to facilitate the directionality of bypassing (10). On the other side of the 30S subunit, the extended 3′ end of the mRNA is bent upward to interact with protein S3 in the 30S head, which may play a role in the unwinding of secondary structure elements as the mRNA enters the channel (12).
Recent single-molecule fluorescence resonance energy transfer (smFRET) studies have suggested that, during pausing at the take-off Gly codon, the ribosome adopts a noncanonical hyper-rotated conformation, which is maintained during bypassing until translation is resumed at the landing codon (11). Inspection of the arrangement of the ribosomal subunits in our take-off complex suggests that the conformation is different from intersubunit rotation and resembles subunit rolling (Fig. 4). This rotation of the 30S around its long axis has been observed in mammalian ribosomes, where it is implicated in the regulation of the elongation cycle (31), and in bacterial SecM-arrested ribosomes (28). Subunit rolling entails a movement of ~5 to 10 Å in the lower part of the 30S, which reshapes the intersubunit space. It generates the closure of the E-site region and the reciprocal opening of the A-site region, which might facilitate hairpin formation. This conformational change causes a slight repositioning of the P site–bound peptidyl-tRNAGly. The elbow and D-arm regions of the tRNA are slightly shifted toward the A site (Fig. 4D). However, the geometry of the codon-anticodon complex is not altered, and the framework of interactions at the tRNA anticodon SL region (32, 33) known to be important to position the tRNA in the P site remains intact (Fig. 4E). Taking into account that the geometry of the codon-anticodon complex is not altered, and that the perturbation of the PTC configuration is also seen in SecM-arrested complexes, we suggest that the observed rolling motion is a hallmark of ribosome stalling, rather than the loss of the interaction with mRNA. The noncanonical intersubunit movement detected by smFRET (11) may take place once the codon-anticodon pairing is disrupted and the P site–bound tRNAGly takes off to initiate sliding or even represent subunit rolling.
Fig. 4. A rolling motion of the 30S subunit.
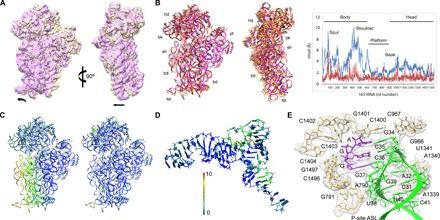
(A) Comparison of the 30S subunit in the take-off complex (yellow) and in the classical pretranslocation state (purple; see Materials and Methods) shown as intersubunit (left) and side (right) views. The 50S subunit was used for the alignment. Arrows indicate directions of movement. (B) Global rearrangements of the 30S subunit. Left: Superposition of the take-off complex with the unrolled 70S–EF-Tu complex [Protein Data Bank (PDB) 5AFI (46)] shown as intersubunit (left) and side (right) views. Right: The graph shows the movements of 16S rRNA (backbone distances) in the take-off complex compared to the unrolled ribosome structure (blue) or the SecM-arrested complex (red) [PDB 3JBU (28)]. 23S rRNA was used as reference for alignment. rmsd, root mean square deviation. (C) The heat map displayed on the take-off complex quantifies the displacement, with respect to the ribosome with EF-Tu bound (left) and the SecM-arrested complex (right). (D) Deviations in the structure of tRNAGly compared to the 70S–EF-Tu complex. 23S rRNA was used as reference for alignment. (E) Interactions of the tRNAGly anticodon SL. The tRNA and mRNA binding the P site are stabilized by the stacking of the ribose and base of nucleotide 34 in the anticodon with G966 and C1400 in 16S rRNA, respectively, and the contacts involving the universally conserved nucleotide A790 with the tRNA nucleotide 38, nucleotides G1338 and A1339 with nucleotide 41, and the G30-C40 base pair in the tRNA, consistent with earlier structural work (32, 33). The cryo-EM density is shown in transparent gray.
