

Abstract
The interplay between Ca2+ and reactive oxygen species (ROS) signaling pathways is well established, with reciprocal regulation occurring at a number of subcellular locations. Many Ca2+ channels at the cell surface and intracellular organelles, including the endoplasmic reticulum and mitochondria are regulated by redox modifications. In turn, Ca2+ signaling can influence the cellular generation of ROS, from sources such as NADPH oxidases and mitochondria. This relationship has been explored in great depth during the process of apoptosis, where surges of Ca2+ and ROS are important mediators of cell death. More recently, coordinated and localized Ca2+ and ROS transients appear to play a major role in a vast variety of pro-survival signaling pathways that may be crucial for both physiological and pathophysiological functions. While much work is required to firmly establish this Ca2+-ROS relationship in cancer, existing evidence from other disease models suggests this crosstalk is likely of significant importance in tumorigenesis. In this review, we describe the regulation of Ca2+ channels and transporters by oxidants and discuss the potential consequences of the ROS-Ca2+ interplay in tumor cells.
Graphical abstract
1. Introduction
The relationship between Calcium (Ca2+) and reactive oxygen/nitrogen species (ROS/RNS) is well established and has been described in numerous disease models. Much of our knowledge has been gained from the cardiovascular system, where this interplay is an important aspect of pathophysiology, a prominent example being ischemia/reperfusion injury, where the Ca2+- ROS interplay is involved in eliciting cell death [1]. Thus, apoptosis is one event where coordinated surges of ROS and Ca2+ have been observed and studied in great depth [2-4]. However, in addition to cell death, emerging evidence reveal that many diverse cellular signaling events are regulated by concomitant and localized increases in ROS and Ca2+ transients [5-8]. This Ca2+ - ROS interaction is evident by the fact that many regulators of Ca2+ signaling are redox modified, and reciprocally Ca2+ signaling is intricately involved in regulating ROS levels. Importantly, the subcellular location of Ca2+ stores and the sites of ROS production are closely linked, prominently the ER-mitochondrial interface and the plasma membrane [9, 10].
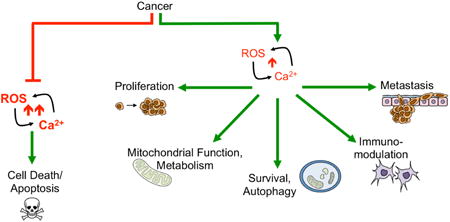
Tight regulation of Ca2+ homeostasis lies at the center of cellular signaling. The type of signaling “output” is dependent on the duration, localization, amplitude and frequency of the Ca2+ signal [11, 12]. Regulation of Ca2+ homeostasis is achieved by a number of ion channels, pumps and exchangers, found on both the cell surface and the organelles that act as primary intracellular Ca2+ stores. Similarly, subcellular regions of ROS/RNS production, such as the leading edge of migrating cells and the ER-mitochondrial interface, are emerging as hubs of signaling, and, as highlighted below, the type of reactive species and signal amplitudes influence the consequential signaling events and cellular responses [13-15]. While many studies have examined the redox control of Ca2+ homeostasis, relatively few studies have investigated this connection specifically as it pertains to carcinogenesis or metastatic progression. This may in part be due to the fact that the role of Ca2+ signaling in cancer is a relatively new field and that Ca2+ signaling mechanisms are complex and do not adhere to a “one size fits all” paradigm in cancer cells [16]. Much like changes in redox balance, this appears to be context and cancer type specific. Underlying genomic differences between tumor types, cellular heterogeneity of individual tumors, and the contribution of the tumor microenvironment likely contribute to this variability. Nevertheless, a number of studies have demonstrated that increased cytosolic Ca2+ is involved in processes such as proliferation, migration, invasion, and anchorage independent survival, clearly demonstrating that Ca2+ signaling is important in cancer progression [16-19]. In the present review, we focus on the interplay between Ca2+ and ROS in cancer, highlighting some of the discoveries pertaining to the redox regulation of Ca2+ transport mechanisms, and how Ca2+ signaling pathways in turn may regulate the cellular redox environment. Although much work is still required to firmly establish this relationship in different cancer types, two themes can be inferred from existing literature. 1) Coordinated ROS and Ca2+ surges are required for apoptosis initiation at the mitochondrial-Endoplasmic Reticulum (ER) interface, with evidence suggesting that this interplay is altered in cancer cells to enhance apoptosis resistance. 2) Localized, sub-lethal changes in both ROS and Ca2+ levels fine-tune signaling cascades that maintain proliferative and metastatic signals (Figure 1).
Figure 1.
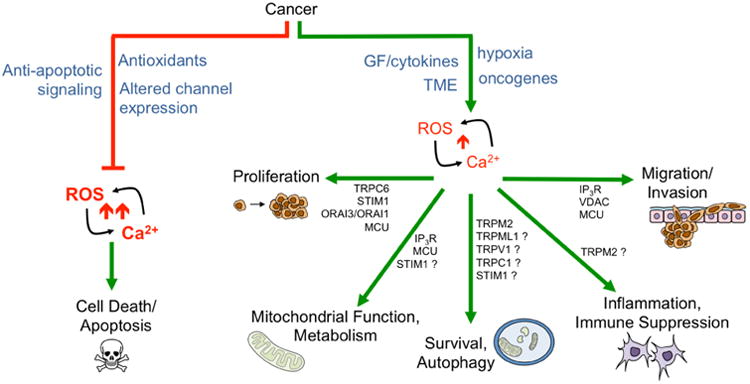
Cancer cells take advantage and manipulate the ROS-Ca2+ interplay in two ways: 1) by inhibiting large ROS-Ca2+ surges that mediate apoptosis (red pathway). Inhibition of Ca2+ ER-mitochondrial transfer by inhibition of receptors and channels such as IP3R and VDAC and subsequent suppression of mitochondrial ROS production are pathways by which cancer cells can evade apoptosis (Figure 9); and 2) by promoting pro-tumorigenic signaling pathways in response to sublethal changes in ROS/Ca2+ levels. Alterations in ROS and Ca2+ levels are often consequences of signaling from Growth factors and cytokines, oncogene expression, and changes in the Tumor microenvironment (TME), including presence of tumor associated fibroblast and macrophages, hypoxia and nutrient stress. ROS are able to directly oxidize or indirectly manipulate activity of Ca2+ channels, pumps and regulators, including plasma membrane and ER and mitochondria localized channels (Figure 3), while Ca2+ signals are known modulators of several ROS generating systems including NADPH oxidases (Nox), NO synthase (NOS) and the mitochondria (Figure 2). In this review we will highlight examples of this crosstalk and how this may relate to pro-tumorigenic signaling. Question marks indicate Ca2+ regulators that have been implicated in driving cellular responses in a ROS dependent manner in other cell models, besides cancer.
2. Oxidants – the importance of what, where and how much
2.1 What and Where?
The terms reactive oxygen species (ROS) and reactive nitrogen species (RNS), are often loosely used to describe a group of very different molecular species that vary in reactivity, half-life, site of production and detoxification reactions (Figure 2). These oxidants can be either free radicals (containing an unpaired electron), such as superoxide anion (O2•-) and hydroxyl radical (•OH), or non-radical oxidants, including hydrogen peroxide (H2O2), hypochlorous acid (HOCl, primarily in neutrophils) and peroxinitrite (ONOO-), the latter being generated in the presence of O2•- and nitric oxide (NO•). NO• is produced by Nitric oxide synthase, of which two isoforms (nNOS/NOS1 and eNOS/NOS3) are regulated by Ca2+ in a calmodulin-dependent manner [20]. It should be noted that these species vary widely in their half-life, reactivity and diffusion rates, and their role on macromolecular oxidation is dependent on amounts and sites of generation, as well as the rate of oxidation and abundance of target moieties [21]. Moreover, the reaction with target molecules, such as other ROS, lipids, proteins and DNA, is dependent on the redox environment of the cell. For example, high abundance of reduced glutathione and fast reaction with more readily oxidized proteins, such as peroxiredoxins, may result in “scavenging” of the oxidant species before these are able to reach their target (Figure 2) [21-23]. The relatively high reactivity of some oxidants limits their diffusion and role as true signaling molecules. This includes the highly reactive •OH (T1/2 10-6-10-9sec). Similarly, O2•- has a half-life of micro to milli seconds, depending on its environment and interactions with cellular and extracellular components such as NO•, transition metals and ascorbic acid; while H2O2 has a half life in the order of seconds [24, 25].
Figure 2.

Examples of common cellular reactive oxygen and nitrogen species. Ca2+ is involved in directly regulating some ROS/RNS “generators” (blue boxes), including NADPH oxidase isoforms Nox5, Duox1 and Duox2, and nitric oxide synthase NOS1 and NOS3. Ca2+ is important for the regulation of Tricarboxylic Acid (TCA) cycle and electron transport chain (ETC) enzymes and may in turn drive superoxide O2•- production in the mitochondria. The short-lived O2•- is likely unable to diffuse far from its site of production, but rather rapidly converted to H2O2. H2O2 can further react with iron to produce highly reactive hydroxyl radical or “scavenged” by Peroxiredoxins (Prx), Glutahtione (GSH) and catalse (Cat; orange boxes).
Oxidants are produced at a number of cellular locations that are important hubs for intracellular Ca2+ regulation, including the plasma membrane- endoplasmic reticulum (ER) junctions, and the interface between the ER and the mitochondria (Figure 2). The primary oxidant produced within cells is O2•-, which is generated enzymatically by membrane-bound NADPH oxidases (Nox) or through electron leakage of the electron transport chain (ETC) within mitochondria [10, 26, 27]. Nox enzymes (Table 1) utilize NADPH as the electron donor to generate O2•- from O2, which is rapidly converted to H2O2. Although Nox-derived O2•- has been implicated in numerous studies in driving redox modifications within the cell, it is unlikely that this short lived oxidant is able to diffuse the plasma membrane and enter the cell. Rather, it likely reacts quickly with extracellular components such as ascorbic acid, or is dismuted to H2O2. H2O2 is able to traverse the plasma membrane, likely through aquaporins [28]. Nox 4 is thought to be able to directly generate H2O2 [29]. Nox enzymes form signaling complexes at cell membranes, and their regulation by growth factor and cytokine receptors, small GTPases, and second messengers, such as Ca2+, illustrates that specific, localized activation of ROS production is an important aspect of cellular signaling [8, 10].
Table 1. Nox Family Members.
| Nox Isoform | Major Cell Type Expression | Cellular Localization | Activation |
|---|---|---|---|
| Nox1 | Colon, Vasculature | Plasma Membrane / Caveolae | Rac/p22phox/Noxa1/Noxo1 |
| Nox2 | Phagocytes | Plasma Membrane / Vesicles | Rac/p22phox/p47phox/p40phox/p67phox |
| Nox3 | Inner Ear | Plasma Membrane | Rac/p22phox/Noxa1/Noxo1 |
| Nox4 | Kidney, Vasculature | Plasma Membrane / ER / Mitochondria | p22phox |
| Nox5 | Lymphoid Tissues | Plasma Membrane | Ca2+ |
| Duox1/2 | Thyroid | Plasma Membrane | Ca2+ |
Nox2, was the first of seven Nox family members to be characterized, and is primarily involved in regulating ROS surges in response to cytokine stimuli in phagocytic cells. Nox enzymes of non-phagocytic cells have different functions based on their compartmentalization and regulation, and are implicated in a number of pathophysiological conditions, including cancer. For example, Nox1-mediated ROS production is necessary for invadipodia formation to drive tumor cell migration [30, 31]. Nox1-3 enzymes are regulated by the small GTPase Rac, which acts as a relay for Nox activation via a number of stimuli, including sheer stress, growth factors and lysophosphatidic acid (For further details on Nox family members and activation mechanisms we refer the reader to reviews by Brandes et al. [10] and Block and Gorin [32]). Further, cytokines and growth factors are able to induce Nox phosphorylation and activity through a variety of kinases and these mechanisms vary depending on the particular receptor ligand and Nox isoform involved. Nox1, 4 and 5 have specifically been implicated in ROS generation in cancer, and shown to be activated by receptor tyrosine kinases, G protein-coupled receptors (GPCRs) and oncogene signaling pathways [32]. Highlighting the evident interplay between ROS and Ca2+, Nox5, Duox1 and Duox2 are regulated by Ca2+, either directly through interactions with Nox-EF hands, or indirectly by calmodulin and protein Kinase C (PKC) [33-37]. Ca2+ channels may also directly interact with Nox at the cell surface to coordinate ROS-Ca2+ signaling. An example being the proposed interaction of the diacylglycerol (DAG)-sensitive transient receptor potential canonical 6 (TRPC6) cation channel with Nox2 in lipid rafts of podocytes [38, 39]. In this example, the authors proposed that TRPC6-associated Nox2 is activated by DAG, and that subsequent Nox2-produced ROS further contribute to TRPC6 activation. As discussed below, the amount and duration of ROS/Ca2+ signals are important determinants for the consequential outcome of tumor cells. Therefore, the expression of ROS generating enzymes, such as Nox, can greatly influence different cellular responses. For example, small increases in Nox5 expression lead to proliferation of several types of cancer cell lines [32]. Conversely, when Nox5 levels reach a specific threshold of expression, O2•- production in response to Ca2+ stimulation can reach toxic levels leading to ROS-mediated apoptosis [40].
Mitochondria are major sources of cellular oxidants [10, 26, 27, 41], and are recognized reservoirs of Ca2+ containing specific channels and transporters that tightly regulate mitochondrial matrix Ca2+ homeostasis [42, 43]. Although it was initially thought that surges of ROS stemming from mitochondria are primarily involved in the process of apoptosis, recent evidence suggest that mitochondrial ROS production contributes to processes such as autophagy and pro-tumorigenic redox-signaling [44-47]. During respiration, electron leakage contributes to O2•- formation from oxygen (O2). Complex I and III of the ETC have been primarily implicated in this process, leading to O2•- production into both the intermembrane space (complex III) and matrix (complex I & III) [26, 27]. Whether or not O2•- is stable enough to elicit cellular signaling outside the mitochondria is still debated. Most likely, it is rapidly converted to the less reactive, more stable and readily diffusible H2O2, either spontaneously or by manganese superoxide dismutases Sod2 in the matrix and Sod1 in the intermembrane space. Due to the much longer half life of H2O2 (seconds), H2O2 may therefore represent a more likely candidate as a “redox second messenger”, rather than O2•-. Major drivers of mitochondrial ROS production during normal and patho-physiological conditions include alterations in mitochondrial electron transport chain complex function, hypoxia and hyperoxia, cytokine and oncogene signaling, including TNFα and the Phosphoinositide 3-kinase (PI3K) – Akt -target of rapamycin (mTOR) pathway [48-51]. Moreover, increased flux of Ca2+ into mitochondria is a driver of mitochondrial ROS production and an integral component of processes such as apoptosis, as discussed in some detail below [52, 53].
The influx of Ca2+ into mitochondria occurs largely at domains termed mitochondria associated membranes (MAMs), where mitochondria are in close vicinity to the ER. The ER is a major intracellular Ca2+ store and is another source of cellular ROS. Protein folding, a primary function of the ER, is dependent on oxidative modification of cysteine thiols and subsequent disulfide bond formation. Therefore, the ER is a more oxidative environment than the cytoplasm and, as a consequence of the protein folding machinery, contributes to the production of H2O2 [54]. Disulfide bonds are exchanged between ER oxidoreductases (Ero1) and Protein disulfide iosmerases (PDIs), and these subsequently target proteins requiring disulfide bond formation for proper folding [54]. This concomitantly results in reverse shuttling of electrons from PDI to Ero1, and the reduction of molecular oxygen to H2O2 (Figure 2) [55], however other pathways of H2O2 production in the ER which are Ero1-independent have been reported [56]. Disulfide formation within Peroxiredoxin 4 (Prx4) in the ER lumen is likely the direct target of ER-produced H2O2, and Prx4 has in turn been shown to transmit this redox signal (i.e. disulfide bond exchange) and to contribute to the protein folding machinery [57, 58]. Another example of a redox sensor and ROS-producing protein localized at MAMs is p66shc. Under normal conditions, this protein is a RAS adaptor protein, but following pro-apoptotic stimuli, such as activation of apoptosis signal-regulating kinase 1 (ASK1), p66shc is translocated to MAMs, where it acts to further induce ROS production by interacting with cytochrome c. This interaction appears to enhance electron transfer to molecular oxygen and the generation of ROS [59].
Evolution has provided the cell with a sophisticated arsenal to prevent the accumulation oxidants (Figure 2). O2•- is either spontaneously or enzymatically (via Sod) dismuted to H2O2. H2O2 is readily transported across cellular membranes [28, 60], including the most likely through aquaporins in the plasma membrane and its levels are regulated by catalase within peroxisomes, glutathione peroxidases, and the peroxiredoxin/thioredoxin system throughout the cell. However, in the presence of transition metals (Fe2+ or Cu+), H2O2 undergoes the Fenton reaction to yield highly reactive •OH. •OH is the major oxidant responsible for DNA oxidation. Therefore, the reactivity and relative abundance of antioxidant scavengers greatly influence the consequential effects of individual oxidants. Studies interrogating the identity of specific oxidants regulating Ca2+ signaling proteins remain few.
The term ROS is often loosely and incorrectly used in the literature to explain a plethora of biological phenomena linked to changes in the redox status of cells [61]. Much of this can be attributed to our limitations in appropriate molecular tools, which are needed to adequately identify the species involved, quantitatively measure their relative amounts, and identify their sites of production, distribution and eventual site of action. While we will not dwell on the limitations of current methods, it should be mentioned that many commonly used redox sensors and ROS scavengers have limitations in sensitivity and accuracy. Moreover, the ability of some of these compounds to react with ROS and consequentially produce reactive species themselves further complicates their use. An example of this is the commonly referenced redox sensitive dye Dichlorodihydrofluorescein (DCF), which has specifically received much criticism for its use [62]. Although it is a useful screening tool, verification of data with appropriate scavenger controls and complementary methods, such as recombinant redox sensors and more sophisticated techniques including electron paramagnetic resonance (EPR) coupled with spin-trapping, are advised [7, 63-65]. As with redox sensitive probes, another challenge in the field is the need for specific scavengers of reactive oxygen species in order to help identify roles of different oxidants. Many selectively designed scavengers have secondary effects and can independently affect the redox status of cells, either by acting as oxidants themselves or by activating transcription of antioxidant enzymes [62, 66].
2.2 How much? - Redox stress versus redox signaling
Similar to Ca2+ signaling, the effects of oxidants are dependent on their relative levels and half-life. It is well known that differences in the frequency and amplitude of Ca2+ signals can have major consequences on eliciting diverse downstream cellular outputs [12]. Fine-tuned regulation of Ca2+ oscillations is thought to be one mechanism that accounts for the large variety of Ca2+-mediated functions emanating from the same Ca2+ channel. An example is the difference in Ca2+ signals resulting from the ER-localized inositol 1,4,5-trisphosphate (IP3) receptor (IP3R). Sustained Ca2+ release from the ER leads to Ca2+ overload in mitochondria that sets off the apoptosis death cascade [67], while controlled oscillations initiate Ca2+ signals leading to numerous Ca2+ regulated signaling pathways, such as NFAT activation via calcineurin, an important transcription factor during proliferation [68, 69]. Similarly, it is appreciated that large ROS surges, or “oxidative stress”, are primarily associated with widespread oxidation of macromolecules and irreversible cellular damage, while sub-lethal changes contribute to redox signaling, as described in more detail below. In the case of immune responses, bolus doses of ROS are generated by phagocytic cells to initiate cell death of the pathogen and infected host cell [70]. In cancer, differences in the levels of ROS and their particular consequences are apparent at different stages of tumor progression. For instance, during tumor initiation/carcinogenesis, exogenous sources of ROS, such as radiation and chemical carcinogens, lead to oxidative damage of macromolecules, including DNA [71]. The resultant genomic instability and incorporation of mutations are well-known drivers of tumor suppressor inhibition and oncogene activation. However, tumor cells readily adapt to and are able to cope with oxidative stress. For example, tumor cells readily activate the Nrf2/Keap1 stress response pathway to increase expression of antioxidant enzymes [72]. This may also provide tumor cells with advantages when faced with changing tumor microenvironment during metastatic progression, including anchorage independent survival, hypoxia and nutrient stress [73, 74]. Similarly, it has been shown that tumor cells can restrict Ca2+ influx from the extracellular milieu and inhibit sustained transfer of Ca2+ from the ER to the mitochondria to inhibit apoptosis [2, 75-78]. Hence, large intrinsic surges of ROS and Ca2+ are generally avoided by tumor cells to prevent cell death.
More recently it has been appreciated that cancer cells inertly are able to adapt to redox stress during tumor progression, and may even thrive with an increased threshold of endogenously generated ROS, utilizing this for redox-mediated signaling [15, 51]. Since cancer cells appear to operate under a higher cellular ROS steady state, there is a precedent to utilize this as an “Achilles Heel” for therapeutic targeting [79, 80]. Many tumor cells are more susceptible to cell death in response to exogenous ROS, or ROS generating agents [40, 81-83]. Hence, an increased cellular redox milieu may place tumor cells closer to the oxidative stress cytotoxic threshold in response to exogenous ROS.
