

Abstract
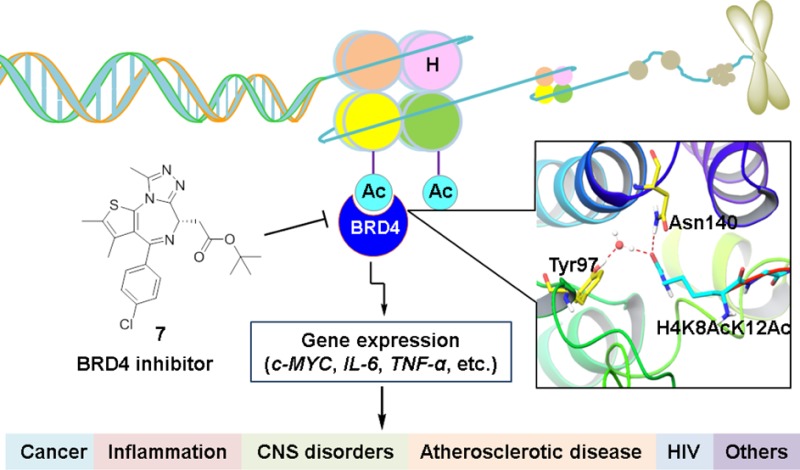
BRD4, the most extensively studied member of the BET family, is an epigenetic regulator that localizes to DNA via binding to acetylated histones and controls the expression of therapeutically important gene regulatory networks through the recruitment of transcription factors to form mediator complexes, phosphorylating RNA polymerase II, and by its intrinsic histone acetyltransferase activity. Disrupting the protein–protein interactions between BRD4 and acetyl-lysine has been shown to effectively block cell proliferation in cancer, cytokine production in acute inflammation, and so forth. To date, significant efforts have been devoted to the development of BRD4 inhibitors, and consequently, a dozen have progressed to human clinical trials. Herein, we summarize the advances in drug discovery and development of BRD4 inhibitors by focusing on their chemotypes, in vitro and in vivo activity, selectivity, relevant mechanisms of action, and therapeutic potential. Opportunities and challenges to achieve selective and efficacious BRD4 inhibitors as a viable therapeutic strategy for human diseases are also highlighted.
1. Introduction
Chemical modifications of DNA (e.g., methylation of cytosine) and the chromosomal DNA-packing histone modifications (e.g., acetylation, methylation, phosphorylation, and ubiquitination) dictate the epigenetic regulation of gene activation and silencing in response to physiological and environmental stimuli.1−3 Histone modification, a covalent posttranslational modification (PTM), has led to a well-established “histone code” hypothesis and an epigenetic mechanism for the regulation of a variety of normal and disease-related processes.4−6 Acetylation of a histone lysine residue7 was historically considered a hallmark of transcriptionally active genes.8 On the one hand, lysine acetylation can neutralize its positive charge leading to reduced affinity of histones for negatively charged DNA or disruption of nucleosome packing and ultimately to an open, accessible chromatin structure that is able to recruit transcriptional machinery.9,10 On the other hand, acetylated lysine provides binding sites for protein recognition modules. The large number (over 24,000) of lysine acetylations in human cells and frequent occurrence indicate that lysine acetylation plays important roles in signal transduction and signaling networks.9 The ε-N-lysine acetylation (KAc) motifs can be recognized by bromodomains11 as well as double plant homeodomain (PHD) fingers12,13 and pleckstrin homology domains.14 Meanwhile, bromodomains exclusively recognize acetylation motifs. The bromodomain-containing proteins (BCPs) can thus act as KAc “readers” of modified histones mediating signaling transduction to changes in gene regulatory networks.15 These readers result in epigenetic modification of target genes through intrinsic histone acetyl transferase or kinase activity or by their ability to serve as scaffolds for assembly of chromatin-modifying enzymes. In the human genome, there are 61 bromodomains found within 46 proteins that can be divided into eight families based on structure/sequence similarity (Figure 1). Among them, bromodomain and extra-terminal domain (BET) family (highlighted in green, Figure 1) bromodomains recognize acetylated lysine residues in histones H3 and H4.16,17 Disrupting the protein–protein interactions between BET protein and acetylated lysine represents a promising target for human diseases including cancer and inflammation.18−20 BET family members have recently attracted increasing attention in drug discovery and are considered to be the most druggable target proteins among BCPs for regulating cellular epigenetics.21
Figure 1.
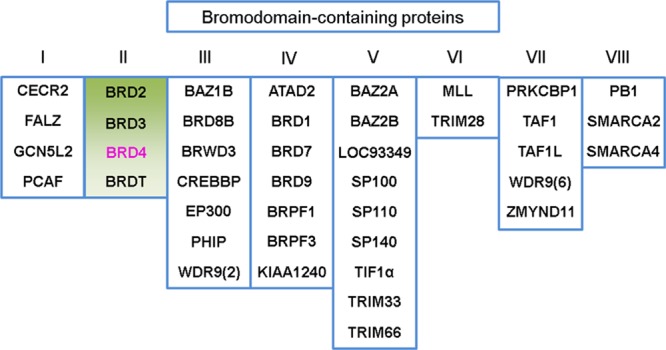
Eight families of bromodomain-containing proteins. Bromodomain-containing protein 4 (BRD4, highlighted in magenta) belongs to the BET family (green).
The BET family is composed of bromodomain-containing protein 2 (BRD2), BRD3, BRD4, and bromodomain testis-specific protein (BRDT). They share two N-terminal bromodomains and an extra C-terminal domain (ET) exhibiting high levels of sequence conservation.22 Normally, BET family proteins all localize in the nucleus23 and recruit transcriptional regulatory complexes to acetylated chromatin, thus being implicated in a number of DNA-centered processes including regulation of gene expression. Unlike other BCPs, which are typically displaced from condensed chromosomes during mitosis, BET proteins are able to associate with mitotic chromosomes.17 BRD2, BRD3, and BRD4 are ubiquitously expressed, whereas BRDT is a tissue-specific isoform expressed in pachytene spermatocytes, diplotene spermatocytes, and round spermatids.24,25 Although their bromodomains share highly similar sequences of amino acids, BET family members recruit different partners. BRD4 recruits positive transcriptional elongation factor complex (P-TEFb), which plays an essential role in the regulation of transcription by RNA polymerase II (RNA Pol II) in eukaryotes.26 In addition, the BRD4 ET domain recruits transcription-modifying factors (e.g., NSD3, a SET domain-containing histone methyltransferase) independently.27 BRD3 was reported to function in the recruitment of hematopoietic transcription factor GATA1 by regulating maturation of erythroid, megakaryocyte, and mast cell lineages.28,29 BRD2 exerts its function via the E2F transcription factor/retinoblastoma pathway in a P-TEFb-independent manner.30 Under normal conditions, BRD2/3/4 perform transcription regulatory functions, and their dysfunction plays critical roles in a variety of human diseases.31 Over the past decade, the number of small-molecule BET inhibitors has expanded dramatically. These inhibitors act as useful chemical probes for exploring the biological functions of BET proteins. Some of them (chemical structures shown in Figure 2) have been enrolled into different phases of human clinical trials (Table 1), including RVX-208/Apabetalone (1),32 I-BET762/GSK-525762A (2),33 OTX-015/MK8628 (3),34 CPI-0610 (4),35 TEN-010 (5),36 and ABBV-075 (6).37 Most of their clinical investigations are focused on cancer therapies.
Figure 2.

