

Abstract
Dimethyl fumarate (DMF; trade name Tecfidera™) is an oral formulation of the fumaric acid ester that is FDA approved for treatment of relapsing-remitting multiple sclerosis (RRMS). To better understand the therapeutic effects of Tecfidera and its rare side-effect of progressive multifocal leukoencephalopathy (PML), we conducted cross-sectional and longitudinal studies by immunophenotyping cells from peripheral blood (particularly T lymphocytes) derived from untreated, 4-6 month and 18-26 month Tecfidera-treated stable RRMS patients using multi-parametric flow cytometry. The absolute numbers of CD4 and CD8 T cells were significantly decreased and the CD4/CD8 ratio was increased with DMF treatment. The proportion of both effector memory (Tem) and central memory T cells (Tcm) were reduced while naïve T cells (Tn) increased in treated patients. T cell activation was reduced with DMF treatment especially among Tem and effector memory RA (Temra) T cells. T helper subsets Th1 (CXCR3+), Th17 (CCR6+), and particularly those expressing both CXCR3 and CD161 were reduced most significantly, while the anti-inflammatory Th2 subset (CCR3+) was increased after DMF treatment. A corresponding increase in IL-4, and decrease in IFNγ and IL-17-expressing CD4+ T cells was observed in DMF-treated patients. DMF in vitro treatment also led to increased T cell apoptosis and decreased activation, proliferation, reactive oxygen species, and CCR7 expression. Our results suggest that DMF acts on specific memory and effector T cell subsets by limiting their survival, proliferation, activation, and cytokine production. Monitoring these subsets could help to evaluate the efficacy and safety of DMF treatment.
Keywords: Dimethyl Fumarate, PBMC, RRMS, Effector memory T cells, T helper cells
Introduction
Multiple sclerosis (MS) is an autoimmune disease of the central nervous system (CNS) that leads to multifocal inflammatory lesions in the brain and spinal cord. MS pathogenesis is thought to involve inflammatory responses against oligodendrocytes mediated by activated myelin-specific T cells, resulting in demyelination of the axon. Investigations using the mouse model of MS, experimental autoimmune encephalomyelitis (EAE), have shown that activation of CD4+ T helper (Th) cells through cognate interactions with myelin peptide-loaded antigen presenting cells (APC) can lead to a breakdown in immune tolerance and the formation of memory T cells capable of transferring disease to naïve recipients. Pathogenesis in EAE has been associated with either activation of Th1- or Th17-type immune responses and involves up-regulation of the surface adhesion molecule VLA-4, which enables Th cells to cross the blood brain barrier (BBB) and infiltrate into the CNS. Additionally, activated CD8+ cytotoxic T lymphocytes (CTL), B lymphocytes, and myeloid cells infiltrate the CNS and participate in inflammation and tissue damage. Both CD8+ and CD4+ T-cells have been visualized in acute inflammatory lesions and to a lesser extent in chronic demyelinated lesions in humans (1).
Immunological memory is an important property of the adaptive immune system. Naive (Tn, CCR7+ CD45RA+) and central memory (Tcm, CCR7+ CD45RA-) T cells exhibit high proliferative potential but lack immediate effector functions, whereas effector memory (Tem, CCR7- CD45RA-) and CD45RA+ effector memory (Temra, CCR7- CD45RA+) T cells have less proliferative potential but produce cytokines and exert competent effector functions, respectively (2, 3). Central memory T (Tcm) cells, marked by co-expression of the memory marker CD45RO and the chemokine receptor CCR7, arise in response to autoantigen exposure. Loss of CCR7 expression facilitates the egress of T cells out of lymphoid organs into the periphery, where they are designated as effector memory T (Tem) cells. Encephalitogenic Tem cells represent a pool of CNS antigen-primed T cells that are capable of extravasation across the endothelial wall of the BBB, migrating into the CNS where they can mediate inflammatory responses (4). In contrast, Tcm cells usually reside in the T cell areas of secondary lymphoid organs. They have little or no direct effector function, but can support development of other effector cells or readily proliferate and differentiate into Tem upon secondary antigenic challenge to generate a new wave of reactions (5). CD45RA+ effector Tmera cells represent the most differentiated type of memory cells; they have high susceptibility to apoptosis and express high levels of cytotoxic molecules such as perforin and Fas ligand (2). As naïve CD4+ T cells (Tn) become activated by APCs and differentiate into memory Th cells, they encounter costimulatory signals that drive their differentiation into subsets: Th1, Th17, Th2, and regulatory T (Treg) cells, with distinct patterns of gene transcription, cytokine production, chemokine receptor expression, and effector functions. A major therapeutic goal in treating MS is to re-establish tolerance toward CNS antigens which in theory, may be achieved through controlling the development, differentiation, and survival of antigen-specific memory Th1 and Th17 cells, or skewing their responses toward Th2 or Treg functions.
Dimethyl fumarate (DMF; also known as BG-12, trade name Tecfidera™) is an oral formulation of the fumaric acid ester that showed remarkable efficacy and low adverse effects in two phase III clinical trials leading to its approval by the FDA for treating relapsing-remitting multiple sclerosis (RRMS) (6, 7). DMF is also an active ingredient of Fumaderm™, a drug which has been used to treat psoriasis in Germany for several decades (8-10). Clinical trials of DMF-treated RRMS patients showed significant reductions in the proportion of patients with relapse, disability progression, and numbers of new magnetic resonance imaging (MRI) lesions (6, 7, 11). We and others have shown that the clinical effects of DMF may involve its direct antioxidant effects on the nervous system and neuroprotection (12, 13). The effectiveness of DMF in psoriasis also suggests an immune regulatory role, which prompted us to study the effects of DMF treatment on human peripheral lymphocytes in DMF-treated RRMS patients. We recently reported that RRMS patients treated with DMF had reduced levels of naïve and memory B cells, while there was an increase in B cells with phenotypes reported to produce IL-10 and to have regulatory capacity (14). Rare cases of progressive multifocal leukoencephalopathy (PML) have been reported to be associated with long-term DMF treatment of psoriasis and MS patients (15-18), which make it important to understand the immunological changes induced by DMF treatment in order to find reliable biomarker(s) that can predict JC virus activation and prevent PML.
In order to assess the balance between memory and effector cells, we analyzed the phenotypes and functional response profile of T cells within various memory and helper subsets in RRMS patients treated with DMF. Collectively, our cross-sectional and prospective longitudinal studies indicate that DMF decreases frequencies and absolute numbers of peripheral T lymphocytes, particularly Tem and Tcm. In addition, T cell activation was reduced with DMF treatment, especially among Tem and effector memory RA (Temra) T cells. Th1 (CXCR3+), Th17 (CCR6+), and particularly those expressing both CXCR3 and CD161 were reduced most significantly, while the anti-inflammatory Th2 subset (CCR3+) was increased after DMF treatment. DMF in vitro treatment also led to increased T cell apoptosis and decreased activation, proliferation, reactive oxidative stress (ROS), and CCR7 expression. These findings implicate T cell involvement in the action of DMF in treating RRMS. Our studies help to establish the link between the efficacy of DMF and its effects on memory T cells and Th subsets. The relevance to therapeutic effects of DMF and possible relationship to rare incidences of PML is also discussed.
Materials and Methods
RRMS Patients and PBMC Isolation
We enrolled all patients in this study from the Multiple Sclerosis Center at the University of Michigan Health System. All subjects had a clinical diagnosis of RRMS and were treated with Tecfidera™. Informed consent was obtained from patients prior to participation in the study, which was approved by the University of Michigan Institutional Review Board. Table I summarizes the demographic characteristics and clinical data of the patients in this study. Prior disease modifying therapies (DMT) for those patients who were treated within a six month time point of starting Tecfidera™ are also listed in Table I. All patients in this study discontinued other DMT while taking Tecfidera™. Blood samples were collected in tubes containing sodium citrate (BD Biosciences) and processed immediately as they were collected. Peripheral blood lymphocytes and monocytes (PBMCs) were isolated from human peripheral blood by density gradient centrifugation according to the manufacturer's suggested protocol.
Table I. Demographic Characteristics of RRMS Patients.
| Untreated (n=18) | DMF 4-6M (n=20) | DMF 18-26M (n=18) | P | Longitudinal Cohort (n=9) | |
|---|---|---|---|---|---|
| Age (year) 1 | 43 ± 8 | 43 ± 8 | 46 ± 9 | 0.601 | 45.7 ± 8.8 |
| Gender2 | |||||
| Female | 15 | 16 | 14 | 0.915 | 7 |
| Male | 3 | 4 | 4 | 2 | |
| Race2 | 1.000 | ||||
| Caucasian | 17 | 19 | 17 | 8 | |
| African American | 1 | 1 | 1 | 1 | |
| Disease duration (year) 3 | 0.7(0.1,3.9) | 1.1(0.5,6.2) | 2.6(2.2,6.9) | 0.738 | 0.7(0.1,5.3) |
| Baseline EDSS1 | 2.4 ± 1.6 | 2.1 ± 1.2 | 2.1 ± 1.3 | 0.782 | 2.5 ± 1.8 |
| Treatment2,4 (Tx) Naive | 8 (44.5%) | 9(45%) | 9 (50%) | 0.828 | 3 (33.3%) |
| Prior Tx5 (> 6 month): | 5(27.8%) Avonex (2), Copaxone(2), Rebif, | 4(20%) Avonex, Copaxone(2), Rebif | 2(11.1%) Avonex, Etanercept6 | 1 (11.1%) Avonex | |
| Prior Tx5 (3-6 month) | 1(5.5%) Avonex | 1(5%) Avonex, | 2(11.1%) Avonex, Tysabri | 1 (11.1%) Avonex | |
| Prior Tx5 (< 3 month) | 4(22.2%) Tysabri, Betaseron, Rebif, Copaxone | 6(30%) Avonex, Betaseron, Copaxone(2), Rebif,Tysabri | 5(27.8%) Avonex, Betaseron, Copaxone, Rebif, Tysabri | 4(44.4%) Avonex, Betaseron, Rebif, Tysabri | |
Data presented as mean ± SD; p-value was from ANOVA test
Data presented as number (%); p-value was from Chi-square or Fisher exact test
Data presented as median (Q1, Q3); p-value was from Kruskal–Wallis ANOVA test
Disease modifying therapy prior to baseline draw
Treatment prior to baseline draw, not time point
Prior treatment for Ankylosing Spondilitis
Flow Cytometry
Isolated PBMCs were stained with panels of monoclonal antibodies against human cell surface and intracellular markers of major cell lineages and lymphocyte subsets. Antibodies and reagents used in this study were summarized in Supplementary Table 1 and the specific antibodies used for each figure are outlined in the legends. 7-Aminoactinomycin D was used to exclude dead cells from the analysis. Only included as a supplementary figure, FoxP3 was stained intracellularly after cell surface staining of CD3, CD4, and CD25 using FoxP3 staining buffer set (eBiosciences, San Diego, CA) according to the manufacturer's suggested protocol. For other intracellular staining, surface stained cells were fixed and permeabilized using BD Cytofix/Cytoperm™ Kit. Stained PBMCs were analyzed on a BD FACSCANTO II Flow Cytometer. Flow cytometry data were analyzed using FlowJo version 7.6.5 software (Flowjo LLC, Ashland, OR. USA). Cell lineage surface markers used in this study are summarized in Table II.