Although the alterations in global redox status appear to be a phenotype of cancer cells, it is important to elucidate how this drives tumorigenesis and metastatic progression through Ca2+ signaling pathways. As evident from other cell model systems, coordinated, localized production of ROS sets up hubs of redox and Ca2+ signaling important for cellular function [84-86]. Like Ca2+ oscillations, or “sparks”, similar observations of ROS flashes have been made with the advent of genetically engineered redox probes, including the redox-sensitive variant of GFP (roGFP) and HyPer [7, 63, 87]. For example, it has been suggested that ROS sparks at the plasma membrane leading edge are necessary for cell migration, and similar observations have been made for Ca2+ flickers. Studies on cardiomyocytes have shown that ROS and Ca2+ sparks are coordinated and are susceptible to stimuli, such as mechanical stretch [88, 89]. Similarly, wounding of C. elegans skin, elicits a localized Ca2+ response that promotes localized mitochondrial ROS sparks, necessary for actin-mediated wound closure [86]. However, it is unknown if these events are similarly coordinated and mechanistically linked during cancer cell migration, an important facet to metastatic spread.
With our understanding that ROS are spatially and temporally regulated within cells and the appreciation that redox modifications of proteins are important regulatory mechanisms, redox signaling has received much attention over the past decades. A number of tumorigenic stimuli such as cytokines and growth factors can initiate O2•- and H2O2 production at the level of Nox enzymes [8, 10]. Examples include epidermal growth factor receptor (EGFR) and platelet-derived growth factor receptor (PDGFR), which mediate ROS-dependent pro-proliferative MAP-kinase signalling via Nox enzyme activation [90]. These stimuli can also lead to mitochondrial ROS production, which can be affected by oncogene expression and changes in metabolic flux and oxygen tension of the tumor microenvironment [91]. These changes are able to elicit ROS production within tumor cells and tumor associated cells such as fibroblast and immune cells. However, it remains difficult to quantify changes in oxidant levels within the tumor microenvironment in vivo. During redox signaling, the changes in ROS/RNS generation and subsequent redox-signaling seem to occur in a spatiotemporal context and appear to be dynamically regulated. Much like other second messengers, it is likely that the type of oxidant species, amount and location are of importance in determining the eventual cellular response elicited [13, 15, 21, 22, 92]. The existence of cellular ROS scavengers and antioxidants means that reactions are largely reversible, a necessary feature of cellular signaling (Figure 2 & 3). While beyond the scope of the present review, it is becoming evident that the levels of certain antioxidants, such as Sod2 and GSH are increased in tumor cells, and potentially necessary to ensure survival during metastatic progression, either to cope with excess redox stress or to contribute to alterations in the redox status and redox signaling of cancer cells [93-100].
Figure 3.
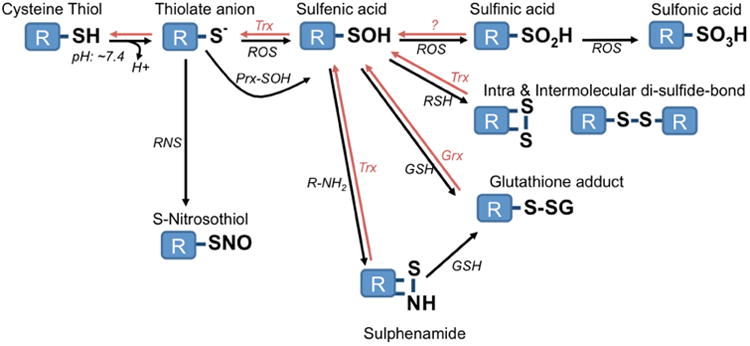
Oxidative modification of cysteine thiols. The reversible nature of most modifications by the Thioredoxin (Trx) and Glutaredoxin (Grx) system highlight their role in cellular signaling. Rather than direct oxidation of cysteine residues by ROS, it is thought that intermediate redox sensors, such as Peroxiredoxins (Prx), may be the first target of ROS and subsequently carry out redox modifications of target proteins.
While several amino acid residues have a higher propensity to react with oxidants (cysteine, selenocysteine, methionine, tyrosine, tryptophan, histidine), one of the most important redox signaling targets and most widely studied are the thiolate anions of cysteine residues (Figure 3). Given their pKa, cysteine residues exist as both the sulfhydryl and thiolate at neutral pH. The thiolate is rapidly oxidized to sulfenic acid in the presence of oxidizing agents. From this, a number of modifications are commonly formed, including the reduction and disulfide bond formation with other thiols, either intra- or inter molecularly [14, 21]. Reaction with •NO to form S-nitrosothiols can also be an important modification that alters protein function. Cysteine residues are central to catalytic function and structural properties of proteins, as they act as nucleophiles in chemical reactions and facilitate disulfide bond formation during protein folding. Hence, cysteine redox modifications and the ability to “reverse” these reactions highlights their importance as cellular signaling intermediates. The degree and reversal of oxidative thiol modifications is often dependent on cellular factors including pH and the levels of enzymes like glutaredoxin (Grx) and thioredoxin (Trx) which reduce disulfides back to the cysteine thiol [14].
Due to its ability to diffuse within the cell, traverse biological membranes and a relatively longer half-life than other ROS, it has been suggested that H2O2 may be a suitable second messenger and major contributor to redox signaling within cells [13]. While H2O2 has been implicated as a ROS second messenger in a manner similar to Ca2+, the role of individual oxidant and reactive nitrogen species as bona fide signaling molecules or second messengers is still under investigation. Many reactive oxygen species, such as O2•- are short lived and therefore either directly oxidize targets within close proximity to their site of production (e.g. near Nox of the plasma membrane or ETC complexes near mitochondrial membranes), or rapidly react to form secondary species, such as ONOO-, or are enzymatically/spontaneously dismuted to H2O2. Interestingly, based on reaction kinetics, it is thought that H2O2 may not be able to directly oxidize cysteine residues of many identified target proteins, such as phosphatases. Instead it has been proposed that reaction with Prx likely quickly consumes most of the H2O2 produced within the cell. This would limit free H2O2 diffusion and direct oxidation of proteins, such as phosphatases, that are potential targets of H2O2 redox signaling (Figure 2). This conclusion is based on the rate constant of the reaction between H2O2 and Prx, and the relatively high abundance of Prx enzymes within cells and compartments such as mitochondria [22, 101]. Instead of direct oxidation of signaling proteins by H2O2, Prx may act as an intermediate or “redox relay” to carry out subsequent redox-mediated signaling, including disulfide exchange [22, 101]. Alternatively, if H2O2 production is very high in specific nano-domains of the cell, irreversible hyperoxidation or other post-translational modifications inactivating the local Prx pool could lead to localized H2O2 build-up that could directly oxidize thiols of target proteins within its vicinity [102].
Redox regulation of Ca2+ homeostasis has been demonstrated in a variety of contexts and shown to occur via direct oxidation of Ca2+ channels and channel regulators, as described in more detail below. While not discussed in depth, indirect modulation of Ca2+ signaling may also occur through the action of redox-mediated activation of either gene transcription or cell signaling pathways, such as oxidation and subsequent inhibition of protein tyrosine phosphatases or activation of kinases [14, 15]. An example of this is the redox regulation of the phosphatases PP2B and PP2A, which are responsible for the dephosphorylation and regulation of the Ca2+/Calmodulin-dependent kinase II (CaMKII). Oxidation leads to PP2A/B inactivation and a subsequent increase in CaMKII phosphorylation, thereby influencing downstream Ca2+ signaling pathways [103]. In contrast kinases are often activated by oxidation. In this context, oxidation and activation of PKC and PKA can further enhance phosphorylation of CaMKII resulting in an overall net effect of redox stimulated CaMKII phosphorylation and activation in response to increases in ROS [104].
Ca2+ channels can be directly affected by oxidation to alter Ca2+ signaling. Cysteine residues within Ca2+ channels and activators of Ca2+ channels such as the Stromal Interacting Molecule-1 (STIM1) are susceptible to oxidation and protein modifications, including glutathionylation and di-sulfide bond formation (Figure 3), which can affect protein conformation and activity. STIM1 is an important Ca2+ sensor located in the ER, which activates ORAI store-operated Ca2+ entry (SOCE) channels. Interestingly, it appears that different redox modifications of STIM1 can elicit divergent consequences on protein function. Although further studies are required to verify these observations, it has been shown that glutathionylation of STIM1 cysteine residues results in store-independent activation of ORAI by STIM1, while STIM1 disulfide formation may decrease Ca2+ entry [105, 106]. Similarly, TRPC5 channel oxidation has been shown to yield different redox modifications and consequential channel function, including S-nitrosothiol formation, glutathionylation and inter- and intra-molecular disulfide bond formation [107-110].
A caveat of many earlier studies examining the role of oxidation on channel function is the lack of attention to the amounts of exogenous ROS applied, and whether these reflect physiologically or pathophysiologically relevant levels. For example, exogenous application of H2O2 in the low mM range is often used to demonstrate the role of oxidants in activating Ca2+ influx, which is a dose that elicits cell death within a matter of hours in most cells, and likely represents redox stress. As it is difficult to mimic the spatio-temporal sub-lethal increases commonly associated with redox-signaling hubs, experimentally applied bolus doses may hence lead to very different oxidation events and cellular outcomes. An example of this is irreversible sulfonic acid formation, which may inactivate proteins indefinitely, while intra- or inter-molecular disulfide formation can be reversed by protein disulfide reductases such as thioredoxins (Figure 3). In addition, the role of each specific reactive species is largely lacking. Reactive oxygen and nitrogen species differ widely in their ability to oxidize proteins and react with other cellular component including metals such as iron. Protein modifications may hence differ based on the cellular redox environment and these differences are observed in studies examining the redox regulation of ion channels, as illustrated below. Although many pathways of Ca2+ regulation and redox signaling have been separately described in cancer cells, their interplay has not been investigated in great detail in the context of this disease. Below we give examples of redox regulation of Ca2+ modulators, as they were discovered in various disease models, and speculate on potential consequences of this interplay in cancer. Examples of studies demonstrating direct cysteine redox modifications of Ca2+ channels and channel regulators, as well as indirect mechanisms of redox regulation are described, as they pertain to Ca2+ regulators of the plasma membrane, ER and mitochondria (Figure 4). From these studies, one can deduct that there are likely two overarching themes that emerge for the redox-Ca2+ interplay in cancer cells: 1. Coordinated ROS and Ca2+ signals are required for apoptosis initiation at the mitochondrial ER interface, with emerging evidence suggesting that this interplay is altered in cancer cells to enhance apoptosis resistance. 2. The interplay between Ca2+ and ROS influences cellular signaling cascades that promote proliferation and metastasis. The latter scenario likely involves coordinated changes in localized, sub-lethal concentrations in both ROS and Ca2+ levels.
Figure 4.
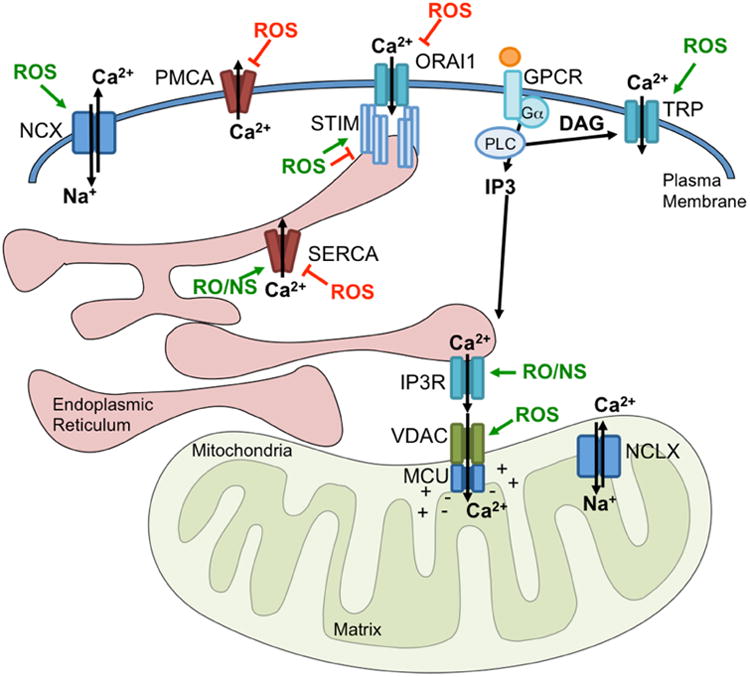
Reactive oxygen and nitrogen species can directly influence the activity of Ca2+ regulators at multiple locations within the cell. Studies have demonstrated either direct or indirect redox regulation of ion channels and pumps at the plasma membrane, ER and Mitochondria (activation indicated in green; inhibition indicated in red). Plasma Membrane channels and pumps represented include Transient Receptor Potential (TRP) Channels, ORAI, Plasma Membrane Ca2+ ATPase (PMCA) and Na+/Ca2+ exchanger (NCX). The ER Ca2+ sensor and ORAI regulator, STIM1 has also been demonstrated to be under redox control. Similarly, ER-localized Inositol 1,4,5-trisphosphate (IP3) receptors (IP3R) and the Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA), as well as mitochondria-localized Voltage-dependent anion channel (VDAC) and the Mitochondrial Ca2+ Uniporter (MCU) activity are influenced by ROS. While much of this information has been gleaned in other model systems, the role of these redox modifications in the context of cancer require further investigation.
3. Redox regulation of cellular Ca2+ homeostasis at the Plasma membrane
Regulation of Ca2+ homeostasis at the cell membrane occurs through a number of channels and pumps that can be activated by a variety of signals including mechanical and chemical stimulants, ligand-dependent receptor activation and subsequent generation of second messengers, intracellular Ca2+ store depletion, and oxidants. It is clear from the literature that the cellular context and differences in the spatio-temporal nature of the oxidant signal influences the activity of plasma membrane channels and subsequent Ca2+ dependent cellular responses. For example, high levels of ROS or “oxidative stress” can inhibit cytoplasmic Ca2+ extrusion [111, 112] and enhance Ca2+ entry through TRP channels located at the plasma membrane [113-118] (Figure 4). This enhances intracellular Ca2+ levels, which are thought to contribute to the induction of cell death. In contrast, it is conceivable that sublethal and localized ROS production, might initiate pro-tumorigenic Ca2+ signaling via activation of cell surface Ca2+ channels, such as proliferation [119], and wound healing [86]. In both oxidative stress and redox signaling, the location of cell surface Ca2+ channels clearly makes these an excellent target for sensing redox changes in the intra and extracellular environment [120].
Many of the stimulatory cues that activate plasma membrane channels are often altered in the tumor microenvironment and it is therefore conceivable that altered activity of plasma membrane associated Ca2+ channels and pumps is a common phenotype of cancer cells[120]. Moreover, remodeling of ion channel expression and alterations in Ca2+ signaling by plasma membrane cation channels, including TRPC1, TRPC3, TRPC6, TRPM2, TRPM7, TRPV6, ORAI1 and ORAI3 have been implicated in enhanced proliferation of tumor cells[16, 121]. Stimuli that activate these channels in cancer cells range from cholesterol (TRPM7) and GPCR agonists (ORAI1 and TRPC channel isoforms) to constitutive activation (TRPV6). In turn, this leads to activation of Ca2+-dependent signaling including the calcineurin/NFAT, CaM Kinase and Akt/Erk pathways to increase cell cycle progression (for reviews see [16-19]). As seen from the examples highlighted below, a common theme in cancer appears to be that tumor cells utilize redox regulated Ca2+ signals to drive proliferation and invasion, while they avoid sustained Ca2+ fluxes associated with apoptosis, which are generally elicited by oxidative stress.
3.1 Transient Receptor Potential (TRP) Cation Channels
Trp refers to a large gene family encoding transient receptor potential (TRP) proteins, which form mostly plasma membrane non-selective cation channels. However, some TRP channels function as calcium release channels in internal organelles [122]. TRP channels have variable activation and gating mechanisms and play a crucial role in a large number of cellular and physiological functions, ranging from sensory signaling to signaling pathways that control contraction, growth, migration and cognition. In mammals, the TRP superfamily consists of 28 TRP genes that can be divided into 6 families based on sequence homology: Canonical TRP (TRPC), Vanilloid TRP (TRPV), Melastatin (TRPM), Ankyrin TRP (TRPA), Mucolipin TRP (TRPML) and Polycystin TRP (TRPP). There are only 27 TRP genes in humans, with TRPC2 (involved downstream pheromone receptor signaling in rodents) being a pseudogene in humans [123]. A number of TRP channels have been shown to alter their activity in response to oxidative stress, either indirectly (e.g. TRPM2) or by direct channel oxidation (e.g. TRPV1, TRPC1, TRPM7) [124] [125]. Studies have demonstrated TRP channel involvement in cell proliferation, migration, angiogenesis, chemokine production and autophagy [17, 18, 120, 126, 127] and there is some evidence that redox regulation of TRP channels may play a role in cancer.
3.1.1 Redox regulation of TRPM2 in cancer
The melastatin TRP subfamily member, TRPM2 (formerly named TRPC7/LTRPC-2) has been a particular focus due to its regulation by oxidants. In other pathologies besides cancer TRPM2 has been linked to mediating Ca2+ influx during apoptosis, including endothelial and neuronal cell death, and male-specific ischemic injury in response to oxidative stress [113, 116, 128, 129]. TRPM2 can also directly influence ROS production at the level of Nox enzymes. It has been shown that TRPM2 regulates Rac1 and Nox activation to mediate ROS production during ischemic kidney injury [130]. TRPM2 was originally identified as a potential tumor suppressor [131]. As such, in an attempt to take advantage of the pro-apoptotic role of TRPM2, forced expression of the channel was shown to enhance cell death in in A172 human glioblastoma cells [132]. However, as detailed below, the role of TRPM2 appears to be complex and multifaceted in cancer (Figure 5).
Figure 5.
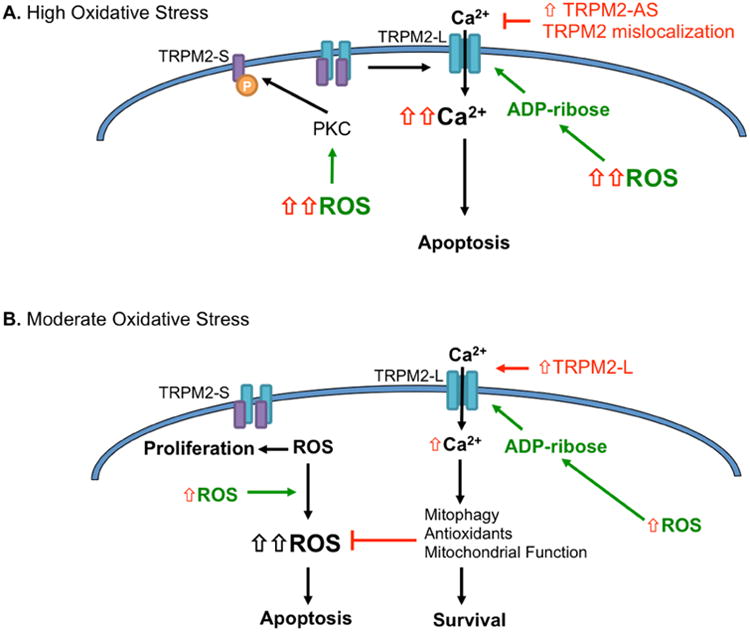
Divergent roles of TRPM2 under redox regulation A. Under high oxidative stress TRPM2 is activated to induce apoptosis. Several mechanisms for this have been proposed, including ROS-dependent increases in ADP-ribose production. Alternatively, ROS-dependent PKC phosphorylation of the TRPM2-S isoform, leads to dissociation of this potential dominant negative splice variant from TRPM2-L to induce TRPM2-L channel activation. Cancer cells have developed several mechanisms to avoid TRPM2-L channel activation in response to high oxidative stress, including TRPM2 mislocalization to the nucleus and high expression of a TRPM2-antisense (AS) mRNA. B. Under moderate oxidative stress, which may be observed in response to pro-oxidant changes, such as nutrient deprivation and hypoxia in the tumor microenvironment, TRPM2-L has a pro-survival function. Cancer cells expressing higher levels of the TRPM2-S isoform were shown to operate under a higher cellular ROS status, which drives pro-proliferative signaling. However, when challenged with moderate ROS stress TRPM2-S cells are unable to elicit necessary Ca2+-dependent pro-survival pathways, unlike TRPM2-L expressing cells. These studies show that expression of TRPM2-L presents cells with a survival advantage under moderate oxidative stress, and highlights the importance of TRPM2 isoform expression and the levels of ROS stress in determining cancer cell fate.
Unlike other TRP family members, the non-selective cation channel TRPM2 is not directly redox modified, as demonstrated by a lack of methionine or cysteine oxidation [116]. A number of studies have shown that H2O2 activates TRPM2 channel activity in an indirect manner. Treatment with a range of H2O2 doses (100μM-3mM) leads to ADP-ribose generation, a known activator of TRPM2, which directly binds its C-terminal domain to cause activation [114, 133, 134]. ADP-ribose is formed in both the mitochondria and in response to cellular stress/DNA damage by poly(ADP-ribose) polymerases (PARP) in a nicotinamide adenine dinucleotide (NAD)-depended manner. Both ADP-ribose generating pathways have been implicated in eliciting indirect H2O2 regulation of TRPM2 [117, 133, 134]. Interestingly, TRPM2-mediated Ca2+ influx during apoptosis elicits caspase activation and PARP cleavage. This suggests a potential negative feedback mechanism, where decreases in PARP-mediated ADP-ribose generation could dampen TRPM2 activity [135]. In addition to TRPM2 activation by ADP-ribose, it was shown that PKCα -mediated phosphorylation of a short TRPM2 isoform (TRPM2-S) is H2O2 dependent and leads to Ca2+ influx in endothelial cells [136]. The authors suggest this to be an alternate mechanism for redox regulation of TRPM2 during the initiation of apoptosis, as TRPM2-S phosphorylation initiates dissociation of TRPM2-S from the full length form TRPM2-L (long isoform). This confirms the potential role of TRPM2-S as a dominant negative regulator [115]. TRPM2-S lacks 4 transmembrane domains, and was previously demonstrated to inhibit ADP-ribose dependent channel activation by binding TRPM2-L [115]. In apparent contrast to Hecquet et al., the investigators noted that TRPM2-S has the ability to blunt H2O2 mediated activation of TRPM2-L at the cell membrane. Here enhanced expression of TRPM2-S inhibited H2O2 mediated apoptosis in cells expressing TRPM2-L, by blunting ADP-ribose binding that is induced in response to H2O2 [115].