Chemical structures of selected BET inhibitors that are currently being evaluated in clinical trials.
Table 1. BET Inhibitors in Clinical Trialsa.
| drug | sponsor | phase | indications | NCT identifier |
|---|---|---|---|---|
| 1(32) | Resverlogix Corp | III | T2DM; CAD; CVDs | NCT02586155 |
| II | diabetes | NCT01728467 | ||
| II | atherosclerosis; CAD | NCT01058018 | ||
| II | CAD; dyslipidemia | NCT01423188 | ||
| II | CAD | NCT01067820 | ||
| I/II | dyslipidemia; atherosclerosis; ACS; CVDs | NCT00768274 | ||
| 2(33) | GlaxoSmithKline | II | ER+ breast cancer | NCT02964507 |
| I | RRHMs | NCT01943851 | ||
| I | NMC and other cancers | NCT01587703 | ||
| I | drug interactions | NCT02706535 | ||
| 3(34) | OncoEthix GmbH | I | AML; DLBCL; ALL; MM | NCT01713582 |
| I | NMC; TNBC; NSCLC; CRPC; PDAC | NCT02259114 | ||
| Merck Sharp and Dohme Corp. | I | AML; DLBCL | NCT02698189 | |
| I | NMC; TNBC; NSCLC; CRPC | NCT02698176 | ||
| 4(35) | Constellation | I | lymphoma | NCT01949883 |
| I | MM | NCT02157636 | ||
| I | AML; MDS; MDS/MPN, U; myelofibrosis | NCT02158858 | ||
| 5(36) | Hoffmann-La Roche | I | solid tumors; AST | NCT01987362 |
| I | MDS; AML | NCT02308761 | ||
| 6(37) | AbbVie | I | advanced cancer; breast cancer; NSCLC; AML; MM | NCT02391480 |
| BI 89499940,b | Boehringer Ingelheim | I | neoplasms | NCT02516553 |
| BMS-98615841,b | Bristol-Myers Squibb | I/II | AST | NCT02419417 |
| FT-110142,b | Forma Therapeutics | I | AML; MDS | NCT02543879 |
| INCB05432943,b | Incyte | I/II | advanced malignancies | NCT02431260 |
| GSK282015144,b | GlaxoSmithKline | I | AST; RST | NCT02630251 |
| ZEN-369445,b | Zenith Epigenetics | I | metastatic CRPC | NCT02711956/NCT02705469 |
| GS-582946,b | Gilead Sciences | I/II | metastatic CRPC | NCT02607228 |
| I | solid tumors; lymphomas | NCT02392611 | ||
| N-methyl-2-pyrrolidone (NMP)47 | Peter MacCallum Cancer Centre | I | RRMM | NCT02468687 |
Data collected from www.clinicaltrials.com on Nov 19, 2016. Abbreviations: ACS, acute coronary syndrome; ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; AST, advanced solid tumors; CAD, coronary artery disease; CRPC, castration-resistant prostate cancer; CVDs, cardiovascular diseases; DLBCL, diffuse large B-cell lymphoma; ER+ breast cancer, estrogen receptor-positive breast cancer; MDS, myelodysplastic syndrome; MM, multiple myeloma; MDS/MPN, U, myelodysplastic/myeloproliferative neoplasm, unclassifiable; NMC, NUT midline carcinoma; NMP, N-methyl-2-pyrrolidone; NSCLC, nonsmall cell lung cancer; PDAC, pancreatic ductal adenocarcinoma; RRHMs, relapsed refractory hematologic malignancies; RRMM, relapsed refractory multiple myeloma; RST, recurrent solid tumors; T2DM, type 2 diabetes mellitus; TNBC, triple negative breast cancer.
Structures are not disclosed.
As the most extensively characterized BET protein, BRD4 has been implicated in a number of human diseases including cancer, inflammation, cardiovascular diseases, central nervous system (CNS) disorders, and human immunodeficiency virus (HIV) infection.38,39 BRD4 represents a promising therapeutic target for various diseases, and targeting BRD4 has attracted significant interest in both pharmaceutical and academic settings. Herein, we mainly focus on summarizing the advances in the drug discovery and development of BRD4 inhibitors, and opportunities and challenges associated with this field are also discussed.
2. BRD4 as a Novel Therapeutic Target: Structures and Disease-Associated Functions
2.1. Structures of BRD4 Bromodomains
BRD4, originally named mitotic chromosomal-associated protein (MCAP) and also called Fshrg4 or Hunk1, was identified in 1988 from studies on the mammalian mediator complex, a multiprotein coactivator that links transcription factors to RNA Pol II activation.48 It has three isoforms of different length: one long isoform (1362 residues) and two shorter forms (722 and 796 residues, respectively).49 BRD4 contains two highly conserved N-terminal bromodomains (BD1 and BD2), an ET domain, and a C-terminal domain (CTD) (Figure 3a). BRD4 BD1 and BRD4 BD2 interact with acetylated chromatin as well as nonhistone proteins to regulate transcription, DNA replication, cell cycle progression, and other cellular activities.50 Despite their sequence similarity (Figure 3b), BD1 and BD2 appear to have distinct functions due to their interactions with different lysine-acetylated histones (e.g., H3 and H4) or with transcriptional proteins. BRD4 BD1 binds to the diacetylated H4K5AcK8Ac mark to anchor its associated proteins to the target gene promoter and enhancer sites in chromatin. BRD4 BD2 does not interact with singly acetylated H3K4ac but displays a strong interaction with diacetylated H3K4AcK9Ac.51 In addition, BRD4 BD2 is associated with the recruitment of nonhistone proteins (e.g., Twist).52 Each BD of BRD4 is composed of a left-handed bundle of four helices (αZ, αA, αB, and αC) linked by the interhelical ZA loop and BC loop, which constitute the active acetyl-lysine binding pocket (Figure 3c).51 The specific residues (e.g., Gln85 of BD1 vs Lys383 of BD2, and Asp144 of BD1 vs His442 of BD2; highlighted by different colors in Figure 3b) within the loops of each BD contribute to determining the acetyl-lysine binding specificity. The cocrystal structure of BRD4 BD1 with histone H4K8Ac12Ac (Figure 3c) indicates that the acetyl-lysine is recognized by a central hydrophobic cavity and anchored by a hydrogen bond with asparagine residue 140 (Asn140).53 Additionally, a second interaction is formed between the acetyl carbonyl oxygen atom and the conserved Tyr97 via a water molecule. Most BRD4 inhibitors block the interactions between BRD4 and acetyl-lysine by mimicking acetyl-lysine and competing with it to bind BRD4. They all have a unique head moiety that can form critical hydrogen bonds with Asn140 and Tyr 97 like the O atom of the acetyl group does. In addition, a small hydrophobic group is usually attached to the moiety that can mimic the methyl group of acetyl to occupy the base of the pocket. Meanwhile, their interactions with “WPF shelf” (W81, P82, F83), a hydrophobic region of the BC loop that includes conserved Trp/Pro/Phe motif present in all of the BET family bromodomains, are also important for BRD4 binding affinities.54
Figure 3.
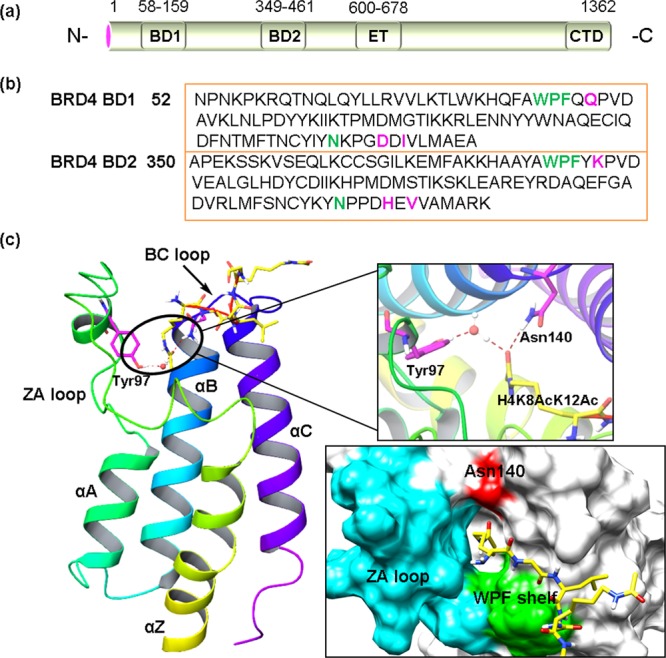
(a) Domain organization of the long form of BRD4. (b) Conserved sequences of BRD4 BD1 and BRD4 BD2. The important critical residues that may contribute to the acetyl-lysine binding specificity are highlighted in different colors. (c) (left panel) Ribbon representation of BRD4 BD1 (PDB ID: 3UW9). (right panel) Surface representation of the KAc binding pocket of BRD4 BD1. Asn140 is highlighted in red, and the WPF shelf is highlighted in green.
2.2. Disease-Related Functions of BRD4
BRD4 plays an important role in cell cycle control of normal mammalian cells, affecting cellular processes including cell proliferation, apoptosis, and transcription.55,56 Microinjection of a BRD4-specific antibody into HeLa cell nuclei can lead to cell cycle arrest, indicating that BRD4 is required for the G2-M phase transition.57 Moreover, BRD4 is essential for the expression of Aurora B kinase, which is responsible for chromosome separation and cytokinesis during mitosis.58 BRD4 recruits transcriptional regulatory complexes to chromatin through various protein–protein interactions (e.g., acetylated histones, transcriptional factors, mediators, P-TEFb) mediated by its bromodomains as well as ET and CTD domains. BET inhibition exhibits effective therapeutic activity in a number of different pathologies, especially in models of cancer and inflammation (Figure 4). Meanwhile, pharmacological BET inhibition also influences other organ systems such as cardiovascular and the CNS.59,60
Figure 4.
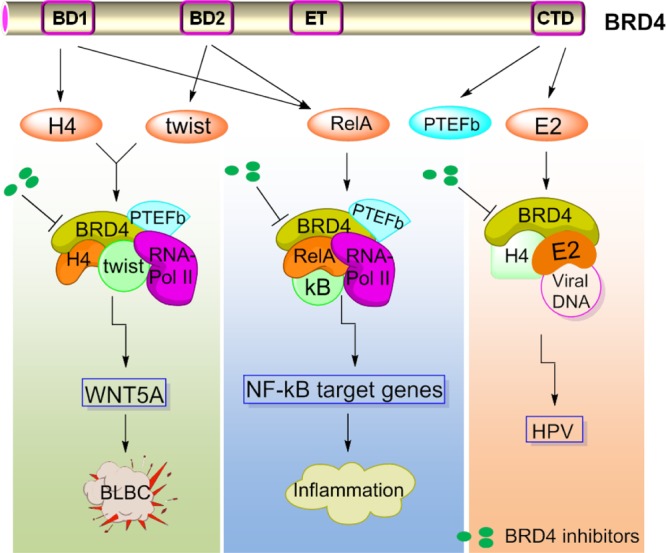
Proposed action modes of BRD4 in selected diseases such as cancer, inflammation, and HPV. (left panel) Interaction of Twist and BRD4 at the enhancer/promoter of WNT5A, leading to the transcriptional activation of WNT5A expression in BLBC. (center panel) Interaction of RelA and BRD4, facilitating the transcription of NF-κB-dependent inflammatory genes. (right panel) BRD4 serves as the receptor of the E2/viral DNA complex on mitotic chromosomes.
Through interactions with cyclin T1 and CDK9 (a validated CLL target),61 BRD4 recruits P-TEFb62,63 to mitotic chromosomes resulting in increased expression of growth-promoting genes.64 Chromosomal translocation of BRD4 to the nuclear protein in the testis (NUT) locus generates a BRD4-NUT fusion protein that results in c-MYC overexpression and NUT midline carcinoma (NMC), an aggressive squamous cell malignancy unresponsive to conventional chemotherapeutics.65 BET inhibition downregulates MYC transcription and subsequent genome-wide MYC-dependent target genes.66 Given the widespread pathogenetic role of MYC in cancers, pharmacological inhibition of MYC through the BET bromodomain holds great promise for the treatment of cancer.67 BRD4 inhibitor (+)-JQ1 (7, Figure 5) is highly efficacious against NMC tumor growth in xenografted mice.68 BRD4 can also physically interact with androgen receptor (AR), and disruption of this interaction by a BET inhibitor can abrogate BRD4 localization to AR target loci and AR-mediated gene transcription. Interestingly, BET inhibition was found to be more efficacious in tumor reduction of CRPC in xenograft mouse models than direct AR antagonism.69 Moreover, the diacetylated Twist protein binds the second domain of BRD4 and recruits the associated P-TEFb/RNA-Pol II to the WNT5A super enhancer to directly activate WNT5A expression, which is required for invasion and maintenance of cancer stem cell-like properties of basal-like breast cancer (BLBC) (Figure 4, left panel).52 Furthermore, BRD4 is amplified and overexpressed in a substantial subset of melanoma specimens and cell lines.70 Treatment with compound 7 attenuates melanoma proliferation in vitro and impairs melanoma tumor growth in vivo, effects that can be mostly recapitulated by individual silencing of BRD4. RNAi screens have also identified BRD4 as a therapeutic target in acute myeloid leukemia (AML) and ovarian carcinoma.71,72 BRD4 is reported to play important roles in various other types of cancer proliferation, such as the activated B-cell-like subtype (ABC) of diffuse large B-cell lymphoma (DLBCL),73 neuroblastoma,74 and lung adenocarcinoma.66,75
Figure 5.
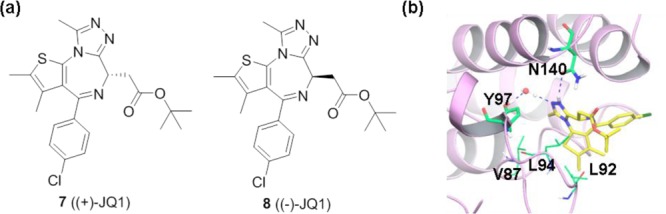
(a) Chemical structures of compounds 7 and 8. (b) Cocrystal structure of 7 with BRD4 BD1 (PDB ID: 3MXF). Residues Asn140 (N140), Tyr97 (Y97), Leu94 (L94), Val87 (V87), and Leu92 (L92) are highlighted.
BRD4 was found to be required for transcriptional coactivation of NF-κB, regulating the transcription of P-TEFb-dependent proinflammatory target genes. Specific binding of BRD4 with acetylated lysine-310 of RelA is proposed as a mechanism for the recruitment of NF-κB (Figure 4, center panel).76,77 BRD4 is highly enriched at enhancers associated with genes involved in multiple profibrotic pathways, where BRD4 is colocalized with profibrotic transcription factors. BRD4 inhibitors can not only abrogate cytokine-induced activation of hepatic stellate cells but also reverse the fibrotic response in carbon tetrachloride-induced fibrosis in mouse models.78 BRD4 inhibition can also attenuate experimental lung fibrosis induced by repetitive TGF-β challenge in a mouse model via the NF-κB/RelA signaling pathway.79 As a synthetic histone mimic, compound 2 was found to suppress inflammation in an LPS-induced C57BL mouse model, suggesting that targeting inflammatory gene expression by interfering with the recognition of acetylated histones by BET inhibitors is a new approach for treating inflammatory conditions.80 siRNA knockdown of BRD4 can induce upregulation of apolipoprotein A (ApoA1), which protects from atherosclerosis progression and other inflammatory processes.54 BRD4 is essential for IL-1β-induced inflammation in human airway epithelial cells, and BRD4 knockdown markedly reduces IL-6 and CXCL8 release, suggesting that BRD4 is a promising target for chronic obstructive pulmonary disease.81 Pharmacological BRD4 inhibition can attenuate the enhanced migration, proliferation, and IL-6 release in lung fibroblasts from patients with rapidly progressing idiopathic pulmonary fibrosis. For example, compound 7 suppresses bleomycin-induced lung fibrosis in mice, indicating that BRD4 inhibitors may hold great promise for the treatment of rapidly progressing idiopathic pulmonary fibrosis.82,83
BET family proteins play a central role in gene control during heart failure pathogenesis. BET inhibition blocks cardiomyocyte hypertrophy in vitro and suppresses pathologic cardiac remodeling in vivo.59 Compound 1 increases ApoA1 and HDL-C in vitro and in vivo, which are potential therapeutic targets for reducing atherosclerotic disease.84,85 In addition, BET family expression is increased during cardiac hypertrophy, which is an independent predictor of adverse outcomes in patients with heart failure. Compound 7 can block agonist-dependent hypertrophy of cultured neonatal rat ventricular myocytes and reverse the prototypical gene program associated with pathological cardiac hypertrophy.86
In the CNS, BRD4 may mediate the transcriptional regulation underlying learning and memory, and the loss of BRD4 function affects critical synaptic proteins, leading to memory deficits in mice but also decreased seizure susceptibility.60 This study suggests that, on one hand, BRD4 inhibitors that can cross the blood-brain barrier (BBB) may pose a risk for neurological side effects, whereas on the other hand, they may possess therapeutic potential for epilepsy. BRD4 is significantly elevated in the nucleus accumbens (NAc) of mice and rats following repeated cocaine injections and self-administration.87 Moreover, BRD4 inhibition by BET inhibitors attenuates transcriptional and behavioral responses to cocaine. Repeated cocaine injections enhance the binding of BRD4 to the promoter region of Bdnf in the NAc, whereas treatment with BRD4 inhibitors or siRNA-mediated knockdown of BRD4 reduces the expression of Bdnf. These findings indicate that BRD4 is a possible therapeutic target for the treatment of drug addiction. Interestingly, selective inhibition of the first domain of BET protein accelerates the progression of mouse primary oligodendrocyte progenitors toward differentiation, whereas blocking both bromodomains hinders differentiation.88 This suggests that selective modulation of BET bromodomains may enhance regenerative strategies in disorders characterized by myelin loss such as aging and neurodegeneration.
Knockdown experiments have implicated BRD4 in the transcriptional regulation of viruses such as HIV89 and Epstein–Barr virus (EBV)90 as well as degradation of human papilloma virus (HPV).91,92 The bovine papillomavirus E2 protein binding to the CTD of BRD4 tethers the viral DNA to host mitotic chromosomes (Figure 4, right panel).93 Disrupting the E2/BRD4 interaction can inhibit viral transformation, providing a new target for the treatment or prevention of HPV infections and related diseases. Bromodomains are linked to diverse aspects of the HIV life cycle, including transcription and integration.94 Binding of the BRD4 CTD domain with P-TEFb disrupts the interaction between the HIV transactivator Tat and P-TEFb, thereby suppressing the ability of Tat to transactivate the HIV promoter.95 As a negative regulator of HIV-1 replication, BRD4 was found to increase proviral transcriptional elongation and alleviate HIV-1 latency in cell-line models. Both compounds 3 and 7 were reported to reactivate latent HIV, suggesting their potential for eliminating latent HIV-1 reservoirs, which is a major hurdle to a complete cure for AIDS.96−101
3. Discovery and Development of BRD4 Inhibitors
Given the extensive disease-related functions of BRD4 and proof-of-concept of disrupting the BRD4–acetyl-lysine interactions as a therapeutic target, significant efforts have thus been made to develop BRD4 inhibitors from both pharmaceutical and academic settings. Various discovery strategies have been utilized including high-throughput screening (HTS), midthroughput screening (MTS), virtual screening (VS), fragment-based drug design (FBDD), and structure-based drug design (SBDD) as well as drug repurposing. On the basis of their core scaffolds, the reported BRD4 inhibitors are chemically classified into several series including azepines, 3,5-dimethylisoxazoles, pyridones, triazolopyrazines, tetrahydroquinolines (THQs), 4-acyl pyrroles, 2-thiazolidinones, and so forth and are discussed below. These compounds show inhibitory effects on BRD4 either in vitro and/or in vivo with selectivity reported at various levels. Given the similarity of bromodomains across over 40 diverse proteins, the selectivity of BRD4 inhibitors can be classified into three categories. The first category is to selectively inhibit BET family proteins over other BCP families. The second category of selectivity is to target BD1 or BD2 of BET proteins. The third category is to specifically inhibit BRD4 BD1 or BRD4 BD2.
3.1. Pan-BET BRD4 Inhibitors
3.1.1. Azepines
Compound 7 (Figure 5a) is one of the first reported BET inhibitors and has now become a widely used chemical probe for exploring the mechanisms and functions of BRD4. Its design is based on molecular modeling and the observation that thienodiazepines possess binding activity for BRD4.68,102 Thienodiazepines (reported by Mitsubishi Tanabe) were originally identified as anti-inflammatory or antiadhesion molecules from phenotypic efforts without prior knowledge of the target.103 In a binding assay based on differential scanning fluorimetry, binding of 7 significantly increases the thermal stability of all bromodomains of BET family with ΔTmobs values between 4.2 °C (BRDT BD1) and 10 °C (BRD4 BD1), whereas no obvious stability shifts are detected for bromodomains outside the BET family. Its stereoisomer (−)-JQ1 (8, Figure 5a) displays no significant interactions with any bromodomains.68 Compound 7 is competitive with a tetra-acetylated histone H4 peptide with IC50 values of 77 nM for BRD4 BD1 and 33 nM for BRD4 BD2. The cocrystal structure of 7 with BRD4 BD1 (Figure 5b) reveals that it binds directly to the KAc binding site and shows extraordinary shape complementarity. One N atom of the triazole moiety mimics the O atom of the acetyl group to form a critical hydrogen bond with Asn140, and the methyl group attached to triazole acts exactly like the methyl group of acetyl-lysine. Ligand binding is stabilized by hydrophobic interactions with conserved residues in the ZA and BC loop (e.g., Leu 92, Leu 94, Val 87, and Tyr 97). Docking of 8 to BRD4 BD1 results in an energetically unfavorable conformation due to steric clashes with Leu92 and Leu94.68 Compound 7 displaces BRD4 from nuclear chromatin and results in differentiation and growth arrest of NMC cells. In vivo proof-of-concept for targeting BRD4 with compound 7 was also established via several xenograft models of NMC including clinically relevant disease models in mice. Although compound 7 is not being evaluated in human clinical trials due to a short half-life, it is widely used as a chemical probe in laboratory applications.104−106 Furthermore, it provides a good lead compound for the subsequent optimization work on azepines.
GlaxoSmithKline identified benzodiazepine (BZD) GW841819 (9, Figure 6) as a potent ApoA1 regulator via a stable human HepG2 hepatocyte cell line containing an ApoA1 luciferase reporter with an EC50 value of 440 nM.80 The BZD core was essential for the activity, and the phenyl group extending from the 6-position on the BZD ring was common to all active analogues. For its direct target to be identified, panels of kinases, ion channels, nuclear receptors, GPCRs, and other drug target proteins were screened using the luciferase assay but failed. Finally, a chemoproteomic approach107,108 was employed and has confirmed that BET proteins are its target and are responsible for the ApoA1 upregulation.54 With an IC50 value of 501 nM for BRD4, compound 9 was considered as a good starting point for medicinal chemistry optimization. Substituents at the meta or para position were introduced (e.g., compound 10(33)) to solve the issue of selectivity for BRDs over GABA receptor, which is usually the target of compounds containing a 1,4-benzodiazepine motif. The benzyl group on carbamate was replaced by alkyl (e.g., compound 11(33)) to reduce both molecular weight (MW) and lipophilicity (cLogP) to a range more desirable for an oral drug (MW < 400 and cLogP < 3). Similar to compound 7, only an enantiomer of 11 was found active. Moreover, a nitrogen atom (colored blue) at the side chain of the BZD ring was removed due to the acidic instability of 11 (t1/2 = 0.23 h at pH 2). All the favored modifications were incorporated into one molecule and led to 2, which showed similar potency to that of 9 but was more suitable for in vivo experiments because of improved physicochemical and pharmacokinetic properties. It has high passive permeability (167 nm/s), excellent solubility in all vehicles (>3 mg/mL), good tissue distributions (6.5 L/kg in mouse), and nice oral bioavailability (44–61% in mouse, dog, and primate).33,109 In vivo studies on 2 have demonstrated significant efficacy in various oncology and immunoinflammatory models.66,67,80 Currently, compound 2 is under Phase I/II clinical trials for the treatment of different cancers.
Figure 6.

Discovery and development of BRD4 inhibitor 2. PBMC: peripheral blood mononuclear cells; BZDR: the central GABA receptor; t1/2, determined by an in vitro assay; T1/2, performed in vivo.
Inspired by the similar crystallographic binding modes of amino-isoxazole fragment 12 and compound 7, Albrecht et al. developed potent BRD4 inhibitor 13 by incorporating the isoxazole motif into an azepine scaffold (Figure 7).110 Compound 13 showed potent inhibitory activity against BRD4 BD1 (IC50 = 26 nM) and significant suppression of MYC expression in Raji cells (IC50 = 140 nM). With the suitable PK profiles in rats, compound 13 was evaluated in the MYC PD model at doses of 10, 30, and 100 mg/kg. A dose-dependent decrease of MYC mRNA expression was observed in vivo after PO dosing. Replacement of the thiophene ring in 13, which has the potential issue of metabolic instability, with a phenyl ring led to compound 4 (Figure 7),111 which is currently undergoing evaluation in multiple Phase I clinical trials. Compound 4 was approximately 6-fold more potent against BET BD2 (BRD4 BD2 IC50 = 18 nM) than against BET BD1 (BRD4 BD2 IC50 = 120 nM). In an MV4-11 (an AML cell line) mouse model, compound 4 was orally dosed as a single agent at 30 mg/kg daily, 30 mg/kg twice daily, or 60 mg/kg once daily. Substantial suppression of tumor growth was observed (tumor growth inhibition (TGI) of 41, 80, and 74%, respectively) with no obvious body weight loss. Preliminary clinical results illustrated that dosing above 230 mg causes substantial modulation of biomarkers linked to BET and other clinically meaningful effects. Antitumor effects have also been observed following the use of 4 for treatment of patients with heavily pretreated DLBCL and follicular lymphoma.111 Various heteroaromatic rings were introduced to the 8-position of 4-(R)-methyl benzoisoxazoleazepine chemotype, which exhibited modest potency in both biochemical and cellular assays. Compound 14 (Figure 7), with an acetamide substituted pyrazole, showed potent BRD4 BD1 inhibition (IC50 = 17 nM) and significant suppression of MYC expression in MV4-11 cells (IC50 = 32 nM).112 Compared to 13, compound 14 has a longer half-life and 3-fold greater bioavailability. It was dosed subcutaneously at 5 and 15 mg/kg twice a day in a MV4-11 tumor xenografts Balb/c nude mouse model, resulting in 50 and 70% reductions in the MYC mRNA level 8 h after the second dose. Substituents on the 8-position were also explored for benzotriazolodiazepine leading to compound 15 (Figure 7).113 Compound 15, with an aminopyridine at the 8-position, displayed a potent BRD4 BD1 inhibitory activity (IC50 = 15 nM) and significant suppression of IL-6 from THP-1 cells (IC50 = 13 nM). The cocrystal structure of 15 with BRD4 BD1 (Figure 7) illustrated that it adopted a cupped shape, filling the space between Asn140 and the WPF shelf and making hydrophobic contacts around gatekeeper residue Ile146. An edge-to-face interaction between aminopyridine and Trp81 explained its increased potency. An in vivo study of compound 15 demonstrated dose- and time-dependent suppression of plasma IL-6 in mice.
Figure 7.

Development of isoxazole azepine analogues as BRD4 inhibitors and complex of 15 (colored magenta) with BRD4 BD1 (PDB ID: 4Z1Q). VSS, volume of distribution at steady state.
Other selected examples of triazoloazepines as BET BRD4 inhibitors are listed in Figure 8. Among them, compounds 16 and 17 from patent WO2016069578 with side chains at the 4-position displayed potent BRD4 BD1 inhibitory activities with IC50 values of 1.3 and 0.43 nM, respectively.114 However, no additional information about selectivity or in vivo efficacy was provided. Benzotriazepine 18,115 thienodiazepine 19,116 and pyrrolodiazepine 20(117) showed promising BRD4 inhibitory activities with IC50 values of 640, 34, and 30 nM, respectively.
Figure 8.
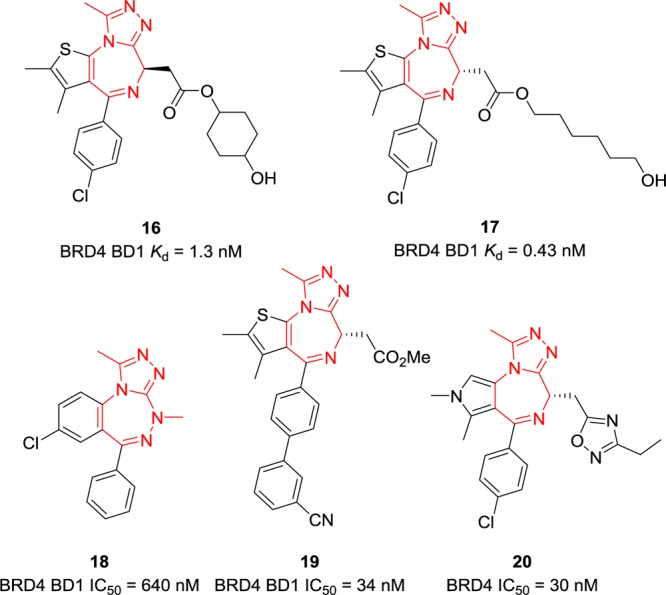
Chemical structures of compounds 16–20.
A number of pyridoazepine derivatives (Figure 9) as potent BET BRD4 inhibitors were reported in patents while having not been published in peer-reviewed journal articles. Both compounds 21 and 22 exhibited single-digit nanomolar BRD4 BD1 activity and potent antiproliferation activity in MV4-11 cell lines with EC50 values of 2 and 13 nM, respectively.118 Compound 23 displayed similar in vitro activity,118 indicating that replacement of the phenyl ring with an aliphatic ring on the 6-position is tolerable. Likely, N-methylpyridin-2(1H)-one is the moiety that can mimic the acetyl group to form the critical hydrogen bonds. Similar to 21–23, compounds 24 and 25 with additional pyrrole rings also showed single-digit nanomolar BRD4 binding activity.119 In an OPM-2 human multiple myeloma cancer xenograft model, compound 24 displayed a TGI of 81% administrated orally at the dose of 5 mg/kg, but 44% of mice were removed due to morbidity or weight loss in excess of 20%. At the dose of 2.5 mg/kg, no mice were removed, but the TGI was decreased to 66%. Compound 25 had a TGI of 79% at the dose of 2.5 mg/kg in the same model, and no mice were removed, indicating a better efficacy and a safer profile. Interestingly, compound 26 without triazoles or isoxazoles retained micromolar BET BRD4 inhibitory activities.120 Compounds 27–29 with novel bicyclic aliphatic rings can also suppress the proliferation of the MV4-11 cell line with IC50 values of less than 100 nM.121
Figure 9.