Table II. Cell Surface Lineage Markers Used.
| Cell type | Marker | |
|---|---|---|
| Lineage | Monocyte | FSC-Ahi, CD14+ |
| CD4 T cells | CD14-TCRαβ+CD4+ | |
| CD8 T cells | CD14-TCRαβ+CD8+ | |
| NKT cells | CD14-TCRαβ+CD56+ | |
| B Cells | CD14-TCRαβ-CD19+ | |
| NK cells | CD14-TCRαβ-CD56+ | |
| T cell subtypes | Memory T cells | CD3+CD4+/-CD45RA-CD45RO+ |
| Non-memory T cells | CD3+CD4+/-CD45RA+CD45RO- | |
| Naïve T cells(Tn) | CD3+CD4+/-CD45RA+CD45RO-CCR7+ | |
| Central Memory T cells (Tcm) | CD3+CD4+/-CD45RA-CD45RO+CCR7+ | |
| Effector Memory T cells (Tem) | CD3+CD4+/-CD45RA-CD45RO+CCR7- | |
| Effector Memory RA T cells (Temra) | CD3+CD4+/-CD45RA+CD45RO-CCR7- | |
| Treg | CD3+CD4+CD25+FoxP3+ | |
| Th1 | CD3+CD4+CXCR3+ | |
| Th17 | CD3+CD4+CCR6+ | |
| Th2 | CD3+CD4+CCR3+ |
Ex vivo T cell Cytokine Analysis
Frozen PBMCs from patients were thawed quickly in 37°C pre-warmed complete RPMI with 5 units/ml of Benzonase (EMD millipore, Billerica, MA. USA). After 5 min incubation at 37°C, cells were washed twice with complete RPMI. The cells were then counted and cultured at 2.5×106 cells/ml with 50 ng/mL phorbol 12-myristate 13-acetate (PMA), 1 μg/mL ionomycin and 1μg/ml of Brefeldin A for 6 hours with 5% CO2 at 37°C. At the end of the culture, cells were collected and washed with PBS containing 2% fetal bovine serum (FBS), and surface-stained with anti-CD3-PE-Cy7 (Tonbo Biosciences) and anti-CD4-Brilliant Violet 510™ (Biolegend). Cells were then fixed for 15 min using BD Cytofix™ fixation buffer at room temperature, washed twice with PBS containing 2% FBS and stored overnight at 4°C. Before intracellular staining, cells were permeabilized using BD Cytofix/Cytoperm™ Perm Wash for 15 min and stained with PE-conjugated antibodies against human IL-17A and IL-17F (clone: eBio64CAP17 and SHLR17, eBiosciences, San Diego, CA. USA) and FITC-conjugated anti-human IFNγ (Clone: B27, Thermo Fisher/Invitrogen, Waltham, MA USA) in BD Cytofix/Cytoperm™ Perm Wash buffer for 30 min with occasional vortex. Cells were then washed with BD Cytofix/Cytoperm™ Perm Wash buffer twice to remove unbound antibodies before analysis using the BD FACS Canto II Flow cytometer in staining buffer (PBS with 2% FBS). To assay the production of IL-4 by ex vivo CD4+ T cells, thawed frozen PBMC derived from patients before or after DMF treatment were rested in complete RPMI at 37°C with 5% CO2 for 18 hr, followed by an additional 6 hours with 1 μg/ml Brefeldin A before being harvested and stained with anti-CD3-PE-Cy7 (Tonbo Biosciences) and anti-CD4-Brilliant Violet 510™ (Biolegend). Intracellular staining of IL-4 was carried out using anti-IL-4-APC (Biolegend) with BD cytofix/cytoperm method as described above.
In vitro DMF treatment assays
PBMCs of healthy donors were prepared from plasmapheresis filters by washing in reverse flow with 30 ml of PBS followed by standard Ficoll-Hypaque™ density centrifugation. After washing with PBS, PBMCs were cultured in complete RPMI (RPMI 1640 with 10% human AB plasma, 2 mM glutamine, 100 IU/ml Penicillin and streptomycin) at 2.5×106 cells/ml for 48 hours in a 6-well plate. DMF (10μM, 100μM) or DMSO (vehicle control) was added at the beginning of the culture.
For apoptosis analysis, unstimulated cells were first stained with anti-CD8-FITC/anti-CD3-PE-Cy7/anti-CD4-BV510 at the end of culture. After washing with AnnexinV-binding buffer (10 mM HEPES, pH 7.4; 140 mM NaCl; 2.5 mM CaCl2), cells were stained with 7AAD/ Annexin V-APC in Annexin V-binding buffer for 10min at room temperature. After diluting 5 times with 1× Annexin V-binding buffer, the cells were analyzed immediately by flow cytometry.
For T cell activation, reactive oxygen species (ROS), cell proliferation, and CCR7 expression analysis, T cells were activated by anti-CD3 and anti-CD28. Briefly, the 6-well plates were pre-coated with 3 μg/mL anti-CD3 for 4 hour at 37°C and washed once with PBS. PBMCs were then seeded at 2.5×106 cells/mL with 5 μg/mL of soluble anti-CD28. After 48 hours, cultured PBMCs were harvested and stained with 7AAD/anti-CD3-PE-Cy7 / Anti-CCR7-APC/anti- CD45RO-pacific blue/ -CD4-BV510. For ROS assay in T cells, stained cells were then washed with complete RPMI, and incubated with 5 μM of CM-H2DCFDA (a chloromethyl derivative of 2′, 7′-dichlorodihydrofluorescein diacetate, Thermo Fisher/Invitrogen, Waltham, MA USA) in complete RPMI at 37°C for 10 min, and washed with staining buffer before being analyzed on flow cytometer. For analyzing cell activation and proliferation, cultured cells were surface-stained with anti-CD3-PE-Cy7/anti-CD45RA-APC-Cy7/anti-CD69-BV510. After washing, cells were fixed and stained intracellularly using anti-Ki67-eF450 with BD Cytofix/Cytoperm™ Kit according to manufacturer's recommended protocol. CCR7 expression was measured using the value of geometric mean fluorescence of CCR7 on gated T cell population calculated by Flowjo.
Statistics
Cross-sectional study of DMF treatment groups: non-treated RRMS, 4-6 month (4-6M) and 18-26 month (>18M) DMF were compared using non-parametric tests and post-hoc Dunn's multiple comparisons to assess the differences between groups. For longitudinal samples, we implemented the Friedman test with Dunn's multiple comparisons to find the pair-wise difference between paired data. For in vitro experiments, Wilcoxon signed-rank test was used to assess the difference between control and DMF groups. All the statistical analyses were performed using GraphPad Prism 6 software (GraphPad Software, Inc. La Jolla, CA. USA ).
Results
DMF treatment reduced circulating lymphocyte count
All RRMS patients treated with Tecfidera™ in this study cohort have been clinically stable without further relapses, new MRI activities, or disease progression measured by EDSS up to 26 months during our study (Table I). Previously, these patients were either treatment naïve (8 of 18 in the baseline cohort), or intolerant to other DMTs (8 of 10 who had been on prior DMT), or had some breakthrough disease activities on other DMTs (5 of 10). More specifically, among the 18 subjects in the baseline cohort, 13 had new MRI lesions and 12 had new symptoms (mostly sensory) in the months to year prior to initiating Tecfidera™. There were no statistical differences in the characteristics of the three study groups in the cross-sectional study (Table I). To determine the effect of DMF on peripheral blood counts, we first compared cross-sectionally RRMS patients treated with DMF for either short-term (4-6 months, n=20) or long-term (18-26 months, n=18) with age and sex-matched untreated RRMS patients (n=18). Furthermore, prospective data from 9 of the RRMS patients from whom a sample was collected at all time points was analyzed and presented in separate graphs. Complete blood counts (CBC) were obtained at the University of Michigan hospital pathology lab and the results were retrieved from the patients' records. As shown in Table III, the total white blood cell (WBC) counts and peripheral blood mononuclear cell (PBMC) counts were reduced significantly with DMF treatment at both short-term and at long-term time points. Among all WBC cell lineages, lymphocyte counts were significantly reduced at 4-6 months; and remained so over more than 18 months with DMF treatment. DMF treatment did not show significant effects on neutrophil, eosinophil, basophil and monocyte populations.