Several lines of evidence suggest that tumor cells are either able to prevent ROS mediated TRPM2 activation during apoptosis initiation, or to use redox regulated TRPM2-dependent Ca2+ signaling to aid in tumor growth and resistance to therapy (Figure 5). An example of the former is highlighted by the interesting observation that expression of a non-coding antisense TRPM2 RNA (TRPM2-AS) is increased in prostate cancer. This appears to protect cells from TRPM2-mediated apoptosis and cellular redox-stress [137, 138]. High expression of TRPM2-AS was also associated with poor patient outcome and increased proliferation, while decreasing expression of TRPM2-AS initiated apoptosis and inhibited tumor growth of prostate cancer cells in vivo. [137]. Although the investigators did not directly measure if this resulted in a change in TRPM2 currents, mRNA levels of TRPM2 were increased in response to TRPM2-AS knockdown [137]. In apparent contrast with this work, high TRPM2 coding mRNA levels were shown to correlate with increased proliferation in another prostate cancer study [139]. However, upon further exploration the investigators noted substantial mislocalization of TRMP2 to the nucleus. This may represent an alternate mechanism by which cancer cells evade pro-apoptotic TRPM2 channel activation at the cell surface. The authors suggest that this enhancesd tumor cell proliferation, although the mechanisms for the latter were not clearly delineated [139].
Given the pro-apoptotic function of TRPM2 it appears counterintuitive that high TRPM2 expression has been observed in a number of cancer types, including neuroblastoma, prostate cancer and melanoma [138-141]. The studies described below suggest that TRPM2 elicits a protective role in response to sublethal increases in oxidative stress, which is dependent on cellular context, amounts of reactive oxygen species, and levels of TRPM2 isoform expression, and highlights the context-dependent nature of the ROS/Ca2+ interplay [142]. Expression of the above mentioned TRPM2 splice variants may influence the Ca2+/ROS interplay mediated by TRPM2 in cancer cells and fine tune signaling to proceed to either a pro-proliferative or pro-apoptotic path (Figure 5). Miller and co-workers showed that expression of both the short TRPM2-S and long TRPM2-L variants are increased in neuroblastoma cells, and that these two isoforms have divergent roles in cancer and in response to variable levels of oxidative stress [140, 141]. TRPM2-S cells were shown to have higher basal levels of cellular ROS than TRPM2-L cells. In turn, this was associated with oxidation and inactivation of the phosphatase and tensin homolog (PTEN) and consequential increases in the PI3K/Akt and Erk pathways, leading to higher proliferative rates in TRPM2-S cells [141]. Interestingly, TRPM2-S cells were unable to cope with the addition of exogenous oxidative stress, which may again point to the fact that an increased in intracellular steady state ROS is deleterious to cancer cells once faced with additional exogenous ROS. In contrast, TRPM2-L expressing cells were protected against exposure to 50-100μM H2O2. Mechanistically, this was shown to be due to TRPM2-L-dependent expression of the transcription factor FOXO3a, and consequential increases in the FOXO3a-regulated mitochondrial antioxidant enzyme Sod2 [141]. Moreover, TRPM2-L expressing cells also expressed high levels of the glucose transporter GLUT-1, a phenotype of glycolytic tumor cells. In a subsequent paper the authors showed that neuroblastoma cells with high TRPM2-L expression were more tumorigenic than cells expressing TRPM2-S using in vivo xenografts model [140]. TRPM2-L expressing cells had increased HIF-1/2α expression and this also conferred chemoresistance to doxorubicin [140]. In essence, these studies suggest that the expression of TRPM2-S may be beneficial to rapidly proliferating cells under optimal conditions, including adequate oxygen and nutrients supply, while under stress conditions the expression of TRPM2-L is essential in initiating pro-survival pathways such as HIF and antioxidant enzyme expression. This was further demonstrated in TRPM2-L CRISPR/Cas9 deleted cells, which were less tumorigenic and more susceptible to doxorubicin cytotoxicity [143]. The pro-tumorigenic action of TRPM-L was again shown to be due to its role in maintaining HIF stabilization, and by maintaining mitochondrial redox balance through expression of FOXO3 and Sod2. This preserved mitochondrial function and ATP production [143]. The complex, yet intriguing, relationship between the TRPM2 isoforms requires further investigation to ascertain if the ratio of TRPM2-L to TRPM2-S is altered in cancers and if this is associated with patient outcome and chemoresistance.
Redox-regulation of TRPM2 may also be important during immune response and inflammation, and shown to be necessary for NLRP3 inflammasome activation, monocyte chemokine production for the recruitment of neutrophils, and bacterial clearance [144-146]. In contrast, it has been shown that activation of TRPM2 which is non-selective and conducts a substantial amount of Na+ ions (in addition to Ca2+) can lead to plasma membrane depolarization in phagocytes, which results in the suppression of Nox-dependent ROS production in response to endotoxin to suppress the inflammatory process [147]. This negative feedback mechanism may ultimately protect cells and tissues against sustained redox stress during inflammation. Melendez and co-workers described an interesting mechanism whereby the Gram-negative bacterium Francisella tularensis utilizes the antioxidant ability of its catalase to limit TRPM2-mediated Ca2+ entry in host macrophages, thus inhibiting actin reorganization and cytokine production by these cells [148]. The role of the TRPM2-redox/Ca2+ axis during tumor clearance by immune cells would be of interest to investigate in future studies.
3.1.2 TRP - Nox interplay
Unlike the indirect regulation of TRPM2 by ROS, other members of the TRP ion channel family have been shown to be directly redox modified [124]. The canonical TRP (TRPC) channel family is primarily regulated by Phospholipase C (PLC) coupled receptors and the downstream actions of Phosphatidyinositol-4,5 bisphosphate (PIP2) hydrolysis, Diacylglycerol (DAG) production and rise in cytosolic Ca2+ [150-152]. ROS/RNS have been shown to regulate a number of TRPC family members, including TRPC3, 4 and 5, which are activated following oxidative stress in a number of cell lines [153-155]. While there are studies to show the importance of TRPC channel activity in regulating cancer cell migration, proliferation, epithelial-to-mesenchymal transition, angiogenesis and chemoresistance [109, 156-161], studies have not focused on the redox regulation of these in the context of an enhanced ROS tumor cell milieu or in response to ROS-generating conditions emanating from the tumor microenvironment. This is clearly an area that requires further attention.
A few divergent examples highlight additional roles for TRP-redox regulation in relation to their interaction with Nox. Again, the resultant cellular consequences are cell type specific. For example, increased TRPC6 activation by insulin initiates kidney podocyte apoptosis in a Nox4-derived H2O2-dependent manner [38, 149], while B-cell lymphoma cell proliferation is dependent on TRPC6 channel activation by Nox2 generation of O2•- [119] (Figure 6). In these cancer cells cholesterol increased expression of the Nox2 subunitsp47-phox and gp91-phox, this led to a concomitant increase in expression and activation of TRPC6, and intracellular Ca2+ increases. This was shown to be dependent on Nox2 mediated ROS production and was abrogated by the cholesterol lowering drug, lovastatin [119]. The interplay between TRP and Nox proteins may also be important for eliciting unwanted side effects of chemotherapeutics, including ototoxicity, which results in damage to the inner ear and hearing loss. For example, the interplay between Nox3 and TRPV1 appears to be involved in initiating apoptosis of outer hair cells of the cochlea in response to Cisplatin treatment [162, 163]. These examples highlight that redox regulation of TRP channel activity can have very different consequences, depending on the cellular context, specific Nox isoform interaction and type of ROS involved.
Figure 6.
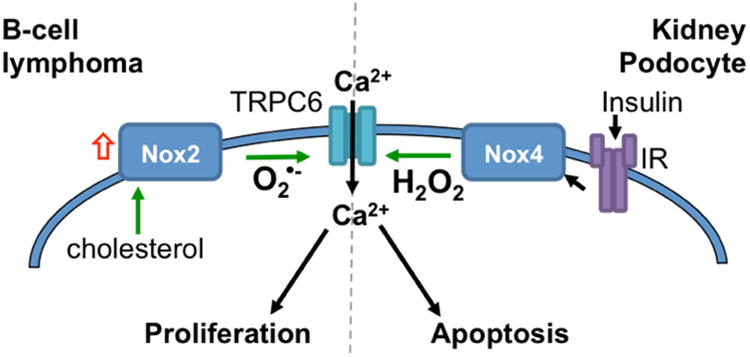
Divergent cellular consequences of Nox interactions with TRPC6 highlights the context dependence in ROS-Ca2+ signaling. Following insulin receptor engagement Nox4 activation leads to H2O2 production in podocytes, which induces TRPC6 activation and Ca2+ influx that drives apoptosis [38, 149]. Conversely, Nox2 expression in response to cholesterol induces redox-dependent expression and activation of TRPC6, required for proliferation of B-cell lymphomas [119].
3.1.3 TRP channels and regulation of autophagy
Recently, attention has been placed on the mucolipin TRPML channel family members in the regulation of autophagy, given their localization to lysosomal membranes and influence on Ca2+ signaling in these organelles [3, 164]. Autophagy is regulated by a number of cellular stressors, including nutrient deprivation, and the unfolded protein response/ER stress, which are closely associated with oxidative stress [44]. TRPML channels are localized to the lysosomal membrane and mediate Fe2+ and Ca2+ release from these organelles. TRPML channels are important for lysosomal pH balance, endo-lysosome formation and trafficking, and can respond to activation by PI(3,5)P2 and changes in pH [165, 166]. TRPML1 was recently shown to be directly activated by H2O2 to elicit lysosomal Ca2+ release. This was shown to increase generation of autophagosomes, as visualized by enhanced accumulation of the autophagy markers Microtubule-associated proteins 1A/1B light chain 3B (LC3-II) and Lysosomal-associated membrane protein 1 (Lamp1). Mechanistically, TRPML1 mediated calcineurin-dependent dephosphorylation of the transcriptional regulator of lysosomal and phagosomal biogenesis, Transcription Factor EB (TFEB), which is usually maintained in an inactive from by mTOR-mediated phosphorylation [167-169]. Interestingly, this regulation was specifically related to redox regulation of autophagy and proposed to represent one mechanism for cells to remove damaged mitochondria in response to oxidative stress [168]. Although TRPML1 can also respond and be upregulated in response to nutrient stress [169], the consequences of this regulation in cancer cells has not been investigated. It is possible that TRPML1 could trigger autophagy more readily in cells with elevated redox thresholds and may be advantageous to cancer cells to enhance survival in situations of nutrient and redox stress. Targeting TRPML1 could present a novel therapeutic strategy, as cutting off various nutrient supplies to tumors is increasingly being explored as a novel mechanism to kill cancer cells.
Non-selective Ca2+-permeable TRP channel isoforms at the plasma membrane are also involved in eliciting Ca2+ signaling that controls autophagy. It was shown that pro-survival induction of autophagy in thymocytes in response to capsaicin was TRPV1-dependent and requires both intracellular Ca2+ rise and ROS generation, which was necessary for 5′ adenosine monophosphate-activated protein kinase (AMPK) activation, Autophagy Related 4C Cysteine Peptidase (Atg4C) expression, and induction of Atg6/Beclin-1-dependent autophagy [170]. Similarly, TRPC1 dependent Ca2+ entry was shown to initiate autophagy in response to hypoxia and nutrient deprivation [171]. A recent report suggests that TRPM2 activation results in Beclin1 phosphorylation via CaMKII to decrease autophagy, making hepatocytes more susceptible to cell death in response to oxidative stress [172]. This contrasts with the role of TRPM2-L mediated induction of HIF-1α discussed above, which was shown to increase autophagy in a BNIP3 dependent manner to enhance survival of neuroblastoma cells [140]. Whether changes in TRP channel expression in cancer cells also alter their ability to initiate autophagy in response to redox activation requires further exploration.
3.2 Store-Operated Calcium Entry (SOCE)
Store operated Calcium entry (SOCE) is a major mechanism for Ca2+ regulation in non-excitable cells, and is increasingly implicated in mediating Ca2+ signals that control a large number of normal physiological processes and its dysfunction contributes to several diseases [173, 174]. SOCE is activated following Ca2+ depletion from ER stores, through phopshtaidylinositol-1,4,5-trisphosphate (IP3) Receptor Ca2+ release channels or Ryanodine receptor (RyR) in the case of muscle. IP3 is produced by activation of plasma membrane receptors to hormones, growth factors and neurotransmitters that couple to phospholipase C (PLC) isoforms leading to hydrolysis of phosphatidylinositiol-4,5-bisphosphate (PIP2) into IP3 and diacylglycerol (DAG). Store depletion activates the store-operated current called Ca2+ release activated Ca2+ (CRAC). CRAC currents are mediated by the Orai protein family (ORAI1, 2 & 3), and their activation is regulated by STIM1 and STIM2 proteins, which are ER-localized Ca2+ sensors [174-177]. In addition to the ER, mitochondria are involved in the regulation of SOCE, either due to their Ca2+ buffering capability [178, 179] or through mitochondria-derived redox-signaling [180]. The plasma membrane, mitochondria and ER are associated with ROS production, and hence it is not surprising that redox regulation of ORAI channels and STIM proteins could be of importance in a variety of cell types. In normal physiology, the SOCE pathway mediates Ca2+ signaling important for maintenance of cell function in non-excitable as well as excitable cells. In cancer, STIM and ORAI isoforms display increased expression in a number of tumor types and have been associated with signaling pathways that positively regulate cancer cell proliferation, migration, invasion, and chemoresistance [181-189]. Further, a number of studies have demonstrated the importance of the SOCE pathway and its regulators in endothelial progenitor cells, VEGF-mediated endothelial tube formation and endothelial cell proliferation [190-192], and this may similarly be important for tumor angiogenesis[193, 194]. Hence, it appears that enhanced SOCE generally supports pro-tumorigenic and pro-metastatic phenotypes.
The interplay between SOCE and ROS is multifaceted and the role of this regulation in cancer is only starting to emerge. However, from other cell model systems it is evident that redox-dependent modifications of ORAI and STIM play a role in the regulation of SOCE (for detailed review see [195]). Again, consequences of the ROS-SOCE Ca2+ interplay depend on the levels of ROS that cells are exposed to. In circumstances of oxidative stress, SOCE leads to the initiation of apoptosis. For example, neuronal cell death in response to ischemia is dependent on STIM2-mediated Ca2+ influx [196], and vascular dysfunction in an acute lung injury model in response to LPA is dependent on Nox2-derived ROS and subsequent redox-mediated activation of STIM1-depedendent SOCE [197]. As illustrated below, oxidation of STIM and ORAI can elicit both positive and negative effects on SOCE. Clearly, the source of ROS, levels and duration of this redox signal will determine the eventual impact on SOCE. For example, domains of localized ROS production at the ER and mitochondria may exclusively affect SOCE via redox activation of STIM1 and hence lead to pro-tumorigenic Ca2+ signaling, while exogenous redox stress sensed by plasma membrane localized ORAI channels may block SOCE and attenuate proliferation of tumor cells and also make tumor cells more susceptible to redox stress (Figure 7) [198]. It is also possible that ROS mediate STIM translocation and SOCE activation indirectly, through redox activation of ER Ca2+ regulators IP3R and SERCA, which are also redox sensitive (See section 4). While detailed studies are required to unravel the complex nature of redox regulation of STIM and ORAI in the context of cancer cells and their tumor microenvironment, the following examples shed light on the potential role of the SOCE-ROS interlay in this context.
Figure 7.
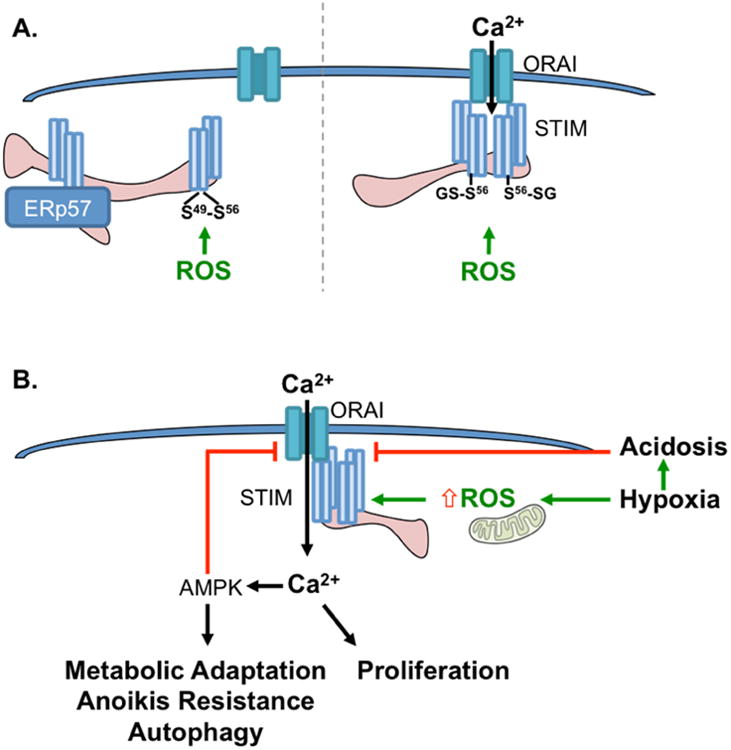
A. Redox Modifications of STIM1. A. STIM1 has the potential to form intra-molecular disulfide bonds (left) at Cysteine 49 and 56 following oxidation, which are also important for ERp57 binding. Both mechanisms are thought to inhibit STIM oligomerization and SOCE [106]. Conversely, glutahionylation (right) of the same cysteine residues is thought to decrease Ca2+ STIM binding to mimic store depletion and facilitate Stim oligometization and Orai interaction to initiate SOCE [105]. enhance inhibition reand glutathione adducts, which may result in either SOCE inhibition or activation. B. Potential consequences of STIM1 redox regulation in tumor cells in response to Hypoxia. Acute hypoxia induces mitochondrial ROS production, resulting in STIM1 puncta formation and SOCE activation. The resultant Ca2+ signal is important for tumor cell proliferation [199], and activates AMPK, a known regulator if metabolic adaptation and autophagy [200, 201]. Prolonged AMPK activation and the development of acidosis in response to long-term hypoxia may represent negative feedback mechanisms to shut-down SOCE [202, 203].
3.2.1 Redox regulation of STIM1
STIM1 has two cysteines (C49, C56) that have been demonstrated to be redox sensitive (Figure 7A) [105, 106]. Redox modification of cysteine 56 was shown to lead to glutathionylation, which resulted in decreased STIM1 Ca2+ binding, thereby mediating STIM1 oligomerization and activation of SOCE, independent of Ca2+ ER store depletion [105]. In somewhat contradictory work, Prins et al. demonstrated that oxidation of cysteine 56 results in intramolecular disulfide bond formation with cysteine 49. These two cysteines were also shown to be necessary for binding ERp57, an ER localized oxidoreductase [106]. ERp57-STIM1 interaction leads to decreased activation of STIM1 oligomerization and a decrease in SOCE. Further work is required to demonstrate that disulfide bond formation is similarly sufficient to block STIM1 puncta formation and activation of ORAI1. Moreover, it is of interest to investigate if the local ER redox status and glutathione pool resembles a potential redox switch for the suppression or activation of SOCE, respectively. These differences in STIM1 cysteine redox modifications may be cell type specific, but highlight that disparities in redox modifications at the same ER-localized cysteine could result in different cellular outputs related to STIM1-regulated SOCE. Redox regulation of STIM1 cysteine residues outside the ER luminal domain have not been investigated, however mutation studies suggest that these could also affect SOCE [195]. Different redox environments and pH between the ER lumen and the cytosol could also influence the difference between oxidation of luminal and cytosolic cysteine residues. Similarly, cysteine residues residing on either the inside or outside of plamsa membrane localized channels may be differentially affected by differences in intracellular and extracellular ROS levels, respectively, which may be influenced by differential antioxidant enzyme expression in subcellular compartments and ROS scavengers such as ascorbate in the extracellular space.
There is also evidence to suggest that STIM2, which is more sensitive to smaller drops in ER Ca2+ [204], may be oxidized at C725 in vitro, and may be important for SOCE regulation in hypoxia [195, 205]. Recent work is starting to unravel the role of STIM2 in regulating SOCE [206-210], and differential expression of STIM2 relative to STIM1 may be a phenotype of certain tumors, where higher STIM1:STIM2 ratio may suggest a worse prognosis in some cancer types [183, 211-213]. However, further studies are needed to illustrate the mechanistic consequences of STIM2 expression and potential redox regulation in cancer.