Chemical structures of compounds 21–29.
Another well-developed compound of this azepine series of BET BRD4 inhibitors is compound 3 (Figure 2). It inhibits binding of BRD2/3/4 to acetylated H4 in a concentration-dependent manner with IC50 values ranging from 92 to 112 nM. Its exposure leads to cell growth inhibition, cell cycle arrest, and apoptosis at submicromolar concentrations in acute leukemia cells and patient-derived leukemic cells.34 Compound 3 is now under a Phase I clinical trial in relapsed/refractory leukemia patients.
3.1.2. 3,5-Dimethyl Isoxazoles
Inspired by the affinity of the solvent N-methylpyrrolidinone (NMP, 30) with bromodomains (e.g., for CREBBP, IC50 = 2.3 mM), Heightman et al. performed subsequent studies on methyl-bearing heterocycles such as dihydroquinazolinone-containing compound 31 (Figure 10).122 The X-ray crystal structure of BRD4 BD1 with 31 was solved, but additional unexplained electron density at the 4-position of the dihydroquinazolinone was observed. Its real structure was interpreted as compound 32, which has a substituent of the ethylene glycol unit arising from oxidation at this position.122 The ethylene glycol unit occupies a hydrophobic groove (formed by Trp81 and Pro82 from the ZA loop and Il46 and Met149 from helix C), which is believed to contribute to the selectivity for BET proteins over other bromodomains. The following modifications are focused on the mono- and dimeta-substituted 3,5-dimethyl-4-phenylisoxazole scaffolds to avoid ready oxidation and retain interactions with that important groove. Compound 33 stood out with IC50 values of 4.8 and 1.6 μM against BRD4 and BRD2, respectively.122 The cocrystal structure of compound 33 with BRD4 BD1 reveals that the dimethylisoxazole moiety acts as a KAc mimic and occupies the KAc binding pocket. The isoxazole oxygen atom of 33 forms the expected hydrogen bond with Asn140, and the nitrogen atom interacts with the phenol group of Tyr97 via a water molecule. The methyl group bound in the WPF shelf was replaced with larger substituents such as aromatic rings leading to compounds 34 and 35 (Figure 10).123 The (R)- and (S)-enantiomers of 34 display similar BRD4 inhibitory activities and identical binding modes. Both 34 and 35 have significant enhancement in BRD4 binding affinity compared to that of 33 with IC50 values of 370–390 nM. Compound 35, with an additional acetyl group, has a 7-fold selectivity over CREBBP, and the acetate carbonyl group was predicted to form a hydrogen bond with Gln85. Additionally, compounds 34 and 35 exhibit cytotoxicity in MV4-11 cells with IC50 values of 794 and 616 nM but no appreciable cytotoxicity (IC50 > 100 μM) in HeLa or U2OS cells, suggesting the effects result mainly from inhibition of the BET proteins.
Figure 10.

Discovery and development of BRD4 inhibitors 33–35.
Compound 36 (Figure 11), developed by GSK via FBDD, has an IC50 value of 500 nM against BRD4 in a TR-FRET assay, but its solubility is very poor (<1 μg/mL).124 Introduction of polar groups to the phenyl ring para to the isoxazole of 36 led to compound 37, which had substantially improved solubility (1125 μg/mL at pH 5).124 Compound 38 (Figure 11) containing an imidazo[1,2-a]pyrazine scaffold binds BRD4 with a Kd value of 550 nM and shows good cellular effects (IC50 = 724 nM) in BRD4-dependent lines.125
Figure 11.

Chemical structures of compounds 36–38.
GW694481 (39, Figure 12) was identified as a potent ApoA1 upregulator (EC170 = 500 nM; EC170 was defined as the concentration of compound resulting in a 70% increase in the luciferase activity) by an HTS approach using a transcriptional cellular assay.126 Optimizations on the 3- and 4-positions led to compound 40,126,127 which was potent in ApoA1 upregulation with an EC170 value of 200 nM and in BET inhibition with an IC50 value of 750 nM against BRD4. However, its cytochrome P450 (CYP450) inhibition was in the low micromolar range. To address this issue, CONH2 was eliminated, and the intramolecular hydrogen bond between C3 and C4 was frozen through cyclization leading to a series of imidazolone analogues. Among them, I-BET151 (41) displayed the most promising potency and properties.126 Compound 41 showed an acceptable CYP450 profile with no observable time-dependent inhibition of CYP2D6 or CYP3A4, nonmutagenicity, significantly reduced PDE4 liability, and good bioavailability (65%) in rats. Additionally, it demonstrated a broad anti-inflammatory profile in an LPS-challenged Balb/C mouse model and efficacy in two distinct mouse models of murine MLL-AF9 and human MLL-AF4 leukemia.128 Recently, Wang et al. introduced a [6,5,6] tricyclic system to mimic the [6,6,5] tricyclic system in compound 41, and compound 42 was obtained after a systematic structure–activity relationship (SAR) study (Figure 12).129 Compound 42 is specific for BET members and has Ki values of 3.2–25 nM with BRD2–4 BD1 and BD2 domain proteins. Moreover, it potently inhibits the viability of MV4-11 and MOLM-13 cells but maintains no obvious inhibition (IC50 > 2 μM) of cell growth in the K562 cell line harboring a BCR-ABL fusion protein.
Figure 12.

Discovery and development of compounds 41 and 42.
Similar tricyclic systems were applied on compounds 43–45 (Figure 13).130 Their IC50 values against BRD4 are in the range of 10–21 nM, but compound 45 is less stable (after being incubated for 30 min in mouse and human liver microsomes, only 3.7% and 1.9% of compound can be recovered) due to the metabolically active CH2 position.130 Moreover, they are nonselective BET BRD4 inhibitors as they display similar inhibitory activities against BRD2 and BRD3. Taking the same [6,5,6] tricyclic system as Wang’s group used, researchers from BMS introduced a 1,2,3-triazole to replace its bioisostere isoxazole. Compounds 46–48 (Figure 13) displayed potent inhibition against BRD4 BD1 with IC50 values of less than 5 nM.131 In vivo efficacy was evaluated in xenograft rodents models derived from H187 human small cell carcinoma or JJN3R multiple myeloma cell lines. At a dose of 1 mg/kg (e.g., compounds 47 and 48), over 90% of TGI was achieved, but no additional data about toxicity were available. The results of compounds 46 and 48 remind us that deuteration may be an effective way of improving drug pharmacodynamics, tolerability, and efficacy as well as getting out of patent protection.132,133
Figure 13.

Chemical structures of compounds 43–48. FL, full length; MLM, mouse liver microsome; HLM, human liver microsome.
Given that the 3,5-dimethyl group plays a critical role in forming hydrogen bonds with Asn and Tyr of BRD4, the main body of this series of BET BRD4 inhibitors are tolerant and so diverse that all single phenyl ring (e.g., compounds 33–38), tricyclic systems (e.g., compounds 41–48), and bicyclic rings134 (Figure 14) work very well with proper substituents. Compounds 49 and 50 with an indoline ring inhibit BRD4 with IC50 values of 35 and 8 nM in a TR-FRET methodology and show promising antiproliferation cellular activity against MV4-11 cell lines.135 Compounds 51 and 52,136 containing a benzoimidazole ring, can displace labeled BRD4 BD1 ligand 2 (K2 was defined based on this ligand) and competitively bind to BRD4 with K2 values of 3 and 2.7 nM, respectively. Both of these significantly suppress the proliferation of the MT-4 cell line with IC50 values around 9 nM. Cyclization of N-methyl with the adjacent carbonyl group of compound 53 led to compound 54.137 Introduction of a bulky piperidine to the N-methyl group of compound 55 gave 56.138 All of them display similar BRD4 inhibitory activities with IC50 values in the range of 14–36 nM.
Figure 14.

Chemical structures of compounds 49–56.
3.1.3. Pyridones
Although few journal articles were previously published on pyridone-derived BET BRD4 inhibitors, this series of compounds have maintained a large proportion of relevant patents. Importantly, compound 6, derived from pyridone, has entered into clinical trials for various cancers. Obviously, the pyridone plays a similar role as that of 3,5-dimethyl isoxazole, forming critical hydrogen bonds with BRD4. Compounds 57 and 58 (Figure 15), developed by AbbVie, display both potent BRD4 BD1 and BD2 inhibitory activity (up to single-digit nanomolar) in vitro, but their cellular activities are not very consistent (submicromolar range).139 Compounds 59 and 60,140 fused with a 12-membered ring, exhibit dramatically improved antiproliferative activity in cellular assays (MX-1 cell lines) with EC50 values of 5.7 and 8.7 nM, respectively. Their ability to inhibit LPS-induced IL-6 production was confirmed (83 and 81%, respectively) in a mouse model. Tricyclic (compounds 61 and 62)141 and bicyclic ring systems (compounds 63 and 64)142 attached to pyridone were investigated by researchers from Boehringer Ingelheim. These compounds show promising BRD4 BD1 inhibition with IC50 values of 5, 2, 12, and 12 nM, respectively.
Figure 15.

Chemical structures of compounds 57–64.
Pyrrole-fused pyridones were also explored by researchers from AbbVie. As depicted in Figure 16, O- or N-substituents at 1-position and 4-sulfonyl substituents of the phenyl ring attached to pyrrolopyridone are their common features. Compounds 65–67 exhibit similar BRD4 BD1 and BD2 inhibitory activities with IC50 values at the single-digit nanomolar level.143 Compared to compound 6, which is in Phase III clinical trials, an additional morpholine was introduced to give compound 68, and its potency indicated that the 5-position of the phenyl ring is tolerable.143 A long side chain was introduced to pyrrole of compounds 69 and 70, which both showed potent BRD4 inhibitory activities with IC50 values in the single-digit nanomolar range.143 They inhibited cell proliferation with EC50 values of 9.4 and 48 nM, respectively, against the MX-1 cell line.
Figure 16.

Chemical structures of compounds 65–70.
3.1.4. Triazolopyrazines
Patent US20160129001 covering [1,2,4]triazolo[4,3-a]pyrazines as BET BRD4 inhibitors was recently highlighted in ACS Med. Chem. Lett.144 Compounds 71–73 (Figure 17) were identified as potent BRD4 BD1 inhibitors by the BRD4-H4 tetra-acetylated peptide inhibition AlphaScreen assay with remarkable IC50 values of 8, 9, and 1 nM, respectively.145 Unfortunately, no binding affinity data toward other close isoforms such as BRD4 BD2 or BRD2/3 were reported; thus, their target specificity remains unclear. Compound 74 expanded the amine on the 8-position of [1,2,4]triazolo[4,3-a]pyrazines, and it retained potent BRD4 BD1 inhibitory activity.145 Compounds 75 and 76 introduced aliphatic side chains to replace the benzyl group without affecting their BRD4 inhibition ability.146
Figure 17.

Chemical structures of compounds 71–76.
Researchers from Novartis introduced unique cyclic rings (dihydropyrrolopyrazoles and dihydropyrrolopyrroles) into bioisosteres of triazolopyrazines including triazolopyridazines and triazolopyridines (Figure 18). Compounds 77 and 78 exhibit IC50 values of less than 11 nM against BRD4 with over 150-fold selectivity against CREBBP.147 Compared to 77, compound 79 used the pyridone as a replacement of triazolopyridazine and its selectivity against CREBBP was decreased to 42-fold.147 Compounds 80 and 81 introduced substituents on the N- position of pyrroles retaining potent BRD4 inhibition.148 The high potency (IC50 < 11 nM) of compound 82 indicates that modifications on the 2-position of pyrrole are tolerable. The MV4-11 cell line was determined to be sensitive to these compounds with IC50 values of less than 10 nM.148
Figure 18.

Chemical structures of compounds 77–82.
3.1.5. Tetrahydroquinolines (THQs)
Compound 83 (Figure 19), a THQ derivative, was identified via HTS as an ApoA1 upregulator in the HepG2 cell luciferase reporter assay.149 Optimization on the N1- and 6-position led to compound 84, which is a potent ApoA1 upregulator (EC170 = 10 nM) and BRD4 (IC50 = 398 nM) inhibitor.149 However, it shows moderate inhibition of multiple CYP450 isoforms. Further modifications on phenyl rings A and B gave compound I-BET726 (85, Figure 19) with an improved CYP450 profile.150 Its affinities with BET proteins (BRD4 IC50 = 4.4–23 nM) were confirmed by different assays. The cocrystal structure of 85 with BRD4 reveals that the N1-acetyl group interacts with Asn140 to form a critical hydrogen bond and with Tyr97 to form a second one through a water molecule. It was further evaluated in mouse xenograft models of human neuroblastoma (orally),151 an acute mouse inflammation model (orally), and mouse septic shock model (intravenously) with promising efficacy.149
Figure 19.

Discovery and development of compound 85 and its complex with BRD4 BD1 (PDB ID: 4BJX).
Compound 86 (Figure 20) developed by GSK was found to inhibit BRD4 BD2 potently with an IC50 value of less than 100 nM and displayed 100-fold selectivity over that of BRD4 BD1.152 However, no data on other BET members were provided. Compounds 87 and 88 focused on modifications of the phenyl ring and displayed promising BRD4 BD1 and BD2 (less than 100 and 50 nM for BD1and BD2, respectively) inhibitory activities.153 Additional N atoms were introduced to compound 89, showing potent BRD4 BD1 and BD2 inhibition with IC50 values of 20 and 43 nM, respectively.154 Variants of THQ such as 3,4-dihydroquinazolin-2(1H)-one in compound 90(155) and benzo[cd]indol-2(1H)-one) in compound 91(156) are capable of mimicking the acetyl-lysine to form hydrogen bonds with conserved residues of BRD4 (e.g., Asn140 and Tyr97). Compound 90 has an IC50 value of 220 nM against BRD4 BD1 and inhibits IL-6 production in human blood mononuclear cells stimulated by LPS with an EC50 value of 1.9 μM. Its oral bioavailability (F = 32% in rats) is relatively low, likely owing to the suboptimal compound solubility in the gut.155 Compound 91 was discovered and developed through structure-based virtual screening, and it had a temperature shift of 9.9 °C at a final concentration of 10 μM of proteins and 200 μM of compounds in the thermal stability shift assay. It exhibits high binding affinity for BRD4 BD1 with a Kd value of 137 nM and good PK property (F = 77%, t1/2 = 4 h, and cellular permeability = 10 × 10–6 cm s–1).156
Figure 20.

Chemical structures of compounds 86–91.
3.1.6. 4-Acyl Pyrroles
Compounds shown in Figure 21 are representatives of 4-acyl pyrrole-derived BET BRD4 inhibitors. Compound 92, discovered by AbbVie, exhibits an IC50 value of 38 nM against BRD4 BD1 and good antiproliferative activity (IC50 = 433 nM) against the MX-1 cell line.157 With an EC50 value of 160 nM against the MX-1 cell line, compound 93 was evaluated in the MX-1 human breast cancer xenograft model, and 80% TGI was achieved at a dose of 100 mg/kg.158 Compound 94 was obtained through high-throughput virtual screening using a library containing more than 7 million small molecules.159 It is the most potent binder in this compound library against BRD4 BD1 with a KD value of 237 nM. Among 56 cell lines (from nine different cancer types), it potently and selectively inhibits leukemia cells.159,160
Figure 21.