Collectively, the data suggest how the ribosome deciphers the regulatory signals in the mRNA to prepare for bypassing. After decoding of the take-off GGA codon and reaching the stop codon, the ribosome stalls in a conformation that obstructs termination by RF1. The mRNA forms a small dynamic hairpin in the A site, which prevents UAG recognition by RF1 or a read-through by near-cognate aminoacyl-tRNAs. However, even if the stop codon becomes occasionally accessible because of the thermal fluctuations of the A-site hairpin, RF1 access to the PTC and peptide release are blocked as a result of the unfavorable conformation of the PTC induced by the interactions with the nascent peptide. The locked inactive conformation of the PTC is common to other translation arrest-mediating peptides and correlates with the subunit rolling. Because the A-site helix becomes more dynamic at higher temperatures, folding and unfolding of the mRNA in the A site may destabilize the mRNA-tRNA match and drag the P-site codon away from tRNAGly, thereby dissolving the codon-anticodon interaction and thus initiating bypassing. Interactions of the nascent peptide anchor the peptidyl-tRNAGly to the 50S subunit and ensure the processivity of ribosome sliding during bypassing. Folding of the mRNA 5′ SL that emerges from the 30S subunit would pull the mRNA further away from the tRNA, ensuring the forward direction of sliding until the landing codon is reached. Future structural studies will decipher still uncharacterized details of the process, such as the dynamics of tRNAGly and the conformation of bypassing ribosomes during sliding.
MATERIALS AND METHODS
Reagents
All experiments were carried out in HiFi buffer [50 mM tris-HCl (pH 7.5), 70 mM NH4Cl, 30 mM KCl, 3.5 mM MgCl2, 8 mM putrescine, and 0.5 mM spermidine] (34). Biochemicals were from Merck, and nucleotide triphosphates were from Jena Bioscience. [3H]methionine and [14C]leucine were from Hartmann Analytics. Bodipy-FL-succinimidyl ester was from Invitrogen. Total E. coli tRNA was from Roche, and oligonucleotides were from IBA. 70S ribosomes, EF-Tu, EF-G, IF1, IF2, IF3, [3H]Met-tRNAfMet, and f[3H]Met-tRNAfMet were prepared from E. coli (35–37). Site-directed mutagenesis of the WT gene 60 construct (9) was performed using the QuikChange polymerase chain reaction protocol with the appropriate oligonucleotide primers. mRNAs were produced by T7 RNA polymerase in vitro transcription and purified by ion-exchange chromatography on a HiTrap Q HP 5-ml column (GE Healthcare). Fluorescence-labeled [3H]Met-tRNAfMet (Bpy-Met-tRNAfMet) was prepared, as previously described (38), with modifications (39).
In vitro translation
Translation was carried out according to the published protocols (10) with the following modifications. Initiation complexes were formed by incubating ribosomes (0.5 μM), mRNA (1.5 μM), IF1, IF2, and IF3 (0.75 μM each), guanosine triphosphate (GTP) (1 mM), and either Bpy-[3H]Met-tRNAfMet or initiator f[3H]Met-tRNAfMet (0.5 μM) in HiFi buffer at different temperatures from 10° to 37°C for 20 min. The ternary complex EF-Tu–GTP–aminoacyl-tRNA was prepared by incubating EF-Tu (60 μM) with GTP (1 mM), phosphoenolpyruvate (3 mM), and pyruvate kinase (0.1 mg ml−1) for 15 min at 37°C, then adding purified total amino acid–tRNA (about 60 μM) and EF-G (1 μM), and incubating for 1 min at 37°C. In vitro translation was started by mixing initiation ribosome complexes (0.08 μM) with the ternary complexes (50 μM) with total amino acid–tRNA and incubated at 37°C. Where indicated, RF1 (nonlabeled or fluorescein-labeled; 1 μM) was added to the translation mixture. Reactions were terminated by shock-freezing aliquots in liquid nitrogen. Translation products were separated by tris-tricine gel electrophoresis (40). Fluorescent peptides were detected after gel electrophoresis using FLA-7000 scanner (FujiFilm) and quantified using the Multi Gauge software. The identity of the stop, bypass, and read-through products was determined using the respective in vitro translated peptides as markers (10). The stop peptide was obtained by translation of the gene 60 mRNA fragment corresponding to amino acids 1 to 46. The size of the bypassing product was verified using a construct in which the two reading frames were fused by omission of the bypassing gap. The size of the read-through product corresponded to the full-length protein in which the gap part was translated. The identity of the products was validated using N- and C-terminal fluorescence reporters and radioactivity counting (10). Bypassing efficiency was calculated as a ratio of the density corresponding to the byp band to the sum of the byp and stop bands (10).