CRAC channel activity appears to be sensitive to stress signals that are associated with changes in cellular ROS and are a common phenotype of adaptations observed in cancer cells, including hypoxia and nutrient stress. Redox modification of SOCE has been implicated in eliciting cellular outcomes in response to hypoxia (Figure 7B). Hypoxia increases mitochondrial ROS production at complex III of the electron transport chain (ETC), and this was shown to cause translocation of STIM1 to the plasma membrane and CRAC channel opening [200, 201]. Hypoxia may also directly regulate STIM1 expression. A recent report demonstrated that STIM1 expression is positively regulated by HIF-1α in response to hypoxia in hepatocarcinoma cells, and that the resultant increase in SOCE is necessary for hypoxic tumor growth [199]. While hypoxia appears to influence the redox activation and expression of STIM1, hypoxia is also able to blunt SOCE via disruption of STIM1-ORAI1 interaction and ORAI1 pore block [202, 203, 214, 215]. Mancarella et al. demonstrated that acidification of the cellular environment in response to hypoxia leads to uncoupling of ORAI1 from STIM1, effectively decreasing SOCE. This sensitivity to acidosis may be an important feedback mechanism to prevent toxic build-up of Ca2+ in response to hypoxia [202, 203]. In essence, these data suggest that hypoxia induces SOCE during early stages of oxygen deprivation, while acidosis, which is a long-term consequence of hypoxia, may shut-down SOCE mediated Ca2+ entry to prevent intracellular Ca2+ accumulation and associated cell death (Figure 7B). These data also highlight the potential differences between chronic and acute hypoxia, which may influence cellular ROS status and signaling pathways, including differences between acute HIF-1α and chronic HIF-2α activation [216]. What role this plays in SOCE inactivation in the context of an acidic tumor microenvironment, which has been generally described as a consequence of increased glycolytic flux of tumor cells, remains to be elucidated.
There is also an interesting link between hypoxic regulation of SOCE and manipulation of nutrient stress response pathways. The redox regulation of STIM1 has been associated with activation of AMPK in conditions of hypoxia (Figure 7B) [200, 201]. As mentioned above, increases in mitochondrial ROS in response to hypoxia are necessary to initiate SOCE [200, 201]. The resultant Ca2+ response was shown to induce AMPK-phosphorylation by Calcium/calmodulin-dependent protein kinase kinase β (CaMKKβ) [201, 217]. Although, it should be noted that oxidative stress has also been demonstrated to increase AMP levels to activate AMPK [218], the above studies demonstrate that Ca2+ activation of AMPK is an important alternate regulatory pathway, independent of the AMPK regulator liver kinase B1 (LKB1). While AMPK activation is commonly associated with stress adaptations, hyperactivation of this pathway via SOCE may also have deleterious consequences on normal cells. For example, in alveolar epithelial cells it was demonstrated that this hypoxia-mediated SOCE regulation of AMPK is deleterious and eventually leads to Na+/K+-ATPase downregulation via endocytosis. This results in the inhibition of alveolar fluid reabsorption and endothelial cell dysfunction [200]. However it appears that cells have the ability to by-pass this damaging pathway. Observations in dendritic cells isolated from AMPK-/- mice suggest that AMPK may also act in a negative feedback loop to blunt Ca2+ influx into cells. For example, loss of AMPK resulted in enhanced SOCE, and in higher expression of ORAI1 and Na+/Ca2+ exchangers [219]. How this hypoxia-ROS-SOCE axis influences AMPK in cancer cells is unclear. AMPK is an important sensor of ATP availability and regulator of metabolism, thereby promoting an increase in catabolism and a decrease in anabolic pathways [220]. AMPK appears to have both pro- and anti-tumorigenic roles, and its positive role in cancer has been associated with eliciting metabolic flexibility of tumor cells under stress conditions [221, 222]. A recent study demonstrated that the ROS-Ca2+-CaMKKβ-AMPK axis plays a role in anoikis resistance of tumor cells. It was previously shown that matrix detachment of cell leads to redox stress due in part to changes in glucose uptake and a decrease in the NAD(P)H/NAD(P)+ ratios [223] and that AMPK can aid tumor cell survival during nutrient stress by resorting NADPH levels through activation of fatty-acid oxidation [224]. Sundararaman et al, showed that SOCE activation in response to matrix detachment precedes redox signaling, leading to AMPK phosphorylation in a CaMKKβ-dependent, LKB1-independent manner. These data suggest that AMPK regulation by SOCE may play an important role in the ability of cancer cells to adapt to anchorage-independence by promoting anoikis resistance and aiding spheroid formation, which are important aspects to tumor metastasis [225]. Whether Ca2+ signals or ROS spikes are the initial signal that drives this AMPK activation may require further investigations, however these studies demonstrate an example of the Ca2+-ROS signaling axis that may be involved in cellular survival and adaptation of tumor cell metabolism in response to stress, such as hypoxia, loss of matrix attachment and nutrient deprivation.
3.2.2 Redox regulation of ORAI1 and ORAI3
The ORAI family consists of 3 isoforms, of which ORAI1 was demonstrated to be inhibited following cellular exposure to H2O2, with and IC50 of 34μM [6]. Reduced channel conductance in response to H2O2 was dependent on C195, which is close to the plasma membrane on the extracellular side. Because the inhibition of ORAI1 channel activity required pre-incubation with H2O2 the authors argued that oxidation of C195 maintains the channel pore in a closed configuration prior to thapsigargin-mediated store depletion and hence prevents activation in response to store depletion. The lesser-studied ORAI2 isoform had a similar inhibition profile to ORAI1 [6]. In subsequent work the authors proposed that pretreatment of cells with H2O2 leads to intramolecular interaction of C195 with S239, locking the channel in a closed conformation [226]. It should be noted that the high concentrations of H2O2 (1mM) used in this work may contribute to the irreversible state of this redox modification [226]. C143 and C126 have also been implicated with electrophilic interactions and inhibition of CRAC currents by curcumin and caffeic acid phenethyl ester [227].
Interestingly, C195 is absent in the ORAI3 isoform, and this difference may play a role in determining redox regulation of ORAI1/ORAI3 heteromultimers in a number of pathophysiological contexts, including cancer. This was first investigated in the context of T Helper (TH) cells, where enhanced expression of ORAI3 were proposed to mediate redox insensitivity of the CRAC channel as TH cells develop from naïve cells to effector cells [6]. Naïve cells, using ORAI1 as their major CRAC channel, were more sensitive to H2O2 mediated cell death and CRAC inhibition by H2O2. In contrast, as cells matured into effector TH cells, they expressed a higher proportion of ORAI3 subunits. Presumably this leads to an increase in the ORAI3/ORAI1 ratio and a decrease in available C195 residues, resulting in channels that are less sensitive to redox-inhibition (Figure 8). Increased ORAI3 expression in effector TH cells correlated with a decrease in redox inhibition of channel conductance and an increase in proliferation and cytokine production [6]. Infection illustrates an important interplay between SOCE activation and ROS regulation, where pathogen associated peptides initiate IP3-mediated Ca2+ store depletion and SOCE activation. The ensuing Ca2+ signals activate Nox2 at the cell surface via PKC to produce O2•- and H2O2 as part of the oxidative burst for the killing of the pathogen. Interestingly, a switch to a redox insensitive ORAI3/ORAI1 heteromultimer was also shown to occur in monocytes following bacterial infection [228]. A higher ORAI3/ORAI1 ratio was shown to reduce the Ca2+ amplitude [229], but resulted in a prolonged Ca2+ signal that was not inhibited by increases in ROS [228]. The authors proposed this to be a mechanism that ensures killing of the pathogen, but avoids extensive tissue damage in response to sustained ROS bursts.
Figure 8.
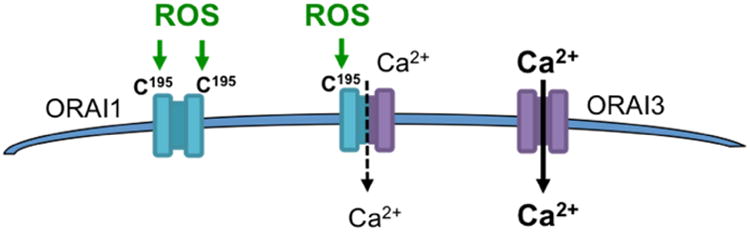
Differential redox regulation between ORAI1 and ORAI3 due to the lack of C195 in ORAI3. Oxidation of ORAI1 leads to channel inhibition. The significance of ORAI1 redox inactivation, the ratio of ORAI1/ORAI3 expression, and the consequences of different ORAI heteromultimer conformations in cancer require further investigation.
Interestingly, the ORAI1/ORAI3 ratio also appears to influence the redox sensitivity of ORAI channels in cancer cells. It was shown that prostate cancer cell lines have a higher ratio of ORAI1/ORAI3 compared to primary human prostate epithelial cells [198]. The authors went on to show that an increased ORAI1/ORAI3 ratio makes prostate cancer cells more redox sensitive to SOCE inactivation by H2O2 [198]. H2O2 exposure of prostate cancer cells and normal prostate epithelial cells led to an initial increase in cytosolic Ca2+, likely as a consequence of ROS-dependent TRP channel activation, which differed between cancer and normal cells. This was followed by H2O2-dependent SOCE inactivation, as tested by store depletion using the SERCA pump blocker thapsigargin. The prostate cancer cell line LNCaP displaying a 10 fold lower H2O2 IC50 value compared to the normal prostate epithelial cell line hPECs. Although the prostate cancer cell DU145 displayed a similar IC50 to hPECs, the authors correlated a change in the ORAI1/ORAI3 expression ratio to increased susceptibility of cells to redox stress [198]. As mentioned previously, tumor cells are often more susceptible to ROS toxicity, This phenotype may be due to some tumor cells having higher intracellular steady state levels of ROS, which in turn places cells closer to the cytotoxic threshold of ROS, an observation that has triggered research into utilizing ROS-agents for chemotherapeutic applications [79-82, 99]. Holzmann et al. proposed that ROS-mediated inactivation of ORAI1 may also contribute to higher sensitivity of prostate cancer cells to ROS, due to dampening of SOCE dependent pro-proliferative Ca2+ signaling [198, 230]. The authors proposed that this altered ORAI1/ORAI3 expression ratio could provide a therapeutic opportunity to target the pro-proliferative actions of SOCE by carefully tuning redox-mediated inactivation to only affect tumor cells [198]. However, it should be pointed out that in all ORAI1/ORAI3 studies described above, it remains uncertain whether ORAI1 and ORAI3 form different quantities of two independent homohexameric channels or form heterohexameric ORAI1/ORAI3 channels with variable stoichiometries. Earlier studies provided evidence for native ORAI1/ORAI3 hetero-multimerization where these channels are not activated by store depletion, but instead by receptor-mediated production of arachidonic acid or its metabolite, LeukotrieneC4 (LTC4) [231-235]. In this context, the work by Holtzmann et al. [198] sharply contrasts with another study of prostate cancer, demonstrating increased levels of ORAI3 expression, in prostate tumor cells, which resulted in ORAI1/ORAI3 heteromultimeric channels that elicited Ca2+ entry in response to receptor stimulation in a store-independent fashion [236]. This was presumed to occur through production of arachidonic acid and/or LTC4. While this study did not investigate the redox regulation of ORAI1/ORAI3 heteromultimeric channels, the authors proposed that these channels elicit Ca2+ signaling to drive proliferation, while a smaller proportion of ORAI1 homomeric channels mediate the classical SOCE pathway, which is pro-apoptotic [236]. Hence, an increase in ORAI3 expression would shift tumor cells towards pro-proliferative Ca2+ signaling. Differences in ORAI subunits expression have also been demonstrated for other tumor types. ORAI3 plays a significant role in mediating SOCE in estrogen receptor positive breast cancer cells, while estrogen receptor negative cells have CRAC channels that are largely composed of ORAI1 [182-184, 189, 237]. Reports of heterogeneity in ORAI1 expression within the same tumor also highlight the potential transient nature of SOCE regulation during tumor progression [213]. How relative ORAI1 and ORAI3 expression levels relate to the redox regulation of SOCE between histological subtypes and different areas of the tumor remains to be elucidated. However, it is reasonable to suspect that this could vary not only in terms of ORAI isoform expression, but also in terms of their exposure to exogenously and endogenously produced ROS, for example in response to hypoxia or enhanced immune cell infiltration.
ORAI1 may also be regulated by ROS in an indirect fashion. Feng et al previously proposed that breast cancer cells express high levels of the Secretory Pathway Ca2+-ATPase, SPCA2, which constitutively activates ORAI1 channels in a STIM and SOCE independent manner [181]. A recent study demonstrated that SPCA2 expression is enhanced in HCT116 colon cancer cells in response to hypoxia, on 3D spheroid growth, after exposure of cells to H2O2, and with agents that induce mitochondrial ROS (Antimycin A) and reactive nitrogen species (DETA NONOate) [238]. Although the investigators did not assess the activation of ORAI1 in this context, SPCA2 regulation by ROS/RNS, may be an indirect mechanism for the redox regulation of ORAI1 in cancer. Moreover, since mitochondrial dysfunction, aberrant mitochondrial redox signaling and ER stress are common occurrences in cancer, it is not unreasonable to suggest that these feed into the redox regulation of SOCE. As described further below, oxidation of the IP3 Receptors has been demonstrated to influence their activity and hence alters the levels of Ca2+ in ER stores. For example, activation of IP3R by H2O2 and consequential ER Ca2+ store depletion was shown to partially contribute to the activation of CRAC channels, indicating that SOCE is influenced at multiple levels by redox regulation [239]. How the oxidation of Ca2+ regulators at internal stores influence SOCE is only starting to be unraveled.
3.3 Plasma Membrane Ca2+ Efflux pumps
3.3.1 Redox regulation of Plasma Membrane Ca2+ ATPase (PMCA)
Contrary to the redox-activation of a number of Ca2+ influx channels, oxidation of the plasma membrane Ca2+ ATPase (PMCA), which extrudes Ca2+ from the cell, is largely inhibitory. This suggests that a global increase in oxidative state could cause enhancement of cytosolic Ca2+ levels. PMCA redox inactivation is thought to arise primarily as a consequence of oxidative stress in pathological conditions, such as neurodegeneration and reperfusion injury, leading to a buildup of cytosolic Ca2+, degradation of the PMCA protein, and cell death [240, 241]. This redox-dependent inhibition of PMCA may also be an avenue that mediates apoptosis in response to cytotoxic agents. In MCF7 breast cancer cells, redox-mediated apoptosis in response to the platinum analog ([Pt(O,O′-acac)(γ-acac)(DMS)]) was primarily initiated by the inhibition of PMCA and the subsequent increase in intracellular Ca2+ [242]. Activity of PMCA is also indirectly affected by oxidation of Calmodulin (CaM). Oxidation of CaM C-terminal methionines inhibits activation of PMCA, while still maintaining the PMCA-CaM interaction [243, 244]. These data suggest that PMCA inactivation is a common consequence of redox stress and is important during apoptosis. It is unknown if tumor cells are more resistant to this inactivation. Studies have shown variable expression patterns of PMCA in cancer cells, which appear to be isoform-, cancer type- and stage-specific [245-250]. Enhanced expression of PMCA in tumor cells may be one mechanism by which tumor cells evade high Ca2+ buildup during apoptosis, and inhibiting this protein may be a way to initiate tumor cell death [248, 250, 251].
3.3.2 Redox regulation of Na+/Ca2+ exchanger (NCX)
Contrary to PMCA, the Na+/Ca2+ exchanger (NCX), which mostly elicits Ca2+ extrusion from the cell but can also work in the reverse mode to cause Ca2+ entry, is activated in response to thiol oxidation by H2O2. NCX activation together with Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA; discussed below) redox inactivation is thought to play a role in sarcoplasmic Ca2+ depletion and a decrease in contractile function of myocytes during heart failure [252]. Further studies are needed to determine if sub-lethal and localized changes in ROS can influence transient alterations in PMCA and NCX activity and if this plays a role in altering Ca2+ homeostasis and Ca2+-mediated signaling in cancer cells.
4. Redox Regulation of ER and mitochondrial Ca2+ modulators
4.1 The ER - Mitochondrial Interface
Controlled Ca2+ homeostasis of mitochondria is imperative for proper mitochondrial function. A number of proteins involved in the TCA cycle and electron transport chain rely on Ca2+ for their activity and can hence determine cellular metabolic flux and redox signaling [253]. However, as discussed below, high mitochondrial Ca2+ import together with increases in ROS are also essential for the onset of apoptotic cell death [254, 255]. This influx of Ca2+ into the mitochondrial matrix is derived primarily from closely apposed ER, although Ca2+ entry channels at the plasma membrane also couple to mitochondrial Ca2+ uptake [256-258]. The importance of mitochondrial-ER cross talk has been emphasized by the discovery of Mitochondria-Associated Membranes (MAMs) as highly regulated hubs of ER-Mitochondria organellar interface. These areas of close proximity between the mitochondrial and ER membranes contain tethering proteins and channels that help facilitate ion exchange between the two organelles, most notably Ca2+. MAMs are characterized by enrichment of ER-localized IP3R/RyR receptors and SERCA pumps, and mitochondrial Voltage-dependent anion channel (VDAC) and the mitochondrial Ca2+ uniporter (MCU) in the outer and inner mitochondrial membrane, respectively (Figure 4). These channels facilitate Ca2+ transfer between the ER and mitochondria and are known redox-activated proteins [52, 89]. While the ER and mitochondrial membranes do not directly fuse, they are closely associated within 10-25 nm, and are tethered by proteins, such as Mitofusin 2 (MFN2), one of two mitochondrial fusion proteins [259, 260]. Ca2+ levels within mitochondria are further regulated by Ca2+ efflux carried out by the mitochondrial Na+/Ca2+ Exchanger (termed NCLX for its ability to exchange both Na+ and Li+ for Ca2+) [261] and the mitochondria Permeability Transition Pore (mPTP) [262]. Mitochondrial Ca2+ homeostasis is fine-tuned by the activity of NCLX which extrudes Ca2+ from mitochondria into the cytosol, and in turn aids in maintaining mitochondrial redox homeostasis [263].
MAMs are increasingly being recognized as domains that are enriched in proteins contributing to both ROS and Ca2+ shuttling between the ER and mitochondria [9, 264]. Interestingly, MAMs are high in oxidoreductase Ero1 levels, which is involved in disulfide bond formation during protein folding and an important regulator of the redox state of the ER [55]. Ero1 was shown to modulate both IP3R-dependent Ca2+ release and the activity of MCU, therefore affecting Ca2+ shuttling from the ER to the mitochondria [265, 266]. This regulation is tightly controlled by redox activation and the relative expression of Ero1. In scenarios of ER stress, the interaction between Ero1 and IP3R and subsequent activation of Ca2+ release is an important mechanism in the initiation of apoptosis [267]. Similarly, Protein kinase RNA-like endoplasmic reticulum kinase (PERK) is localized to MAMs and shown to be an important mediator of ER-mitochondrial Ca2+ transfer in response to ROS-mediated ER stress [268]. Altered ER stress response pathways have been reported in cancer [269, 270], but these have not been specifically linked with redox-sensitive Ca2+ regulatory pathways at the MAMs. However, one could speculate that this may be an important aspect to cancer cell adaptations in response to ER stress and the unfolded protein response.
4.1.1 Ca2+-ROS interplay during Apoptosis
The consequence of the Ca2+-ROS interplay at the ER-mitochondrial interface is dependent on the amplitude and frequency of these signals. Most commonly, studies have focused on the large fluxes of Ca2+ and surges in ROS associated with apoptosis, which results in the opening of the mitochondrial Permeability Transition Pore (mPTP), mitochondrial membrane potential collapse, H2O influx, mitochondrial swelling, and cytochrome c release (Figure 9A) [271, 272]. SERCA, IP3R and VDAC are important mediators of Ca2+ transfer from the ER to the mitochondria during apoptosis [52, 273, 274]. Interestingly, a number of tumor suppressors have also been shown to associate with MAMs and been demonstrated to directly influence this Ca2+ shuttling [275-277]. While it has been shown that Ca2+ can directly initiate mitochondrial membrane permeabilization, through calcineurin-dependent dephosphorylation of BAD and its association with Bcl-xL [278], the coordinated, yet complex interaction of both Ca2+ and ROS, appears to be necessary for mPTP opening and apoptotic (and necrotic) cell death activation [279]. Madesh and Hajnoczky showed that O2•- promotes mPTP opening in a Ca2+-dependent manner. VDAC is an important mediator of Ca2+ influx into the mitochondria in this context, as inhibition of VDAC inhibited O2•--dependent cytochorome c release [52]. While the exact mechanisms of redox regulation of mPTP are still being elucidated, adenine nucleotide translocase and cyclophilin D have been shown to have redox active cysteines, which control the opening of mPTP [280-282]. Cyclophilin D sensitizes the mPTP to Ca2+, due to its direct interaction with F0F1-ATP synthase, dimers of which are thought to constitute the mPTP channel [283].
Figure 9.