Chemical structures of compounds 92–94.
3.1.7. 2-Thiazolidinones
Shen et al. took fragment 95 (Figure 22), which bears a 2-thiazolidinone core, as the starting point for integrated lead optimization.161 The 2-thiazolidinone motif of 95 can mimic the 3,5-dimethylisoxazole fragment that is mentioned above to occupy the KAc binding pocket. Similar modifications were performed at the meta or para position of the phenyl ring of 95. Compound 96, bearing only one thiophene sulfonamide at the meta position of the phenyl ring, displayed BRD4 inhibition with an IC50 value of 4.1 μM in a fluorescence anisotropy assay.161 Introducing an additional substituent at the meta position gave compound 97, which is 17-fold more potent than that of 96 and more stable in liver microsomes.161 However, none of them showed acceptable proliferation inhibitory activity in human colon cancer HT-29 cell line (GI50 of the best one is 37 μM). In the follow-up study, the sulfonamide group was reversed, and further modifications on the phenyl ring were explored. Compound 98 is the most potent one in this series using the cellular antiproliferation assay with IC50 values of 860 nM in HT-29 cells and 180 nM in the MV4-11 cell line.162
Figure 22.
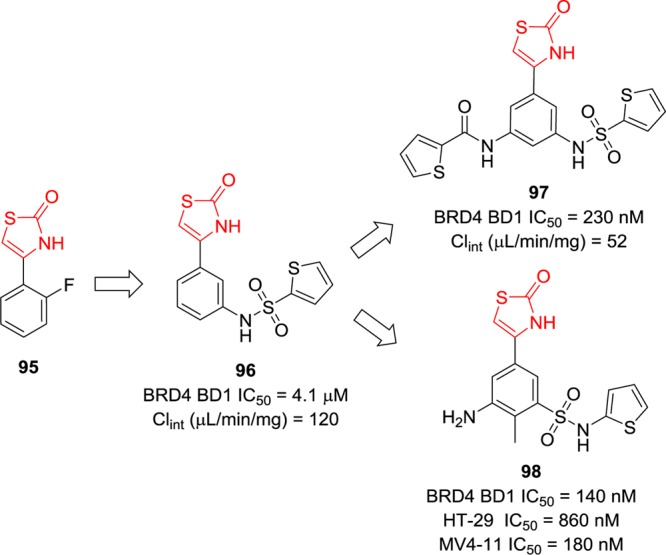
Discovery and development of 2-thiazolidinones as BRD4 inhibitors.
3.1.8. Proteolysis Targeting Chimera (PROTAC)
Different from traditional small molecule drugs, proteolysis targeting chimera (PROTAC) is an emerging novel technology that takes advantage of a small molecule to control intracellular protein levels through recruiting target proteins to the ubiquitin/proteasome system for selective degradation.163−166 It is a combination of small molecule and genetic knockdown techniques, and recently, this method was applied to BRD4. DBET1 (99, Figure 23) was designed by conjugating compound 7 with thalidomide whose target is cereblon (CRBN), a component of a cullin-RING ubiquitin ligase.167 Significant loss of BRD4 (>80%) was observed for the treatment of 99 at a concentration of 100 nM in MV4-11, whereas 7 or thalidomide alone was not sufficient to induce BRD4 degradation. Treatment of CRBN-deficient human MM cell line (MM1.S-CRBN–/–) with 99 was ineffectual, suggesting that proteasomal degradation of BRD4 by 99 is CRBN dependent. In a human leukemia xenograft mouse model, 99 also displayed improved antitumor efficacy compared with the effects of 7. ARV-825 (100)168 and MZ1 (101)169 were designed based on a similar hypothesis. Compound 100 suppressed c-MYC levels, inhibited cell proliferation, and induced apoptosis in Burkitt’s lymphoma more effectively than using BRD4 inhibitors alone.168 Compound 101, by tethering 7 to a ligand for the E3 ubiquitin ligase VHL, induced selective removal of BRD4 over BRD3 and BRD2.169 Given that compound 7 has no preference for binding BRD4 over BRD2/3, the observed selectivity was attributed to the more efficient polyubiquitination of lysine residues on the BRD4 surface or more productive formation of the VHL:101:BRD4 complex due to preferential direct interaction or reduced steric constraints between VHL and BRD4.169
Figure 23.

Chemical structures of compounds 99–101.
3.1.9. Bivalent Inhibitors
Recently, bivalent BET BRD4 inhibitors have emerged. Compound 102 (MT1, Figure 24) is the first one reported given that all previously reported BET BRD4 inhibitors bind in a monovalent fashion.170 A homodimer of compound 7 with a long PEG linker led to 102, which has an IC50 value of 3.1 nM against BRD4 BD1. Size-exclusion chromatography results indicate that it binds to tandem bromodomains in an intramolecular fashion and that both BRD4 bromodomains are directly involved. In cellular assays, it displayed a 100-fold higher potency over compound 7 (0.17 vs 72 nM in the MV4-11 cell line). Compound 102 significantly reduced leukemia burden in the aggressive disseminated leukemia mouse model at a dose of 25 mg/kg compared to vehicle and compound 7. Compound 103 is a bivalent and potent BRD4 inhibitor with IC50 values of 5 nM against full length BRD4 and 1.6 μM against BRD4 BD1.171 In a xenograft model of MV4-11, a dose-dependent TGI was observed for 1 (TGI = 72%), 2.5, and 5 mg/kg (regression) daily oral doses of compound 103.
Figure 24.

Chemical structures of compounds 102 and 103.
3.2. Dual BRD4 and Kinase Inhibitors
Because the nonspecific CDK inhibitor Dinaciclib was reported to interact with the acetyl-lysine binding site of BRDT,172 the vast chemical space of kinase inhibitors as bromodomain modulators is attracting more and more attention. Schönbrunn et al. evaluated the binding potential of 581 diverse kinase inhibitors toward BETs (taking BRD4 BD1 as a representative) via a robotic cocrystallization screening campaign.173 Among the 14 identified kinase inhibitors, BI2536 (104), which was developed as a potent and selective PLK1 inhibitor (IC50 = 0.83 nM) and is currently in clinical trials,174 showed the most potent BRD4 inhibition with an IC50 value of 25 nM. Cocrystal structure of 104 with BRD4 BD1 reveals that the dihydropteridine oxygen of 104 interacts with Asn140, and the aminopyrimidine moiety forms a network of hydrogen bonds with residues Glu85, Pro82, and water molecules of the ZA loop (Figure 25, left panel).173 In the cocrystal structure of 104 with PLK1, it binds to the hinge region through the aminopyrimidine moiety while the carbonyl oxygen of the 2-amino-6-oxo-dihydropteridine moiety is involved in H2O-mediated interactions around the gatekeeper residue Leu130. TG-101209 (105), a JAK2 inhibitor, has an IC50 value of 130 nM against BRD4.173 It was reported to form the critical hydrogen bonds with both BRD4 and JAK2 by the same fragment.
Figure 25.

Chemical structure of dual kinase-bromodomain inhibitors 104–106 and their cocrystal structures with BRD4 BD1 as well as their target kinases. (left panel) Cocomplex of 104 with BRD4 (PDB ID: 4OGI) and PLK1 (PDB ID: 2RKU). (center panel) Crystal structures of 105 with BRD4 (PDB ID: 4O76) and JAK2 (PDB ID: 4JI9). (right panel) Cocomplex of 106 with BRD4 (PDB ID: 4O77) and SB2 (4-(4-(4-fluorophenyl)-2-(4-(methylsulfinyl)phenyl)-1H-imidazol-5-yl)pyridine) with p38α (PDB ID: 3ZS5).
Compound 105 utilizes the aminopyrimidine moiety to interact with Asn140 of BRD4 directly, whereas it forms hydrogen bonds with the hinge region residue Leu932 in JAK2 (Figure 25, center panel). SB202190 (106),173 a p38α/β inhibitor, takes two different binding modes with BRD4 and p38α/β. Its cocrystal structure with BRD4 displays that imidazole nitrogen interacts with Asn140 and that it forms hydrogen bonds via the hydroxyl group with Tyr97 and Met132. It is suggested to interact with the hinge region through the pyridine ring (Figure 25, right panel). Compounds 104 and 105 were also reported to be able to displace BRD4 from chromatin and suppress c-MYC expression in MM.1S cells.175 These dual inhibitors remind us of potential off-target concerns but also provide us the opportunity to seek an efficient therapy by developing one single drug molecule to disrupt both transcriptional and cell signaling events. Meanwhile, with the binding modes in hand, re-exploring the SAR on these dual inhibitors is attractive for tuning the selectivity and achieving BRD4-selective inhibitors.176
Other dual BRD4/kinase inhibitors such as dual PI3K/BET BD1 inhibitor LY294002 (107),177 dual EGFR/BRD4 inhibitor Z118332870 (108),178 and dual HDAC/BRD4 inhibitor 109(179) (Figure 26) are also explored and provide good starting points for developing cancer therapeutics via an approach of polypharmacology.
Figure 26.

Chemical structures of compounds 107–109.
3.3. Relatively Selective BRD4 Inhibitors
3.3.1. BD Domain Selective Inhibitors
Compound 1 (Figure 27), a derivative of the plant polyphenol resveratrol, was originally developed by Resverlogix Corporation for the treatment of cardiovascular disease associated with atherosclerosis given that it can increase the plasma level of the high-density lipid protein ApoA1.84 After entering clinical trials, the function of 1 as a bromodomain inhibitor was explored.32 Temperature-shift assays show that 1 selectively targets BD2s of the BET subfamily. The KD values of 1 by isothermal titration calorimetry (ITC) are 135 nM against BRD4 BD2 and 1142 nM against BRD4 BD1 with almost 10-fold selectivity. However, the selectivity of individual BET family members was not achieved. Cocrystal structures of 1 with the first and second domain of BET proteins display that the phenyl ring of 1 packs against the BD2 unique residue His433 (Asp in BD1 domain) to form a π–π interaction, which may explain its higher affinity for BD2 domains. Currently, a phase III clinical study of 1 is recruiting participants to explore whether this treatment in high-risk type 2 diabetes mellitus patients with coronary artery disease can increase the time to major adverse cardiovascular events.
Figure 27.

(left panel) Structure of compound 1 and KD values with different BD domains of BET members. (right panel) Overlap of cocrystal structures of 1 with BRD4 BD1 (yellow; PDB ID: 4MR4) and BRD2 BD2 (turquoise; PDB ID: 4MR6). The labeled residue is His433 (magenta) in the BRD2 BD2 domain and Asp144 (green) in the BRD4 BD1 domain.
Starting from CREBBP inhibitor MS120 (110, Figure 28) with a modest inhibitory activity against BRD4 BD1 (Ki = 11 μM) and BRD4 BD2 (Ki = 20 μM) in a fluorescent polarization assay, diazobenzene compound MS436 (111) was developed with a potent affinity (Ki = 30–50 nM) for BRD4 BD1, i.e., 10-fold selectivity over that for BRD4 BD2.180 However, this selectivity is limited only for BRD4, it has similar affinity with BRD3 BD1 (Ki = 0.1 μM) and BRD3 BD2 (Ki = 0.14 μM). Compound 111 blocks LPS-induced proinflammatory cytokine IL-6 expression in macrophage cells and NF-κB-directed NO production in RAW264.7 cells with IC50 values of 4.9 and 3.8 μM, respectively. Little cytotoxicity on cell growth or proliferation was observed in an MTT assay at concentrations up to 100 μM.180
Figure 28.