To assess the rate of the Pmn reaction, we incubated the translation mixture (25 μl) for 20 min at 10°C (until the bypassing complex was formed). Then, Pmn (10 mM) was added to the translation mixture, and the reactions were stopped at different time points by reducing the pH using 3 M sodium acetate (pH 5.0). Peptidyl-tRNAs were separated from Pmn-released peptides by bis-tris gel electrophoresis (41). In addition to the main band representing tRNAGly attached to the stop peptide, we identified a minor band with a higher mobility on the gel representing a peptidyl-tRNA attached to a peptide shorter than the stop peptide. From the time-resolved translation experiments and peptide product analysis (see above), we identified the shorter peptide as a residual product of the transient ribosome pausing upon translation of gene 60 mRNA before the take-off codon was reached. The pausing was reminiscent of the ribosome slowdown observed by smFRET (11). Although the exact identity of the shorter peptidyl-tRNA is not known, the fact that it accumulates indicates that this peptidyl-tRNA is the least reactive peptidyl-tRNA during ongoing translation, providing a lower limit for the rate of the Pmn reaction for peptidyl-tRNAs during elongation.
To check the binding of RF1 to the ribosome during take-off, we added RF1 labeled with fluorescein in a 10-fold excess over the ribosomes. The translation mixture (25 μl) was incubated for 20 min at different temperatures (10°, 22°, and 37°C), and the resulting ribosome complexes were purified by gel filtration on a BioSuite 450 HR 8-μm column (Waters) at 4°C. The amount of the nascent peptide bound to the ribosome was calculated from the f[3H]Met radioactivity in the ribosome fraction. Fractions with ribosome complexes were collected, concentrated using a vacuum concentrator, and analyzed by tris-tricine gel electrophoresis (40).
Cryo-EM and image processing
The translation mixture was incubated at 10°C for 20 min and purified by gel filtration chromatography on a BioSuite 450 HR 8-μm column (Waters) at 4°C. Fractions that contained ribosomes and radioactivity (corresponding to f[3H]Met-tRNAfMet) were collected and frozen in liquid nitrogen. The ribosome samples were applied to Quantifoil R 2/2 holey carbon grids coated with a thin layer of carbon. Grids vitrified in a FEI Vitrobot were then transferred to a Titan Krios (FEI) electron microscope operated at 300 kV. Images were acquired using a K2 detector at a magnification of 47,170 (yielding a pixel size of 1.06 Å). Initial beam-induced motion correction was performed at the level of entire micrographs (42). Contrast transfer function parameters were estimated using CTFFIND3 (43). Data were processed using the Relion software package (44).
An initial data set of 150,569 particles was manually picked and subjected to a round of two-dimensional (2D) class averaging. The selected 140,985 particles gave rise to a refined 3.2 Å map after particle movement correction (that is, “polishing”). The final accumulative electron dose was ~26 e−/Å2. These particles were used for treatment of structural heterogeneity (fig. S1): First, 3D classification with exhaustive angular search (0.9°) into five classes yielded three distinct recognizable classes—classical state–like (12.2%), hybrid state–like (19.1%), and the state with the P-site tRNA only (60%). The two smaller classes gave suboptimal reconstructions, and the particles from these classes were discarded.
To identify the complexes in a better-defined conformation, we performed additional rounds of classification on the isolated subpopulations. This approach allowed us to further clean the classical-state subset (the selected particles yielded a 5.1 Å map); however, despite the fragmented appearance of the A/P tRNA, they were unsuccessful in the attempt to find different subpopulations in the hybrid-state subset (these particles yielded a 4.65 Å map).