Table III. Effects of DMF on Complete Blood Counts of RRMS.
| Untreated | 4-6M DMF | 18-26M DMF | |||
|---|---|---|---|---|---|
|
| |||||
| Median (IQR) | Median (IQR) | p-value | Median (IQR) | p-value | |
| WBC | 7.2 (3.27) | 5.4 (1.1)* | 0.022 | 5.6 (2.6) * | 0.015 |
| Neutrophil | 4.4 (2.33) | 3.7 (1.8) | NS | 3.6 (1.82) | NS |
| Lymphocytes | 1.8 (1.3) | 1.3 (0.6)** | 0.002 | 1.1 (0.8)** | 0.001 |
| Monocytes | 0.60 (0.35) | 0.6 (0.3) | NS | 0.55 (0.4) | NS |
| Eosinophil | 0.20 (0.18) | 0.10 (0.1) | NS | 0.10 (0.1) | NS |
| PBMC | 2.6 (1.33) | 1.7 (0.7)** | 0.003 | 1.8 (0.9)** | 0.002 |
Numbers represent cell counts (×106)/ml of blood. The absolute counts of PBMCs are deduced from the CBC value by subtracting absolute counts of neutrophils, eosinophils and basophils from that of WBCs. PBMC values are the sum of lymphocytes, monocytes and early granulocytes. Abbreviations: WBC, white blood cell; IQR, interquartile range; NS, not significant
P<0.05;
P<0.01, compared to RRMS by Kruskal-Wallis ANOVA with Dunn multiple comparisons.
DMF treatment reduced both circulating CD4+ T cell and CD8+ T cell counts
To further understand the effects of DMF treatment on subtypes of lymphocytes, PBMCs were isolated and stained with fluorochrome-conjugated antibodies against cell surface markers for T cells, B cells, monocytes, and NK cells, followed by multi-color flow cytometric analysis. Representative staining profiles are shown in Fig 1A and the gating was consistent for all of the samples analyzed in the study. As shown in Fig 1B, compared to the untreated group, CD4+ T lymphocyte counts were significantly reduced in the 4-6 months treated group (P= 0.002) and in the over 18 months treated group (P= 0.002). Absolute numbers of CD8+ lymphocytes were lower in the 4-6M treatment group (median=0.22×106/mL), compared to the untreated group (median=0.40×106/mL, P =0.045), but these were even lower in the >18M treated group (median=0.17×106/mL, P=0.0002). With more extensive reductions of CD8+ T cells, the CD4/CD8 T cell ratio was significantly higher for the >18M group (median=6.3) than that of the untreated group (median=3.9, P=0.010). Moreover, NK-like T cells (NKT), identified as FSClowCD14-TCRαβ+CD56+, were progressively decreased and the differences were significant between the long term >18M treatment group and the untreated group (P=0.0007). B cell counts were significantly decreased after >18 months of treatment and the detailed B cell subtype changes were described in our recent paper (14). Total NK cell counts showed no significant differences among the three groups.
Figure 1. DMF treatment reduced circulating CD4+ T cells, CD8+ T cells, NKT cells and B cells.
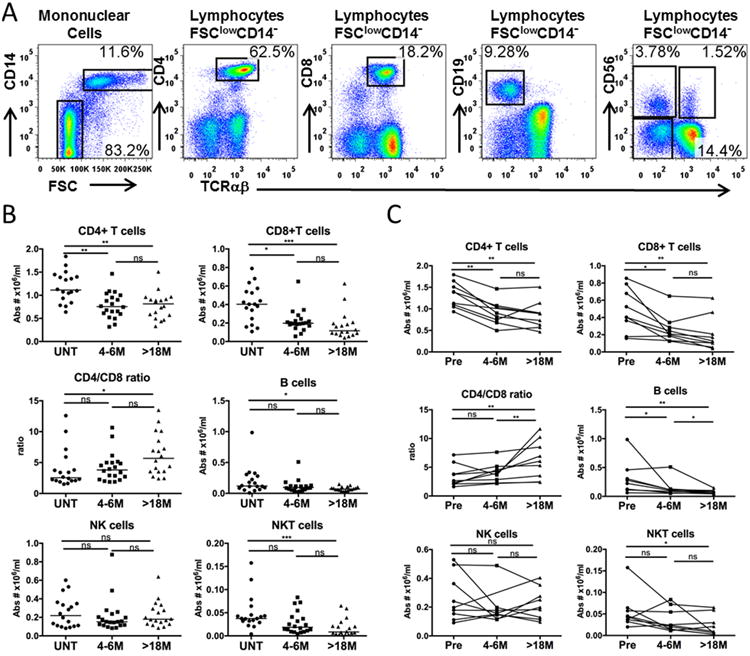
PBMCs were isolated from peripheral blood of patients with RRMS. Cells were stained with 7AAD and antibodies against TCRαβ, CD4, CD14, CD8, CD19, CD56, washed and then analyzed on a flow cytometer. (A) Typical staining profile of an RRMS patient without DMF treatment. Labels above FACS profiles indicate the gated population being analyzed. Percentages noted on the dot plots are in relation to the gated population. (B) Total absolute cell numbers (Abs #) /mL of blood of the indicated subsets were calculated based on clinical complete blood cell counts and the percentages of the indicated subsets as determined by flow cytometry. Absolute numbers of different lineages were compared between untreated (UNT) RRMS patients (n=18), and patients after 4-6 months (4-6M, n=20) or patients after 18-26 months (>18M) of DMF treatment (n=18). Scatter plots show individual patient data as dots and the median of each group as a line. (C) Longitudinal samples (n=9) were used to track changes in lineages over time within the same patient. Samples were obtained before (Pre), after 4-6 months (4-6M) and after 18-26 M (>18M) months of DMF treatment. P values from Friedman test with Dunn's multiple comparisons for pair-wise time points are shown above the data for each cell type group. *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant.
To further validate the changes to the lymphocyte profiles noted in the cross-sectional cohort with DMF treatment over time, we analyzed the longitudinal data from 9 out of these 18 RRMS patients who were able to be followed at all three time points before and after DMF treatment (Fig. 1C). The results for individual patients showed similar trends as were observed in the cross-sectional analysis. CD4+ T, CD8+ T, B and NKT cell counts were significantly decreased, especially with long term treatment. More significant reductions were seen of CD8+ T cells compared with CD4+ T cells, resulting in a significant increase of the CD4/CD8 ratio after >18 months of treatment with DMF.
DMF treatment reduced memory T cells, particularly effector memory T cells
To determine the effect of DMF on subsets of T cells with memory or non-memory phenotypes, PBMCs from patients before and after DMF treatment were analyzed for their expression of CD45RA, CD45RO and CCR7 on T cells. A representative flow cytometry staining profile for one patient before and after treatment is shown in Fig. 2A. Compared with the untreated group, the memory fractions (CD45RA-CD45RO+) of T cells were both significantly reduced from a median of 45.2% (CD3+CD4+) and 37.7% (CD3+CD4-) in the untreated group to 28.4% (p=0.0007) and 19.2% (p=0.0093) respectively after 4-6 months of DMF treatment. These subsets were further reduced to 24.7% (p=0.0006) and 17.5% (p=0.0004) respectively after 18 months of treatment (Fig. 2B). Using analysis of chemokine receptor CCR7 expression, the central memory fraction of CCR7+CD45RO+CD3+CD4+ T cells (CD4+ Tcm) was found to be significantly lower in both the 4-6 month group (p=0.028) and >18 month group (p=0.009) than in the untreated patient group (Fig. 2C). In contrast, the CD4- Tcm fraction was not altered significantly with DMF treatment. Compared to the untreated group, the fraction of CD45RO+ CCR7- (Tem) of both CD4+ and CD4- T cells were decreased from a median of 15.5% and 30.0% in the untreated group to 6.8% (p=0.0008) and 14.0% (p=0.002) respectively in the 4-6 month group, and were sustained at a low level in the long-term (>18 month) treatment group (p<0.0001 and p=0.0007, respectively). Reciprocally, we found that the naïve fractions (CCR7+CD45RA+ Tn) of both CD4+ and CD4- T cells were relatively higher in both short-term and long-term treatment groups than in the untreated control group. Effector Memory RA+ T cells (Temra, CD45RO-CCR7-) fractions were less affected in both CD4+ and CD4- T cells by DMF treatment.
Figure 2. DMF Treatment reduced memory T cells.
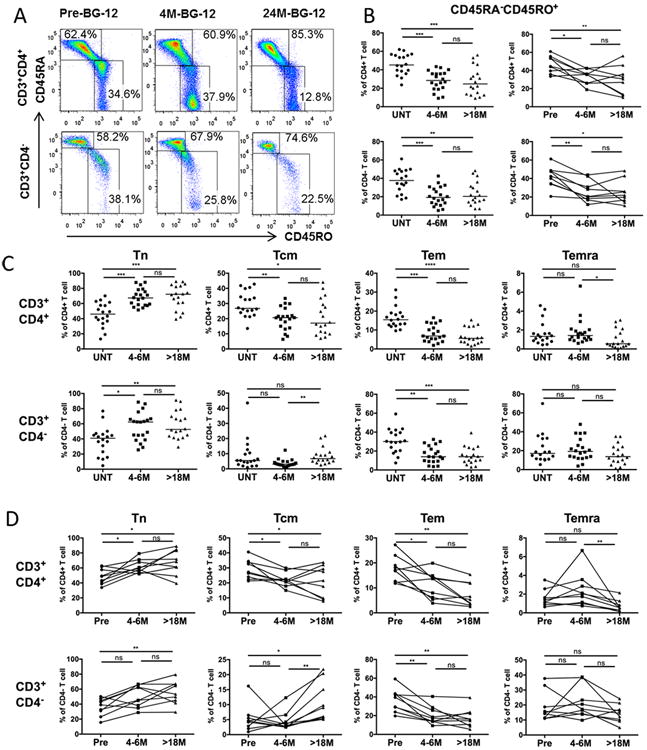
PBMCs were isolated from blood of patients and stained with antibodies specific to surface markers: CD3, CD4, CD45RO, CD45RA and CCR7 in order to analyze subsets of memory cells. (A) Representative profile depicting the frequencies of memory and non-memory phenotypes among CD4+CD3+ T cells (top panels) and CD3+CD4- T cells (lower panels) from an RRMS patient prior to DMF treatment, and after 4-6 months and >18 months of continuous DMF treatment (B) Frequencies of memory (CD45RA-/CD45RO+) fractions as a percentage of CD3+CD4+ (upper panels) and CD3+CD4- (lower panels) T cells in cross-sectional (left-hand panel) untreated (UNT, n=18), 4-6 month DMF-treated (4-6M, n=20), and 18-26 month (>18M, n=18) DMF-treated groups; as well as longitudinal cohort (n=9, right hand panel) (C) Frequencies of naïve Tn (CD45RO-CCR7+), central memory Tcm (CD45RO+CCR7+), effector memory Tem (CD45RO+CCR7-), and CD45RA+ effector memory Temra (CD45RO-CCR7-) of CD3+ CD4+ (upper panels) and CD3+CD4- (lower panels) T cells in three cross-sectional patient groups. Scatter plots show data for individual patients as dots and the median of each group as a line. P-values from Kruskal-Wallis ANOVA with Dunn's multiple comparison tests are shown above the data for each cell type. (D) Tn, Tcm, Tem, and Temra cells as a percentage of CD3+CD4+ (upper panels) or CD3+CD4- (lower panels) T cells in the longitudinal cohort at the three time points. P values from Friedman test with Dunn's multiple comparisons for pair-wise time points are shown above the data for each cell type. *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant.