(a) Development of compound 111. (b) Development of compound 113. (c) Cocrystal structure of 113 with BRD4 BD1 (PDB ID: 4QB3).
Inspired by the structural insights of tetrahydropyrido indole MS7972 (112, Figure 28) bound to the CREBBP BD domain, Olinone (113) was developed.88 Compound 113 displays preferred BD1 binding over BD2 for all three BET proteins BRD4, BRD3, and BRD2, while exhibiting nearly no detectable binding to other BCPs. Cocrystal structure of 113 with BRD4 BD1 (Figure 28c) reveals that, besides the critical hydrogen bond with Asn140, compound 113 interacts with Asp144, which is one of a few residues that is distinct between BRD4 BD1 and BD2 in the acetyl-lysine binding site.
3.3.2. BRD4 Selective Inhibitors
BAY1238097 (structure not disclosed), developed by Bayer, displays potent inhibition against BRD4 with an IC50 value of 63 nM, i.e., 10-fold selectivity over BRD3 and 39-fold selectivity over BRD2.181 It was enrolled into human clinical trials (NCT02369029), but the development is currently terminated due to unknown reasons.
3.3.3. BRD4 BD1 Selective Inhibitors
Compound 114 (Figure 29, left panel), an acetyl-lysine xanthine derivative inhibitor, was identified via an MTS follow-up program targeting BRD4 BD1.182 It displays inhibitory activity against BRD4 BD1 with an IC50 value of 5 μM in a homogeneous time-resolved fluorescence assay, i.e., 10-fold selectivity of BD1 over its relatives, and no detectable inhibition against BD2 counterparts. From the superimposition of BRD4 with and without 114 (Figure 29, right panel), it is suggested that the ZA loop shift, which was not observed with the pan-BET inhibitors, is critical for selectivity.
Figure 29.
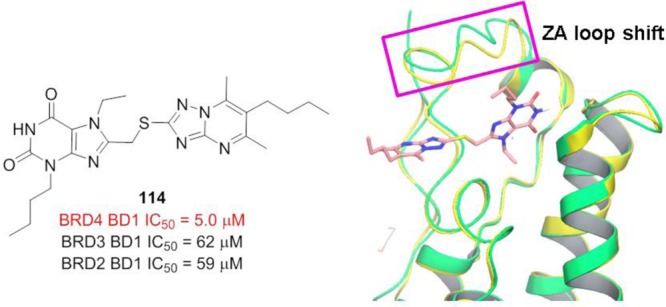
(left panel) Structure of compound 114 and IC50 values for different BD1 domains of BET members. (right panel) Superimposition of BRD4 alone (green; PDB ID: 2OSS) and BRD4 with compound 114 (yellow; PDB ID: 5EGU). The ZA loop shift is highlighted by the magenta box.
4. Concluding Remarks: Challenges and Opportunities
As epigenetic readers of the histone code, BET family members play a critical role in a number of human diseases. Among them, BRD4 is the most extensively studied member. BRD4 recruits transcriptional regulatory complexes to acetylated chromatin via recognition of acetylated lysine. In this regard, BRD4 plays an important role as a serine kinase of RNA Pol II and an atypical histone acetyltransferase controlling gene expression through chromatin relaxation, transcriptional elongation, and ejection of histones from coding regions.10,183 These activities make BRD4 an integral component of many disease-associated gene regulatory networks. BRD4 is thus considered as a promising therapeutic target for a variety of human diseases including cancer, inflammation, HIV infection, CNS disorders, and cardiovascular diseases. BET inhibition displays efficacy against various different pathologies especially cancer and inflammation. These effects have been attributed to the specific set of downstream target genes whose expression is disproportionately sensitive to pharmacological targeting of BET proteins. Over the past decade, the biological elucidation of BRD4 functions and the development of BRD4 inhibitors have made great progress along with the emergence of new technologies, such as PROTAC and the bump-and-hole approach.184 Various series of BRD4 inhibitors have emerged such as azepines, 3,5-dimethyl isoxazoles, pyridones, triazolopyrazines, THQs, 4-acyl pyrroles, 2-thizalidinones, and so forth,185 and some of them exhibit potent BRD4 inhibition in vitro and effective therapeutic activity in vivo with no obvious toxicity. The common feature of these molecules is that they all have a unique head moiety that can form critical hydrogen bonds with residues of the BRD4 binding pocket (e.g., Asn140 and Tyr 97). In addition, almost all of them contain a small hydrophobic group around the hydrogen bond, mostly a methyl group. Currently, more than a dozen diverse BET BRD4 inhibitors have been advanced into human clinical trials for the treatment of cancer, inflammation, and other diseases. Notably, compound 1, a relatively selective inhibitor of BET BD2, is now in Phase III human clinical trials.
Both challenges and opportunities remain regarding the drug development of BRD4 inhibitors. Primarily, more potent and specific BRD4 inhibitors are in urgent need including BRD4 BD1- and BRD4 BD2-selective compounds over other BET family members along with further exploration of the specific transcriptional effects and therapeutic end points. Although a number of BET BRD4 inhibitors described above are highly potent with single-digit nanomolar BRD4 inhibitory activity, very few of them exhibit excellent selectivity among BET family members or sub-bromodomains. For example, compound 80 has an IC50 value of 12 nM against BRD4 as well as IC50 values of 14 nM against BRD3 and 14 nM against BRD2. In contrast, compounds 60 and 61 are BRD4 selective, but the potency is only in the micromolar range. Several different approaches may contribute to developing inhibitors that are more BRD4 specific. The first is to take advantage of the crystallography and the subtle amino acid differences of BET members to design new inhibitors with improved BRD4 selectivity.186 The second is to develop inhibitors of BRD4 interacting proteins. Inhibiting the PPI interface can be achieved by targeting either of the proteins involved in the interaction. Inhibition of BRD4 interacting partners may be specific to one signaling pathway and affect only the desired genes’ expression. The third is to screen and repurpose known kinase inhibitors to target BRD4, which may accelerate the drug approval process. Chemical optimizations on dual kinase/BRD4 inhibitors to obtain kinase selective or BRD4 selective appear to be feasible.176 Given that a number of kinase inhibitors were found effective on BET inhibition, using them to target BRD4 by selectivity tuning is thus less time-consuming because their physicochemical properties and metabolism profiles have been extensively explored. Moreover, evaluation of BRD4 selectivity and specificity should include the panel assays on kinases as well as BCPs and BET proteins. Although different chemical scaffolds are being explored, the privileged scaffold diazepine appears to dominate the BET BRD4 inhibitors in clinical trials. Many novel fragments have been discovered by fragment screening, rational design, bioisosterism, and cocrystal analyses and may be worth further exploration.187−191 Clearly, there exists a large chemical space to develop structurally diverse BRD4 inhibitors. With the assistance of traditional drug discovery methods (HTS, VS, crystallography, SBDD, FBDD, etc.),192 it is the opinion of the authors that more potent and specific BRD4 inhibitors with different chemotypes will enter human clinical trials in the near future.
Along with the promising results of early clinical trials using BET inhibitors in hematologic malignancies,193 resistance mechanisms were evaluated to optimize the clinical efficacy of these drugs. In human and mouse leukemia cells, increased WNT/β-catenin signaling is considered to be an alternative mechanism that regulates transcription and promotes resistance to the BET inhibitor 27.194 Negative modulation of this signaling can restore sensitivity to 27 in vitro and in vivo. Other studies identified WNT signaling as a driver and candidate biomarker of primary and acquired BET resistance in leukemia.195 In BET inhibition of resistant triple-negative breast cancer cells, a bromodomain-independent recruitment mechanism was facilitated by decreased protein phosphatase 2 activity and subsequent hyperphosphorylated BRD4, which binds more strongly to MED1, a mediator of RNA Pol II transcription subunit 1.196 Resistance to BET inhibitors reported to be mediated by kinome reprogramming and cotargeting BET protein and RTK or PI3K signaling enhance growth inhibition in ovarian cancer cells.197 In this situation, combination of BET inhibitors and corresponding signaling negative modulator may overcome the BET inhibition resistance.198 In addition, the aforementioned one drug with polypharmacology holds great promise for mitigating resistance in cancer therapy and may guide the next generation of efficacious BRD4 inhibitors as anticancer agents.
Given that BRD4 is involved in a variety of gene regulatory networks, signaling pathways, and disease-associated function explorations, more detailed molecular and mechanistic studies as well as establishment of novel in vivo animal models of disease are requisite for investigating the unique characteristics/partners of BRD4. Although targeting BET inhibition to combat cancer/inflammation with no obvious toxicity has been demonstrated as a proof-of-concept, attention should be paid to the major dose-limiting toxicity (e.g., thrombocytopenia observed in clinical trials of compound 3).199 Whether the side effects are induced by global BET inhibition remains to be extensively explored and evaluated for drug development. The downstream target genes have different levels of sensitivity toward BET inhibition, and it is still challenging to block some of them while leaving others intact. Utilizing proper administration strategies, taking advantage of physicochemical properties of compounds and identifying different pharmacological mechanisms via divergent biological pathways may be of value for facilitating therapeutic development. Drug properties including physicochemical parameters should be considered in the early drug discovery of BRD4 inhibitors depending on the intended therapeutic applications. For example, compound 7 can cause memory deficits in mice, indicating its potential neurological side effects for patients with cancer. Thus, developing compounds that do not cross the BBB may benefit cancer patients with fewer side effects, whereas BRD4 inhibitors designed for the treatment of CNS disorders are required to be capable of penetrating the BBB. Thus, it is important to understand the tissue distribution of drug candidates. To this end, developing properly radiolabeled positron emission tomography imaging ligands of BRD4 inhibitors may be very powerful for this research field. Limited distribution to target cells, tissues, and organs may contribute to mitigating unwanted side effects. Different administration approaches may also be used to produce the desired distribution, elimination rate, and efficacy. For example, targeted delivery of drug candidates to specific tissues and organ systems can be achieved by encapsulating them into polymeric nanoparticles.200
Currently, the majority of drug development efforts have been focused on disrupting protein interaction networks between BET and KAc-modified proteins. However, BRD4 is a multifunctional protein that controls transcriptional elongation and also maintains intrinsic serine kinase activity directed toward the RNA Pol II CTD and CDK9.89,183 Additionally, more recent work has shown that BRD4 is a previously unrecognized atypical histone acetyl-transferase whose activity is directed toward Lys residues on the periphery of the nucleosome, enabling RNA Pol II to more easily displace nucleosomes during the process of transcription.10 Small molecule therapeutics developed to target these activities may also have very interesting actions and distinct toxicity profiles.
Taken together, targeting BRD4 with small molecules holds promise as a viable therapeutic strategy for various human diseases, as aforementioned. There remains a long road ahead toward market approval, and this effort will require interdisciplinary collaborations. Discovery of potent and specific acetyl-lysine competitive BRD4 inhibitors with high isoform or bromodomain selectivity will fill roles in multiple applications as useful pharmacological probes for elucidating BRD4 biological functions and in the development of potential medications to benefit patients in the near future.
Acknowledgments
This work was supported by grants P30 DA028821, R01 DA038446, and T32 DA07287 from the National Institutes of Health, Sanofi Innovation Awards (iAwards) Program, Breast Cancer Research Program Breakthrough Award (BC160038) from the Department of Defense, Cancer Prevention Research Institute of Texas award, R. A. Welch Foundation Chemistry and Biology Collaborative Grant from the Gulf Coast Consortia, John Sealy Memorial Endowment Fund, Institute for Translational Sciences, Sealy Center for Molecular Medicine, and the Center for Addiction Research at UTMB.
Glossary
Abbreviations
- ABC
activated B-cell-like subtype
- ACS
acute coronary syndrome
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- ApoA1
apolipoprotein A
- AST
advanced solid tumors
- BBB
blood-brain barrier
- BCPs
bromodomain-containing proteins
- BET
bromodomain and extra-terminal domain
- BLBC
basal-like breast cancer
- BRD4
bromodomain-containing protein 4
- BZD
benzodiazepine
- CAD
coronary artery disease
- CDKs
cyclin-dependent kinases
- CNS
central nervous system
- CRPC
castration-resistant prostate cancer
- CTD
C-terminal domain
- CVDs
cardiovascular diseases
- DLBCL
diffuse large B-cell lymphoma
- FBDD
fragment-based drug design
- HPV
human papillomavirus
- HTS
high-throughput screening
- KAc
lysine acetylation
- MDS
myelodysplastic syndrome
- MM
multiple myeloma
- MPN, U
myeloproliferative neoplasm, unclassifiable
- MM1.S-CRBN–/–
CRBN-deficient human MM cell line
- MTS
midthroughput screening
- MW
molecular weight
- NAc
nucleus accumbens
- NMC
NUT midline carcinoma
- NMP
N-methylpyrrolidinone
- NSCLC
nonsmall cell lung cancer
- NUT
nuclear protein in testis
- PHD
plant homeodomain
- PROTAC
proteolysis targeting chimera
- P-TEFb
positive transcriptional elongation factor complex
- RNA Pol II
RNA polymerase II
- RRHMs
relapsed/refractory hematologic malignancies
- RRMM
relapsed/refractory multiple myeloma
- RST
recurrent solid tumors
- T2DM
type 2 diabetes mellitus
- TGI
tumor growth inhibition
- THQ
tetrahydroquinoline
- WPF
Trp-Pro-Phe
- VS
virtual screening
Biographies
Zhiqing Liu received her B.S. degree in Pharmaceutical Engineering from Shandong Normal University in 2009. She obtained her Ph.D. degree from Shanghai Institute of Materia Medica, Chinese Academy of Sciences in 2014 under the supervision of Professor Ao Zhang. She is currently a postdoctoral research fellow in Professor Jia Zhou’s chemical biology program at the University of Texas Medical Branch (UTMB). Her research interests include the rational design and chemical synthesis of small molecules as novel pharmacological probes and therapeutics for inflammation, human cancer, and CNS disorders.
Pingyuan Wang received his Ph.D. in Pesticide Science and Medicinal Chemistry from Central China Normal University (CCNU) in 2016 under the joint supervision of Professor Guang-Fu Yang from CCNU and Professor Ao Zhang from Shanghai Institute of Materia Medica, Chinese Academy of Sciences. He is currently pursuing his postdoctoral training under the supervision of Professor Jia Zhou at the Chemical Biology Program, Department of Pharmacology and Toxicology at UTMB. His research interests focus on the design and synthesis of novel small molecules as chemical probes and drug candidates for neurological disorders, cancer, and other human diseases.
Haiying Chen received her B.S. degree in Engineering from Tianjin University (Branch) in 1995. She worked as an engineer in Tianjin Research Institute of Construction Machinery associated with designing and programming computer testing systems. She joined Professor Jia Zhou’s drug discovery program in 2014 as a Research Associate. Her research interests focus on computer-assisted rational drug design of small molecules and understanding of the ligand–target interactions for CNS disorders, cancer/inflammation, and infectious diseases.
Eric A. Wold received his B.S. degrees in Biotechnology and Biology from the University of Houston and completed his undergraduate thesis on microbial expression systems and enzymatic organophosphate hydrolysis under the guidance of Dr. Rupa Iyer. He is pursuing a Ph.D. in the Pharmacology program at UTMB under the training of Professor Jia Zhou. His research interests include the rational design and chemical synthesis of small molecules as novel pharmacological probes and therapeutics for CNS disorders and cancer.
Bing Tian obtained his Ph.D. degree in Pharmacology from UTMB in 1999. He is now Assistant Professor in Internal Medicine and a member of the Sealy Center for Molecular Medicine at UTMB. He is an expert on innate immune signaling pathways with extensive experience in molecular biology, cell biology, pharmacology, and animal experimental methodology. He has been working on viral and cytokine-induced innate immune responses in airway epithelium and has made substantial contributions with more than 40 publications as the primary author or coauthor. He also holds two patents.
Allan R. Brasier received his M.D. in 1983 from the University of California, San Francisco. After completing a research fellowship in the Laboratory of Molecular Endocrinology at the Massachusetts General Hospital, he assumed a faculty position at UTMB. There, he directs the Sealy Center for Molecular Medicine and the Institute for Translational Sciences. As an expert in gene expression control by NF-κB in innate inflammation, he has published 150 peer reviewed manuscripts, 40 books and invited reviews, and holds seven patents.
Jia Zhou received his Ph.D. in organic chemistry in 1997 from Nankai University, China. He joined the chemistry faculty there and was promoted to Associate Professor. In 1999, he started his postdoctoral training in organic chemistry with Dr. Sidney M. Hecht at the University of Virginia. After further training in medicinal chemistry with Dr. Alan P. Kozikowski at Georgetown University, he conducted research at Acenta Discovery and PsychoGenics as a Senior Principal Scientist for seven years. He is currently a tenured Professor and also a faculty member of the Center for Addiction Research, Center for Biodefense and Emerging Infectious Diseases, and Sealy Center for Molecular Medicine at UTMB. He is an author of more than 100 papers, seven book chapters, and an inventor of 16 patents.
The authors declare no competing financial interest.
References