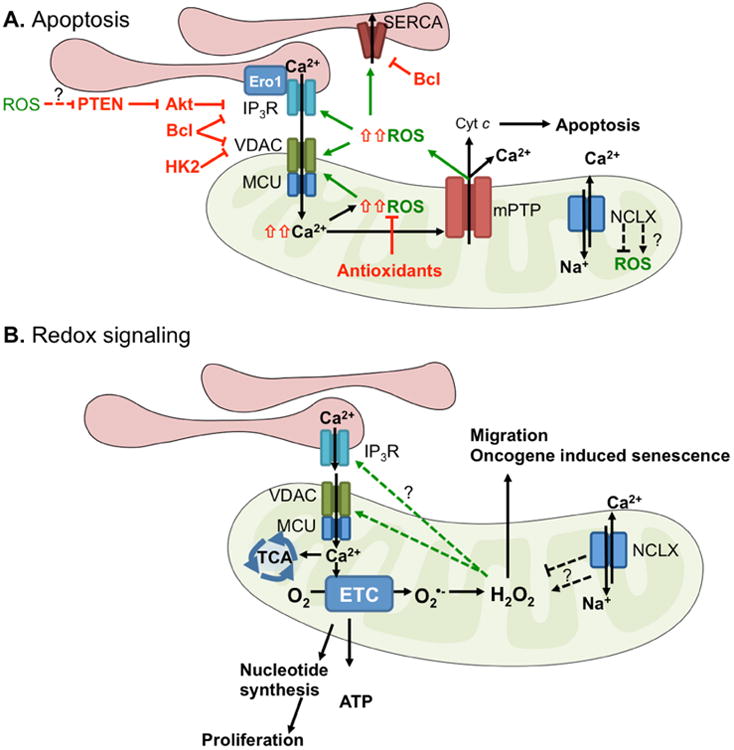
Cancer cells have developed several mechanism to evade the pro-apoptotic ROS-Ca2+ cross talk at the ER-mitochondrial interface (A), while maintaining regulated mitochondrial Ca2+ influx that can regulate mitochondrial redox signaling to support tumor survival, growth and metastasis (B). A. Tumor cells have been shown to express anti-apoptotic proteins such as Bcl-2, Bcl-XL and HK2 to bind and inhibit IP3R and VCDA, and suppressing the Ca2+transfer from the ER to the mitochondria, inhibiting Ca2+-ROS dependent apoptosis [75-78] [300-302] [303]. B. The transfer of Ca2+ to the mitochondria is also important in maintaining ETC and TCA function and the ability of tumor cells to synthesize adequate levels of nucleotides for cell growth [299]. There are preliminary reports that this increase in Ca2+ ER- mitochondrial shuttling may be important for mitochondrial ROS production and redox mediated signaling to drive migration [304] and oncogene induced senescence [305]. Whether or not this increase in mitochondrial H2O2 exerts a positive feedback on IP3R [85], VDAC and MCU in tumor cells remains to be determined. Studies have shown that activation of NCLX may increase mitochondrial ROS production, by decreasing Ca2+ levels in mitochondria [306] [263]. However, NCLX knock down can also result in ROS generation, presumably due to its important role in fine tuning Na+/Ca2+ balance [180]. The consequences of NCLX –mediated mitochondrial ROS production on cellular signaling are still unclear, but could have influence redox regulation of the CRAC channel ORAI [180].
A number of mechanisms for the Ca2+ induced O2•- production in mitochondria have been proposed, including Ca2+ mediated increase in the activity of components of the TCA cycle and ETC, thereby accelerating electron leakage and O2•- production, and the role of Ca2+ on mPTP opening which may lead to further ROS production due to transient mitochondrial membrane potential depolarization and resultant increases in ETC O2•- production (for reviews see [5, 271, 282]). Moreover, ROS release from mitochondria appears to perpetuate mitochondrial ROS surges during apoptosis. This may be necessary for transmittal to adjacent mitochondria, eventually resulting in global activation of apoptotic cell death [284]. In contrast to the above, a decrease in mitochondrial Ca2+ may also interfere with normal function of the ETC to increase mitochondrial ROS production. An example is a recent study demonstrating that Ca2+ overload in mitochondria leads to Complex II disintegration from the ETC. While complex II is uncoupled from the ETC it remains active and produces ROS [53]. The investigators show that cardiolipin can interact with and sequester Complex II into protein aggregates, and that this is dependent on Ca2+.
Tumor cells adapt to evade apoptosis, with several reports demonstrating direct manipulation of mPTP opening in cancer cells [271]. These include modulation of cellular signaling to regulate mPTP opening, such as the dampening of PTEN mediated regulation of Ca2+ influx via IP3R, as described below (reviewed in [271, 279]). Tumor cells exhibit changes in Bcl apoptotic protein expression, and it is known that these can also alter the function of a number of Ca2+ channels including IP3R (Figure 9A) [285]. An example, is the interaction of the anti-apoptotic protein Bcl-2 with IP3R. Binding of Bcl-2 to IP3R slows IP3-mediated release of Ca2+ from the ER, which results in a lowering of mitochondrial Ca2+ pools in cancer cells. This is sufficient to inhibit the initiation of apoptosis [75-78]. Increasing Ca2+ ER-mitochondrial shuttling may therefore be a therapeutic approach to enhance apoptosis initiation in cancer cells, and recent work has explored the use of a Bcl-2/IP3R disrupter (BIRD-2) peptide to induce apoptosis in a number of hematological cancers and lung cancer studies [286-288]. In addition to the effects of dampening Ca2+ flux into mitochondria, cancer cells likely evade apoptosis by enhanced mitochondrial ROS scavenging, which could further inhibit mPTP opening.
4.1.2 Ca2+-ROS signaling at the ER – Mitochondrial Interface
In comparison to ROS/Ca2+ surges during apoptosis, tightly controlled ROS and Ca2+ oscillations appear to be important in regulating cell signaling and the maintenance of energy production by mitochondria [289]. As such, small increases in H2O2 can initiate Ca2+ release from ER stores. For example, it was shown that H2O2-dependent oxidation and activation of PLCγ1 mediates IP3R-dependent Ca2+ oscillations [290]. The investigators further demonstrated that EGF treatment of rat cortical astrocytes stimulated H2O2 production to initiate these oscillations. This may have implications for growth factor signaling and ROS-Ca2+ crosstalk in cancer, where EGF is a major pro-proliferative pathway.
In general, it is accepted that physiologically relevant mitochondrial Ca2+ uptake occurs through shuttling of Ca2+ from the ER to the mitochondria and that this process is of importance for the TCA cycle generation of NADH, increase mitochondrial respiration and ATP production (Figure 9B) [291-293]. A number of metabolic enzymes, electron transport chain proteins, substrate transporters, as well as changes in mitochondrial membrane potential are regulated by mitochondrial Ca2+ [253, 294, 295]. For example, Pyruvate dehydrogenase (PDH) activity is tightly regulated by Ca2+. PDH-Phosphatase is activated by Ca2+, which in turn dephosphorylates PDH, leading to an increase in PDH enzyme activity [296, 297]. Ca2+-dependent regulation of mitochondrial metabolic pathways is clearly an important aspect of normal cell function, and inhibition of Ca2+ transfer by blocking IP3R results in decreased ATP production and activation of AMPK and autophagy [298]. In a recent study it was suggested that this switch to autophagy is not sufficient to ensure survival of tumor cells under stress conditions. It was proposed that mitochondrial Ca2+ uptake is required for maintenance of cancer cell nucleotide synthesis [299]. Thus Ca2+ transfer from the ER to mitochondria is an important pro-survival signal that is required for the synthesis of cellular building blocks by cancer cells, even if they rely less on mitochondrial ATP production. As described below, increased mitochondrial Ca2+, as well as low mitochondrial Ca2+ have both been implicated with an increase in mitochondrial ROS production. This may occur through different mechanisms, including Ca2+ driven increases in ETC flux, which enhances electron leakage and O2•- production. Alternatively, a decline in mitochondrial Ca2+ could lead to a decrease in TCA cycle generation of reducing equivalents, thereby inhibiting the ROS scavenging ability of mitochondria [253]. The observation that mitochondrial Ca2+ can also regulate ROS production may be one mechanism by which Ca2+ initiates a subsequent mitochondrial redox-signaling loop to further ensure tumor cell survival. As with plasma membrane channels, the levels and duration of Ca2+ mitochondrial influx determine the cellular consequences of the ER-mitochondrial ROS-Ca2+ cross talk (Figure 9).
4.2 Redox regulation of Inositol 1,4,5-trisphosphate (IP3) receptors
As mentioned above, maintenance of physiologically relevant Ca2+ levels in the mitochondria is important for proper mitochondrial function. The IP3R and the related RyR are the primary regulators of ER/SR Ca2+ release into the cytoplasm and Ca2+ transfer from the ER (or SR) to the mitochondria. The latter is important in facilitating large surges of Ca2+ transfer during apoptosis, as well as more transient Ca2+ oscillations to maintain ATP production. Like RyR, IP3R is a demonstrated target for redox-mediated regulation [307, 308]. Different redox modifications have also been observed for IP3R. Glutathionylation induced by Diamide and H2O2 treatment enhances IP3R Ca2+ release in response to Ca2+ [309]. In addition, disulfide bond formation of ER luminal cysteines (C2496, C2504, or C2527) was demonstrated to lead to IP3R mediated Ca2+ release, due to the dissociation of IP3R1 from the thioredoxin ERp44, which inhibits IP3R under reducing conditions [310], Moreover, direct action of O2•- on IP3R activity was demonstrated using a xanthine-xanthine oxidase delivery system, that presumably resulted in the oxidation of cytosolic residues of the receptor [311]. The authors argued that any mitochondria-derived O2•- may not have the ability to reach the IP3R and instead needs to be first dismuted to H2O2. Indeed a follow-up study demonstrated that IP3R Ca2+ oscillations yield localized increases in H2O2 at MAMs and that this results in a positive redox feedback on IP3R activation [85]. Interestingly, these localized H2O2 “nano-domains” are thought to originate as a consequence of Ca2+ induced H2O and K+ influx into the matrix, which results in mitochondrial cristae deformation and potential rearrangement of ETC components into supercomplexes that could be involved in the observed localized H2O2 increases [85, 312]. It would be of great interest to determine if such Ca2+-dependent mitochondrial cristae shape changes are a phenotype of cancer cells that regulate redox and Ca2+ signaling, or if these changes are altered to prevent Ca2+ influx into mitochondria during the process of apoptosis. Moreover, this may have implications for ETC function and metabolism of cancer cells. As such, it has been shown that IP3R-dependent Ca2+ transfer from the ER to the mitochondria sustains ATP production while maintenance of ATP levels ensures that AMPK-dependent autophagy is suppressed in nutrient-rich environment [298]. IP3R-mediated Ca2+ transfer to mitochondria is also linked to the Ca2+-dependent activation of PDH phosphatase, which in turn activates PDH, leading to increased Acetyl CoA flow into the TCA cycle and respiration [298]. Conversely, when this Ca2+ flow from the ER to the mitochondria is suppressed, ATP levels fall and autophagy is initiated as a survival mechanism. It should be pointed out that there are conflicting reports on the role of IP3R on both promoting and suppressing autophagy, which is likely dependent on nutrient availability (i.e. culture conditions), cell type and feedback mechanisms of Ca2+ signaling that control autophagy regulators such as Beclin I [164]. Many tumor cells are thought to survive without oxidative phosphorylation, relying primarily on aerobic glycolysis. As mentioned above, a recent study found that tumor cells are exquisitely sensitive to a block in ER to mitochondrial Ca2+ transfer. While this resulted in phosphorylation of AMPK and initiation of autophagy in both normal and cancer cells, lack of Ca2+ release through IP3R or inhibition of mitochondrial Ca2+ uptake by blocking MCU led to increased cell death specifically in tumor cells. This was shown to be due to initiation of necrosis, and the authors concluded that inhibition of ER-mitochondrial Ca2+ transfer leads to decreased mitochondrial function and a lack of nucleotide production, important building blocks for proliferating cancer cells [299]. With the recognition that metabolic pathways besides aerobic glycolysis are important for the maintenance of tumor cell macromolecules, the importance of altered mitochondrial function in tumors is of great interest. The role for the IP3R in maintaining adequate Ca2+ balance within mitochondria of cancer cells is starting to emerge. The interplay between ROS and IP3R activity will no doubt be an important regulatory mechanism in this context.
In addition to direct oxidation of IP3R, ROS can influence IP3R activity indirectly. This was demonstrated in breast cancer cells, where cells with mitochondrial dysfunction and increased ROS production displayed enhanced expression of the chemokine CXCL14, as a consequence of ROS and AP1-dependent regulation of the CXCL14 promoter. In turn, CXCL14 induced IP3R-mediated Ca2+ release, which was necessary for ROS-dependent breast cancer cell migration [313]. IP3R was shown to interact and be regulated by a number of kinases and phosphatases, including the Akt and PTEN signaling axis, where IP3R phosphorylation by Akt has been shown to decrease IP3R3 activity and suppress pro-apoptotic Ca2+ release [275, 276, 314]. Conversely, during induction of apoptosis association of IP3R with the phosphatase PTEN is enhanced at MAMs and leads to de-phosphorylation of IP3R, resulting in increased mitochondrial Ca2+ [276]. PTEN is a known tumor suppressor commonly lost in a number of cancer types and re-expression of PTEN has been shown to restore IP3R mediated apoptosis [315]. Interestingly, PTEN is a direct target for redox mediated signaling, as cysteine oxidation leads to decreased PTEN activity, and redox mediated inactivation has been observed in cancer cells with higher redox status [98, 316, 317]. Whether PTEN oxidation and a concomitant decrease in IP3R mediated Ca2+ release into mitochondria are involved in apoptosis resistance of tumor cells, remains to be elucidated.
4.3 Redox regulation of Voltage-dependent anion channel (VDAC)
VDACs are localized to the outer mitochondrial membrane, are commonly localized in MAMs, facilitate shuttling of metabolites and ions across the outer mitochondrial membrane and may serve as a channel that allows O2•- release from the mitochondria [5, 318]. Although VDACs are implicated in mPTP opening, studies from knockout cells suggest that VDACs do not constitute the pore forming unit of the mPTP. Cysteine residues of VDAC face the intermembrane space, are located in the relatively accessible pore region and are potentially oxidized by O2•- generated from electron leakage from the ETC at complex III [318, 319]. Screens for oxidized thiols have identified VDACs as potential redox modified proteins, however the consequences of specific redox modifications of different VDAC isoforms are only starting to be interrogated and it is unclear if these play a role in VDAC activity or participate in altered VDAC function in a pathophysiological situation like cancer. As described briefly below, the role of VDAC may be primarily by facilitating mitochondrial Ca2+ uptake to enable mitochondrial ROS production, which is particularly evident during the process of apoptosis and necrosis. For example, pharmacological and antibody blockade of VDAC inhibited O2•--dependent cytochorome c release [52]. Of the three VDAC isoforms, VDAC1 is thought to be the primary channel involved in Ca2+ loading of mitochondria during apoptosis [274]. Similar to IP3R, VDAC1 activity is inhibited by binding to anti-apoptotic proteins like hexokinase I and Bcl-2 family members, with Bcl-XL being more important to VDAC1 inhibition than Bcl-2 [300-302]. To increase apoptotic cell death in cancer cells, studies have focused on inhibiting the interaction between anti-apoptotic proteins and VDAC using specific peptides to prevent protein-protein interactions. In addition, it has been shown that forced VDAC expression and pharmacological approaches that enhance VDAC activity can enhance apoptosis of cancer cells [320-322]. Interaction of VDAC with Hexokinase (HK) may decrease apoptosis. This may be particularly important in cancers demonstrating high glycolytic flux and overexpression of HK2. Dissociation of HK2 lifts its inhibitory action on the channel, leads to VDAC oligomerization and allows for initiation of apoptosis [303]. Again, disrupting this interaction has been proposed as a therapeutic strategy. However, in addition to prevention of VDAC-mediated apoptosis, the interactions of VDAC with anti-apoptotic proteins may also serve to fine tune VDAC-mediated Ca2+ mitochondrial uptake thus regulating mitochondrial ROS signaling. For example, in non-small cell lung carcinoma the antiapoptotic protein Mcl-1, was shown to bind VDAC1 and 3 isoforms and this interaction was necessary for Ca2+-driven ROS production that was important for lung cancer cell migration, but not proliferation [304]. Therein, VDAC facilitates non-canonical signaling of anti-apoptotic proteins and connects Ca2+ to mitochondrial redox signaling to regulate pro-metastatic phenotypes.
4.4 Redox regulation of Mitochondrial Ca2+ Uniporter (MCU)
The MCU is a Ca2+-selective channel localized in the inner mitochondrial membrane and mediates Ca2+ uptake into the mitochondrial matrix, which is dependent on mitochondrial membrane potential (ΔΨm) and pH [323, 324]. MCU comprises the pore-forming unit of a complex of proteins that includes regulatory proteins involved in gating of MCU (for review see [325]). The role of MCU-regulated mitochondrial Ca2+ uptake on maintaining mitochondrial bioenergetics is dependent on the relative metabolic activity of specific tissues, as demonstrated in MCU knockout animals, where loss of MCU has little effect on Oxidative Phosphorylation of mouse embryonic fibroblasts. On the contrary, pronounced effect on mitochondrial bioenergetics are observed following MCU loss in skeletal and cardiac tissues under stress condition [326, 327]. MCU channel activity is regulated by a number of accessory proteins, including Mitochondrial Ca2+ Uptake 1 (MICU1), a Ca2+ regulated gate keeper that modulates mitochondrial Ca2+ uptake by Ca2+ dependent inhibition of MCU [324, 328]. Loss of MICU1, results in sustained Ca2+ influx into mitochondria leading to increased mitochondrial ROS production [328]. Depending on the levels of Ca2+ influx and ROS production this can either lead to redox-mediated signaling, inhibition of Oxidative Phosphorylation, or increased susceptibility to apoptosis [328]. It appears that the MCU regulatory proteins may also determine MCU activation in a cell type specific manner [329]. Loss of the positive MCU Regulator 1 (MCUR1), disrupts mitochondrial function in vascular endothelial cells and leads to induction of autophagy, as an apparent compensatory mechanism [329]. The function of MCU and its regulators in cancer is only starting to be unraveled. In one study, IP3R2 and MCU were identified as two important mediators of oncogene-induced senescence and replicative senescence using an shRNA screen [305]. Knockdown of these proteins caused tumor cells to exit senescence, and caused a concomitant decrease in mitochondrial Ca2+ influx. It was proposed that the maintenance of mitochondrial Ca2+ and ROS by MCU and IP3R2 are required for cells to stay senescent [305].
There is conflicting data on the changes in MCU expression between cancer types and the respective involvement of MCU in regulating mitochondrial function in this context. Decreased MCU expression in colon and prostate cancer was associated with high levels of the micro RNA miR-25. Low MCU levels correlated with reduced susceptibility to apoptosis, due to lack of MCU-mediated Ca2+ mitochondrial uptake [330]. Conversely, increased MCU and low MICU1 expression has been associated with poorer survival outcomes in breast cancer patients [331]. Interestingly, changing expression of either protein did not significantly alter clonogenic survival of MDA-MB-231 breast cancer cells in response to various stressors, such as nutrient deprivation or chemotherapy, while normal breast epithelial cells were dependent on the presence of MCU and MICU1 for cell survival [331]. Other investigators similarly demonstrated that high MCU expression correlates with increasing stage of breast cancer and an invasive phenotype, yet that decreasing MCU expression has little effect on inhibiting cell proliferation or viability [332, 333]. It was shown that loss of MCU does not influence caspase-dependent apoptosis, but that ionomycin-induced caspase-independent cell death is increased in response to MCU loss in estrogen receptor negative and basal-like breast cancers [332]. This may be related to histological subtype as, triple negative cancers appear to require MCU for tumor growth, as described below [334].
Interestingly, recent studies suggest that MCU may control the interplay between Ca2+ and redox signaling emanating at the mitochondria. Reducing either MCU or MICU1 expression diminishes mitochondrial O2•- flashes in response to hyperosmotic stress [335]. This study highlights that changes in these signaling flashes are initiated and dependent on synergistic, physiologically relevant increases in mitochondrial Ca2+ and ROS. These flashes were shown to be important for activating the MAP Kinase pathway as both Jnk and Erk phosphorylation were inhibited with the use of a mitochondrial ROS scavenger [335]. MCU was also shown to be important for the generation of mitochondrial ROS flashes that drive oxidation and inactivation of Rho-1 and consequential actin mediated wound-closure in a C.elegans wound healing model [86]. The investigators showed rapid increase in intracellular Ca2+ following wounding, which is buffered by the mitochondria, specifically through the MCU. MCU was necessary to elicit the production of mitochondrial ROS sparks, and the investigators concluded that the primary reactive species eliciting redox signaling is O2•-, as mutants lacking SOD isoform expression displayed enhanced wound healing. Due to its short-lived nature, this would imply that mitochondria are closely associated with Rho-1 at the actin cytoskeleton. It is unclear if MCU plays a similar role in regulating mitochondrial ROS flashes to drive proliferative and migratory cell signaling in cancer cells. A recent study investigating the role of MCU in triple negative breast cancer progression indicates that this may indeed be relevant in cancer. Knock-down of MCU expression decreased cell migration and invasion and inhibited tumor formation and metastatic spread of triple negative breast cancer cells in an in vivo xenograft model [334]. Moreover, MCU-mediated mitochondrial Ca2+ influx and concomitant ROS-increases were shown to be necessary for HIF-1α activation, indicating an important role of MCU in regulating mitochondrial redox signaling in cancer cells [334]. In addition, it was shown that MCU is necessary for breast cancer cell migration and that this is dependent on SOCE activation [333].
While it appears that MCU can initiate redox changes at the mitochondria, reciprocal regulation of MCU channel activation by ROS has not been investigated in great detail. Direct oxidation of MCU or its interacting partner has not been described. The redox sensitive CaMKII has been shown to activate MCU-mediated Ca2+ currents in response to ischemia reperfusion injury, leading to myocardial cell death [336]. This could represent one indirect mechanism of MCU redox regulation in response to oxidative stress.