Longitudinal data derived from 9 patients were analyzed to validate our cross-sectional findings on memory T cells (Fig. 2D). Overall, both CD4+ and CD4- memory T cell frequencies were decreased after short term treatment and remained decreased after long term treatment. The Tem and Tcm fractions of CD4+ T cells, and the Tem fraction of CD4- T cells were decreased persistently after DMF treatment, while Tcm fractions of CD4- T cells were decreased slightly after 4-6 months of treatment, but then increased notably after >18 months of DMF treatment. The Tn frequency of both CD4 + and CD4- T cells were increased over time, while the percentages of CD3+CD4+CCR7-CD45RA+ (CD4+ Temra) and CD4- Temra cells were not changed over the duration of the study (Fig. 2D).
DMF inhibited T cell activation
CD69 is a marker commonly used to evaluate T cell activation (19, 20). Its expression on T cells is induced by T cell receptor engagement, and correlated with 3H-thymidine incorporation (21). To study whether T cell activation could be affected by DMF treatment, we analyzed the frequency of CD69+ cells among T cells. The percentage of CD69+ of total CD4+ and CD4- T cells were decreased significantly after >18 months of treatment as compared with the untreated group (P=0.004 and P=0.03 respectively, Fig. 3), suggesting DMF inhibits T cell activation. Furthermore, the percentage of CD69+ cells in Tem and Temra cells were decreased significantly after 18 months of treatment with DMF, particularly for CD4+ T cells (Fig. 3). Though the proportion of reduction of Temra was not as significant comparing Tcm and Tem (Figure 2), there was a significant reduction of the percentage of CD69+ Temra (Figure 3). This indicated that DMF reduced functional activation of Temra.
Figure 3. DMF reduced the T cell activation status.
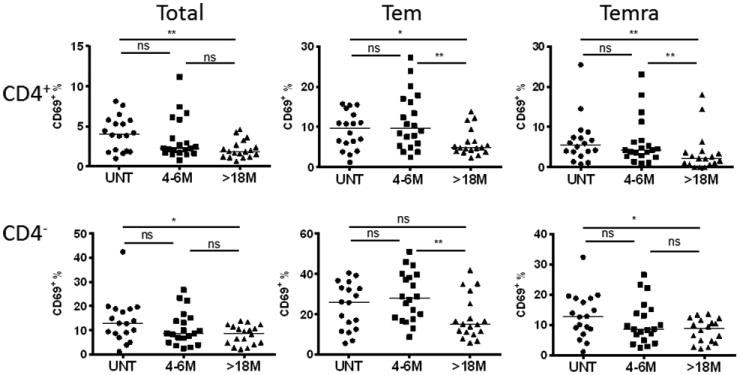
PBMCs were isolated from blood of patients and stained with antibodies against CD3, CD4, CD45RO, CCR7 and CD69 to assess activation status in T cells. From left to right, activation was shown as frequencies of CD69+ cells of total and corresponding Tem, and Temra subsets at three different time points (UNT: n=18; 4-6M: n=20, >18M: n=18) for CD4+ (upper panel) and CD4- (lower panel) T cells. Scatter plots show data for individual patients as dots and median of each group as a line. P-values from Kruskal-Wallis ANOVA with Dunn's multiple comparison tests are shown above the data for each cell type. *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant.
DMF treatment reduced Th1, Th17 and Th1-Th17 cells and increased Th2 cells, but did not alter frequencies of regulatory T cells
Th1 and Th17 subsets have been found to be pathogenic in MS, while Th2 cells and regulatory T cells may suppress autoimmune responses and have protective effects in MS (22, 23). Peripheral T helper subsets from patients before and after treatment with DMF were analyzed using antibodies against CXCR3, CCR6, CCR3, CD161, CD25 and FoxP3 that mark specific Th cell functional subsets (Table II). CCR3, CCR4 and CRTh2 have all been used as surface markers of Th2 cells, and we found in preliminary studies that the majority of IL-4+ Th cells ex vivo were enriched in the CCR3+ fraction (data not shown) (24-26). Compared to the untreated group, there was a significant reduction of the proportion of CXCR3+ CD4+ (Th1), CCR6+ CD4+ (Th17); while the CCR3+CD4+ (Th2) frequency was significantly higher in the long-term treatment group but not in the 4-6 month group (Fig.4A and 4B). Since CD161 was also found to be associated with the IL-17-producing T cell subset (27-29), its expression was also analyzed. With DMF treatment, we found that the fraction of CD161+ cells among CD4+ T cells was reduced significantly after more than 18 months of treatment with DMF (data not shown). Significant reductions were observed in those CD4+ T cells that were triple positive for CXCR3, CCR6 and CD161 (cross-sectional: p=0.0052; longitudinal: P=0.0066 Fig 4C) and in double positive cells expressing CXCR3 and CD161, but not CCR6 (cross-sectional: P=0.0004; longitudinal: P=0.0029, (Fig. 4D). Based on these findings, the functional capacity of CD4+ T cells to produce pathogenic cytokines IFNγ and IL-17 upon ex vivo stimulation was evaluated (Figure 4E, Suppl. Fig. 1A). The percentages of CD4+ T cells producing IFNγ or IL-17 were gradually reduced and more significantly reduced over 18 months of treatment when compared to baseline, while the frequency of ex vivo CD4+ T cell producing IL-4 without stimulation was increased after DMF treatment (Fig. 4E and Suppl. 1B). Interestingly, the frequencies of the two CD4+ T cell subsets (CD161+/CCR6+/CXCR3+ and CD161+/CCR6-/CXCR3+) that were preferentially reduced by DMF treatment were found closely correlated with the frequency of CD4+ T cells producing interferon γ, but not IL-17 (Fig 4C and 4D).
Figure 4. DMF specifically reduced Th1 and Th17, particularly those expressing CXCR3 and CD161, but increased Th2 cells.
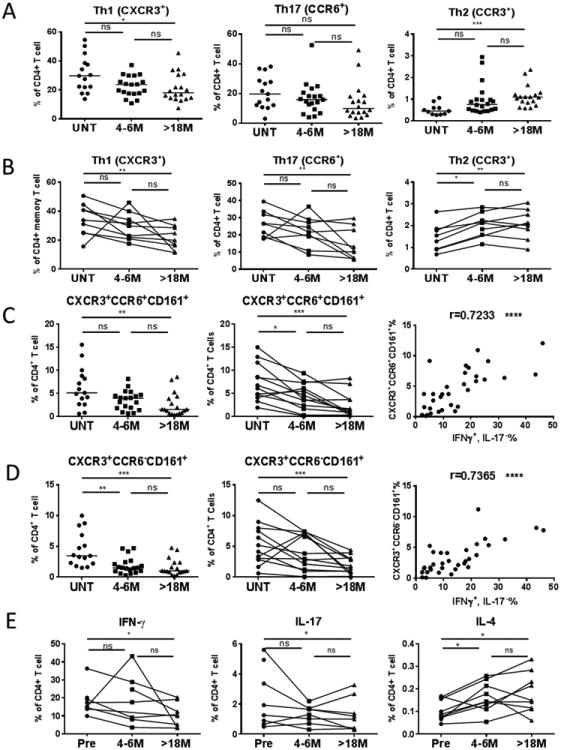
PBMCs from patients were stained with antibodies against CD3, CD4, CXCR3, CCR6, CCR3, and CD161 to evaluate frequencies of CXCR3+ (Th1), CCR6+ (Th17) of CD4+ T cells (UNT: n=15; 4-6M: n=19; >18M: n=18). The CCR3+ (Th2) of total CD4+ T cells were also analyzed (UNT: n=11, 4-6M: n=20, >18M: n=18). These frequencies were plotted as dots at three time points with the median of each group as a line in three cross-sectional groups (A) or in the longitudinal cohort (B). DMF effects on the frequencies of CD4+ T cells expressing CD161and CXCR3 with (C) or without (D) CCR6 in cross sectional (left-hand panels) and longitudinal (middle panels) studies, and corresponding correlation (right-hand panels) with frequencies of CD4+ T cells producing IFNγ but not IL-17. (E) IFNγ (left-hand panel) and IL-17 (middle panel) production by CD4+ T cells was analyzed in our longitudinal study following stimulation of PBMCs with PMA and ionomycin for 6 hours in the presence of brefeldin A. Cells were then stained with fluorochrome-conjugated antibodies against CD3, CD4, followed by intracellular staining of antibodies against IFNγ, IL-17A and IL-17F. IL-4 (right-hand panel) production was measured from ex vivo PBMC without further in vitro stimulation since it was clearly detectable (Suppl Fig 1B). PBMC were incubated in complete RPMI for 24hrs with brefeldin A added in the last 6 hours of culture. Cells were then stained with fluorochrome-conjugated antibodies against CD3, CD4, followed by intracellular staining with antibodies against human IL-4. P-values from Kruskal-Wallis ANOVA with Dunn's multiple comparison tests are shown above the data for each cell type group. *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant.