- Kouzarides T. Chromatin modifications and their function. Cell 2007, 128, 693–705. 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Li B.; Carey M.; Workman J. L. The role of chromatin during transcription. Cell 2007, 128, 707–719. 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Qiu J. Epigenetics: unfinished symphony. Nature 2006, 441, 143–145. 10.1038/441143a. [DOI] [PubMed] [Google Scholar]
- Bannister A. J.; Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrowsmith C. H.; Bountra C.; Fish P. V.; Lee K.; Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat. Rev. Drug Discovery 2012, 11, 384–400. 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- Strahl B. D.; Allis C. D. The language of covalent histone modifications. Nature 2000, 403, 41–45. 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Choudhary C.; Kumar C.; Gnad F.; Nielsen M. L.; Rehman M.; Walther T. C.; Olsen J. V.; Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Hewings D. S.; Rooney T. P.; Jennings L. E.; Hay D. A.; Schofield C. J.; Brennan P. E.; Knapp S.; Conway S. J. Progress in the development and application of small molecule inhibitors of bromodomain-acetyl-lysine interactions. J. Med. Chem. 2012, 55, 9393–9413. 10.1021/jm300915b. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation?. EMBO. J. 2000, 19, 1176–1179. 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaiah B. N.; Case-Borden C.; Gegonne A.; Hsu C. H.; Chen Q.; Meerzaman D.; Dey A.; Ozato K.; Singer D. S. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat. Struct. Mol. Biol. 2016, 23, 540–548. 10.1038/nsmb.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L.; Zhou M. M. Bromodomain: an acetyl-lysine binding domain. FEBS Lett. 2002, 513, 124–128. 10.1016/S0014-5793(01)03309-9. [DOI] [PubMed] [Google Scholar]
- Lange M.; Kaynak B.; Forster U. B.; Tonjes M.; Fischer J. J.; Grimm C.; Schlesinger J.; Just S.; Dunkel I.; Krueger T.; Mebus S.; Lehrach H.; Lurz R.; Gobom J.; Rottbauer W.; Abdelilah-Seyfried S.; Sperling S. Regulation of muscle development by DPF3, a novel histone acetylation and methylation reader of the BAF chromatin remodeling complex. Genes Dev. 2008, 22, 2370–2384. 10.1101/gad.471408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L.; Zhang Q.; Li S.; Plotnikov A. N.; Walsh M. J.; Zhou M. M. Mechanism and regulation of acetylated histone binding by the tandem PHD finger of DPF3b. Nature 2010, 466, 258–262. 10.1038/nature09139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su D.; Hu Q.; Li Q.; Thompson J. R.; Cui G.; Fazly A.; Davies B. A.; Botuyan M. V.; Zhang Z.; Mer G. Structural basis for recognition of H3K56-acetylated histone H3-H4 by the chaperone Rtt106. Nature 2012, 483, 104–107. 10.1038/nature10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discovery 2014, 13, 337–356. 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Chiang C. M. Brd4 engagement from chromatin targeting to transcriptional regulation: selective contact with acetylated histone H3 and H4. F1000Prime Rep. 2009, 1, 98. 10.3410/B1-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A.; Chitsaz F.; Abbasi A.; Misteli T.; Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 8758–8763. 10.1073/pnas.1433065100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero F. A.; Taylor A. M.; Crawford T. D.; Tsui V.; Cote A.; Magnuson S. Disrupting acetyl-lysine recognition: Progress in the development of bromodomain inhibitors. J. Med. Chem. 2016, 59, 1271–1298. 10.1021/acs.jmedchem.5b01514. [DOI] [PubMed] [Google Scholar]
- Ghoshal A.; Yugandhar D.; Srivastava A. K. BET inhibitors in cancer therapeutics: a patent review. Expert Opin. Ther. Pat. 2016, 26, 505–522. 10.1517/13543776.2016.1159299. [DOI] [PubMed] [Google Scholar]
- Yu L.; Wang Z.; Zhang Z.; Ren X.; Lu X.; Ding K. Small-molecule BET inhibitors in clinical and preclinical development and their therapeutic potential. Curr. Top. Med. Chem. 2015, 15, 776–794. 10.2174/1568026615666150302110135. [DOI] [PubMed] [Google Scholar]
- Vidler L. R.; Brown N.; Knapp S.; Hoelder S. Druggability analysis and structural classification of bromodomain acetyl-lysine binding sites. J. Med. Chem. 2012, 55, 7346–7359. 10.1021/jm300346w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhalluin C.; Carlson J. E.; Zeng L.; He C.; Aggarwal A. K.; Zhou M.-M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- Fukazawa H.; Masumi A. The conserved 12-amino acid stretch in the inter-bromodomain region of BET family proteins functions as a nuclear localization signal. Biol. Pharm. Bull. 2012, 35, 2064–2068. 10.1248/bpb.b12-00527. [DOI] [PubMed] [Google Scholar]
- Jones M. H.; Numata M.; Shimane M. Identification and characterization of BRDT: A testis-specific gene related to the bromodomain genes RING3 and Drosophila fsh. Genomics 1997, 45, 529–534. 10.1006/geno.1997.5000. [DOI] [PubMed] [Google Scholar]
- Shang E.; Nickerson H. D.; Wen D.; Wang X.; Wolgemuth D. J. The first bromodomain of Brdt, a testis-specific member of the BET sub-family of double-bromodomain-containing proteins, is essential for male germ cell differentiation. Development 2007, 134, 3507–3515. 10.1242/dev.004481. [DOI] [PubMed] [Google Scholar]
- Zhou Q.; Li T.; Price D. H. RNA polymerase II elongation control. Annu. Rev. Biochem. 2012, 81, 119–143. 10.1146/annurev-biochem-052610-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S.; Sowa M. E.; Ottinger M.; Smith J. A.; Shi Y.; Harper J. W.; Howley P. M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 2011, 31, 2641–2652. 10.1128/MCB.01341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamonica J. M.; Deng W.; Kadauke S.; Campbell A. E.; Gamsjaeger R.; Wang H.; Cheng Y.; Billin A. N.; Hardison R. C.; Mackay J. P.; Blobel G. A. Bromodomain protein Brd3 associates with acetylated GATA1 to promote its chromatin occupancy at erythroid target genes. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, E159–168. 10.1073/pnas.1102140108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamsjaeger R.; Webb S. R.; Lamonica J. M.; Billin A.; Blobel G. A.; Mackay J. P. Structural basis and specificity of acetylated transcription factor GATA1 recognition by BET family bromodomain protein Brd3. Mol. Cell. Biol. 2011, 31, 2632–2640. 10.1128/MCB.05413-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J.; Dong W.; Chen L.; Zou T.; Qi Y.; Liu Y. Brd2 is a TBP-associated protein and recruits TBP into E2F-1 transcriptional complex in response to serum stimulation. Mol. Cell. Biochem. 2007, 294, 45–54. 10.1007/s11010-006-9223-6. [DOI] [PubMed] [Google Scholar]
- Prinjha R. K.; Witherington J.; Lee K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol. Sci. 2012, 33, 146–153. 10.1016/j.tips.2011.12.002. [DOI] [PubMed] [Google Scholar]
- Picaud S.; Wells C.; Felletar I.; Brotherton D.; Martin S.; Savitsky P.; Diez-Dacal B.; Philpott M.; Bountra C.; Lingard H.; Fedorov O.; Muller S.; Brennan P. E.; Knapp S.; Filippakopoulos P. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 19754–19759. 10.1073/pnas.1310658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirguet O.; Gosmini R.; Toum J.; Clement C. A.; Barnathan M.; Brusq J. M.; Mordaunt J. E.; Grimes R. M.; Crowe M.; Pineau O.; Ajakane M.; Daugan A.; Jeffrey P.; Cutler L.; Haynes A. C.; Smithers N. N.; Chung C. W.; Bamborough P.; Uings I. J.; Lewis A.; Witherington J.; Parr N.; Prinjha R. K.; Nicodeme E. Discovery of epigenetic regulator I-BET762: lead optimization to afford a clinical candidate inhibitor of the BET bromodomains. J. Med. Chem. 2013, 56, 7501–7515. 10.1021/jm401088k. [DOI] [PubMed] [Google Scholar]
- Coude M. M.; Braun T.; Berrou J.; Dupont M.; Bertrand S.; Masse A.; Raffoux E.; Itzykson R.; Delord M.; Riveiro M. E.; Herait P.; Baruchel A.; Dombret H.; Gardin C. BET inhibitor OTX015 targets BRD2 and BRD4 and decreases c-MYC in acute leukemia cells. Oncotarget 2015, 6, 17698–17712. 10.18632/oncotarget.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu K. T.; Eda H.; Santo L.; Ramachandran J.; Koulnis M.; Mertz J.; Sims R. J.; Cooper M.; Raje N. S. Effect of the BET inhibitor, Cpi-0610, alone and in combination with lenalidomide in multiple myeloma. Blood 2015, 126, 4255–4255. [Google Scholar]
- Shapiro G. I.; Dowlati A.; LoRusso P. M.; Eder J. P.; Anderson A.; Do K. T.; Kagey M. H.; Sirard C.; Bradner J. E.; Landau S. B. Abstract A49: Clinically efficacy of the BET bromodomain inhibitor TEN-010 in an open-label substudy with patients with documented NUT-midline carcinoma (NMC). Mol. Cancer Ther. 2015, 14, A49–A49. 10.1158/1535-7163.TARG-15-A49. [DOI] [Google Scholar]
- Sarthy A.; Li L.; Albert D. H.; Lin X.; Scott W.; Faivre E.; Bui M. H.; Huang X.; Wilcox D. M.; Magoc T.; Buchanan F. G.; Tapang P.; Sheppard G. S.; Wang L.; Fidanze S. D.; Pratt J.; Liu D.; Hasvold L.; Hessler P.; Uziel T.; Lam L.; Rajaraman G.; Fang G.; Elmore S. W.; Rosenberg S. H.; McDaniel K.; Kati W.; Shen Y. Abstract 4718: ABBV-075, a novel BET family bromodomain inhibitor, represents a promising therapeutic agent for a broad spectrum of cancer indications. Cancer Res. 2016, 76, 4718–4718. 10.1158/1538-7445.AM2016-4718. [DOI] [Google Scholar]
- Belkina A. C.; Denis G. V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan B.; Mathur S.; Manjula R.; Tripathi S. Bromodomain and extra-terminal (BET) family proteins: New therapeutic targets in major diseases. J. Biosci. 2016, 41, 295–311. 10.1007/s12038-016-9600-6. [DOI] [PubMed] [Google Scholar]
- Tontsch-Grunt U.; Savarese F.; Gerlach D.; Gianni D.; Baum A.; Scharn D.; Engelhardt H.; Kaya O.; Schweifer N.; Gerstberger T.; Kraut N. Abstract B79: BI 894999, a novel BET inhibitor: Treatment of hematological malignancies by repression of super-enhancer driven oncogenes. Mol. Cancer Ther. 2015, 14, B79. 10.1158/1535-7163.TARG-15-B79. [DOI] [Google Scholar]
- Study of BMS-986158 in subjects with select advanced solid tumors (BET). Clinical trials, 2 April, 2015; https://clinicaltrials.gov/ct2/show/NCT02419417.
- Millan D. S.; Alvarez Morales M. A.; Barr K. J.; Cardillo D.; Collis A.; Dinsmore C. J.; Escobedo J. A.; Foley K. P.; Herbertz T.; Hubbs S.; Kauffman G. S.; Kayser-Bricker K. J.; Kershaw M. T.; Luke G. P.; Martin M. W.; McKinnon C.; Mendes R. L.; Muskiewicz K. R.; Schiller S. E. R.; Soulard P.; Talbot A. C.; Walker D.; Yao L.; Williams G. L. FT-1101: A structurally distinct pan-BET bromodomain inhibitor with activity in preclinical models of hematologic malignancies. Blood 2015, 126, 1367.26224646 [Google Scholar]
- Liu P. C. C.; Liu X. M.; Stubbs M. C.; Maduskuie T.; Sparks R.; Zolotarjova N.; Li J.; Wen X.; Favata M.; Feldman P.; Volgina A.; DiMatteo D.; Collins R.; Falahatpisheh N.; Polam P.; Li Y.; Covington M.; Diamond-Fosbenner S.; Wynn R.; Burn T.; Vaddi K.; Yeleswaram S.; Combs A. P.; Yao W.; Huber R.; Scherle P.; Hollis G. Abstract 3523: Discovery of a novel BET inhibitor INCB054329. Cancer Res. 2015, 75, 3523. 10.1158/1538-7445.AM2015-3523. [DOI] [Google Scholar]
- Dose escalation study of GSK2820151 in subjects with advanced or recurrent solid tumors. Clinical trials, 30 November, 2015; https://clinicaltrials.gov/ct2/show/NCT02630251.
- Attwell S.; Campeau E.; Jahagirdar R.; Kharenko O.; Norek K.; Tsujikawa L.; Calosing C.; Patel R.; Gesner E.; Lakhotia S.; Hansen H. Abstract C86: The clinical candidate ZEN-3694, a novel BET bromodomain inhibitor, is efficacious in the treatment of a variety of solid tumor and hematological malignancies, alone or in combination with several standard of care and targeted therapies. Mol. Cancer Ther. 2015, 14, C86. 10.1158/1535-7163.TARG-15-C86. [DOI] [Google Scholar]
- Bates J.; Kusam S.; Tannheimer S.; Chan J.; Li Y.; Breckenridge D.; Tumas D. The combination of a BET inhibitor (GS-5829) and a BTK Inhibitor (GS-4059) potentiates DLBCL cell line cell death and reduces expression of MYC, IL-10, and IL-6 in vitro. Blood 2016, 128, 5116. [Google Scholar]
- Gjoksi B.; Ghayor C.; Bhattacharya I.; Zenobi-Wong M.; Weber F. E. The bromodomain inhibitor N-methyl pyrrolidone reduced fat accumulation in an ovariectomized rat model. Clin. Epigenet. 2016, 8, 42. 10.1186/s13148-016-0209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. W.; Veschambre P.; Erdjument-Bromage H.; Tempst P.; Conaway J. W.; Conaway R. C.; Kornberg R. D. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 8538–8543. 10.1073/pnas.95.15.8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd S. R.; Pacold M. E.; Huang Q.; Clarke S. M.; Lam F. C.; Cannell I. G.; Bryson B. D.; Rameseder J.; Lee M. J.; Blake E. J.; Fydrych A.; Ho R.; Greenberger B. A.; Chen G. C.; Maffa A.; Del Rosario A. M.; Root D. E.; Carpenter A. E.; Hahn W. C.; Sabatini D. M.; Chen C. C.; White F. M.; Bradner J. E.; Yaffe M. B. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature 2013, 498, 246–250. 10.1038/nature12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. Y.; Chiang C. M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 2007, 282, 13141–13145. 10.1074/jbc.R700001200. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J. P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Muller S.; Pawson T.; Gingras A. C.; Arrowsmith C. H.; Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Wang Y.; Zeng L.; Wu Y.; Deng J.; Zhang Q.; Lin Y.; Li J.; Kang T.; Tao M.; Rusinova E.; Zhang G.; Wang C.; Zhu H.; Yao J.; Zeng Y. X.; Evers B. M.; Zhou M. M.; Zhou B. P. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 2014, 25, 210–225. 10.1016/j.ccr.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmuth F.; Blankenfeldt W.; Geyer M. Structures of the dual bromodomains of the P-TEFb-activating protein Brd4 at atomic resolution. J. Biol. Chem. 2009, 284, 36547–36556. 10.1074/jbc.M109.033712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C. W.; Coste H.; White J. H.; Mirguet O.; Wilde J.; Gosmini R. L.; Delves C.; Magny S. M.; Woodward R.; Hughes S. A.; Boursier E. V.; Flynn H.; Bouillot A. M.; Bamborough P.; Brusq J. M.; Gellibert F. J.; Jones E. J.; Riou A. M.; Homes P.; Martin S. L.; Uings I. J.; Toum J.; Clement C. A.; Boullay A. B.; Grimley R. L.; Blandel F. M.; Prinjha R. K.; Lee K.; Kirilovsky J.; Nicodeme E. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med. Chem. 2011, 54, 3827–3838. 10.1021/jm200108t. [DOI] [PubMed] [Google Scholar]
- Kanno T.; Kanno Y.; Siegel R. M.; Jang M. K.; Lenardo M. J.; Ozato K. Selective recognition of acetylated histones by bromodomain proteins visualized in living cells. Mol. Cell 2004, 13, 33–43. 10.1016/S1097-2765(03)00482-9. [DOI] [PubMed] [Google Scholar]
- Dey A.; Nishiyama A.; Karpova T.; McNally J.; Ozato K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol. Biol. Cell 2009, 20, 4899–4909. 10.1091/mbc.E09-05-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A.; Ellenberg J.; Farina A.; Coleman A. E.; Maruyama T.; Sciortino S.; Lippincott-Schwartz J.; Ozato K. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G(2)-to-M transition. Mol. Cell. Biol. 2000, 20, 6537–6549. 10.1128/MCB.20.17.6537-6549.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J.; Li Q.; Wu C.; Kim J.; Ottinger M.; Howley P. M. Regulation of aurora B expression by the bromodomain protein Brd4. Mol. Cell. Biol. 2009, 29, 5094–5103. 10.1128/MCB.00299-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand P.; Brown J. D.; Lin C. Y.; Qi J.; Zhang R.; Artero P. C.; Alaiti M. A.; Bullard J.; Alazem K.; Margulies K. B.; Cappola T. P.; Lemieux M.; Plutzky J.; Bradner J. E.; Haldar S. M. BET bromodomains mediate transcriptional pause release in heart failure. Cell 2013, 154, 569–582. 10.1016/j.cell.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb E.; Herre M.; Zucker-Scharff I.; Darnell R. B.; Allis C. D. BET protein Brd4 activates transcription in neurons and BET inhibitor Jq1 blocks memory in mice. Nat. Neurosci. 2015, 18, 1464–1473. 10.1038/nn.4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J.; Zhu Y.; Milton J. T.; Price D. H. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998, 12, 755–762. 10.1101/gad.12.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Yik J. H.; Chen R.; He N.; Jang M. K.; Ozato K.; Zhou Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- Jang M. K.; Mochizuki K.; Zhou M.; Jeong H. S.; Brady J. N.; Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- Yang Z.; He N.; Zhou Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell. Biol. 2008, 28, 967–976. 10.1128/MCB.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French C. A.; Miyoshi I.; Kubonishi I.; Grier H. E.; Perez-Atayde A. R.; Fletcher J. A. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Res. 2003, 63, 304–307. [PubMed] [Google Scholar]