The visual inspection of the reconstruction for blurry parts of density indicated the existence of remaining structural heterogeneity in the main subset. The heterogeneity was particularly well seen in the mRNA structure. To improve the density, we took a new approach: subtraction of part of the signal from the particles, creating two new data sets of experimental images, each of them without one of the subunits. We then performed classification on the new set of particles with no signal for the 50S subunit while keeping all orientations fixed at the values determined in the refinement of the consensus 30S subunit reconstruction. Sorting into six groups yielded two major classes, one of which showed an improved density for the mRNA, particularly for the A-site secondary structure. The remaining classes were ignored. To obtain meaningful subclasses, it was necessary to limit the resolution (using the“-strict_highres_exp” Relion1.4 command) to 10 Å. Because the information about the interface between the subunits was not well defined in the separately masked reconstructions, the entire ribosome was refined to generate the final map at 3.6 Å resolution. Local resolution variability was estimated using ResMap (Fig. 1E and fig. S3) (45).
The take-off complexes represented approximately 60% of the total ribosome population in the sample, which matched the maximal percentage of bypassing of the in vitro system. The other two subpopulations were ribosomes that resembled hybrid-state (A/P, P/E, and rotated 30S) and classical-state forms (less than 20 and 10%, respectively). The quality/resolution of these 3D maps was low, and there were no structural details that might support the assignment of the peptidyl-tRNA or the identity of the tRNAs in A and P sites. At this point, we cannot specify whether these complexes were by-products of the sample preparation/purification or ribosomes stalled at previous translocation stages.
Note that multiple additional runs (not shown in fig. S1) with varying numbers of classes were performed at different stages of sorting. During these runs, orientation searches were performed exhaustively, that is, every 0.9° (restricted to local searches) or kept fixed at the orientations from the consensus refinements. Separate 3D autorefinements were also performed for each of the corresponding subsets after each round of classification. The visual inspection of the resulting 3D reconstructions, in conjunction with the local resolution assessment, helped in planning the best strategy to deal with the structural heterogeneity of the sample.
Model building and refinement
The atomic model was generated starting with the structure of the E. coli ribosome–EF-Tu complex [PDB 5AFI (46)] and an RNA hairpin SL [PDB 1F7Y (47)]. The initial rigid-body fitting of the model was carried out manually via Chimera (48), and the rest of the structure was built using Coot (49). The geometry of the mRNA was further improved using the Rossetta Erraser tool (50). The model was improved by iterative cycles of manual model rebuilding and refinement. The real space procedure in Phenix (51) was used to refine the model and to improve the stereochemistry (fig. S2). The final model was evaluated with MolProbity (52). Chimera was used to generate the figures and to color the structures by root mean square deviation values (Fig. 4) (48).
Supplementary Material
Acknowledgments
We thank W. Wintermeyer for critical reading; P. Kharki and C. Maracci for providing fluorescence-labeled RF1; M. Thommen for the modified protocol for bis-tris SDS-PAGE; and A. Bursy, O. Geintzer, S. Kappler, C. Kothe, T. Niese, T. Wiles, and M. Zimmermann (all from the Max Planck Institute for Biophysical Chemistry, Göttingen, Germany), as well as J. Cifuente (CIC bioGUNE), for expert technical assistance. We acknowledge Diamond for access and support of the cryo-EM facilities at the UK national electron bio-imaging centre (eBIC), proposal EM14264, funded by the Wellcome Trust, Medical Research Council, and UK Biotechnology and Biological Sciences Research Council. We thank A. Siebert and D. Clare for assistance during EM data collection. Funding: This work was supported by grants from the Spanish Ministry of Economy and Competitiveness to X.A. (CTQ2015-73560-JIN) and to M.V. (BFU2015-66326-P) and a grant from the Deutsche Forschungsgemeinschaft to M.V.R. (SFB860). M.V. acknowledges funding from the Human Science Frontiers Program grant (RGP0062/2016). Author contributions: E.S. and M.K. prepared the samples and performed the assays. X.A., D.G.-C., and M.Z. performed preliminary electron microscopy and image processing. X.A. processed cryo-EM data and generated the atomic models. X.A., E.S., M.V.R., and M.V. designed the study and interpreted the data. All authors participated in the writing or technical editing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The cryo-EM map and atomic model are deposited under accession codes EMD-3618 and PDB-5NP6, respectively.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1700147/DC1
fig. S1. Overview of particle classification and structure determination.