4.5 Redox regulation of Mitochondrial Na+/Ca2+ Exchanger (NCLX)
The molecular identity of the mitochondrial Na+/Ca2+ Exchanger (NCLX) was recently identified and shown to be an important regulator of Ca2+ efflux from the mitochondria [261]. Since NCLX mediated Ca2+ efflux is dependent on cytosolic Na+ levels, cytosolic Na+ homeostasis can influence mitochondrial Ca2+ levels. Interestingly, under certain pathological conditions an increase in cytosolic Na+ has been shown to mediate decreased mitochondrial Ca2+ levels that in turn yield increases in mitochondrial ROS production. This is in apparent contrast to the above mentioned studies that show synergistic increases between mitochondrial Ca2+ influx and ROS generation. In a heart disease model it was shown that reduced mitochondrial Ca2+ as a consequence of increased cytosolic Na+ build-up, results in decreased mitochondrial bioenergetics and TCA cycle activation [306, 337]. This effectively decreased NAD(P)H:NA(P)D+ ratios and abrogated the reducing potential of the mitochondria, leading to a build-up of mitochondrial ROS [306]. These data implicate NCLX as a regulator of mitochondrial ROS generation in pathophysiological contexts. Indeed, it was demonstrated that forced expression of NCLX could increase mitochondrial oxidation of the redox sensitive RoGFP probe and abrogate histamine-induced increases in mitochondrial Ca2+ and NAD(P)H, presumably through its role in decreasing mitochondrial Ca2+ levels and hence inhibiting matrix dehydrogenase activity [263]. Recently, the consequences of this mitochondrial ROS-NCLX interplay were further explored using a model of NCLX knock-down [180]. In this study, it was demonstrated that knockdown of NCLX increases mitochondrial ROS production by enhancing mitochondrial Ca2+ levels and that this mediates inhibition of the CRAC channel ORAI1. Interestingly, ER Ca2+ store depletion resulted in both SOCE activation and cytosolic Na+ accumulation, suggesting an important interplay between SOCE and NCLX regulation [180]. While knock-down of NCLX had no effect on ORAI1-STIM1 interaction, it blunted CRAC channel activity, which was demonstrated to be dependent on an increase in mitochondrial ROS generation. Scavenging of mitochondrial H2O2 with a mitochondrial targeted catalase construct could rescue SOCE. It was shown that in response to NCLX knockdown, increased mitochondrial H2O2 inactivates ORAI1 through oxidation of C195, as the ORAI1 C195S was insensitive to NCLX expression loss [180]. The above two studies demonstrate that regulation of Ca2+ transients by NCLX is important in modulating mitochondrial ROS production, and that both NCLX-dependent decreases or increases in mitochondrial Ca2+ can result in enhanced mitochondrial ROS production. This may have implications for pathological conditions where NCLX activity is altered. Whether or not NCLX expression or Na+-mediated NCLX activity is altered in a majority of tumor cells remains to be investigated. However, this could have direct consequences on pro-tumorigenic redox signaling or through redox control of SOCE-regulated pathways. The potential role of NCLX in fine-tuning the cross talk between ER-mitochondria Ca2+ transfer and SOCE regulation were studied in a model of malignant melanoma cells [338]. Enhanced trans-mitochondrial Ca2+ flux was observed in metastatic cells compared to non-metastatic clones, and this was correlated with enhanced SOCE and activation of the pro-survival Akt pathway [338]. Pharmacological inhibition of NCLX blocked this trans-mitochondrial flux and SOCE activation. This study also highlighted previous work demonstrating that nano-domains of close interactions between mitochondria and the plasma membrane play a central role in the regulation of plasma membrane CRAC channels by supporting mitochondrial Ca2+ buffering at the plasma membrane thus relieving Ca2+-dependent inhibition of CRAC channels [256-258].
4.6 Redox regulation of Sarco/endoplasmic reticulum Ca2+-ATPase (SERCA)
The SERCA pump is an important regulator of ER Ca2+ homeostasis and involved in rapidly refilling stores in response to receptor-mediated store depletion. Like the IP3Rs, SERCA pump family (SERCA1-3) members are important regulators of ER Ca2+ during apoptosis and integral in maintaining Ca2+ levels to support the protein synthesis and folding machinery of the ER, including calreticulin and calnexin [339]. Hence, ER Ca2+ imbalance is a feature of ER-stress and the unfolded protein response [340, 341]. The expression of SERCA isoforms during tumorigenesis appears to be dependent on tumor stage and type. Several studies have demonstrated direct redox modification of this pump, and the consequence on SERCA activity appears to be dependent on the specific modification present as well as the level of oxidant stress. While these redox changes in cancers have not been explored in depth, there is precedent to suggest that redox mediated inactivation of SERCA could be a phenotype of cancer cells to avoid Ca2+ mediated cell death. Screening for SERCA oxidation-sensitive cysteines revealed that a number of thiol residues susceptible to redox modification in response to aging and peroxinitrite exposure, with C344 and C349 forming intra-molecular disulfide bonds [342]. Age related cysteine modifications were associated with a decrease in SERCA activity during this process.
Illustrating that different modifications have diverse actions on SERCA activity, S-glutahtionylation of C669 and C674 activated SERCA in the carotid artery in response to NO. This was shown to be mediated by the action of ONOO-, and dependent on the presence of adequate levels of GSH [343]. This highlights that the GSH/GSSH pool within cells is an important determinant for protein redox modification. S-glutahtionlylation of SERCA was also recently shown to occur during the unfolded protein response, with loss of the ER glutathione S-transferase Pi expression resulting in cells being more sensitive to ER-stress causing agents [344]. SERCA Tyrosine nitration was also demonstrated in models of cardiovascular disease [345, 346]. In this case nitration may be associated with decreases in SERCA activity. Disruption of these oxidative modifications has been proposed as a potential therapeutic strategy to alter SERCA activity in disease models [347]. This could have potential use to either induce activity for promotion of apoptosis in cancer cells, or inhibit SERCA mediated apoptosis in cases of cardiovascular disease.
Like IP3R, SERCA1 and SERCA2b have also been shown to interact with a number of anti-apoptotic factors, including Bcl-2, which inhibit SERCA-mediated Ca2+ ER uptake by eliciting a conformational change in the SERCA proteins [348, 349]. This may also result in protein degradation. Similarly, in cells expressing mutant K-Ras, decreased IP3R3 and SERCA2b protein expression were observed [350], suggesting an overall dampening of the ER-mitochondrial Ca2+ transfer through oncogenic inhibition that may provide anti-apoptotic benefits to cancer cells. The tumor suppressor p53 was recently shown to initiate cell death in response to cytotoxic agent Adriamycin, ROS and ER stress, by directly interacting with SERCA at MAMs, and activating SERCA to increase ER Ca2+ uptake [351, 352]. This non-canonical action of p53 outside the nucleus in turn led to mitochondrial Ca2+ accumulation and initiation of apoptosis. This p53-mediated activation was shown to correlate with a decrease in SERCA oxidation, although the mechanisms behind this loss of oxidation are unclear [352]. Given that a number of cancers display high frequency p53 mutations, this may be one mechanism by which apoptosis is avoided in tumors. Indeed the investigators demonstrated that common p53 mutants are not able to increase SERCA mediated mitochondrial Ca2+ accumulation and cell death.
5. Conclusion
The interplay between Ca2+ and oxidants in cancer is only starting to be unraveled. From the above work we can conclude that cancer cells successfully utilize this crosstalk to initiate pro-tumorigenic signaling pathways that may be influenced by oncogene expression, growth factor signaling and changes in the tumor microenvironment (e.g. hypoxia, nutrient deprivation). Moreover, tumor cells appear to blunt the large surges in ROS and Ca2+ associated with apoptosis. As described in several examples above, the spatio-temporal nature of the ROS-Ca2+ interplay is of importance to the eventual cellular response, and future studies will need to focus on these important aspects to gain a better picture of the complex nature of this interplay in cancer. While methods for fast, precise and accurate measurements of Ca2+ levels in different organelles are fast improving, similar strategies for spatio-temporal ROS measurements are lagging behind highlighting the need of developing high resolution imaging tools to track ROS changes in specific organelles and specific regions of the cell. In addition, studies will need to focus on the role of the tumor microenvironment in manipulating these changes in Ca2+ and ROS signaling. These will include the influence of tumor-associated cells including fibroblasts and macrophages, and further exploration of the role of chemical and physical cues of the tumor environment. Since many modulators of Ca2+ signaling are potential “druggable” targets, understanding their regulation by altered redox-signaling is imperative in the context of cancer. Utilizing the knowledge gained by understanding the relative alterations in redox-Ca2+ homeostasis in tumor cells compared to normal cells, will hopefully allow us to exploit these changes to promote pro-apoptotic pathways for the purpose of cancer therapy.
Highlights.
The interplay between Ca2+ and reactive oxygen species (ROS) signaling pathways is an important determinant in normal physiology and in several pathophysiologies
Several Ca2+ channels at the plasma membrane and intracellular organelles are regulated by ROS. Reciprocally, Ca2+ signaling regulates the cellular generation of ROS from NADPH oxidases and mitochondria.
With direct relevance to cancer, coordinated and localized Ca2+ and ROS transients appear to play a major role in a vast variety of pro-survival signaling pathways to promote tumorigenesis.
Cancer cells appear to skew the ROS-Ca2+ interplay to their advantage in two ways (Graphical Abstract): 1) by inhibiting large ROS-Ca2+ surges that mediate apoptosis; and 2) by promoting pro-tumorigenic signaling in response to sublethal changes in ROS/Ca2+ levels.
Acknowledgments
Research in the authors' laboratories are funded by by grants R01HL123364, R01HL097111, R21AG050072 from the NIH, grant NPRP8-110-3-021 from the Qatar National Research Fund (GNRF) and grant 14GRNT18880008 from the AHA to MT, and by grant W81XWH-16-1-0117 from the Department of Defense (DoD) to NH. The authors have no conflict of interests to declare.
Abbreviations
- AMPK
5′ adenosine monophosphate-activated protein kinase
- ASK1
apoptosis signal-regulating kinase 1
- Atg
Autophagy Related Cysteine Peptidase
- BIRD-2
Bcl-2/IP3R disrupter
- CaM
Calmodulin
- CaMKII
Ca2+/Calmodulin-dependent kinase II
- Cat
Catalase
- CRAC
Ca2+ release activated Ca2+
- DAG
Diacylglycerol
- ER
Endoplasmic Reticulum
- Ero
ER oxidoreductase
- ETC
Electron Transport Chain
- GPCR
G-protein coupled receptor
- GPx
Glutahione Peroxidase
- GR
Glutathione Reductase
- Grx
Glutaredoxin
- GSH
Glutathione
- HK
Hexokinase
- H2O2
hydrogen peroxide
- HOCl
hypochlorous acid
- HIF
Hypoxia Inducible Factor
- IP3
inositol 1,4,5-trisphosphate
- IP3R
IP3 receptor
- Lamp1
Lysosomal-associated membrane protein 1
- LC3-II
Microtubule-associated proteins 1A/1B light chain 3B
- MAM
mitochondria associated membrane
- MCUR1
MCU Regulator 1
- MICU
Mitochondrial Ca2+ Uptake
- mPTP
mitochondria Permeability Transition Pore
- mTOR
mechanistic target of rapamycin
- NCLX
Na+/Ca2+ Li+ Exchanger
- NCX
Na+/Ca2+ exchanger
- NAD
nicotinamide adenine dinucleotide
- NO•
nitric oxide
- NOS
Nitric Oxide Synthase
- Nox
NADPH oxidase
- O2•-
superoxide
- •OH
hydroxyl radical
- ONOO-
peroxinitrite
- PARP
poly(ADP-ribose) polymerases
- PDH
Pyruvate dehydrogenase
- PDI
protein disulfide isomerase
- PERK
RNA-dependent protein kinase (PKR)-like ER kinase
- PI3K
Phosphoinositide 3-kinase
- PLC
Phospholipase C
- PKC
Protein kinase C
- PTEN
phosphatase and tensin homolog
- Prx
Peroxidase
- PMCA
plasma membrane Ca2+ ATPase
- RNS
Reactive nitrogen Species
- ROS
Reactive Oxygen Species
- RNS
Reactive Nitrogen Species
- RyR
Ryanodine Receptor
- SERCA
Sarco/endoplasmic reticulum Ca2+-ATPase
- SOCE
Store operated Calcium entry
- Sod
Superoxide dismutase
- TFEB
Transcription Factor EB
- TRP
Transient Receptor Potential
- Trx
Thioredoxin
- VDAC
Voltage-dependent anion channel
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Webster KA. Mitochondrial membrane permeabilization and cell death during myocardial infarction: roles of calcium and reactive oxygen species. Future Cardiol. 2012;8:863–884. doi: 10.2217/fca.12.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, Missiroli S, Patergnani S, Rimessi A, Suski JM, Wieckowski MR, Pinton P. Mitochondrial Ca(2+) and apoptosis. Cell calcium. 2012;52:36–43. doi: 10.1016/j.ceca.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.La Rovere RM, Roest G, Bultynck G, Parys JB. Intracellular Ca(2+) signaling and Ca(2+) microdomains in the control of cell survival, apoptosis and autophagy. Cell calcium. 2016;60:74–87. doi: 10.1016/j.ceca.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 4.Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti F, Rimessi A, Duszynski J, Wieckowski MR, Pinton P. Mitochondriaros crosstalk in the control of cell death and aging. J Signal Transduct. 2012;2012:329635. doi: 10.1155/2012/329635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uchi JO, Ryu SY, Jhun BS, Hurst S, Sheu SS. Mitochondrial ion channels/transporters as sensors and regulators of cellular redox signaling. Antioxidants & redox signaling. 2014;21:987–1006. doi: 10.1089/ars.2013.5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bogeski I, Kummerow C, Al-Ansary D, Schwarz EC, Koehler R, Kozai D, Takahashi N, Peinelt C, Griesemer D, Bozem M, Mori Y, Hoth M, Niemeyer BA. Differential redox regulation of ORAI ion channels: a mechanism to tune cellular calcium signaling. Science signaling. 2010;3:ra24. doi: 10.1126/scisignal.2000672. [DOI] [PubMed] [Google Scholar]
- 7.Booth DM, Joseph SK, Hajnoczky G. Subcellular ROS imaging methods: Relevance for the study of calcium signaling. Cell calcium. 2016;60:65–73. doi: 10.1016/j.ceca.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diaz B, Courtneidge SA. Redox signaling at invasive microdomains in cancer cells. Free radical biology & medicine. 2012;52:247–256. doi: 10.1016/j.freeradbiomed.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giorgi C, De Stefani D, Bononi A, Rizzuto R, Pinton P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. The international journal of biochemistry & cell biology. 2009;41:1817–1827. doi: 10.1016/j.biocel.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brandes RP, Weissmann N, Schroder K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free radical biology & medicine. 2014;76:208–226. doi: 10.1016/j.freeradbiomed.2014.07.046. [DOI] [PubMed] [Google Scholar]
- 11.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 12.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 13.Forman HJ, Maiorino M, Ursini F. Signaling functions of reactive oxygen species. Biochemistry. 2010;49:835–842. doi: 10.1021/bi9020378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paulsen CE, Carroll KS. Orchestrating redox signaling networks through regulatory cysteine switches. ACS chemical biology. 2010;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–990. doi: 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stewart TA, Yapa KT, Monteith GR. Altered calcium signaling in cancer cells. Biochimica et biophysica acta. 2015;1848:2502–2511. doi: 10.1016/j.bbamem.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 17.Deliot N, Constantin B. Plasma membrane calcium channels in cancer: Alterations and consequences for cell proliferation and migration. Biochimica et biophysica acta. 2015;1848:2512–2522. doi: 10.1016/j.bbamem.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 18.Monteith GR, Davis FM, Roberts-Thomson SJ. Calcium channels and pumps in cancer: changes and consequences. The Journal of biological chemistry. 2012;287:31666–31673. doi: 10.1074/jbc.R112.343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roderick HL, Cook SJ. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer. 2008;8:361–375. doi: 10.1038/nrc2374. [DOI] [PubMed] [Google Scholar]
- 20.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. The Biochemical journal. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davies MJ. Protein oxidation and peroxidation. The Biochemical journal. 2016;473:805–825. doi: 10.1042/BJ20151227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Randall LM, Ferrer-Sueta G, Denicola A. Peroxiredoxins as preferential targets in H2O2-induced signaling. Methods in enzymology. 2013;527:41–63. doi: 10.1016/B978-0-12-405882-8.00003-9. [DOI] [PubMed] [Google Scholar]
- 23.Winterbourn CC. The biological chemistry of hydrogen peroxide. Methods in enzymology. 2013;528:3–25. doi: 10.1016/B978-0-12-405881-1.00001-X. [DOI] [PubMed] [Google Scholar]
- 24.Saran M, Bors W. Signalling by O2-. and NO.: how far can either radical, or any specific reaction product, transmit a message under in vivo conditions? Chem Biol Interact. 1994;90:35–45. doi: 10.1016/0009-2797(94)90109-0. [DOI] [PubMed] [Google Scholar]
- 25.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. The American journal of physiology. 1996;271:C1424–1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 26.Brand MD. The sites and topology of mitochondrial superoxide production. Exp Gerontol. 2010;45:466–472. doi: 10.1016/j.exger.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murphy MP. How mitochondria produce reactive oxygen species. The Biochemical journal. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bienert GP, Moller AL, Kristiansen KA, Schulz A, Moller IM, Schjoerring JK, Jahn TP. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. The Journal of biological chemistry. 2007;282:1183–1192. doi: 10.1074/jbc.M603761200. [DOI] [PubMed] [Google Scholar]
- 29.Takac I, Schroder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, Shah AM, Morel F, Brandes RP. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. The Journal of biological chemistry. 2011;286:13304–13313. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diaz B, Shani G, Pass I, Anderson D, Quintavalle M, Courtneidge SA. Tks5-dependent, nox-mediated generation of reactive oxygen species is necessary for invadopodia formation. Science signaling. 2009;2:ra53. doi: 10.1126/scisignal.2000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gianni D, Diaz B, Taulet N, Fowler B, Courtneidge SA, Bokoch GM. Novel p47(phox)-related organizers regulate localized NADPH oxidase 1 (Nox1) activity. Science signaling. 2009;2:ra54. doi: 10.1126/scisignal.2000370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Block K, Gorin Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat Rev Cancer. 2012;12:627–637. doi: 10.1038/nrc3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banfi B, Tirone F, Durussel I, Knisz J, Moskwa P, Molnar GZ, Krause KH, Cox JA. Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5) The Journal of biological chemistry. 2004;279:18583–18591. doi: 10.1074/jbc.M310268200. [DOI] [PubMed] [Google Scholar]
- 34.Rigutto S, Hoste C, Grasberger H, Milenkovic M, Communi D, Dumont JE, Corvilain B, Miot F, De Deken X. Activation of dual oxidases Duox1 and Duox2: differential regulation mediated by camp-dependent protein kinase and protein kinase C-dependent phosphorylation. The Journal of biological chemistry. 2009;284:6725–6734. doi: 10.1074/jbc.M806893200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tirone F, Cox JA. NADPH oxidase 5 (NOX5) interacts with and is regulated by calmodulin. FEBS letters. 2007;581:1202–1208. doi: 10.1016/j.febslet.2007.02.047. [DOI] [PubMed] [Google Scholar]
- 36.Jagnandan D, Church JE, Banfi B, Stuehr DJ, Marrero MB, Fulton DJ. Novel mechanism of activation of NADPH oxidase 5. calcium sensitization via phosphorylation. The Journal of biological chemistry. 2007;282:6494–6507. doi: 10.1074/jbc.M608966200. [DOI] [PubMed] [Google Scholar]
- 37.Ha EM, Lee KA, Park SH, Kim SH, Nam HJ, Lee HY, Kang D, Lee WJ. Regulation of DUOX by the Galphaq-phospholipase Cbeta-Ca2+ pathway in Drosophila gut immunity. Developmental cell. 2009;16:386–397. doi: 10.1016/j.devcel.2008.12.015. [DOI] [PubMed] [Google Scholar]
- 38.Kim EY, Anderson M, Dryer SE. Insulin increases surface expression of TRPC6 channels in podocytes: role of NADPH oxidases and reactive oxygen species. Am J Physiol Renal Physiol. 2012;302:F298–307. doi: 10.1152/ajprenal.00423.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim EY, Anderson M, Wilson C, Hagmann H, Benzing T, Dryer SE. NOX2 interacts with podocyte TRPC6 channels and contributes to their activation by diacylglycerol: essential role of podocin in formation of this complex. Am J Physiol Cell Physiol. 2013;305:C960–971. doi: 10.1152/ajpcell.00191.2013. [DOI] [PubMed] [Google Scholar]