- Delmore J. E.; Issa G. C.; Lemieux M. E.; Rahl P. B.; Shi J.; Jacobs H. M.; Kastritis E.; Gilpatrick T.; Paranal R. M.; Qi J.; Chesi M.; Schinzel A. C.; McKeown M. R.; Heffernan T. P.; Vakoc C. R.; Bergsagel P. L.; Ghobrial I. M.; Richardson P. G.; Young R. A.; Hahn W. C.; Anderson K. C.; Kung A. L.; Bradner J. E.; Mitsiades C. S. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz J. A.; Conery A. R.; Bryant B. M.; Sandy P.; Balasubramanian S.; Mele D. A.; Bergeron L.; Sims R. J. 3rd Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 16669–16674. 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asangani I. A.; Dommeti V. L.; Wang X.; Malik R.; Cieslik M.; Yang R.; Escara-Wilke J.; Wilder-Romans K.; Dhanireddy S.; Engelke C.; Iyer M. K.; Jing X.; Wu Y. M.; Cao X.; Qin Z. S.; Wang S.; Feng F. Y.; Chinnaiyan A. M. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. 10.1038/nature13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura M. F.; Fontanals-Cirera B.; Gaziel-Sovran A.; Guijarro M. V.; Hanniford D.; Zhang G.; Gonzalez-Gomez P.; Morante M.; Jubierre L.; Zhang W.; Darvishian F.; Ohlmeyer M.; Osman I.; Zhou M. M.; Hernando E. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013, 73, 6264–6276. 10.1158/0008-5472.CAN-13-0122-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J.; Shi J.; Wang E.; Rappaport A. R.; Herrmann H.; Sison E. A.; Magoon D.; Qi J.; Blatt K.; Wunderlich M.; Taylor M. J.; Johns C.; Chicas A.; Mulloy J. C.; Kogan S. C.; Brown P.; Valent P.; Bradner J. E.; Lowe S. W.; Vakoc C. R. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baratta M. G.; Schinzel A. C.; Zwang Y.; Bandopadhayay P.; Bowman-Colin C.; Kutt J.; Curtis J.; Piao H.; Wong L. C.; Kung A. L.; Beroukhim R.; Bradner J. E.; Drapkin R.; Hahn W. C.; Liu J. F.; Livingston D. M. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 232–237. 10.1073/pnas.1422165112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceribelli M.; Kelly P. N.; Shaffer A. L.; Wright G. W.; Xiao W.; Yang Y.; Mathews Griner L. A.; Guha R.; Shinn P.; Keller J. M.; Liu D.; Patel P. R.; Ferrer M.; Joshi S.; Nerle S.; Sandy P.; Normant E.; Thomas C. J.; Staudt L. M. Blockade of oncogenic IkappaB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 11365–11370. 10.1073/pnas.1411701111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puissant A.; Frumm S. M.; Alexe G.; Bassil C. F.; Qi J.; Chanthery Y. H.; Nekritz E. A.; Zeid R.; Gustafson W. C.; Greninger P.; Garnett M. J.; McDermott U.; Benes C. H.; Kung A. L.; Weiss W. A.; Bradner J. E.; Stegmaier K. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discovery 2013, 3, 308–323. 10.1158/2159-8290.CD-12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood W. W.; Zejnullahu K.; Bradner J. E.; Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 19408–19413. 10.1073/pnas.1216363109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B.; Yang X. D.; Zhou M. M.; Ozato K.; Chen L. F. Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol. Cell. Biol. 2009, 29, 1375–1387. 10.1128/MCB.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. F.; Mu Y.; Greene W. C. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. EMBO. J. 2002, 21, 6539–6548. 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding N.; Hah N.; Yu R. T.; Sherman M. H.; Benner C.; Leblanc M.; He M.; Liddle C.; Downes M.; Evans R. M. BRD4 is a novel therapeutic target for liver fibrosis. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 15713–15718. 10.1073/pnas.1522163112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B.; Zhao Y.; Sun H.; Zhang Y.; Yang J.; Brasier A. R. BRD4 mediates NFkB-dependent epithelial-mesenchymal transition and pulmonary fibrosis via transcriptional elongation. Am. J. Physiol. 2016, 311, L1183. 10.1152/ajplung.00224.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C. W.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan Y. M.; Kirkham P.; Barnes P. J.; Adcock I. M. Brd4 is essential for IL-1beta-induced inflammation in human airway epithelial cells. PLoS One 2014, 9, e95051. 10.1371/journal.pone.0095051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Peng R.; Phillips J. E.; Deguzman J.; Ren Y.; Apparsundaram S.; Luo Q.; Bauer C. M.; Fuentes M. E.; DeMartino J. A.; Tyagi G.; Garrido R.; Hogaboam C. M.; Denton C. P.; Holmes A. M.; Kitson C.; Stevenson C. S.; Budd D. C. Assessment of Brd4 inhibition in idiopathic pulmonary fibrosis lung fibroblasts and in vivo models of lung fibrosis. Am. J. Pathol. 2013, 183, 470–479. 10.1016/j.ajpath.2013.04.020. [DOI] [PubMed] [Google Scholar]
- Tang X.; Peng R.; Ren Y.; Apparsundaram S.; Deguzman J.; Bauer C. M.; Hoffman A. F.; Hamilton S.; Liang Z.; Zeng H.; Fuentes M. E.; Demartino J. A.; Kitson C.; Stevenson C. S.; Budd D. C. BET bromodomain proteins mediate downstream signaling events following growth factor stimulation in human lung fibroblasts and are involved in bleomycin-induced pulmonary fibrosis. Mol. Pharmacol. 2013, 83, 283–293. 10.1124/mol.112.081661. [DOI] [PubMed] [Google Scholar]
- Bailey D.; Jahagirdar R.; Gordon A.; Hafiane A.; Campbell S.; Chatur S.; Wagner G. S.; Hansen H. C.; Chiacchia F. S.; Johansson J.; Krimbou L.; Wong N. C.; Genest J. RVX-208: a small molecule that increases apolipoprotein A-I and high-density lipoprotein cholesterol in vitro and in vivo. J. Am. Coll. Cardiol. 2010, 55, 2580–2589. 10.1016/j.jacc.2010.02.035. [DOI] [PubMed] [Google Scholar]
- Jahagirdar R.; Zhang H.; Azhar S.; Tobin J.; Attwell S.; Yu R.; Wu J.; McLure K. G.; Hansen H. C.; Wagner G. S.; Young P. R.; Srivastava R. A.; Wong N. C.; Johansson J. A novel BET bromodomain inhibitor, RVX-208, shows reduction of atherosclerosis in hyperlipidemic ApoE deficient mice. Atherosclerosis 2014, 236, 91–100. 10.1016/j.atherosclerosis.2014.06.008. [DOI] [PubMed] [Google Scholar]
- Spiltoir J. I.; Stratton M. S.; Cavasin M. A.; Demos-Davies K.; Reid B. G.; Qi J.; Bradner J. E.; McKinsey T. A. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2013, 63, 175–179. 10.1016/j.yjmcc.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartor G. C.; Powell S. K.; Brothers S. P.; Wahlestedt C. Epigenetic readers of lysine acetylation regulate cocaine-induced plasticity. J. Neurosci. 2015, 35, 15062–15072. 10.1523/JNEUROSCI.0826-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gacias M.; Gerona-Navarro G.; Plotnikov A. N.; Zhang G.; Zeng L.; Kaur J.; Moy G.; Rusinova E.; Rodriguez Y.; Matikainen B.; Vincek A.; Joshua J.; Casaccia P.; Zhou M. M. Selective chemical modulation of gene transcription favors oligodendrocyte lineage progression. Chem. Biol. 2014, 21, 841–854. 10.1016/j.chembiol.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou M.; Huang K.; Jung K. J.; Cho W. K.; Klase Z.; Kashanchi F.; Pise-Masison C. A.; Brady J. N. Bromodomain protein Brd4 regulates human immunodeficiency virus transcription through phosphorylation of CDK9 at threonine 29. J. Virol. 2009, 83, 1036–1044. 10.1128/JVI.01316-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A.; Wang S.; Nguyen T.; Shire K.; Frappier L. The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. J. Virol. 2008, 82, 12009–12019. 10.1128/JVI.01680-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon D.; Joubert S.; Senechal H.; Fradet-Turcotte A.; Torre S.; Archambault J. Proteasomal degradation of the papillomavirus E2 protein is inhibited by overexpression of bromodomain-containing protein 4. J. Virol. 2009, 83, 4127–4139. 10.1128/JVI.02468-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. Y.; Lee A. Y.; Hou S. Y.; Kemper J. K.; Erdjument-Bromage H.; Tempst P.; Chiang C. M. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006, 20, 2383–2396. 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J.; Croyle J. L.; Nishimura A.; Ozato K.; Howley P. M. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 2004, 117, 349–360. 10.1016/S0092-8674(04)00402-7. [DOI] [PubMed] [Google Scholar]
- Boehm D.; Conrad R. J.; Ott M. Bromodomain proteins in HIV infection. Viruses 2013, 5, 1571–1586. 10.3390/v5061571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgrove D. A.; Mahmoudi T.; Henklein P.; Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 13690–13695. 10.1073/pnas.0705053104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Gaiha G. D.; John S. P.; Pertel T.; Chin C. R.; Gao G.; Qu H.; Walker B. D.; Elledge S. J.; Brass A. L. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012, 2, 807–816. 10.1016/j.celrep.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P.; Qu X.; Shen Y.; Jiang Z.; Wang P.; Zeng H.; Ji H.; Deng J.; Yang X.; Li X.; Lu H.; Zhu H. The BET inhibitor OTX015 reactivates latent HIV-1 through P-TEFb. Sci. Rep. 2016, 6, 24100. 10.1038/srep24100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee C.; Archin N.; Michaels D.; Belkina A. C.; Denis G. V.; Bradner J.; Sebastiani P.; Margolis D. M.; Montano M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukocyte Biol. 2012, 92, 1147–1154. 10.1189/jlb.0312165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm D.; Calvanese V.; Dar R. D.; Xing S.; Schroeder S.; Martins L.; Aull K.; Li P. C.; Planelles V.; Bradner J. E.; Zhou M. M.; Siliciano R. F.; Weinberger L.; Verdin E.; Ott M. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. 10.4161/cc.23309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Guo J.; Wu Y.; Zhou Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287. 10.1093/nar/gks976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archin N. M.; Liberty A. L.; Kashuba A. D.; Choudhary S. K.; Kuruc J. D.; Crooks A. M.; Parker D. C.; Anderson E. M.; Kearney M. F.; Strain M. C.; Richman D. D.; Hudgens M. G.; Bosch R. J.; Coffin J. M.; Eron J. J.; Hazuda D. J.; Margolis D. M. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi S.; Ooike S.; Iwata K.; Hikawa H.; Sugahara K.. Antitumor agents. WO2009084693, 2009.
- Nasveschuk C. G.; Audia J. E.; Leblanc Y.; Hewitt M. C.; Côté A.; Vaswani R. G.; Albrecht B. K.; Taylor A. M.; Harmange J. C.; Gehling V. S.. Bromodomain inhibitors and uses thereof. US08796261, 2010.
- Jang J. E.; Eom J. I.; Jeung H. K.; Cheong J. W.; Lee J. Y.; Kim J. S.; Min Y. H. AMPK-ULK1-mediated autophagy confers resistance to BET inhibitor JQ1 in acute myeloid leukemia stem cells. Clin. Cancer Res. 2016, 1–39. 10.1158/1078-0432.CCR-16-1903. [DOI] [PubMed] [Google Scholar]
- Yokoyama Y.; Zhu H.; Lee J. H.; Kossenkov A. V.; Wu S. Y.; Wickramasinghe J. M.; Yin X.; Palozola K. C.; Gardini A.; Showe L. C.; Zaret K. S.; Liu Q.; Speicher D.; Conejo-Garcia J. R.; Bradner J. E.; Zhang Z.; Sood A. K.; Ordog T.; Bitler B. G.; Zhang R. BET inhibitors suppress ALDH activity by targeting ALDH1A1 super-enhancer in ovarian cancer. Cancer Res. 2016, 76, 6320–6330. 10.1158/0008-5472.CAN-16-0854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Motta L. L.; Ledaki I.; Purshouse K.; Haider S.; De Bastiani M. A.; Baban D.; Morotti M.; Steers G.; Wigfield S.; Bridges E.; Li J. L.; Knapp S.; Ebner D.; Klamt F.; Harris A. L.; McIntyre A. The BET inhibitor JQ1 selectively impairs tumour response to hypoxia and downregulates CA9 and angiogenesis in triple negative breast cancer. Oncogene 2017, 36, 122. 10.1038/onc.2016.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krugmann S.; Anderson K. E.; Ridley S. H.; Risso N.; McGregor A.; Coadwell J.; Davidson K.; Eguinoa A.; Ellson C. D.; Lipp P.; Manifava M.; Ktistakis N.; Painter G.; Thuring J. W.; Cooper M. A.; Lim Z. Y.; Holmes A. B.; Dove S. K.; Michell R. H.; Grewal A.; Nazarian A.; Erdjument-Bromage H.; Tempst P.; Stephens L. R.; Hawkins P. T. Identification of ARAP3, a novel PI3K effector regulating both Arf and Rho GTPases, by selective capture on phosphoinositide affinity matrices. Mol. Cell 2002, 9, 95–108. 10.1016/S1097-2765(02)00434-3. [DOI] [PubMed] [Google Scholar]
- Conway S. J.; Gardiner J.; Grove S. J.; Johns M. K.; Lim Z. Y.; Painter G. F.; Robinson D. E.; Schieber C.; Thuring J. W.; Wong L. S.; Yin M. X.; Burgess A. W.; Catimel B.; Hawkins P. T.; Ktistakis N. T.; Stephens L. R.; Holmes A. B. Synthesis and biological evaluation of phosphatidylinositol phosphate affinity probes. Org. Biomol. Chem. 2010, 8, 66–76. 10.1039/B913399B. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Yang C. Y.; Wang S. The making of I-BET762, a BET bromodomain inhibitor now in clinical development. J. Med. Chem. 2013, 56, 7498–7500. 10.1021/jm4014407. [DOI] [PubMed] [Google Scholar]
- Gehling V. S.; Hewitt M. C.; Vaswani R. G.; Leblanc Y.; Cote A.; Nasveschuk C. G.; Taylor A. M.; Harmange J. C.; Audia J. E.; Pardo E.; Joshi S.; Sandy P.; Mertz J. A.; Sims R. J. 3rd; Bergeron L.; Bryant B. M.; Bellon S.; Poy F.; Jayaram H.; Sankaranarayanan R.; Yellapantula S.; Bangalore Srinivasamurthy N.; Birudukota S.; Albrecht B. K. Discovery, design, and optimization of isoxazole azepine BET inhibitors. ACS Med. Chem. Lett. 2013, 4, 835–840. 10.1021/ml4001485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht B. K.; Gehling V. S.; Hewitt M. C.; Vaswani R. G.; Cote A.; Leblanc Y.; Nasveschuk C. G.; Bellon S.; Bergeron L.; Campbell R.; Cantone N.; Cooper M. R.; Cummings R. T.; Jayaram H.; Joshi S.; Mertz J. A.; Neiss A.; Normant E.; O’Meara M.; Pardo E.; Poy F.; Sandy P.; Supko J.; Sims R. J. 3rd; Harmange J. C.; Taylor A. M.; Audia J. E. Identification of a benzoisoxazoloazepine inhibitor (CPI-0610) of the bromodomain and extra-terminal (BET) Family as a candidate for human clinical trials. J. Med. Chem. 2016, 59, 1330–1339. 10.1021/acs.jmedchem.5b01882. [DOI] [PubMed] [Google Scholar]
- Hewitt M. C.; Leblanc Y.; Gehling V. S.; Vaswani R. G.; Cote A.; Nasveschuk C. G.; Taylor A. M.; Harmange J. C.; Audia J. E.; Pardo E.; Cummings R.; Joshi S.; Sandy P.; Mertz J. A.; Sims R. J. 3rd; Bergeron L.; Bryant B. M.; Bellon S.; Poy F.; Jayaram H.; Tang Y.; Albrecht B. K. Development of methyl isoxazoleazepines as inhibitors of BET. Bioorg. Med. Chem. Lett. 2015, 25, 1842–1848. 10.1016/j.bmcl.2015.03.045. [DOI] [PubMed] [Google Scholar]
- Taylor A. M.; Vaswani R. G.; Gehling V. S.; Hewitt M. C.; Leblanc Y.; Audia J. E.; Bellon S.; Cummings R. T.; Cote A.; Harmange J. C.; Jayaram H.; Joshi S.; Lora J. M.; Mertz J. A.; Neiss A.; Pardo E.; Nasveschuk C. G.; Poy F.; Sandy P.; Setser J. W.; Sims R. J. 3rd; Tang Y.; Albrecht B. K. Discovery of benzotriazolo[4,3-d][1,4]diazepines as orally active inhibitors of BET bromodomains. ACS Med. Chem. Lett. 2016, 7, 145–150. 10.1021/ml500411h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau S. B.; Kagey M.. Bromodomain inhibitors. WO2016069578, 2016.
- Filippakopoulos P.; Picaud S.; Fedorov O.; Keller M.; Wrobel M.; Morgenstern O.; Bracher F.; Knapp S. Benzodiazepines and benzotriazepines as protein interaction inhibitors targeting bromodomains of the BET family. Bioorg. Med. Chem. 2012, 20, 1878–1886. 10.1016/j.bmc.2011.10.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo J.; Hikawa H.; Hamada M.; Ishibuchi S.; Fujie N.; Sugiyama N.; Tanaka M.; Kobayashi H.; Sugahara K.; Oshita K.; Iwata K.; Ooike S.; Murata M.; Sumichika H.; Chiba K.; Adachi K. A phenotypic drug discovery study on thienodiazepine derivatives as inhibitors of T cell proliferation induced by CD28 co-stimulation leads to the discovery of a first bromodomain inhibitor. Bioorg. Med. Chem. Lett. 2016, 26, 1365–1370. 10.1016/j.bmcl.2016.01.084. [DOI] [PubMed] [Google Scholar]
- Schmees N.; Buchmann B.; Haendler B.; Neuhaus R.; Lejeune P.; Kruger M.; Fernandez-Montalvan A.; Kunzer H.; Rehwinkel H.. 4-Substituted pyrrolo- and pyrazolo- diazepines. WO2014128111, 2014.
- Vadivelu S.; Rajagopal S.; Chinnapattu M.; Gondrala P. K.; Sivanandhan D.; Mulakala C.. Tricyclic fused derivatives of 1-(cyclo)alkyl pyridin-2-one useful for the treatment of cancer. WO2016157221, 2016.
- Liu D.; Pratt J.; Wang L.; Hasvold L. A.; Bogdan A.. Bromodomain inhibitors. US20140256710, 2014.
- Li J.; Wang P.; Zhou B.; Shi J.; Liu J.; Li X.; Fan L.; Zheng Y.; Ouyang L. Development of 4,5-dihydro-benzodiazepinone derivatives as a new chemical series of BRD4 inhibitors. Eur. J. Med. Chem. 2016, 121, 294–299. 10.1016/j.ejmech.2016.05.057. [DOI] [PubMed] [Google Scholar]
- Siegel S.; Baurle S.; Cleve A.; Haendler B.; Fernandez-Montalvan A.; Monning U.; Krause S.; Lejeune P.; Busemann M.; Kuhnke J.. Bicyclo 2,3-benzodiazepines and spirocyclically substituted 2,3-benzodiazepines. WO2014128067, 2014.
- Hewings D. S.; Wang M.; Philpott M.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Vuppusetty C.; Marsden B.; Knapp S.; Conway S. J.; Heightman T. D. 3,5-dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J. Med. Chem. 2011, 54, 6761–6770. 10.1021/jm200640v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewings D. S.; Fedorov O.; Filippakopoulos P.; Martin S.; Picaud S.; Tumber A.; Wells C.; Olcina M. M.; Freeman K.; Gill A.; Ritchie A. J.; Sheppard D. W.; Russell A. J.; Hammond E. M.; Knapp S.; Brennan P. E.; Conway S. J. Optimization of 3,5-dimethylisoxazole derivatives as potent bromodomain ligands. J. Med. Chem. 2013, 56, 3217–3227. 10.1021/jm301588r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamborough P.; Diallo H.; Goodacre J. D.; Gordon L.; Lewis A.; Seal J. T.; Wilson D. M.; Woodrow M. D.; Chung C. W. Fragment-based discovery of bromodomain inhibitors part 2: optimization of phenylisoxazole sulfonamides. J. Med. Chem. 2012, 55, 587–596. 10.1021/jm201283q. [DOI] [PubMed] [Google Scholar]
- McKeown M. R.; Shaw D. L.; Fu H.; Liu S.; Xu X.; Marineau J. J.; Huang Y.; Zhang X.; Buckley D. L.; Kadam A.; Zhang Z.; Blacklow S. C.; Qi J.; Zhang W.; Bradner J. E. Biased multicomponent reactions to develop novel bromodomain inhibitors. J. Med. Chem. 2014, 57, 9019–9027. 10.1021/jm501120z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirguet O.; Lamotte Y.; Donche F.; Toum J.; Gellibert F.; Bouillot A.; Gosmini R.; Nguyen V. L.; Delannee D.; Seal J.; Blandel F.; Boullay A. B.; Boursier E.; Martin S.; Brusq J. M.; Krysa G.; Riou A.; Tellier R.; Costaz A.; Huet P.; Dudit Y.; Trottet L.; Kirilovsky J.; Nicodeme E. From ApoA1 upregulation to BET family bromodomain inhibition: discovery of I-BET151. Bioorg. Med. Chem. Lett. 2012, 22, 2963–2967. 10.1016/j.bmcl.2012.01.125. [DOI] [PubMed] [Google Scholar]
- Seal J.; Lamotte Y.; Donche F.; Bouillot A.; Mirguet O.; Gellibert F.; Nicodeme E.; Krysa G.; Kirilovsky J.; Beinke S.; McCleary S.; Rioja I.; Bamborough P.; Chung C. W.; Gordon L.; Lewis T.; Walker A. L.; Cutler L.; Lugo D.; Wilson D. M.; Witherington J.; Lee K.; Prinjha R. K. Identification of a novel series of BET family bromodomain inhibitors: binding mode and profile of I-BET151 (GSK1210151A). Bioorg. Med. Chem. Lett. 2012, 22, 2968–2972. 10.1016/j.bmcl.2012.02.041. [DOI] [PubMed] [Google Scholar]
- Dawson M. A.; Prinjha R. K.; Dittmann A.; Giotopoulos G.; Bantscheff M.; Chan W. I.; Robson S. C.; Chung C. W.; Hopf C.; Savitski M. M.; Huthmacher C.; Gudgin E.; Lugo D.; Beinke S.; Chapman T. D.; Roberts E. J.; Soden P. E.; Auger K. R.; Mirguet O.; Doehner K.; Delwel R.; Burnett A. K.; Jeffrey P.; Drewes G.; Lee K.; Huntly B. J.; Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran X.; Zhao Y.; Liu L.; Bai L.; Yang C. Y.; Zhou B.; Meagher J. L.; Chinnaswamy K.; Stuckey J. A.; Wang S. Structure-based design of gamma-carboline analogues as potent and specific BET bromodomain inhibitors. J. Med. Chem. 2015, 58, 4927–4939. 10.1021/acs.jmedchem.5b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slassi A.; Dove P.. Fluorinated imidazo[4,5-c]quinoline derivatives as inhibitors of bromodomain containing proteins. WO2016123709, 2016.
- Norris D. J.; Dellucca G. V.; Gavai A. V.; Quesnelle C. A.; Gill P.; O’malley D.; Vaccaro W.; lEE F. Y.; Debenedetto M. V.; Degnan A. P.; Fang H.; Hill M. D.; Huang H.; Schmitz W. D.; Starrett J. E.; Han W.; Tokarski J. S.; Mandal S. K.. Tricyclic compounds as anticancer agents. WO2015100282, 2015.
- Foster A. B. Deuterium isotope effects in studies of drug metabolism. Trends Pharmacol. Sci. 1984, 5, 524–527. 10.1016/0165-6147(84)90534-0. [DOI] [Google Scholar]
- Timmins G. S. Deuterated drugs: where are we now?. Expert Opin. Ther. Pat. 2014, 24, 1067–1075. 10.1517/13543776.2014.943184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D.; Fedorov O.; Filippakopoulos P.; Martin S.; Philpott M.; Picaud S.; Hewings D. S.; Uttakar S.; Heightman T. D.; Conway S. J.; Knapp S.; Brennan P. E. The design and synthesis of 5- and 6-isoxazolylbenzimidazoles as selective inhibitors of the BET bromodomains. MedChemComm 2013, 4, 140–144. 10.1039/C2MD20189E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B.; Zhou C.; Wang H.. Substituted 5-(3,5-dimethylisoxazole-4-yl)indoline-2-ones. WO2014173241, 2014.