Frequencies of CD25+FoxP3+CD4+ regulatory T (Treg) cells were not changed significantly at both 4-6 months and at >18 months of DMF treatment (Suppl. Fig. 2A). Since the balance between Th1 and Th17 to Th2 is important for the pathogenesis of RRMS, the ratio of Th1 (CXCR3+), Th17 (CCR6+) or (CD161+) to Th2 (CCR3+) were calculated. All of these ratios were found significantly reduced with DMF treatment (Suppl. Fig. 2B). However, the ratio of these Th1/Th17 subsets to Treg was unchanged with DMF treatment (Suppl. Fig. 2C).
Effects of DMF on T cell apoptosis, activation, ROS production and proliferation in vitro
To further investigate potential mechanisms underlying the treatment effects of DMF in MS patients, we cultured fresh unstimulated or anti-CD3/anti-CD28 stimulated peripheral blood mononuclear cells (PBMCs) derived from healthy individuals and treated the cells with DMF. As shown in Fig. 5A, DMF treatment significantly increased apoptosis (%Annexin V+ 7AAD-) of both unstimulated CD4+ T cells (10μM: P=0.0156, 100μM: P=0.0156) and CD4- T cells (10μM: P=0.0078, 100μM: P=0.0078).
Figure 5. Direct Effect of DMF on T cells in vitro.
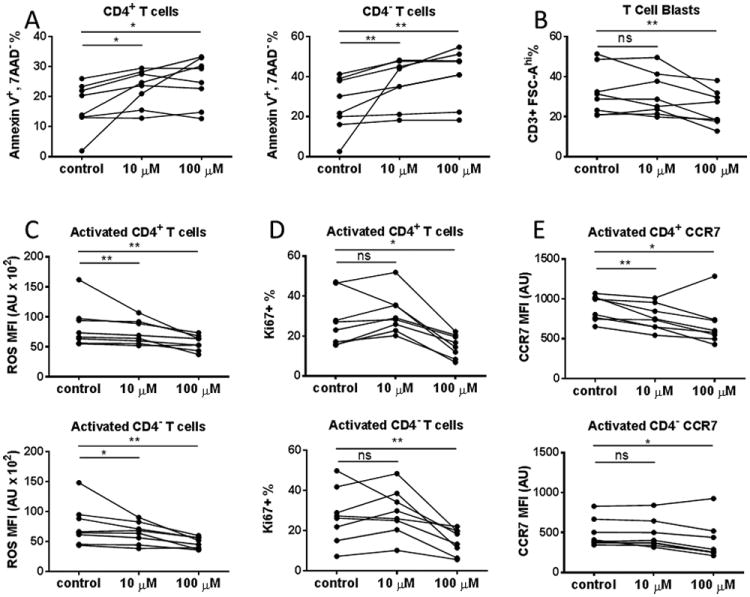
PBMCs derived from 8 healthy donors were isolated and cultured in the absence or presence of 10μM or 100μM DMF for 48 hours. (A) DMF-induced T cell apoptosis in vitro. Cultured PBMCs were harvested, washed and surface stained with fluorochrome-conjugated antibodies against CD3, CD8, CD4, as well as 7AAD and Annexin V. The frequency of apoptotic cells (AnnexinV+7AAD-) among total CD3+ CD4+ (left) and CD3+ CD4- (right) T cells in the vehicle control and10μM or 100μM DMF-treated groups are shown. (B) DMF suppressed T cell activation in vitro. PBMCs from healthy donors were cultured in the presence or absence of DMF for 48 hours on plates coated with anti-CD3 and soluble anti-CD28 antibodies to activate T cells. The percentage of activated T cell blast (CD3+ FSC-Ahi) was shown. (C) DMF-induced reduction of ROS level in activated T cells. Plate-bound anti-CD3/soluble anti-CD28 activated PBMC cultures from healthy donors with or without DMF were harvested and stained with antibodies against CD3, CD4, followed by staining of ROS with CM-H2DCFDA. The level of ROS on CD4+ (upper) and CD4- T cell (lower) blasts (CD3+ FSC-Ahi) are expressed as arbitrary units (AU) of mean fluorescence intensity. We also repeated the statistical calculations with the patient showing most reduction removed, the data still showed significant reductions (p<0.05). (D) DMF inhibited T cell proliferation in vitro. T cells from healthy donors were activated in the presence or absence of DMF for 48hours on plates with immobilized anti-CD3 and soluble anti-CD28 antibodies. After culture, cells were surfaced stained with CD3, CD4 and CD69, followed by intracellular staining of Ki67 to measure cell proliferation. The frequency of Ki67+ cells among CD69+ population of CD4+ (upper panel) and CD4- (lower panel) T cells are shown. (E) DMF reduced CCR7 expression on activated T cells in vitro. T cells were activated in the presence or absence of DMF for 48 hours on plates with immobilized anti-CD3 and soluble anti-CD28 antibodies. After culture, cells were surfaced stained with fluorochrome-conjugated antibodies against CD3, CD4 and CCR7. CD3+ T blasts were gated and CCR7 geometric mean fluorescence intensity (MFI; expressed in arbitrary units (A. U.)) was compared among vehicle control, 10μM DMF and 100μM DMF groups, respectively. P values from Wilcoxon signed rank test for the comparisons between control vs. 10 μM and control vs. 100 μM are shown above the data for each cell type group.*P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant.
To investigate whether DMF will have direct effects on T cell activation, we analyzed the percentage of enlarged blasts (CD3+, FSC-Ahi) in the PBMC culture activated by anti-CD3 and anti-CD28. T cell blasts are activated T cells expressing CD69. Few of these activated T cells (CD69+) were positive for 7AAD staining, confirming that they were alive (Suppl. Fig. 3). A significant reduction of the percentage T cell blasts was observed in cultures treated with 100μM DMF (Median= 23.05% P=0.0156) compared with the vehicle control group (Median=30.15%). (Figure 5B).
T cell activation triggers the production of superoxides that can alter the behavior of the cells in positive or negative ways. Since DMF was reported as an anti-oxidant, we measured the effect of DMF on ROS level in T cells by staining anti-CD3/anti-CD28 stimulated PBMCs with CM-H2DCFDA which can penetrate into live cells and get oxidized by ROS to produce fluorescence. As shown in Fig. 5C, compared with vehicle control, the mean fluorescence intensity (MFI) of ROS of both CD4+ and CD4- activated T cells was reduced at 10μM DMF (CD4+: P=0.0078, CD4-: P=0.0391), and significantly at 100μM concentrations (CD4+: P=0.0078; CD4-: P=0.0078). Reduction of ROS further confirmed that DMF played an anti-oxidant role in activated T cells.
The effect of DMF on proliferation of activated T cells was determined by Ki67 expression (Fig.5D). The percentage of Ki67+ cells among activated CD4+ and CD4- T cells were significantly reduced in the culture with 100μM DMF treatment (CD4+ T cells: P=0.0156; CD4- T cells: P=0.0078), but not with 10 μM DMF.
Since CCR7 is important for lymphocyte migration into secondary lymphoid organs, we analyzed the effects of DMF on CCR7 surface expression on T cells. Anti-CD3 and anti-CD28 stimulated PBMCs were treated with DMF for 48 hours. As shown in Figure 5E, 10 μM DMF exerts suppressive effect on the expression of CCR7, particularly on CD4+ T cells (P=0.0181 for 10μM, and P=0.0181 for 100μM). There was also a reduction of CCR7 expression for CD4- T cells at 100μM DMF treatment (P=0.0199).
Discussion
Understanding the lymphocyte subpopulation changes associated with DMF treatment is important for monitoring treatment efficacy and potential long-term side effects. Such studies could also lead to discovery of surrogate markers for evaluating MS disease activity and progression. We thus conducted a cross-sectional study of RRMS patients on DMF treatment for up to two years, along with a smaller longitudinal study within the same cohort of patients. Utilizing multi-parametric flow cytometry, we recorded comprehensive immunophenotyping results following short-term (4-6 months) and long-term (18-26 months) DMF treatment in RRMS patients. There were reductions of multiple peripheral immune cell subtypes, including CD4+ T cells, CD8+ T cells, B lymphocytes, and NKT cells. Compared with CD4+ T cells, we observed a more significant reduction of CD8+ T cells. Among all T cells, memory T cells (especially effector memory T cells) were the most affected by DMF treatment, both short term and long term. In addition to the reduction of both frequency and absolute numbers, the activation potential of Tem and Temra was also reduced. Furthermore, DMF treatment was also shown to increase the abundance of anti-inflammatory Th2 cells while decreasing pro-inflammatory Th1 and Th17 cells. Correspondingly, DMF also led to an increase in IL-4 and a decrease in IFNγ and IL-17 positive Th cells.
Our study followed RRMS patients on DMF treatment for over 2 years, contrasting with an earlier report that followed DMF treatment for only 6 months (30). In our study of DMF-treated patients, approximately 15% of the RRMS patients (3 out of 20) had grade 2-3 lymphopenia (absolute lymphocyte count < 800 cells/μl) at 4-6 months, while at 18-24 months only 1 out of 18 patients had grade 2-3 lymphopenia; none of our patients had grade 4 lymphopenia (lymphocyte count < 200 cells/μl, Fig. 1 and Table II). This differs from the patient cohort in a previous study, where 17 of 41 (41%) patients were lymphopenic (31). Our cohort did not show such a significant degree of lymphopenia overall. As initially reported by Spencer et al, there was an increased CD4/CD8 ratio at 12 months with DMF treatment (32). We found that there was a more pronounced reduction of CD8+ T cells than CD4+ T cells after 18 months of DMF treatment as well, resulting in an elevation of the CD4:CD8 ratio in the 18-26M group but not in the 4-6M group. A recent cross-sectional study (31) demonstrated that only in patients with Grade 2-3 lymphopenia were CD4+ and CD8+ T cells decreased and the CD4/CD8 ratio increased. In our group of patients without significant lymphopenia, a significant reduction of CD4+ T cells was observed as early as 4-6 months, while more significant reduction of CD8+ T cells and an increase of the CD4/CD8 ratio became more compelling with longer term (>18 month) DMF treatment (Fig. 1). DMF showed more profound suppressive effects on CD4- T cells compared with CD4+ T cells in our in vitro study (Fig. 5), which was consistent with the in vivo DMF treatment effect in RRMS patients (as a majority of CD4- T cells are CD8 cells). The findings in the cross-sectional study were consistently confirmed in individual patients that were followed longitudinally. Thus, a longer and more detailed follow up of DMF-treated MS patients, as was shown here, could be beneficial in detecting differences in the effect of DMF on the immunological profile in individual patients, especially for those without marked lymphopenia.