- 40.Dho SH, Kim JY, Kwon ES, Lim JC, Park SS, Kwon KS. NOX5-L can stimulate proliferation and apoptosis depending on its levels and cellular context, determining cancer cell susceptibility to cisplatin. Oncotarget. 2015;6:39235–39246. doi: 10.18632/oncotarget.5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drose S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol. 2012;748:145–169. doi: 10.1007/978-1-4614-3573-0_6. [DOI] [PubMed] [Google Scholar]
- 42.Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+ network. The Journal of cell biology. 1997;136:833–844. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rizzuto R, De Stefani D, Raffaello A, Mammucari C. Mitochondria as sensors and regulators of calcium signalling. Nat Rev Mol Cell Biol. 2012;13:566–578. doi: 10.1038/nrm3412. [DOI] [PubMed] [Google Scholar]
- 44.Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell death and differentiation. 2015;22:377–388. doi: 10.1038/cdd.2014.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collins Y, Chouchani ET, James AM, Menger KE, Cocheme HM, Murphy MP. Mitochondrial redox signalling at a glance. Journal of cell science. 2012;125:801–806. doi: 10.1242/jcs.098475. [DOI] [PubMed] [Google Scholar]
- 46.Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–664. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 47.Sharma LK, Fang H, Liu J, Vartak R, Deng J, Bai Y. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum Mol Genet. 2011;20:4605–4616. doi: 10.1093/hmg/ddr395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. The Journal of biological chemistry. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 50.Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, Chandel NS. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. The Journal of cell biology. 2007;177:1029–1036. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sabharwal SS, Schumacker PT. Mitochondrial ROS in cancer: initiators, amplifiers or an Achilles' heel? Nat Rev Cancer. 2014;14:709–721. doi: 10.1038/nrc3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madesh M, Hajnoczky G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. The Journal of cell biology. 2001;155:1003–1015. doi: 10.1083/jcb.200105057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hwang MS, Schwall CT, Pazarentzos E, Datler C, Alder NN, Grimm S. Mitochondrial Ca(2+) influx targets cardiolipin to disintegrate respiratory chain complex II for cell death induction. Cell death and differentiation. 2014;21:1733–1745. doi: 10.1038/cdd.2014.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxidants & redox signaling. 2014;21:396–413. doi: 10.1089/ars.2014.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Enyedi B, Varnai P, Geiszt M. Redox state of the endoplasmic reticulum is controlled by Ero1L-alpha and intraluminal calcium. Antioxidants & redox signaling. 2010;13:721–729. doi: 10.1089/ars.2009.2880. [DOI] [PubMed] [Google Scholar]
- 56.Konno T, Pinho Melo E, Lopes C, Mehmeti I, Lenzen S, Ron D, Avezov E. ERO1-independent production of H2O2 within the endoplasmic reticulum fuels Prdx4-mediated oxidative protein folding. The Journal of cell biology. 2015;211:253–259. doi: 10.1083/jcb.201506123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tavender TJ, Bulleid NJ. Peroxiredoxin IV protects cells from oxidative stress by removing H2O2 produced during disulphide formation. Journal of cell science. 2010;123:2672–2679. doi: 10.1242/jcs.067843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zito E, Melo EP, Yang Y, Wahlander A, Neubert TA, Ron D. Oxidative protein folding by an endoplasmic reticulum-localized peroxiredoxin. Molecular cell. 2010;40:787–797. doi: 10.1016/j.molcel.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 60.Bienert GP, Schjoerring JK, Jahn TP. Membrane transport of hydrogen peroxide. Biochimica et biophysica acta. 2006;1758:994–1003. doi: 10.1016/j.bbamem.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 61.Forman HJ, Augusto O, Brigelius-Flohe R, Dennery PA, Kalyanaraman B, Ischiropoulos H, Mann GE, Radi R, Roberts LJ, 2nd, Vina J, Davies KJ. Even free radicals should follow some rules: a guide to free radical research terminology and methodology. Free radical biology & medicine. 2015;78:233–235. doi: 10.1016/j.freeradbiomed.2014.10.504. [DOI] [PubMed] [Google Scholar]
- 62.Kalyanaraman B, Darley-Usmar V, Davies KJ, Dennery PA, Forman HJ, Grisham MB, Mann GE, Moore K, Roberts LJ, 2nd, Ischiropoulos H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free radical biology & medicine. 2012;52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gibhardt CS, Zimmermann KM, Zhang X, Belousov VV, Bogeski I. Imaging calcium and redox signals using genetically encoded fluorescent indicators. Cell calcium. 2016;60:55–64. doi: 10.1016/j.ceca.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 64.Mason RP. Imaging free radicals in organelles, cells, tissue, and in vivo with immuno-spin trapping. Redox Biol. 2016;8:422–429. doi: 10.1016/j.redox.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mason RP, Hanna PM, Burkitt MJ, Kadiiska MB. Detection of oxygen-derived radicals in biological systems using electron spin resonance. Environ Health Perspect. 1994;102(10):33–36. doi: 10.1289/ehp.94102s1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Batinic-Haberle I, Tovmasyan A, Spasojevic I. An educational overview of the chemistry, biochemistry and therapeutic aspects of Mn porphyrins--From superoxide dismutation to H2O2-driven pathways. Redox Biol. 2015;5:43–65. doi: 10.1016/j.redox.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Szalai G, Krishnamurthy R, Hajnoczky G. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. The EMBO journal. 1999;18:6349–6361. doi: 10.1093/emboj/18.22.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lipskaia L, Pourci ML, Delomenie C, Combettes L, Goudouneche D, Paul JL, Capiod T, Lompre AM. Phosphatidylinositol 3-kinase and calcium-activated transcription pathways are required for VLDL-induced smooth muscle cell proliferation. Circ Res. 2003;92:1115–1122. doi: 10.1161/01.RES.0000074880.25540.D0. [DOI] [PubMed] [Google Scholar]
- 69.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- 70.Dupre-Crochet S, Erard M, Nubetae O. ROS production in phagocytes: why, when, and where? J Leukoc Biol. 2013;94:657–670. doi: 10.1189/jlb.1012544. [DOI] [PubMed] [Google Scholar]
- 71.Klaunig JE, Wang Z, Pu X, Zhou S. Oxidative stress and oxidative damage in chemical carcinogenesis. Toxicol Appl Pharmacol. 2011;254:86–99. doi: 10.1016/j.taap.2009.11.028. [DOI] [PubMed] [Google Scholar]
- 72.Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, Elia A, Berger T, Cescon DW, Adeoye A, Brustle A, Molyneux SD, Mason JM, Li WY, Yamamoto K, Wakeham A, Berman HK, Khokha R, Done SJ, Kavanagh TJ, Lam CW, Mak TW. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27:211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 74.Pani G, Galeotti T, Chiarugi P. Metastasis: cancer cell's escape from oxidative stress. Cancer Metastasis Rev. 2010;29:351–378. doi: 10.1007/s10555-010-9225-4. [DOI] [PubMed] [Google Scholar]
- 75.Pinton P, Ferrari D, Magalhaes P, Schulze-Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. The Journal of cell biology. 2000;148:857–862. doi: 10.1083/jcb.148.5.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. The Journal of cell biology. 2004;166:193–203. doi: 10.1083/jcb.200309146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rong YP, Aromolaran AS, Bultynck G, Zhong F, Li X, McColl K, Matsuyama S, Herlitze S, Roderick HL, Bootman MD, Mignery GA, Parys JB, De Smedt H, Distelhorst CW. Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2's inhibition of apoptotic calcium signals. Molecular cell. 2008;31:255–265. doi: 10.1016/j.molcel.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rong YP, Bultynck G, Aromolaran AS, Zhong F, Parys JB, De Smedt H, Mignery GA, Roderick HL, Bootman MD, Distelhorst CW. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14397–14402. doi: 10.1073/pnas.0907555106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schumacker PT. Reactive oxygen species in cancer cells: live by the sword, die by the sword. Cancer Cell. 2006;10:175–176. doi: 10.1016/j.ccr.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 80.Wang J, Yi J. Cancer cell killing via ROS: to increase or decrease, that is the question. Cancer Biol Ther. 2008;7:1875–1884. doi: 10.4161/cbt.7.12.7067. [DOI] [PubMed] [Google Scholar]
- 81.Hemachandra LP, Shin DH, Dier U, Iuliano JN, Engelberth SA, Uusitalo LM, Murphy SK, Hempel N. Mitochondrial Superoxide Dismutase Has a Protumorigenic Role in Ovarian Clear Cell Carcinoma. Cancer research. 2015;75:4973–4984. doi: 10.1158/0008-5472.CAN-14-3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hileman EO, Liu J, Albitar M, Keating MJ, Huang P. Intrinsic oxidative stress in cancer cells: a biochemical basis for therapeutic selectivity. Cancer Chemother Pharmacol. 2004;53:209–219. doi: 10.1007/s00280-003-0726-5. [DOI] [PubMed] [Google Scholar]
- 83.Trachootham D, Zhou Y, Zhang H, Demizu Y, Chen Z, Pelicano H, Chiao PJ, Achanta G, Arlinghaus RB, Liu J, Huang P. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by beta-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 84.Davidson SM, Duchen MR. Calcium microdomains and oxidative stress. Cell calcium. 2006;40:561–574. doi: 10.1016/j.ceca.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 85.Booth DM, Enyedi B, Geiszt M, Varnai P, Hajnoczky G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Molecular cell. 2016;63:240–248. doi: 10.1016/j.molcel.2016.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xu S, Chisholm AD. C. elegans epidermal wounding induces a mitochondrial ROS burst that promotes wound repair. Developmental cell. 2014;31:48–60. doi: 10.1016/j.devcel.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459:996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wei C, Wang X, Chen M, Ouyang K, Song LS, Cheng H. Calcium flickers steer cell migration. Nature. 2009;457:901–905. doi: 10.1038/nature07577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yan Y, Liu J, Wei C, Li K, Xie W, Wang Y, Cheng H. Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovascular research. 2008;77:432–441. doi: 10.1093/cvr/cvm047. [DOI] [PubMed] [Google Scholar]
- 90.Mesquita FS, Dyer SN, Heinrich DA, Bulun SE, Marsh EE, Nowak RA. Reactive oxygen species mediate mitogenic growth factor signaling pathways in human leiomyoma smooth muscle cells. Biol Reprod. 2010;82:341–351. doi: 10.1095/biolreprod.108.075887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sullivan LB, Chandel NS. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014;2:17. doi: 10.1186/2049-3002-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cox AG, Winterbourn CC, Hampton MB. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. The Biochemical journal. 2010;425:313–325. doi: 10.1042/BJ20091541. [DOI] [PubMed] [Google Scholar]
- 93.Davison CA, Durbin SM, Thau MR, Zellmer VR, Chapman SE, Diener J, Wathen C, Leevy WM, Schafer ZT. Antioxidant enzymes mediate survival of breast cancer cells deprived of extracellular matrix. Cancer research. 2013;73:3704–3715. doi: 10.1158/0008-5472.CAN-12-2482. [DOI] [PubMed] [Google Scholar]
- 94.Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, Morrison SJ. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–191. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, Dalin MG, Akyurek LM, Lindahl P, Nilsson J, Bergo MO. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med. 2015;7:308re308. doi: 10.1126/scitranslmed.aad3740. [DOI] [PubMed] [Google Scholar]
- 96.Hawk MA, McCallister C, Schafer ZT. Antioxidant Activity during Tumor Progression: A Necessity for the Survival of Cancer Cells? Cancers (Basel) 2016;8 doi: 10.3390/cancers8100092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hempel N, Carrico PM, Melendez JA. Manganese superoxide dismutase (Sod2) and redox-control of signaling events that drive metastasis. Anticancer Agents Med Chem. 2011;11:191–201. doi: 10.2174/187152011795255911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hempel N, Melendez JA. Intracellular redox status controls membrane localization of pro-and anti-migratory signaling molecules. Redox Biol. 2014;2:245–250. doi: 10.1016/j.redox.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hempel N, Ye H, Abessi B, Mian B, Melendez JA. Altered redox status accompanies progression to metastatic human bladder cancer. Free radical biology & medicine. 2009;46:42–50. doi: 10.1016/j.freeradbiomed.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hemachandra LP, Chandrasekeran A, Melendez JA, Hempel N. Regulation of the cellular redox environment by superoxide dismutases, catalase and glutathione peroxidases during tumor metastasis. In: Batinic-Haberle I, Spasojevic I, Reboucas JS, editors. Redox-Active Theraputics. Springer; Switzerland: 2016. pp. 11–50. [Google Scholar]
- 101.Sobotta MC, Liou W, Stocker S, Talwar D, Oehler M, Ruppert T, Scharf AN, Dick TP. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat Chem Biol. 2015;11:64–70. doi: 10.1038/nchembio.1695. [DOI] [PubMed] [Google Scholar]
- 102.Woo HA, Yim SH, Shin DH, Kang D, Yu DY, Rhee SG. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell. 2010;140:517–528. doi: 10.1016/j.cell.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 103.Howe CJ, Lahair MM, McCubrey JA, Franklin RA. Redox regulation of the calcium/calmodulin-dependent protein kinases. The Journal of biological chemistry. 2004;279:44573–44581. doi: 10.1074/jbc.M404175200. [DOI] [PubMed] [Google Scholar]
- 104.Hongpaisan J, Winters CA, Andrews SB. Strong calcium entry activates mitochondrial superoxide generation, upregulating kinase signaling in hippocampal neurons. J Neurosci. 2004;24:10878–10887. doi: 10.1523/JNEUROSCI.3278-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hawkins BJ, Irrinki KM, Mallilankaraman K, Lien YC, Wang Y, Bhanumathy CD, Subbiah R, Ritchie MF, Soboloff J, Baba Y, Kurosaki T, Joseph SK, Gill DL, Madesh M. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. The Journal of cell biology. 2010;190:391–405. doi: 10.1083/jcb.201004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Prins D, Groenendyk J, Touret N, Michalak M. Modulation of STIM1 and capacitative Ca2+ entry by the endoplasmic reticulum luminal oxidoreductase ERp57. EMBO Rep. 2011;12:1182–1188. doi: 10.1038/embor.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y, Mori Y. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- 108.Hong C, Kwak M, Myeong J, Ha K, Wie J, Jeon JH, So I. Extracellular disulfide bridges stabilize TRPC5 dimerization, trafficking, and activity. Pflugers Archiv : European journal of physiology. 2015;467:703–712. doi: 10.1007/s00424-014-1540-0. [DOI] [PubMed] [Google Scholar]
- 109.Hong C, Seo H, Kwak M, Jeon J, Jang J, Jeong EM, Myeong J, Hwang YJ, Ha K, Kang MJ, Lee KP, Yi EC, Kim IG, Jeon JH, Ryu H, So I. Increased TRPC5 glutathionylation contributes to striatal neuron loss in Huntington's disease. Brain. 2015;138:3030–3047. doi: 10.1093/brain/awv188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xu SZ, Sukumar P, Zeng F, Li J, Jairaman A, English A, Naylor J, Ciurtin C, Majeed Y, Milligan CJ, Bahnasi YM, Al-Shawaf E, Porter KE, Jiang LH, Emery P, Sivaprasadarao A, Beech DJ. TRPC channel activation by extracellular thioredoxin. Nature. 2008;451:69–72. doi: 10.1038/nature06414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zaidi A, Barron L, Sharov VS, Schoneich C, Michaelis EK, Michaelis ML. Oxidative inactivation of purified plasma membrane Ca2+-ATPase by hydrogen peroxide and protection by calmodulin. Biochemistry. 2003;42:12001–12010. doi: 10.1021/bi034565u. [DOI] [PubMed] [Google Scholar]
- 112.Schwab BL, Guerini D, Didszun C, Bano D, Ferrando-May E, Fava E, Tam J, Xu D, Xanthoudakis S, Nicholson DW, Carafoli E, Nicotera P. Cleavage of plasma membrane calcium pumps by caspases: a link between apoptosis and necrosis. Cell death and differentiation. 2002;9:818–831. doi: 10.1038/sj.cdd.4401042. [DOI] [PubMed] [Google Scholar]
- 113.Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, Minami T, Takada Y, Kume T, Katsuki H, Mori Y, Akaike A. A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. Journal of pharmacological sciences. 2006;101:66–76. doi: 10.1254/jphs.fp0060128. [DOI] [PubMed] [Google Scholar]
- 114.Kuhn FJ, Heiner I, Luckhoff A. TRPM2: a calcium influx pathway regulated by oxidative stress and the novel second messenger ADP-ribose. Pflugers Archiv : European journal of physiology. 2005;451:212–219. doi: 10.1007/s00424-005-1446-y. [DOI] [PubMed] [Google Scholar]
- 115.Zhang W, Chu X, Tong Q, Cheung JY, Conrad K, Masker K, Miller BA. A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. The Journal of biological chemistry. 2003;278:16222–16229. doi: 10.1074/jbc.M300298200. [DOI] [PubMed] [Google Scholar]
- 116.Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Molecular cell. 2002;9:163–173. doi: 10.1016/s1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- 117.Perraud AL, Takanishi CL, Shen B, Kang S, Smith MK, Schmitz C, Knowles HM, Ferraris D, Li W, Zhang J, Stoddard BL, Scharenberg AM. Accumulation of free ADP-ribose from mitochondria mediates oxidative stress-induced gating of TRPM2 cation channels. The Journal of biological chemistry. 2005;280:6138–6148. doi: 10.1074/jbc.M411446200. [DOI] [PubMed] [Google Scholar]
- 118.Shin CY, Shin J, Kim BM, Wang MH, Jang JH, Surh YJ, Oh U. Essential role of mitochondrial permeability transition in vanilloid receptor 1-dependent cell death of sensory neurons. Mol Cell Neurosci. 2003;24:57–68. doi: 10.1016/s1044-7431(03)00121-0. [DOI] [PubMed] [Google Scholar]
- 119.Song X, Liu BC, Lu XY, Yang LL, Zhai YJ, Eaton AF, Thai TL, Eaton DC, Ma HP, Shen BZ. Lovastatin inhibits human B lymphoma cell proliferation by reducing intracellular ROS and TRPC6 expression. Biochimica et biophysica acta. 2014;1843:894–901. doi: 10.1016/j.bbamcr.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nielsen N, Lindemann O, Schwab A. TRP channels and STIM/ORAI proteins: sensors and effectors of cancer and stroma cell migration. Br J Pharmacol. 2014;171:5524–5540. doi: 10.1111/bph.12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Vashisht A, Trebak M, Motiani RK. STIM and Orai proteins as novel targets for cancer therapy. A Review in the Theme: Cell and Molecular Processes in Cancer Metastasis. Am J Physiol Cell Physiol. 2015;309:C457–469. doi: 10.1152/ajpcell.00064.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Nilius B, Szallasi A. Transient receptor potential channels as drug targets: from the science of basic research to the art of medicine. Pharmacol Rev. 2014;66:676–814. doi: 10.1124/pr.113.008268. [DOI] [PubMed] [Google Scholar]
- 123.Smyth JT, Dehaven WI, Jones BF, Mercer JC, Trebak M, Vazquez G, Putney JW., Jr Emerging perspectives in store-operated Ca2+ entry: roles of Orai, Stim and TRP. Biochimica et biophysica acta. 2006;1763:1147–1160. doi: 10.1016/j.bbamcr.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 124.Ogawa N, Kurokawa T, Mori Y. Sensing of redox status by TRP channels. Cell calcium. 2016;60:115–122. doi: 10.1016/j.ceca.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 125.Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- 126.Hall DP, Cost NG, Hegde S, Kellner E, Mikhaylova O, Stratton Y, Ehmer B, Abplanalp WA, Pandey R, Biesiada J, Harteneck C, Plas DR, Meller J, Czyzyk-Krzeska MF. TRPM3 and miR-204 establish a regulatory circuit that controls oncogenic autophagy in clear cell renal cell carcinoma. Cancer Cell. 2014;26:738–753. doi: 10.1016/j.ccell.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kondratskyi A, Yassine M, Kondratska K, Skryma R, Slomianny C, Prevarskaya N. Calcium-permeable ion channels in control of autophagy and cancer. Front Physiol. 2013;4:272. doi: 10.3389/fphys.2013.00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sun L, Yau HY, Wong WY, Li RA, Huang Y, Yao X. Role of TRPM2 in H(2)O(2)-induced cell apoptosis in endothelial cells. PLoS One. 2012;7:e43186. doi: 10.1371/journal.pone.0043186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nakayama S, Vest R, Traystman RJ, Herson PS. Sexually dimorphic response of TRPM2 inhibition following cardiac arrest-induced global cerebral ischemia in mice. J Mol Neurosci. 2013;51:92–98. doi: 10.1007/s12031-013-0005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gao G, Wang W, Tadagavadi RK, Briley NE, Love MI, Miller BA, Reeves WB. TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J Clin Invest. 2014;124:4989–5001. doi: 10.1172/JCI76042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Duncan LM, Deeds J, Hunter J, Shao J, Holmgren LM, Woolf EA, Tepper RI, Shyjan AW. Down-regulation of the novel gene melastatin correlates with potential for melanoma metastasis. Cancer research. 1998;58:1515–1520. [PubMed] [Google Scholar]