- Aktoudianakis E.; Chin G.; Corkey B. K.; Elbel J. D.; Jiang K.; Kobayashi R.; Martinez T.; Metobo T. R.; Mish S. E.; Shevick M.; Sperandio S.; Yang D.; Zablocki H.. J. Benzimidazolone derivatives as bromodomain inhibitors. WO2014160873, 2014.
- Engelhardt H.; Gianni D.; Mantoulidis A.; Smethurst C.. Indolinone analogues as BRD4 inhibitors. WO2014154760, 2014.
- Smethurst C.; Engelhardt H.; Gianni D.; Reiser U.. Dihydroquinazolinone analogues as BRD4 inhibitors. WO2014154762, 2014.
- Bogdan A.; Kati W. M.; Mcdaniel K. F.; Park C. H.; Sheppard G. S.; Wang L.. Bromodomain inhibitors. US20150158873, 2015.
- Wang L.; Frey R. R.; Hansen T. M.; Liu D.; Mcclellan W. J.; Mcdaniel K. F.; Pratt J. K.; Wada C. K.. Bromodomain inhibitors. WO2015085925, 2015.
- Engelhardt H.Benzimidazole derivatives. WO2015169962, 2015.
- Engelhardt H.; Martin L.; Smethurst C.. Pyridinones. WO2015022332, 2015.
- Wang L.; Dai Y.; Holms J.; Liu D.; Mcclellan W.; Mcdaniel K.; Hasvold L.; Fidanze S. D.; Sheppard G.; Marjanovic J.. Bromodomain inhibitors. WO2014206150, 2014.
- Abdel-Magid A. F. Inhibitors of BRD4 as potential cancer therapy. ACS Med. Chem. Lett. 2016, 7, 728–729. 10.1021/acsmedchemlett.6b00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt H.; Gianni D.; Smethurst C.. Substituted [1,2,4]triazolo[4,3-a]pyrazines as BRD4 inhibitors. US20160129001, 2016.
- Engelhardt H.Triazolopyrazine derivatives as BRD4 inhibitors. WO2015067770, 2015.
- Bordas V.; Cotesta S.; Guagnano V.; Rueeger H.; Vaupel A.. Pyrazolopyrrolidine derivatives and their use in the treatment of disease. US20140349990, 2014.
- Blank J.; Bold G.; Bordas V.; Cotesta S.; Guagnano V.; Rueger H.; Vaupel A.. Pyrrolopyrrolone derivatives and their use as BET inhibitors. WO2015075665, 2015.
- Gosmini R.; Nguyen V. L.; Toum J.; Simon C.; Brusq J. M.; Krysa G.; Mirguet O.; Riou-Eymard A. M.; Boursier E. V.; Trottet L.; Bamborough P.; Clark H.; Chung C. W.; Cutler L.; Demont E. H.; Kaur R.; Lewis A. J.; Schilling M. B.; Soden P. E.; Taylor S.; Walker A. L.; Walker M. D.; Prinjha R. K.; Nicodeme E. The discovery of I-BET726 (GSK1324726A), a potent tetrahydroquinoline ApoA1 up-regulator and selective BET bromodomain inhibitor. J. Med. Chem. 2014, 57, 8111–8131. 10.1021/jm5010539. [DOI] [PubMed] [Google Scholar]
- Chung C. W.; Dean A. W.; Woolven J. M.; Bamborough P. Fragment-based discovery of bromodomain inhibitors part 1: inhibitor binding modes and implications for lead discovery. J. Med. Chem. 2012, 55, 576–586. 10.1021/jm201320w. [DOI] [PubMed] [Google Scholar]
- Wyce A.; Ganji G.; Smitheman K. N.; Chung C. W.; Korenchuk S.; Bai Y.; Barbash O.; Le B.; Craggs P. D.; McCabe M. T.; Kennedy-Wilson K. M.; Sanchez L. V.; Gosmini R. L.; Parr N.; McHugh C. F.; Dhanak D.; Prinjha R. K.; Auger K. R.; Tummino P. J. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLoS One 2013, 8, e72967. 10.1371/journal.pone.0072967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amans D.; Atkinson S. J.; Harrison D. J.; Hirst D. J.; Law R. P.; Lindon M.; Preston A.; Seal J. T.; Wellaway C. R.. 2,3-Disubstituted 1-acyl-4-amino-1,2,3,4-tetrahydroquinoline derivatives and their use as bromodomain inhibitors. WO2014140076, 2014.
- Bair K. W.; Herbertz T.; Kauffman G. S.; Kayser-Bricker K. J.; Luke G. P.; Martin M. W.; Millan D. S.; Schiller S. R.; Talbot A. C.. Tetrahydroquinoline composition as BET bromodomain inhibitors. WO2015074064, 2015.
- Schmees N.; Haendler B.; Stockigt D.; Gallenkamp D.; Bissell R. A.; Bouglas R. A.. Modified BET-protein-inhibiting dihydroquinoxalinones and dihydropyridopyrazinones. WO2015004075, 2015.
- Fish P. V.; Filippakopoulos P.; Bish G.; Brennan P. E.; Bunnage M. E.; Cook A. S.; Federov O.; Gerstenberger B. S.; Jones H.; Knapp S.; Marsden B.; Nocka K.; Owen D. R.; Philpott M.; Picaud S.; Primiano M. J.; Ralph M. J.; Sciammetta N.; Trzupek J. D. Identification of a chemical probe for bromo and extra C-terminal bromodomain inhibition through optimization of a fragment-derived hit. J. Med. Chem. 2012, 55, 9831–9837. 10.1021/jm3010515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X.; Zhang Y.; Liu Z.; Song M.; Xing Y.; Xiang Q.; Wang Z.; Tu Z.; Zhou Y.; Ding K.; Xu Y. Discovery of benzo[cd]indol-2(1H)-ones as potent and specific BET bromodomain inhibitors: Structure-based virtual screening, optimization, and biological evaluation. J. Med. Chem. 2016, 59, 1565–1579. 10.1021/acs.jmedchem.5b01511. [DOI] [PubMed] [Google Scholar]
- Hasvold L. A.; Pratt J. K.; Mcdaniel K. F.; Sheppard G. S.; Liu D.; Elmore S. W.; Hubbard R. D.. Pyrrole-3-carboxamide bromodomain inhibitors. US20140275079, 2014.
- Hasvold L. A.; Liu D.; Park C. H.; Pratt J. K.; Sheppard G. S.; Wang L.. Isoindolone derivatives. WO2013158952, 2013.
- Lucas X.; Wohlwend D.; Hugle M.; Schmidtkunz K.; Gerhardt S.; Schule R.; Jung M.; Einsle O.; Gunther S. 4-Acyl pyrroles: mimicking acetylated lysines in histone code reading. Angew. Chem., Int. Ed. 2013, 52, 14055–14059. 10.1002/anie.201307652. [DOI] [PubMed] [Google Scholar]
- Hugle M.; Lucas X.; Weitzel G.; Ostrovskyi D.; Breit B.; Gerhardt S.; Einsle O.; Gunther S.; Wohlwend D. 4-Acyl pyrrole derivatives yield novel vectors for designing inhibitors of the acetyl-lysine recognition site of BRD4(1). J. Med. Chem. 2016, 59, 1518–1530. 10.1021/acs.jmedchem.5b01267. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Cao D.; Chen T.; Wang Y.; Miao Z.; Xu Y.; Chen W.; Wang X.; Li Y.; Du Z.; Xiong B.; Li J.; Xu C.; Zhang N.; He J.; Shen J. Fragment-based drug discovery of 2-thiazolidinones as inhibitors of the histone reader BRD4 bromodomain. J. Med. Chem. 2013, 56, 3833–3851. 10.1021/jm301793a. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Wang Y.; Cao D.; Chen T.; Wang Q.; Li Y.; Xu Y.; Zhang N.; Wang X.; Chen D.; Chen L.; Chen Y. L.; Xia G.; Shi Z.; Liu Y. C.; Lin Y.; Miao Z.; Shen J.; Xiong B. Fragment-based drug discovery of 2-thiazolidinones as BRD4 inhibitors: 2. Structure-based optimization. J. Med. Chem. 2015, 58, 1281–1297. 10.1021/jm501504k. [DOI] [PubMed] [Google Scholar]
- Toure M.; Crews C. M. Small-molecule PROTACS: new approaches to protein degradation. Angew. Chem., Int. Ed. 2016, 55, 1966–1973. 10.1002/anie.201507978. [DOI] [PubMed] [Google Scholar]
- Buckley D. L.; Crews C. M. Small-molecule control of intracellular protein levels through modulation of the ubiquitin proteasome system. Angew. Chem., Int. Ed. 2014, 53, 2312–2330. 10.1002/anie.201307761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raina K.; Crews C. M. Chemical inducers of targeted protein degradation. J. Biol. Chem. 2010, 285, 11057–11060. 10.1074/jbc.R109.078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulatov E.; Ciulli A. Targeting Cullin–RING E3 ubiquitin ligases for drug discovery: structure, assembly and small-molecule modulation. Biochem. J. 2015, 467, 365–386. 10.1042/BJ20141450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G. E.; Buckley D. L.; Paulk J.; Roberts J. M.; Souza A.; Dhe-Paganon S.; Bradner J. E. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 2015, 348, 1376–1381. 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Qian Y.; Altieri M.; Dong H.; Wang J.; Raina K.; Hines J.; Winkler J. D.; Crew A. P.; Coleman K.; Crews C. M. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015, 22, 755–763. 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengerle M.; Chan K. H.; Ciulli A. Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 2015, 10, 1770–1777. 10.1021/acschembio.5b00216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M.; Roberts J. M.; Seo H. S.; Souza A.; Paulk J.; Scott T. G.; DeAngelo S. L.; Dhe-Paganon S.; Bradner J. E. Design and characterization of bivalent BET inhibitors. Nat. Chem. Biol. 2016, 12, 1089–1096. 10.1038/nchembio.2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhyasen G. W.; Hattersley M. M.; Yao Y.; Dulak A.; Wang W.; Petteruti P.; Dale I. L.; Boiko S.; Cheung T.; Zhang J.; Wen S.; Castriotta L.; Lawson D.; Collins M.; Bao L.; Ahdesmaki M. J.; Walker G.; O’Connor G.; Yeh T. C.; Rabow A. A.; Dry J. R.; Reimer C.; Lyne P.; Mills G. B.; Fawell S. E.; Waring M. J.; Zinda M.; Clark E.; Chen H. AZD5153: A novel bivalent BET bromodomain inhibitor highly active against hematologic malignancies. Mol. Cancer Ther. 2016, 15, 2563–2574. 10.1158/1535-7163.MCT-16-0141. [DOI] [PubMed] [Google Scholar]
- Martin M. P.; Olesen S. H.; Georg G. I.; Schonbrunn E. Cyclin-dependent kinase inhibitor dinaciclib interacts with the acetyl-lysine recognition site of bromodomains. ACS Chem. Biol. 2013, 8, 2360–2365. 10.1021/cb4003283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ember S. W.; Zhu J. Y.; Olesen S. H.; Martin M. P.; Becker A.; Berndt N.; Georg G. I.; Schonbrunn E. Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem. Biol. 2014, 9, 1160–1171. 10.1021/cb500072z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steegmaier M.; Hoffmann M.; Baum A.; Lenart P.; Petronczki M.; Krssak M.; Gurtler U.; Garin-Chesa P.; Lieb S.; Quant J.; Grauert M.; Adolf G. R.; Kraut N.; Peters J. M.; Rettig W. J. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007, 17, 316–322. 10.1016/j.cub.2006.12.037. [DOI] [PubMed] [Google Scholar]
- Ciceri P.; Muller S.; O’Mahony A.; Fedorov O.; Filippakopoulos P.; Hunt J. P.; Lasater E. A.; Pallares G.; Picaud S.; Wells C.; Martin S.; Wodicka L. M.; Shah N. P.; Treiber D. K.; Knapp S. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat. Chem. Biol. 2014, 10, 305–312. 10.1038/nchembio.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Yap J. L.; Yoshioka M.; Lanning M. E.; Fountain R. N.; Raje M.; Scheenstra J. A.; Strovel J. W.; Fletcher S. BRD4 structure-activity relationships of dual PLK1 kinase/BRD4 bromodomain inhibitor BI-2536. ACS Med. Chem. Lett. 2015, 6, 764–769. 10.1021/acsmedchemlett.5b00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmann A.; Werner T.; Chung C. W.; Savitski M. M.; Falth Savitski M.; Grandi P.; Hopf C.; Lindon M.; Neubauer G.; Prinjha R. K.; Bantscheff M.; Drewes G. The commonly used PI3-kinase probe LY294002 is an inhibitor of BET bromodomains. ACS Chem. Biol. 2014, 9, 495–502. 10.1021/cb400789e. [DOI] [PubMed] [Google Scholar]
- Allen B. K.; Mehta S.; Ember S. W.; Schonbrunn E.; Ayad N.; Schurer S. C. Large-scale computational screening identifies first in class multitarget inhibitor of EGFR kinase and BRD4. Sci. Rep. 2015, 5, 16924. 10.1038/srep16924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Hou S.; Chen H.; Ran T.; Jiang F.; Bian Y.; Zhang D.; Zhi Y.; Wang L.; Zhang L.; Li H.; Zhang Y.; Tang W.; Lu T.; Chen Y. Targeting epigenetic reader and eraser: Rational design, synthesis and in vitro evaluation of dimethylisoxazoles derivatives as BRD4/HDAC dual inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 2931–2935. 10.1016/j.bmcl.2016.04.034. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Plotnikov A. N.; Rusinova E.; Shen T.; Morohashi K.; Joshua J.; Zeng L.; Mujtaba S.; Ohlmeyer M.; Zhou M. M. Structure-guided design of potent diazobenzene inhibitors for the BET bromodomains. J. Med. Chem. 2013, 56, 9251–9264. 10.1021/jm401334s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galdeano C.; Ciulli A. Selectivity on-target of bromodomain chemical probes by structure-guided medicinal chemistry and chemical biology. Future Med. Chem. 2016, 8, 1655–1680. 10.4155/fmc-2016-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raux B.; Voitovich Y.; Derviaux C.; Lugari A.; Rebuffet E.; Milhas S.; Priet S.; Roux T.; Trinquet E.; Guillemot J. C.; Knapp S.; Brunel J. M.; Fedorov A. Y.; Collette Y.; Roche P.; Betzi S.; Combes S.; Morelli X. Exploring selective inhibition of the first bromodomain of the human bromodomain and extra-terminal domain (BET) proteins. J. Med. Chem. 2016, 59, 1634–1641. 10.1021/acs.jmedchem.5b01708. [DOI] [PubMed] [Google Scholar]
- Devaiah B. N.; Lewis B. A.; Cherman N.; Hewitt M. C.; Albrecht B. K.; Robey P. G.; Ozato K.; Sims R. J. 3rd; Singer D. S. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 6927–6932. 10.1073/pnas.1120422109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud M. G.; Lin-Shiao E.; Cardote T.; Tallant C.; Pschibul A.; Chan K. H.; Zengerle M.; Garcia J. R.; Kwan T. T.; Ferguson F. M.; Ciulli A. Chemical biology. A bump-and-hole approach to engineer controlled selectivity of BET bromodomain chemical probes. Science 2014, 346, 638–641. 10.1126/science.1249830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier J. M.; Sharp P. P.; Burns C. J. BET bromodomain inhibitors: a patent review. Expert Opin. Ther. Pat. 2014, 24, 185–199. 10.1517/13543776.2014.859244. [DOI] [PubMed] [Google Scholar]
- Jung M.; Philpott M.; Muller S.; Schulze J.; Badock V.; Eberspacher U.; Moosmayer D.; Bader B.; Schmees N.; Fernandez-Montalvan A.; Haendler B. Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J. Biol. Chem. 2014, 289, 9304–9319. 10.1074/jbc.M113.523019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp P. P.; Garnier J.-M.; Huang D. C. S.; Burns C. J. Evaluation of functional groups as acetyl-lysine mimetics for BET bromodomain inhibition. MedChemComm 2014, 5, 1834–1842. 10.1039/C4MD00182F. [DOI] [Google Scholar]
- Duffy B. C.; Liu S.; Martin G. S.; Wang R.; Hsia M. M.; Zhao H.; Guo C.; Ellis M.; Quinn J. F.; Kharenko O. A.; Norek K.; Gesner E. M.; Young P. R.; McLure K. G.; Wagner G. S.; Lakshminarasimhan D.; White A.; Suto R. K.; Hansen H. C.; Kitchen D. B. Discovery of a new chemical series of BRD4(1) inhibitors using protein-ligand docking and structure-guided design. Bioorg. Med. Chem. Lett. 2015, 25, 2818–2823. 10.1016/j.bmcl.2015.04.107. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Gartenmann L.; Dong J.; Spiliotopoulos D.; Caflisch A. Discovery of BRD4 bromodomain inhibitors by fragment-based high-throughput docking. Bioorg. Med. Chem. Lett. 2014, 24, 2493–2496. 10.1016/j.bmcl.2014.04.017. [DOI] [PubMed] [Google Scholar]
- Noguchi-Yachide T.; Sakai T.; Hashimoto Y.; Yamaguchi T. Discovery and structure-activity relationship studies of N6-benzoyladenine derivatives as novel BRD4 inhibitors. Bioorg. Med. Chem. 2015, 23, 953–959. 10.1016/j.bmc.2015.01.022. [DOI] [PubMed] [Google Scholar]
- Vidler L. R.; Filippakopoulos P.; Fedorov O.; Picaud S.; Martin S.; Tomsett M.; Woodward H.; Brown N.; Knapp S.; Hoelder S. Discovery of novel small-molecule inhibitors of BRD4 using structure-based virtual screening. J. Med. Chem. 2013, 56, 8073–8088. 10.1021/jm4011302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Zhou X.; Wang A.; Zheng Y.; Gao Y.; Zhou J. Evolutions in fragment-based drug design: the deconstruction-reconstruction approach. Drug Discovery Today 2015, 20, 105–113. 10.1016/j.drudis.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herait P. E.; Berthon C.; Thieblemont C.; Raffoux E.; Magarotto V.; Stathis A.; Thomas X.; Leleu X.; Gomez-Roca C.; Odore E.; Roumier C.; Bourdel F.; Quesnel B.; Zucca E.; Michallet M.; Recher C.; Cvitkovic E.; Rezai K.; Preudhomme C.; Facon T.; Palumbo A.; Dombret H. Abstract CT231: BET-bromodomain inhibitor OTX015 shows clinically meaningful activity at nontoxic doses: interim results of an ongoing phase I trial in hematologic malignancies. Cancer Res. 2014, 74, CT231–CT231. 10.1158/1538-7445.AM2014-CT231. [DOI] [Google Scholar]
- Fong C. Y.; Gilan O.; Lam E. Y.; Rubin A. F.; Ftouni S.; Tyler D.; Stanley K.; Sinha D.; Yeh P.; Morison J.; Giotopoulos G.; Lugo D.; Jeffrey P.; Lee S. C.; Carpenter C.; Gregory R.; Ramsay R. G.; Lane S. W.; Abdel-Wahab O.; Kouzarides T.; Johnstone R. W.; Dawson S. J.; Huntly B. J.; Prinjha R. K.; Papenfuss A. T.; Dawson M. A. BET inhibitor resistance emerges from leukaemia stem cells. Nature 2015, 525, 538–542. 10.1038/nature14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P.; Roth M.; Neumann T.; Muerdter F.; Roe J. S.; Muhar M.; Deswal S.; Cerny-Reiterer S.; Peter B.; Jude J.; Hoffmann T.; Boryn L. M.; Axelsson E.; Schweifer N.; Tontsch-Grunt U.; Dow L. E.; Gianni D.; Pearson M.; Valent P.; Stark A.; Kraut N.; Vakoc C. R.; Zuber J. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 2015, 525, 543–547. 10.1038/nature14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu S.; Lin C. Y.; He H. H.; Witwicki R. M.; Tabassum D. P.; Roberts J. M.; Janiszewska M.; Huh S. J.; Liang Y.; Ryan J.; Doherty E.; Mohammed H.; Guo H.; Stover D. G.; Ekram M. B.; Peluffo G.; Brown J.; D’Santos C.; Krop I. E.; Dillon D.; McKeown M.; Ott C.; Qi J.; Ni M.; Rao P. K.; Duarte M.; Wu S. Y.; Chiang C. M.; Anders L.; Young R. A.; Winer E. P.; Letai A.; Barry W. T.; Carroll J. S.; Long H. W.; Brown M.; Liu X. S.; Meyer C. A.; Bradner J. E.; Polyak K. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature 2016, 529, 413–417. 10.1038/nature16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimchak A. M.; Shelton C.; Duncan K. E.; Johnson K. J.; Brown J.; O’Brien S.; Gabbasov R.; Fink L. S.; Li Y.; Lounsbury N.; Abou-Gharbia M.; Childers W. E.; Connolly D. C.; Chernoff J.; Peterson J. R.; Duncan J. S. Resistance to BET bromodomain inhibitors is mediated by kinome reprogramming in ovarian cancer. Cell Rep. 2016, 16, 1273–1286. 10.1016/j.celrep.2016.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muralidharan S. V.; Bhadury J.; Nilsson L. M.; Green L. C.; McLure K. G.; Nilsson J. A. BET bromodomain inhibitors synergize with ATR inhibitors to induce DNA damage, apoptosis, senescence-associated secretory pathway and ER stress in Myc-induced lymphoma cells. Oncogene 2016, 35, 4689–4697. 10.1038/onc.2015.521. [DOI] [PubMed] [Google Scholar]
- Amorim S.; Stathis A.; Gleeson M.; Iyengar S.; Magarotto V.; Leleu X.; Morschhauser F.; Karlin L.; Broussais F.; Rezai K.; Herait P.; Kahatt C.; Lokiec F.; Salles G.; Facon T.; Palumbo A.; Cunningham D.; Zucca E.; Thieblemont C. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol. 2016, 3, e196–204. 10.1016/S2352-3026(16)00021-1. [DOI] [PubMed] [Google Scholar]
- Amarnath Praphakar R.; Munusamy M. A.; Sadasivuni K. K.; Rajan M. Targeted delivery of rifampicin to tuberculosis-infected macrophages: design, in-vitro, and in-vivo performance of rifampicin-loaded poly(ester amide)s nanocarriers. Int. J. Pharm. 2016, 513, 628–635. 10.1016/j.ijpharm.2016.09.080. [DOI] [PubMed] [Google Scholar]