Earlier studies have shown that RRMS patients had fewer peripheral naïve T cells and more effector memory T cells than healthy controls (33, 34), emphasizing the potential importance of effector memory T cells in the pathogenesis of RRMS. In our study, DMF suppressed both Tcm and Tem cells as early as 4-6 months of treatment, supporting previous results that showed >6 month treatment of DMF decreased the frequency of Tcm and Tem with relative expansion of naïve T cells in DMF-treated patients with or without lymphopenia (31). Compared to previous publications, our study provided a more long-term follow up of DMF treatment that in some cases was more than 2 years. Our data showed interesting kinetics of CD4- memory T cells: the percentage of CD4- Tcm cells decreased after 4-6 months and then recovered significantly after 18 months of treatment, while the percentage of CD4- Tem stayed low with long-term DMF treatment (>18 month) (Fig. 2). Though the absolute number and proportion of reduction of Temra was not as significant comparing Tcm and Tem, there was a significant reduction of percent CD69+ Temra. This indicates that DMF reduced functional activation of Temra. Temra is a very interesting subset of memory T cells which has not been previously studied following DMF treatment. Temra represent the most differentiated type of memory cells and are highly susceptible to apoptosis. They also express high levels of cytotoxic molecules such as perforin and Fas ligand (2). Similar to Tem, Temra in particular are more competent to carry out effector functions than Tcm; they exhibit potent ex vivo cytotoxicity and produce Th1 cytokines upon stimulation (2). Therefore, the reduction of CD69+ frequency among CD4+ Temra is consistent with reductions of Th1 cells observed for patients treated with DMF.
Considering the significant decrease of the absolute CD8 count, the recovery of CD4- Tcm (which contains mostly CD8+ T cells) may reflect lymphopenia-induced homeostatic proliferation (35). CD8+ Tcm cells were found to be at low frequencies but with high proliferative ability, playing an important role in self-renewal and long-term survival (36, 37). Thus, the recovery of CD8+ Tcm found in this study may suggest that DMF at current dosing does not completely shut off homeostatic proliferation, which is usually driven by a relative overabundance of IL-7 and self Ag-MHC complexes (35). CD4+ Tcm was not increased, possibly due to the fact that they were not depleted as much as CD8+ T cells by DMF treatment.
Th17 and Th1 cells play a significant role in the pathogenesis of MS (38, 39), and some MS DMTs have been shown to down-regulate Th1 and Th17 and up-regulate Th2 cell differentiation. Surface expression of the chemokine receptor CXCR3 is commonly used to identify the Th1 population, while CCR6 is commonly used to define the Th17 population. CD161+ Th17 cells were closely associated with autoimmune diseases (40-43), and the CD161+ fraction of CD4+ T cells was capable of producing IFNγ, IL-17, or both (44). We analyzed the frequencies of individual CXCR3+, CCR6+ or CD161+ within CD4+ T cell populations in our study, and found that all of these subsets were significantly reduced in patients after >18 months of DMF treatment compared to those in the untreated group (Fig. 4). Consistent with surface marker findings, we showed that there was a significant reduction of IFNγ and IL-17 producing T cells following DMF treatment.
The reduction of CD4+ T cells expressing both CXCR3 and CD161 suggests that this cell population was also preferentially depleted with DMF treatment. Since this population is more proinflammatory in nature compared with other CD4+ T cells (42), its preferential depletion may play an important therapeutic role for the success of DMF as an RRMS disease modifying medication. With the knowledge that IL-17 producing cells are also within the CD161+ fraction of CD4 T cells, and CD161+ cells could also produce IFNγ (44), the analysis of double marker CXCR3+ CD161+ and triple marker CXCR3+CCR6+CD161+ expression of Th cells might further refine the Th1-Th17 subsets. The fact that DMF treated MS patients have both reduced level of Th1 and Th17 cytokines produced by CD4+ T cells and reduced T helper subset defined by CXCR3, CD161 and CCR6, provided us a unique opportunity to correlate these cell surface markers with the level of IFNγ and IL-17 production by CD4+ T cells. We found that CXCR3+CD161+ with or without CCR6, correlate closely with the frequency of CD4+ T cells producing IFNγ, but not IL-17, suggesting that these markers more closely define Th1 cells.
The phenotypical and functional reduction of Th1, Th17 as well as Th1-Th17 population, accompanied by the boosting effects on Th2, might play pivotal anti-inflammatory roles contributing to the therapeutic effects of DMF in MS. Although earlier in vitro studies suggested that DMF mainly suppresses Th1 responses in psoriasis (45), a more recent study of DMF-treated psoriasis patients has shown that DMF modulates Th17 cells in addition to Th1/Th2 cells (46). DMF seems to exert similar multifaceted effects on T helper cells in MS, as we show in this study. More research is needed to address the question of whether DMF is capable of reducing numbers of myelin-specific Th17 cells, especially those within the central nervous system.
CD69 is a marker commonly used to evaluate T cell activation (19, 20). We found that the percentages of CD69+CD4+ and CD69+CD4- T cells were both reduced after 4-6 months of DMF treatment. The inhibitory effects on T cell activation were found most on memory T cells and the effects sustained beyond 18 months (Fig. 3), suggesting there were fewer newly activated T cells in circulation with DMF treatment.
DMF was found to be able to modestly reduce CCR7 expression on T lymphocytes in vitro (Figure 5E). The CCR7 ligands CCL19 and CCL20 have been detected in venules surrounded by inflammatory cells at the BBB (47). CCR7+ central memory T cells were found to be enriched in the cerebrospinal fluid (CSF) of subjects with inflammatory neurological diseases (IND) including MS (48). Down-regulation of CCR7 by DMF may thereby affect Tcm cell trafficking to the CNS and could be another mechanism that contributes to the therapeutic effect of DMF in MS. A recent study by Kihara et.al has demonstrated that DMF inhibits integrin α4 expression on CD3+ T cells and B220+ B cells in EAE mice, as well as on activated human Jurkat T cells (49). The adhesion molecule VLA4, which is the α4β1 integrin, has been shown to be involved in the mechanism of T lymphocytes crossing the BBB. By down-regulating VLA4 on T cells, DMF would be expected to limit lymphocyte infiltration through the BBB into CNS, therefore curtailing inflammation in CNS.
We and others have shown that DMF treatment could be associated with varying degrees of lymphopenia (32, 50). The mechanism underlying lymphopenia is not entirely clear. Our study was consistent with a previous report showing DMF can induce T cell apoptosis (51). By performing an in vitro assay using PBMCs from healthy controls treated with DMF, we showed that there was increased T cell apoptosis and decreased proliferation after DMF treatment. Furthermore, we showed that CD4- T cells were more susceptible to these effects than CD4+ T cells (Fig. 5). These results are consistent with our in vivo data, which demonstrate that over time DMF suppresses CD8+ T cell counts more than CD4+ T cells. DMF has been shown to activate the Nrf2-antioxidant response element signaling pathway (12, 52). Consistent with this, we showed that ROS level in activated T cells declined with the increasing concentration of DMF. DMF suppressed T cell activation both in vivo and in vitro, which is consistent with an earlier study by Turley et.al. who determined that tert-Butylhydroquinone (tBHQ), an Nrf2 activator, inhibited primary human CD4+ T cell activation (53). However, DMF was unable to intercept T cell activation in Tcm of CD8+ T cells, which seemed to have undergone homeostatic proliferation induced by lymphocyte depletion (Fig. 2) (54). A separate study demonstrated that DMF can induce apoptosis in hematopoietic cells by inhibition of NF-κB nuclear translocation and down regulation of Bcl-XL and XIAP (55). Our group recently showed that DMF prevented apoptosis in neural stem/progenitor cells by activating the Nrf2-antioxidant response element signaling pathway (13), suggesting that DMF's effects on apoptosis are perhaps cell-type specific and may involve different signaling pathways in different cell types. A recent mouse study comparing DMF effects on EAE induced in WT and Nrf2 knockout mice showed that DMF induced both adaptive and innate immune modulation independent of Nrf2 (56). Further studies are needed to demonstrate the exact signaling pathway(s) and mechanism for this differential inhibitory effect of DMF on selected subsets of T cells.
Several cases of PML have been reported in psoriasis and MS patients who have been treated with DMF (57). PML is a potentially lethal viral disease caused by JC virus, a type of polyomavirus. JC virus infects a substantial fraction of the general population, initially as an asymptomatic kidney infection. However, a mutated form of JC virus can become pathogenic in immunocompromised individuals by invading the CNS and eliciting progressive damage to white matter in the brain (58). DMF could facilitate JC virus reactivation by reducing the memory and cytotoxic CD8+ T cells targeting the mutated JC virus, potentially reducing their ability to infiltrate into the CNS to fight against the virus. The suppressive effects of DMF on peripheral lymphocytes and particularly effector memory CD8+ T cells were seen as early as 4 months, and sustained after 18-26 months of treatment in our study. The depletion of CD8 Tem, as well as the diminished activation of potential of Temra by DMF may be particularly devastating, because it would weaken the normal immune surveillance against JC virus. Careful monitoring of peripheral T cells, particularly JC-virus-specific CD8+ as well as CD4+ T cells could be clinically important for making therapeutic decisions regarding continual and long-term DMF usage for treating RRMS. Further studies should also be conducted to investigate the correlation of these specific changes in the immunological profile with clinical efficacy after long term treatment, and how it might play a role in relapse and potential side effects. Since we only tested circulating lymphocytes in this study, the effects of DMF on immune cells in the CSF or within the CNS still remain to be addressed.