fig. S2. Fourier shell correlation curves and model statistics.
fig. S3. Local resolution.
fig. S4. Effect of the A-site hairpin on the stability of peptidyl-tRNA binding and on the action of RF1.
fig. S5. The reactivity of the PTC with Pmn at 10°C.
fig. S6. Conformation of the PTC.
REFERENCES AND NOTES
- 1.Baranov P. V., Atkins J. F., Yordanova M. M., Augmented genetic decoding: Global, local and temporal alterations of decoding processes and codon meaning. Nat. Rev. Genet. 16, 517–529 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Lang B. F., Jakubkova M., Hegedusova E., Daoud R., Forget L., Brejova B., Vinar T., Kosa P., Fricova D., Nebohacova M., Griac P., Tomaska L., Burger G., Nosek J., Massive programmed translational jumping in mitochondria. Proc. Natl. Acad. Sci. U.S.A. 111, 5926–5931 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang W. M., Ao S. Z., Casjens S., Orlandi R., Zeikus R., Weiss R., Winge D., Fang M., A persistent untranslated sequence within bacteriophage T4 DNA topoisomerase gene 60. Science 239, 1005–1012 (1988). [DOI] [PubMed] [Google Scholar]
- 4.Weiss R. B., Huang W. M., Dunn D. M., A nascent peptide is required for ribosomal bypass of the coding gap in bacteriophage T4 gene 60. Cell 62, 117–126 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herr A. J., Gesteland R. F., Atkins J. F., One protein from two open reading frames: Mechanism of a 50 nt translational bypass. EMBO J. 19, 2671–2680 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herr A. J., Nelson C. C., Wills N. M., Gesteland R. F., Atkins J. F., Analysis of the roles of tRNA structure, ribosomal protein L9, and the bacteriophage T4 gene 60 bypassing signals during ribosome slippage on mRNA. J. Mol. Biol. 309, 1029–1048 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Herr A. J., Wills N. M., Nelson C. C., Gesteland R. F., Atkins J. F., Drop-off during ribosome hopping. J. Mol. Biol. 311, 445–452 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Herr A. J., Wills N. M., Nelson C. C., Gesteland R. F., Atkins J. F., Factors that influence selection of coding resumption sites in translational bypassing: Minimal conventional peptidyl-tRNA:mRNA pairing can suffice. J. Biol. Chem. 279, 11081–11087 (2004). [DOI] [PubMed] [Google Scholar]
- 9.Wills N. M., O’Connor M., Nelson C. C., Rettberg C. C., Huang W. M., Gesteland R. F., Atkins J. F., Translational bypassing without peptidyl-tRNA anticodon scanning of coding gap mRNA. EMBO J. 27, 2533–2544 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Samatova E., Konevega A. L., Wills N. M., Atkins J. F., Rodnina M. V., High-efficiency translational bypassing of non-coding nucleotides specified by mRNA structure and nascent peptide. Nat. Commun. 5, 4459 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Chen J., Coakley A., O’Connor M., Petrov A., O’Leary S. E., Atkins J. F., Puglisi J. D., Coupling of mRNA structure rearrangement to ribosome movement during bypassing of non-coding regions. Cell 163, 1267–1280 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Todd G. C., Walter N. G., Secondary structure of bacteriophage T4 gene 60 mRNA: Implications for translational bypassing. RNA 19, 685–700 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bornemann T., Jöckel J., Rodnina M. V., Wintermeyer W., Signal sequence-independent membrane targeting of ribosomes containing short nascent peptides within the exit tunnel. Nat. Struct. Mol. Biol. 15, 494–499 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Schmeing T. M., Huang K. S., Kitchen D. E., Strobel S. A., Steitz T. A., Structural insights into the roles of water and the 2′ hydroxyl of the P site tRNA in the peptidyl transferase reaction. Mol. Cell 20, 437–448 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Schmeing T. M., Huang K. S., Strobel S. A., Steitz T. A., An induced-fit mechanism to promote peptide bond formation and exclude hydrolysis