In conclusion, our results suggest that efforts should be made to monitor the immune modulating effects of DMF on the CD4+ and CD8+ memory T cells, Th1, Th17, Th1-Th17 cells, as well as NKT and B cell compartments. These could be an important link in understanding how depletion of these cell lineages might be associated with drug efficacy as well as the risk of PML. Future longitudinal studies with larger patient cohorts should allow for teasing out the predictors of drug response and rare incidence of PML.
Supplementary Material
Acknowledgments
Author Disclosures: We would like to acknowledge Lisa Le for her technical assistance for this study.
Drs. Wu, Wang, Dowling and Mao have no funding disclosures to report.
No commercial funding was received to support this work. The study was funded with institutional support from the University of Michigan Medical School.
Dr. Lundy was supported by grants from NIH NIAID: R03-AI105029, R21-AI115117 and Autoimmune Center of Excellence grant: UM1-AI110557; and has received additional research support from the Edward T. and Ellen K. Dryer Foundation, the Merck Corporation and Chugai Pharmaceuticals.
Dr. Mao-Draayer has served as a consultant and/or received grant support from: Acorda, Bayer Pharmaceutical, Biogen Idec, EMD Serono, Genzyme, Novartis, Questor, Chugai Pharmaceuticals, and Teva Neuroscience. Dr. Mao-Draayer is currently supported by grants from NIH NIAID Autoimmune Center of Excellence: UM1-AI110557; NIH NINDS R01-NS080821 and the University of Michigan Neurology Department.
References
- 1.Stadelmann C, Brück W. Interplay between mechanisms of damage and repair in multiple sclerosis. Journal of neurology. 2008;255:12–18. doi: 10.1007/s00415-008-1003-7. [DOI] [PubMed] [Google Scholar]
- 2.Geginat J, Lanzavecchia A, Sallusto F. Proliferation and differentiation potential of human CD8+ memory T-cell subsets in response to antigen or homeostatic cytokines. Blood. 2003;101:4260–4266. doi: 10.1182/blood-2002-11-3577. [DOI] [PubMed] [Google Scholar]
- 3.Teixeira AM, Rama L, Carvalho HM, Borges G, Carvalheiro T, Gleeson M, Alves F, Trindade H, Paiva A. Changes in naive and memory T-cells in elite swimmers during a winter training season. Brain, behavior, and immunity. 2014;39:186–193. doi: 10.1016/j.bbi.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Lyck R, Engelhardt B. Going against the tide–how encephalitogenic T cells breach the blood-brain barrier. Journal of vascular research. 2012;49:497–509. doi: 10.1159/000341232. [DOI] [PubMed] [Google Scholar]
- 5.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 6.Gold R, Kappos L, Arnold DL, Bar-Or A, Giovannoni G, Selmaj K, Tornatore C, Sweetser MT, Yang M, Sheikh SI, Dawson KT. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. The New England journal of medicine. 2012;367:1098–1107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- 7.Fox RJ, Miller DH, Phillips JT, Hutchinson M, Havrdova E, Kita M, Yang M, Raghupathi K, Novas M, Sweetser MT, Viglietta V, Dawson KT. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. The New England journal of medicine. 2012;367:1087–1097. doi: 10.1056/NEJMoa1206328. [DOI] [PubMed] [Google Scholar]
- 8.Kolbach DN, Nieboer C. Fumaric acid therapy in psoriasis: results and side effects of 2 years of treatment. Journal of the American Academy of Dermatology. 1992;27:769–771. doi: 10.1016/s0190-9622(08)80228-9. [DOI] [PubMed] [Google Scholar]
- 9.Altmeyer PJ, Matthes U, Pawlak F, Hoffmann K, Frosch PJ, Ruppert P, Wassilew SW, Horn T, Kreysel HW, Lutz G, et al. Antipsoriatic effect of fumaric acid derivatives. Results of a multicenter double-blind study in 100 patients. Journal of the American Academy of Dermatology. 1994;30:977–981. doi: 10.1016/s0190-9622(94)70121-0. [DOI] [PubMed] [Google Scholar]
- 10.Mrowietz U, Christophers E, Altmeyer P. Treatment of psoriasis with fumaric acid esters: results of a prospective multicentre study. German Multicentre Study. The British journal of dermatology. 1998;138:456–460. doi: 10.1046/j.1365-2133.1998.02124.x. [DOI] [PubMed] [Google Scholar]
- 11.Schimrigk S, Brune N, Hellwig K, Lukas C, Bellenberg B, Rieks M, Hoffmann V, Pohlau D, Przuntek H. Oral fumaric acid esters for the treatment of active multiple sclerosis: an open-label, baseline-controlled pilot study. European journal of neurology. 2006;13:604–610. doi: 10.1111/j.1468-1331.2006.01292.x. [DOI] [PubMed] [Google Scholar]
- 12.Scannevin RH, Chollate S, Jung MY, Shackett M, Patel H, Bista P, Zeng W, Ryan S, Yamamoto M, Lukashev M, Rhodes KJ. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. The Journal of pharmacology and experimental therapeutics. 2012;341:274–284. doi: 10.1124/jpet.111.190132. [DOI] [PubMed] [Google Scholar]
- 13.Wang Q, Chuikov S, Taitano S, Wu Q, Rastogi A, Tuck SJ, Corey JM, Lundy SK, Mao-Draayer Y. Dimethyl fumarate protects neural stem/progenitor cells and neurons from oxidative damage through Nrf2-ERK1/2 MAPK pathway. International journal of molecular sciences. 2015;16:13885–13907. doi: 10.3390/ijms160613885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lundy SK, Wu Q, Wang Q, Dowling CA, Taitano SH, Mao G, Mao-Draayer Y. Dimethyl fumarate treatment of relapsing-remitting multiple sclerosis influences B-cell subsets. Neurology(R) neuroimmunology & neuroinflammation. 2016;3:e211. doi: 10.1212/NXI.0000000000000211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nieuwkamp DJ, Murk JL, van Oosten BW, Cremers CH, Killestein J, Viveen MC, Van Hecke W, Frijlink DW, Wattjes MP. PML in a patient without severe lymphocytopenia receiving dimethyl fumarate. The New England journal of medicine. 2015;372:1474–1476. doi: 10.1056/NEJMc1413724. [DOI] [PubMed] [Google Scholar]
- 16.Rosenkranz T, Novas M, Terborg C. PML in a patient with lymphocytopenia treated with dimethyl fumarate. The New England journal of medicine. 2015;372:1476–1478. doi: 10.1056/NEJMc1415408. [DOI] [PubMed] [Google Scholar]
- 17.Bartsch T, Rempe T, Wrede A, Leypoldt F, Bruck W, Adams O, Rohr A, Jansen O, Wuthrich C, Deuschl G, Koralnik IJ. Progressive neurologic dysfunction in a psoriasis patient treated with dimethyl fumarate. Annals of neurology. 2015;78:501–514. doi: 10.1002/ana.24471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dammeier N, Schubert V, Hauser TK, Bornemann A, Bischof F. Case report of a patient with progressive multifocal leukoencephalopathy under treatment with dimethyl fumarate. BMC neurology. 2015;15:108. doi: 10.1186/s12883-015-0363-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hara T, Jung LK, Bjorndahl JM, Fu SM. Human T cell activation. III. Rapid induction of a phosphorylated 28 kD/32 kD disulfide-linked early activation antigen (EA 1) by 12-o-tetradecanoyl phorbol-13-acetate, mitogens, and antigens. J Exp Med. 1986;164:1988–2005. doi: 10.1084/jem.164.6.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simms PE, Ellis TM. Utility of flow cytometric detection of CD69 expression as a rapid method for determining poly- and oligoclonal lymphocyte activation. Clinical and diagnostic laboratory immunology. 1996;3:301–304. doi: 10.1128/cdli.3.3.301-304.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caruso A, Licenziati S, Corulli M, Canaris AD, De Francesco MA, Fiorentini S, Peroni L, Fallacara F, Dima F, Balsari A, Turano A. Flow cytometric analysis of activation markers on stimulated T cells and their correlation with cell proliferation. Cytometry. 1997;27:71–76. doi: 10.1002/(sici)1097-0320(19970101)27:1<71::aid-cyto9>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 22.Yura M, Takahashi I, Serada M, Koshio T, Nakagami K, Yuki Y, Kiyono H. Role of MOG-stimulated Th1 type “light up” (GFP+) CD4+ T cells for the development of experimental autoimmune encephalomyelitis (EAE) J Autoimmun. 2001;17:17–25. doi: 10.1006/jaut.2001.0520. [DOI] [PubMed] [Google Scholar]
- 23.Hofstetter H, Gold R, Hartung HP. Th17 Cells in MS and Experimental Autoimmune Encephalomyelitis. International MS journal / MS Forum. 2009;16:12–18. [PubMed] [Google Scholar]
- 24.Andalib A, Doulabi H, Maracy MR, Rezaei A, Hasheminia SJ. CCR3, CCR4, CCR5, and CXCR3 expression in peripheral blood CD4+ lymphocytes in gastric cancer patients. Adv Biomed Res. 2013;2:2277–9175. doi: 10.4103/2277-9175.108770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sallusto F, Mackay CR, Lanzavecchia A. Selective expression of the eotaxin receptor CCR3 by human T helper 2 cells. Science. 1997;277:2005–2007. doi: 10.1126/science.277.5334.2005. [DOI] [PubMed] [Google Scholar]
- 26.Venet F, Lepape A, Debard AL, Bienvenu J, Bohe J, Monneret G. The Th2 response as monitored by CRTH2 or CCR3 expression is severely decreased during septic shock. Clinical immunology (Orlando, Fla) 2004;113:278–284. doi: 10.1016/j.clim.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 27.Annunziato F, Cosmi L, Romagnani S. Human and murine Th17. Current opinion in HIV and AIDS. 2010;5:114–119. doi: 10.1097/COH.0b013e32833647c2. [DOI] [PubMed] [Google Scholar]
- 28.Cosmi L, De Palma R, Santarlasci V, Maggi L, Capone M, Frosali F, Rodolico G, Querci V, Abbate G, Angeli R, Berrino L, Fambrini M, Caproni M, Tonelli F, Lazzeri E, Parronchi P, Liotta F, Maggi E, Romagnani S, Annunziato F. Human interleukin 17-producing cells originate from a CD161+CD4+ T cell precursor. J Exp Med. 2008;205:1903–1916. doi: 10.1084/jem.20080397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maggi L, Santarlasci V, Capone M, Peired A, Frosali F, Crome SQ, Querci V, Fambrini M, Liotta F, Levings MK, Maggi E, Cosmi L, Romagnani S, Annunziato F. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. European journal of immunology. 2010;40:2174–2181. doi: 10.1002/eji.200940257. [DOI] [PubMed] [Google Scholar]
- 30.Gross CC, Schulte-Mecklenbeck A, Klinsing S, Posevitz-Fejfár A, Wiendl H, Klotz L. Dimethyl fumarate treatment alters circulating T helper cell subsets in multiple sclerosis. Neurology-Neuroimmunology Neuroinflammation. 2016;3:e183. doi: 10.1212/NXI.0000000000000183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Longbrake EE, Ramsbottom MJ, Cantoni C, Ghezzi L, Cross AH, Piccio L. Dimethyl fumarate selectively reduces memory T cells in multiple sclerosis patients. Multiple sclerosis (Houndmills, Basingstoke, England) 2015 doi: 10.1177/1352458515608961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spencer CM, Crabtree-Hartman EC, Lehmann-Horn K, Cree BA, Zamvil SS. Reduction of CD8+ T lymphocytes in multiple sclerosis patients treated with dimethyl fumarate. Neurology-Neuroimmunology Neuroinflammation. 2015;2:e76. doi: 10.1212/NXI.0000000000000076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsui M, Araya SI, Wang HY, Matsushima K, Saida T. Immunomonitoring measures in relapsing-remitting multiple sclerosis. Journal of neuroimmunology. 2004;148:192–199. doi: 10.1016/j.jneuroim.2003.11.020. [DOI] [PubMed] [Google Scholar]
- 34.Mikulková Z, Praksová P, Stourac P, Bednarik J, Michálek J. Imbalance in T-cell and cytokine profiles in patients with relapsing-remitting multiple sclerosis. Journal of the neurological sciences. 2011;300:135–141. doi: 10.1016/j.jns.2010.08.053. [DOI] [PubMed] [Google Scholar]
- 35.Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29:848–862. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 36.Neuenhahn M, Busch DH. Whole-body anatomy of human T cells. Immunity. 2013;38:10–12. doi: 10.1016/j.immuni.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 37.Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJ, Bickham KL, Lerner H, Goldstein M, Sykes M. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity. 2013;38:187–197. doi: 10.1016/j.immuni.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grigoriadis N, van Pesch V. A basic overview of multiple sclerosis immunopathology. European journal of neurology. 2015;22(2):3–13. doi: 10.1111/ene.12798. [DOI] [PubMed] [Google Scholar]
- 39.Sie C, Korn T, Mitsdoerffer M. Th17 cells in central nervous system autoimmunity. Experimental neurology. 2014;262(Pt A):18–27. doi: 10.1016/j.expneurol.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 40.Poggi A, Canevali P, Contatore M, Ciprandi G. Higher frequencies of CD161+ circulating T lymphocytes in allergic rhinitis patients compared to healthy donors. International archives of allergy and immunology. 2012;158:151–156. doi: 10.1159/000330903. [DOI] [PubMed] [Google Scholar]
- 41.Basdeo SA, Moran B, Cluxton D, Canavan M, McCormick J, Connolly M, Orr C, Mills KH, Veale DJ, Fearon U, Fletcher JM. Polyfunctional, Pathogenic CD161+ Th17 Lineage Cells Are Resistant to Regulatory T Cell-Mediated Suppression in the Context of Autoimmunity. Journal of immunology (Baltimore, Md : 1950) 2015;195:528–540. doi: 10.4049/jimmunol.1402990. [DOI] [PubMed] [Google Scholar]
- 42.Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA, McCauley JL, Abreu MT, Unutmaz D, Sundrud MS. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. The Journal of experimental medicine. 2014;211:89–104. doi: 10.1084/jem.20130301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fergusson JR, Smith KE, Fleming VM, Rajoriya N, Newell EW, Simmons R, Marchi E, Bjorkander S, Kang YH, Swadling L, Kurioka A, Sahgal N, Lockstone H, Baban D, Freeman GJ, Sverremark-Ekstrom E, Davis MM, Davenport MP, Venturi V, Ussher JE, Willberg CB, Klenerman P. CD161 defines a transcriptional and functional phenotype across distinct human T cell lineages. Cell reports. 2014;9:1075–1088. doi: 10.1016/j.celrep.2014.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gonzalez Y, Herrera MT, Juarez E, Salazar-Lezama MA, Bobadilla K, Torres M. CD161 Expression Defines a Th1/Th17 Polyfunctional Subset of Resident Memory T Lymphocytes in Bronchoalveolar Cells. PloS one. 2015;10:e0123591. doi: 10.1371/journal.pone.0123591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moed H, Stoof TJ, Boorsma DM, von Blomberg BM, Gibbs S, Bruynzeel DP, Scheper RJ, Rustemeyer T. Identification of anti-inflammatory drugs according to their capacity to suppress type-1 and type-2 T cell profiles. Clinical and experimental allergy : journal of the British Society for Allergy and Clinical Immunology. 2004;34:1868–1875. doi: 10.1111/j.1365-2222.2004.02124.x. [DOI] [PubMed] [Google Scholar]
- 46.Tahvili S, Zandieh B, Amirghofran Z. The effect of dimethyl fumarate on gene expression and the level of cytokines related to different T helper cell subsets in peripheral blood mononuclear cells of patients with psoriasis. International journal of dermatology. 2015;54:e254–260. doi: 10.1111/ijd.12834. [DOI] [PubMed] [Google Scholar]
- 47.Alt C, Laschinger M, Engelhardt B. Functional expression of the lymphoid chemokines CCL19 (ELC) and CCL 21 (SLC) at the blood-brain barrier suggests their involvement in G-protein-dependent lymphocyte recruitment into the central nervous system during experimental autoimmune encephalomyelitis. European journal of immunology. 2002;32:2133–2144. doi: 10.1002/1521-4141(200208)32:8<2133::AID-IMMU2133>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 48.Giunti D, Borsellino G, Benelli R, Marchese M, Capello E, Valle MT, Pedemonte E, Noonan D, Albini A, Bernardi G, Mancardi GL, Battistini L, Uccelli A. Phenotypic and functional analysis of T cells homing into the CSF of subjects with inflammatory diseases of the CNS. Journal of leukocyte biology. 2003;73:584–590. doi: 10.1189/jlb.1202598. [DOI] [PubMed] [Google Scholar]
- 49.Kihara Y, Groves A, Rivera RR, Chun J. Dimethyl fumarate inhibits integrin alpha4 expression in multiple sclerosis models. Annals of clinical and translational neurology. 2015;2:978–983. doi: 10.1002/acn3.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Longbrake EE, Naismith RT, Parks BJ, Wu GF, Cross AH. Dimethyl fumarate-associated lymphopenia: Risk factors and clinical significance. Multiple sclerosis journal - experimental, translational and clinical. 2015;1 doi: 10.1177/2055217315596994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Treumer F, Zhu K, Glaser R, Mrowietz U. Dimethylfumarate is a potent inducer of apoptosis in human T cells. The Journal of investigative dermatology. 2003;121:1383–1388. doi: 10.1111/j.1523-1747.2003.12605.x. [DOI] [PubMed] [Google Scholar]
- 52.A S, R RK. Role of dimethyl fumarate in oxidative stress of multiple sclerosis: A review. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2016 doi: 10.1016/j.jchromb.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 53.Turley AE, Zagorski JW, Rockwell CE. The Nrf2 activator tBHQ inhibits T cell activation of primary human CD4 T cells. Cytokine. 2015;71:289–295. doi: 10.1016/j.cyto.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boyman O, Letourneau S, Krieg C, Sprent J. Homeostatic proliferation and survival of naive and memory T cells. European journal of immunology. 2009;39:2088–2094. doi: 10.1002/eji.200939444. [DOI] [PubMed] [Google Scholar]
- 55.Tsubaki M, Ogawa N, Takeda T, Sakamoto K, Shimaoka H, Fujita A, Itoh T, Imano M, Satou T, Nishida S. Dimethyl fumarate induces apoptosis of hematopoietic tumor cells via inhibition of NF-kappaB nuclear translocation and down-regulation of Bcl-xL and XIAP. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2014;68:999–1005. doi: 10.1016/j.biopha.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 56.Schulze-Topphoff U, Varrin-Doyer M, Pekarek K, Spencer CM, Shetty A, Sagan SA, Cree BA, Sobel RA, Wipke BT, Steinman L. Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proceedings of the National Academy of Sciences. 2016;113:4777–4782. doi: 10.1073/pnas.1603907113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Faulkner M. Risk of progressive multifocal leukoencephalopathy in patients with multiple sclerosis. Expert opinion on drug safety. 2015;14:1737–1748. doi: 10.1517/14740338.2015.1093620. [DOI] [PubMed] [Google Scholar]
- 58.Bellizzi A, Anzivino E, Rodio DM, Palamara AT, Nencioni L, Pietropaolo V. New insights on human polyomavirus JC and pathogenesis of progressive multifocal leukoencephalopathy. Clinical & developmental immunology. 2013;2013:839719. doi: 10.1155/2013/839719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.