of peptidyl-tRNA. Nature 438, 520–524 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Laurberg M., Asahara H., Korostelev A., Zhu J., Trakhanov S., Noller H. F., Structural basis for translation termination on the 70S ribosome. Nature 454, 852–857 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Voorhees R. M., Weixlbaumer A., Loakes D., Kelley A. C., Ramakrishnan V., Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat. Struct. Mol. Biol. 16, 528–533 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin H., Kelley A. C., Loakes D., Ramakrishnan V., Structure of the 70S ribosome bound to release factor 2 and a substrate analog provides insights into catalysis of peptide release. Proc. Natl. Acad. Sci. U.S.A. 107, 8593–8598 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Youngman E. M., Brunelle J. L., Kochaniak A. B., Green R., The active site of the ribosome is composed of two layers of conserved nucleotides with distinct roles in peptide bond formation and peptide release. Cell 117, 589–599 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Weixlbaumer A., Jin H., Neubauer C., Voorhees R. M., Petry S., Kelley A. C., Ramakrishnan V., Insights into translational termination from the structure of RF2 bound to the ribosome. Science 322, 953–956 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Korostelev A., Asahara H., Lancaster L., Laurberg M., Hirschi A., Zhu J., Trakhanov S., Scott W. G., Noller H. F., Crystal structure of a translation termination complex formed with release factor RF2. Proc. Natl. Acad. Sci. U.S.A. 105, 19684–19689 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frolova L. Y., Tsivkovskii R. Y., Sivolobova G. F., Oparina N. Y., Serpinsky O. I., Blinov V. M., Tatkov S. I., Kisselev L. L., Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis. RNA 5, 1014–1020 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaw J. J., Green R., Two distinct components of release factor function uncovered by nucleophile partitioning analysis. Mol. Cell 28, 458–467 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bischoff L., Berninghausen O., Beckmann R., Molecular basis for the ribosome functioning as an L-tryptophan sensor. Cell Rep. 9, 469–475 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Arenz S., Meydan S., Starosta A. L., Berninghausen O., Beckmann R., Vázquez-Laslop N., Wilson D. N., Drug sensing by the ribosome induces translational arrest via active site perturbation. Mol. Cell 56, 446–452 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arenz S., Ramu H., Gupta P., Berninghausen O., Beckmann R., Vázquez-Laslop N., Mankin A. S., Wilson D. N., Molecular basis for erythromycin-dependent ribosome stalling during translation of the ErmBL leader peptide. Nat. Commun. 5, 3501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sohmen D., Chiba S., Shimokawa-Chiba N., Innis C. A., Berninghausen O., Beckmann R., Ito K., Wilson D. N., Structure of the Bacillus subtilis 70S ribosome reveals the basis for species-specific stalling. Nat. Commun. 6, 6941 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang J., Pan X., Yan K., Sun S., Gao N., Sui S.-F., Mechanisms of ribosome stalling by SecM at multiple elongation steps. eLife 4, e09684 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson D. N., Arenz S., Beckmann R., Translation regulation via nascent polypeptide-mediated ribosome stalling. Curr. Opin. Struct. Biol. 37, 123–133 (2016). [DOI] [PubMed] [Google Scholar]
- 30.N. M. Wills, Translational bypassing–peptidyl-tRNA re-pairing at non-overlapping sites, in Recoding: Expansion of Decoding Rules Enriches Gene Expression, J. F. Atkins, R. F. Gesteland, Eds. (Springer, 2010), pp. 365–381. [Google Scholar]
- 31.Budkevich T. V., Giesebrecht J., Behrmann E., Loerke J., Ramrath D. J. F., Mielke T., Ismer J., Hildebrand P. W., Tung C.-S., Nierhaus K. H., Sanbonmatsu K. Y., Spahn C. M. T., Regulation of the mammalian elongation cycle by subunit rolling: A eukaryotic-specific ribosome rearrangement. Cell 158, 121–131 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jenner L. B., Demeshkina N., Yusupova G., Yusupov M., Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nat. Struct. Mol. Biol. 17, 555–560 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Berk V., Zhang W., Pai R. D., Cate J. H. D., Structural basis for mRNA and tRNA positioning on the ribosome. Proc. Natl. Acad. Sci. U.S.A. 103, 15830–15834 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gromadski K. B., Rodnina M. V., Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol. Cell 13, 191–200 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Milon P., Konevega A. L., Peske F., Fabbretti A., Gualerzi C. O., Rodnina M. V., Transient kinetics, fluorescence, and FRET in studies of initiation of translation in bacteria. Methods Enzymol. 430, 1–30 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Rodnina M. V., Wintermeyer W., GTP consumption of elongation factor Tu during translation of heteropolymeric mRNAs. Proc. Natl. Acad. Sci. U.S.A. 92, 1945–1949 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodnina M. V., Savelsbergh A., Matassova N. B., Katunin V. I., Semenkov Y. P., Wintermeyer W., Thiostrepton inhibits the turnover but not the GTPase of elongation factor G on the ribosome. Proc. Natl. Acad. Sci. U.S.A. 96, 9586–9590 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gite S., Mamaev S., Olejnik J., Rothschild K., Ultrasensitive fluorescence-based detection of nascent proteins in gels. Anal. Biochem. 279, 218–225 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Mittelstaet J., Konevega A. L., Rodnina M. V., A kinetic safety gate controlling the delivery of unnatural amino acids to the ribosome. J. Am. Chem. Soc. 135, 17031–17038 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Schägger H., von Jagow G., Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166, 368–379 (1987). [DOI] [PubMed] [Google Scholar]
- 41.Varshney U., Lee C.-P., RajBhandary U. L., Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J. Biol. Chem. 266, 24712–24718 (1991). [PubMed] [Google Scholar]
- 42.Li X., Mooney P., Zheng S., Booth C. R., Braunfeld M. B., Gubbens S., Agard D. A., Cheng Y., Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mindell J. A., Grigorieff N., Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 142, 334–347 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Scheres S. H. W., RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kucukelbir A., Sigworth F. J., Tagare H. D., Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fischer N., Neumann P., Konevega A. L., Bock L. V., Ficner R., Rodnina M. V., Stark H., Structure of the E. coli ribosome–EF-Tu complex at <3 Å resolution by Cs-corrected cryo-EM. Nature 520, 567–570 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Ennifar E., Nikulin A., Tishchenko S., Serganov A., Nevskaya N., Garber M., Ehresmann B., Ehresmann C., Nikonov S., Dumas P., The crystal structure of UUCG tetraloop. J. Mol. Biol. 304, 35–42 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chou F.-C., Sripakdeevong P., Dibrov S. M., Hermann T., Das R., Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat. Methods 10, 74–76 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Svidritskiy E., Madireddy R., Korostelev A. A., Structural basis for translation termination on a pseudouridylated stop codon. J. Mol. Biol. 428, 2228–2236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1700147/DC1
fig. S1. Overview of particle classification and structure determination.
fig. S2. Fourier shell correlation curves and model statistics.
fig. S3. Local resolution.
fig. S4. Effect of the A-site hairpin on the stability of peptidyl-tRNA binding and on the action of RF1.
fig. S5. The reactivity of the PTC with Pmn at 10°C.
fig. S6. Conformation of the PTC.