- 132.Ishii M, Oyama A, Hagiwara T, Miyazaki A, Mori Y, Kiuchi Y, Shimizu S. Facilitation of H2O2-induced A172 human glioblastoma cell death by insertion of oxidative stress-sensitive TRPM2 channels. Anticancer Res. 2007;27:3987–3992. [PubMed] [Google Scholar]
- 133.Fonfria E, Marshall IC, Benham CD, Boyfield I, Brown JD, Hill K, Hughes JP, Skaper SD, McNulty S. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br J Pharmacol. 2004;143:186–192. doi: 10.1038/sj.bjp.0705914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Buelow B, Song Y, Scharenberg AM. The Poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. The Journal of biological chemistry. 2008;283:24571–24583. doi: 10.1074/jbc.M802673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zhang W, Hirschler-Laszkiewicz I, Tong Q, Conrad K, Sun SC, Penn L, Barber DL, Stahl R, Carey DJ, Cheung JY, Miller BA. TRPM2 is an ion channel that modulates hematopoietic cell death through activation of caspases and PARP cleavage. Am J Physiol Cell Physiol. 2006;290:C1146–1159. doi: 10.1152/ajpcell.00205.2005. [DOI] [PubMed] [Google Scholar]
- 136.Hecquet CM, Zhang M, Mittal M, Vogel SM, Di A, Gao X, Bonini MG, Malik AB. Cooperative interaction of trp melastatin channel transient receptor potential (TRPM2) with its splice variant TRPM2 short variant is essential for endothelial cell apoptosis. Circ Res. 2014;114:469–479. doi: 10.1161/CIRCRESAHA.114.302414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Orfanelli U, Jachetti E, Chiacchiera F, Grioni M, Brambilla P, Briganti A, Freschi M, Martinelli-Boneschi F, Doglioni C, Montorsi F, Bellone M, Casari G, Pasini D, Lavorgna G. Antisense transcription at the TRPM2 locus as a novel prognostic marker and therapeutic target in prostate cancer. Oncogene. 2015;34:2094–2102. doi: 10.1038/onc.2014.144. [DOI] [PubMed] [Google Scholar]
- 138.Orfanelli U, Wenke AK, Doglioni C, Russo V, Bosserhoff AK, Lavorgna G. Identification of novel sense and antisense transcription at the TRPM2 locus in cancer. Cell Res. 2008;18:1128–1140. doi: 10.1038/cr.2008.296. [DOI] [PubMed] [Google Scholar]
- 139.Zeng X, Sikka SC, Huang L, Sun C, Xu C, Jia D, Abdel-Mageed AB, Pottle JE, Taylor JT, Li M. Novel role for the transient receptor potential channel TRPM2 in prostate cancer cell proliferation. Prostate Cancer Prostatic Dis. 2010;13:195–201. doi: 10.1038/pcan.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen SJ, Hoffman NE, Shanmughapriya S, Bao L, Keefer K, Conrad K, Merali S, Takahashi Y, Abraham T, Hirschler-Laszkiewicz I, Wang J, Zhang XQ, Song J, Barrero C, Shi Y, Kawasawa YI, Bayerl M, Sun T, Barbour M, Wang HG, Madesh M, Cheung JY, Miller BA. A splice variant of the human ion channel TRPM2 modulates neuroblastoma tumor growth through hypoxia-inducible factor (HIF)-1/2alpha. The Journal of biological chemistry. 2014;289:36284–36302. doi: 10.1074/jbc.M114.620922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chen SJ, Zhang W, Tong Q, Conrad K, Hirschler-Laszkiewicz I, Bayerl M, Kim JK, Cheung JY, Miller BA. Role of TRPM2 in cell proliferation and susceptibility to oxidative stress. Am J Physiol Cell Physiol. 2013;304:C548–560. doi: 10.1152/ajpcell.00069.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Miller BA, Cheung JY. TRPM2 protects against tissue damage following oxidative stress and ischaemia-reperfusion. J Physiol. 2016;594:4181–4191. doi: 10.1113/JP270934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bao L, Chen SJ, Conrad K, Keefer K, Abraham T, Lee JP, Wang J, Zhang XQ, Hirschler-Laszkiewicz I, Wang HG, Dovat S, Gans B, Madesh M, Cheung JY, Miller BA. Depletion of the Human Ion Channel TRPM2 in Neuroblastoma Demonstrates Its Key Role in Cell Survival through Modulation of Mitochondrial Reactive Oxygen Species and Bioenergetics. The Journal of biological chemistry. 2016;291:24449–24464. doi: 10.1074/jbc.M116.747147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, Kaneko S, Lange I, Fleig A, Penner R, Nishi M, Takeshima H, Mori Y. TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med. 2008;14:738–747. doi: 10.1038/nm1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, Qiao L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun. 2013;4:1611. doi: 10.1038/ncomms2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Qian X, Numata T, Zhang K, Li C, Hou J, Mori Y, Fang X. Transient receptor potential melastatin 2 protects mice against polymicrobial sepsis by enhancing bacterial clearance. Anesthesiology. 2014;121:336–351. doi: 10.1097/ALN.0000000000000275. [DOI] [PubMed] [Google Scholar]
- 147.Di A, Gao XP, Qian F, Kawamura T, Han J, Hecquet C, Ye RD, Vogel SM, Malik AB. The redox-sensitive cation channel TRPM2 modulates phagocyte ROS production and inflammation. Nat Immunol. 2012;13:29–34. doi: 10.1038/ni.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shakerley NL, Chandrasekaran A, Trebak M, Miller BA, Melendez JA. Francisella tularensis Catalase Restricts Immune Function by Impairing TRPM2 Channel Activity. The Journal of biological chemistry. 2016;291:3871–3881. doi: 10.1074/jbc.M115.706879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Liu BC, Song X, Lu XY, Li DT, Eaton DC, Shen BZ, Li XQ, Ma HP. High glucose induces podocyte apoptosis by stimulating TRPC6 via elevation of reactive oxygen species. Biochimica et biophysica acta. 2013;1833:1434–1442. doi: 10.1016/j.bbamcr.2013.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Trebak M, Vazquez G, Bird GS, Putney JW., Jr The TRPC3/6/7 subfamily of cation channels. Cell calcium. 2003;33:451–461. doi: 10.1016/s0143-4160(03)00056-3. [DOI] [PubMed] [Google Scholar]
- 151.Trebak M, JB St G, McKay RR, Birnbaumer L, Putney JW., Jr Signaling mechanism for receptor-activated canonical transient receptor potential 3 (TRPC3) channels. The Journal of biological chemistry. 2003;278:16244–16252. doi: 10.1074/jbc.M300544200. [DOI] [PubMed] [Google Scholar]
- 152.Vazquez G, Wedel BJ, Aziz O, Trebak M, Putney JW., Jr The mammalian TRPC cation channels. Biochimica et biophysica acta. 2004;1742:21–36. doi: 10.1016/j.bbamcr.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 153.Rybarczyk P, Gautier M, Hague F, Dhennin-Duthille I, Chatelain D, Kerr-Conte J, Pattou F, Regimbeau JM, Sevestre H, Ouadid-Ahidouch H. Transient receptor potential melastatin-related 7 channel is overexpressed in human pancreatic ductal adenocarcinomas and regulates human pancreatic cancer cell migration. Int J Cancer. 2012;131:E851–861. doi: 10.1002/ijc.27487. [DOI] [PubMed] [Google Scholar]
- 154.Sun Y, Selvaraj S, Varma A, Derry S, Sahmoun AE, Singh BB. Increase in serum Ca2+/Mg2+ ratio promotes proliferation of prostate cancer cells by activating TRPM7 channels. The Journal of biological chemistry. 2013;288:255–263. doi: 10.1074/jbc.M112.393918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Roedding AS, Gao AF, Au-Yeung W, Scarcelli T, Li PP, Warsh JJ. Effect of oxidative stress on TRPM2 and TRPC3 channels in B lymphoblast cells in bipolar disorder. Bipolar Disord. 2012;14:151–161. doi: 10.1111/j.1399-5618.2012.01003.x. [DOI] [PubMed] [Google Scholar]
- 156.Antigny F, Girardin N, Frieden M. Transient receptor potential canonical channels are required for in vitro endothelial tube formation. The Journal of biological chemistry. 2012;287:5917–5927. doi: 10.1074/jbc.M111.295733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kuang CY, Yu Y, Wang K, Qian DH, Den MY, Huang L. Knockdown of transient receptor potential canonical-1 reduces the proliferation and migration of endothelial progenitor cells. Stem Cells Dev. 2012;21:487–496. doi: 10.1089/scd.2011.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Song HB, Jun HO, Kim JH, Fruttiger M, Kim JH. Suppression of transient receptor potential canonical channel 4 inhibits vascular endothelial growth factor-induced retinal neovascularization. Cell calcium. 2015;57:101–108. doi: 10.1016/j.ceca.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 159.Veliceasa D, Ivanovic M, Hoepfner FT, Thumbikat P, Volpert OV, Smith ND. Transient potential receptor channel 4 controls thrombospondin-1 secretion and angiogenesis in renal cell carcinoma. FEBS J. 2007;274:6365–6377. doi: 10.1111/j.1742-4658.2007.06159.x. [DOI] [PubMed] [Google Scholar]
- 160.Ma X, Cai Y, He D, Zou C, Zhang P, Lo CY, Xu Z, Chan FL, Yu S, Chen Y, Zhu R, Lei J, Jin J, Yao X. Transient receptor potential channel TRPC5 is essential for P-glycoprotein induction in drug-resistant cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:16282–16287. doi: 10.1073/pnas.1202989109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Stewart TA, Azimi I, Thompson EW, Roberts-Thomson SJ, Monteith GR. A role for calcium in the regulation of ATP-binding cassette, sub-family C, member 3 (ABCC3) gene expression in a model of epidermal growth factor-mediated breast cancer epithelial-mesenchymal transition. Biochem Biophys Res Commun. 2015;458:509–514. doi: 10.1016/j.bbrc.2015.01.141. [DOI] [PubMed] [Google Scholar]
- 162.Mukherjea D, Jajoo S, Kaur T, Sheehan KE, Ramkumar V, Rybak LP. Transtympanic administration of short interfering (si)RNA for the NOX3 isoform of NADPH oxidase protects against cisplatin-induced hearing loss in the rat. Antioxidants & redox signaling. 2010;13:589–598. doi: 10.1089/ars.2010.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mukherjea D, Jajoo S, Whitworth C, Bunch JR, Turner JG, Rybak LP, Ramkumar V. Short interfering RNA against transient receptor potential vanilloid 1 attenuates cisplatin-induced hearing loss in the rat. J Neurosci. 2008;28:13056–13065. doi: 10.1523/JNEUROSCI.1307-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sukumaran P, Schaar A, Sun Y, Singh BB. Functional role of TRP channels in modulating ER stress and Autophagy. Cell calcium. 2016;60:123–132. doi: 10.1016/j.ceca.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Dong XP, Shen D, Wang X, Dawson T, Li X, Zhang Q, Cheng X, Zhang Y, Weisman LS, Delling M, Xu H. PI(3,5)P(2) controls membrane trafficking by direct activation of mucolipin Ca(2+) release channels in the endolysosome. Nat Commun. 2010;1:38. doi: 10.1038/ncomms1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Miao Y, Li G, Zhang X, Xu H, Abraham SN. A TRP Channel Senses Lysosome Neutralization by Pathogens to Trigger Their Expulsion. Cell. 2015;161:1306–1319. doi: 10.1016/j.cell.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang W, Gao Q, Yang M, Zhang X, Yu L, Lawas M, Li X, Bryant-Genevier M, Southall NT, Marugan J, Ferrer M, Xu H. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E1373–1381. doi: 10.1073/pnas.1419669112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, Hu X, Gao Q, Yang M, Lawas M, Delling M, Marugan J, Ferrer M, Xu H. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016;7:12109. doi: 10.1038/ncomms12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, Ballabio A. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–299. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Farfariello V, Amantini C, Santoni G. Transient receptor potential vanilloid 1 activation induces autophagy in thymocytes through ROS-regulated AMPK and Atg4C pathways. J Leukoc Biol. 2012;92:421–431. doi: 10.1189/jlb.0312123. [DOI] [PubMed] [Google Scholar]
- 171.Sukumaran P, Sun Y, Vyas M, Singh BB. TRPC1-mediated Ca(2)(+) entry is essential for the regulation of hypoxia and nutrient depletion-dependent autophagy. Cell Death Dis. 2015;6:e1674. doi: 10.1038/cddis.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang Q, Guo W, Hao B, Shi X, Lu Y, Wong CW, Ma VW, Yip TT, Au JS, Hao Q, Cheung KH, Wu W, Li GR, Yue J. Mechanistic study of TRPM2-Ca(2+)-CAMK2-BECN1 signaling in oxidative stress-induced autophagy inhibition. Autophagy. 2016;12:1340–1354. doi: 10.1080/15548627.2016.1187365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Putney JW., Jr A model for receptor-regulated calcium entry. Cell calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 174.Prakriya M, Lewis RS. Store-Operated Calcium Channels. Physiol Rev. 2015;95:1383–1436. doi: 10.1152/physrev.00020.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW., Jr Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. The Journal of biological chemistry. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet JP. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Deak AT, Blass S, Khan MJ, Groschner LN, Waldeck-Weiermair M, Hallstrom S, Graier WF, Malli R. IP3-mediated STIM1 oligomerization requires intact mitochondrial Ca2+ uptake. Journal of cell science. 2014;127:2944–2955. doi: 10.1242/jcs.149807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Singaravelu K, Nelson C, Bakowski D, de Brito OM, Ng SW, Di Capite J, Powell T, Scorrano L, Parekh AB. Mitofusin 2 regulates STIM1 migration from the Ca2+ store to the plasma membrane in cells with depolarized mitochondria. The Journal of biological chemistry. 2011;286:12189–12201. doi: 10.1074/jbc.M110.174029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ben-Kasus Nissim T, Zhang X, Elazar A, Roy S, Stolwijk JA, Zhou Y, Motiani RK, Gueguinou M, Hempel N, Hershfinkel M, Gill DL, Trebak M, Sekler I. Mitochondria control store-operated Ca2+ entry through Na+ and redox signals. The EMBO journal. 2016 doi: 10.15252/embj.201592481. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Feng M, Grice DM, Faddy HM, Nguyen N, Leitch S, Wang Y, Muend S, Kenny PA, Sukumar S, Roberts-Thomson SJ, Monteith GR, Rao R. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell. 2010;143:84–98. doi: 10.1016/j.cell.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15:124–134. doi: 10.1016/j.ccr.2008.12.019. [DOI] [PubMed] [Google Scholar]
- 183.Motiani RK, Abdullaev IF, Trebak M. A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. The Journal of biological chemistry. 2010;285:19173–19183. doi: 10.1074/jbc.M110.102582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Motiani RK, Hyzinski-Garcia MC, Zhang X, Henkel MM, Abdullaev IF, Kuo YH, Matrougui K, Mongin AA, Trebak M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Archiv : European journal of physiology. 2013;465:1249–1260. doi: 10.1007/s00424-013-1254-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Umemura M, Baljinnyam E, Feske S, De Lorenzo MS, Xie LH, Feng X, Oda K, Makino A, Fujita T, Yokoyama U, Iwatsubo M, Chen S, Goydos JS, Ishikawa Y, Iwatsubo K. Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration. PLoS One. 2014;9:e89292. doi: 10.1371/journal.pone.0089292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhu H, Zhang H, Jin F, Fang M, Huang M, Yang CS, Chen T, Fu L, Pan Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget. 2014;5:3455–3471. doi: 10.18632/oncotarget.1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kondratska K, Kondratskyi A, Yassine M, Lemonnier L, Lepage G, Morabito A, Skryma R, Prevarskaya N. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochimica et biophysica acta. 2014;1843:2263–2269. doi: 10.1016/j.bbamcr.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 188.Wei J, Zhang J, Si Y, Kanada M, Zhang Z, Terakawa S, Watanabe H. Blockage of LMP1-modulated store-operated Ca(2+) entry reduces metastatic potential in nasopharyngeal carcinoma cell. Cancer Lett. 2015;360:234–244. doi: 10.1016/j.canlet.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 189.Motiani RK, Zhang X, Harmon KE, Keller RS, Matrougui K, Bennett JA, Trebak M. Orai3 is an estrogen receptor alpha-regulated Ca(2)(+) channel that promotes tumorigenesis. FASEB J. 2013;27:63–75. doi: 10.1096/fj.12-213801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Li J, Cubbon RM, Wilson LA, Amer MS, McKeown L, Hou B, Majeed Y, Tumova S, Seymour VA, Taylor H, Stacey M, O'Regan D, Foster R, Porter KE, Kearney MT, Beech DJ. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ Res. 2011;108:1190–1198. doi: 10.1161/CIRCRESAHA.111.243352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sanchez-Hernandez Y, Laforenza U, Bonetti E, Fontana J, Dragoni S, Russo M, Avelino-Cruz JE, Schinelli S, Testa D, Guerra G, Rosti V, Tanzi F, Moccia F. Store-operated Ca(2+) entry is expressed in human endothelial progenitor cells. Stem Cells Dev. 2010;19:1967–1981. doi: 10.1089/scd.2010.0047. [DOI] [PubMed] [Google Scholar]
- 192.Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ Res. 2008;103:1289–1299. doi: 10.1161/01.RES.0000338496.95579.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lodola F, Laforenza U, Bonetti E, Lim D, Dragoni S, Bottino C, Ong HL, Guerra G, Ganini C, Massa M, Manzoni M, Ambudkar IS, Genazzani AA, Rosti V, Pedrazzoli P, Tanzi F, Moccia F, Porta C. Store-operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS One. 2012;7:e42541. doi: 10.1371/journal.pone.0042541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Moccia F, Poletto V. May the remodeling of the Ca(2)(+) toolkit in endothelial progenitor cells derived from cancer patients suggest alternative targets for anti-angiogenic treatment? Biochimica et biophysica acta. 2015;1853:1958–1973. doi: 10.1016/j.bbamcr.2014.10.024. [DOI] [PubMed] [Google Scholar]
- 195.Bhardwaj R, Hediger MA, Demaurex N. Redox modulation of STIM-ORAI signaling. Cell calcium. 2016;60:142–152. doi: 10.1016/j.ceca.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 196.Berna-Erro A, Braun A, Kraft R, Kleinschnitz C, Schuhmann MK, Stegner D, Wultsch T, Eilers J, Meuth SG, Stoll G, Nieswandt B. STIM2 regulates capacitive Ca2+ entry in neurons and plays a key role in hypoxic neuronal cell death. Science signaling. 2009;2:ra67. doi: 10.1126/scisignal.2000522. [DOI] [PubMed] [Google Scholar]
- 197.Gandhirajan RK, Meng S, Chandramoorthy HC, Mallilankaraman K, Mancarella S, Gao H, Razmpour R, Yang XF, Houser SR, Chen J, Koch WJ, Wang H, Soboloff J, Gill DL, Madesh M. Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J Clin Invest. 2013;123:887–902. doi: 10.1172/JCI65647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Holzmann C, Kilch T, Kappel S, Dorr K, Jung V, Stockle M, Bogeski I, Peinelt C. Differential Redox Regulation of Ca(2)(+) Signaling and Viability in Normal and Malignant Prostate Cells. Biophysical journal. 2015;109:1410–1419. doi: 10.1016/j.bpj.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Li Y, Guo B, Xie Q, Ye D, Zhang D, Zhu Y, Chen H, Zhu B. STIM1 Mediates Hypoxia-Driven Hepatocarcinogenesis via Interaction with HIF-1. Cell Rep. 2015;12:388–395. doi: 10.1016/j.celrep.2015.06.033. [DOI] [PubMed] [Google Scholar]
- 200.Gusarova GA, Trejo HE, Dada LA, Briva A, Welch LC, Hamanaka RB, Mutlu GM, Chandel NS, Prakriya M, Sznajder JI. Hypoxia leads to Na,K-ATPase downregulation via Ca(2+) release-activated Ca(2+) channels and AMPK activation. Mol Cell Biol. 2011;31:3546–3556. doi: 10.1128/MCB.05114-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol. 2011;31:3531–3545. doi: 10.1128/MCB.05124-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Mancarella S, Wang Y, Deng X, Landesberg G, Scalia R, Panettieri RA, Mallilankaraman K, Tang XD, Madesh M, Gill DL. Hypoxia-induced acidosis uncouples the STIM-Orai calcium signaling complex. The Journal of biological chemistry. 2011;286:44788–44798. doi: 10.1074/jbc.M111.303081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Thompson MA, Pabelick CM, Prakash YS. Role of STIM1 in regulation of store-operated Ca2+ influx in pheochromocytoma cells. Cell Mol Neurobiol. 2009;29:193–202. doi: 10.1007/s10571-008-9311-0. [DOI] [PubMed] [Google Scholar]
- 204.Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131:1327–1339. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Lu W, Wang J, Peng G, Shimoda LA, Sylvester JT. Knockdown of stromal interaction molecule 1 attenuates store-operated Ca2+ entry and Ca2+ responses to acute hypoxia in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2009;297:L17–25. doi: 10.1152/ajplung.00063.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.El Boustany C, Katsogiannou M, Delcourt P, Dewailly E, Prevarskaya N, Borowiec AS, Capiod T. Differential roles of STIM1, STIM2 and Orai1 in the control of cell proliferation and SOCE amplitude in HEK293 cells. Cell calcium. 2010;47:350–359. doi: 10.1016/j.ceca.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 207.Miederer AM, Alansary D, Schwar G, Lee PH, Jung M, Helms V, Niemeyer BA. A STIM2 splice variant negatively regulates store-operated calcium entry. Nat Commun. 2015;6:6899. doi: 10.1038/ncomms7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ong HL, de Souza LB, Zheng C, Cheng KT, Liu X, Goldsmith CM, Feske S, Ambudkar IS. STIM2 enhances receptor-stimulated Ca(2)(+) signaling by promoting recruitment of STIM1 to the endoplasmic reticulum-plasma membrane junctions. Science signaling. 2015;8:ra3. doi: 10.1126/scisignal.2005748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Rana A, Yen M, Sadaghiani AM, Malmersjo S, Park CY, Dolmetsch RE, Lewis RS. Alternative splicing converts STIM2 from an activator to an inhibitor of store-operated calcium channels. The Journal of cell biology. 2015;209:653–669. doi: 10.1083/jcb.201412060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Wang X, Wang Y, Zhou Y, Hendron E, Mancarella S, Andrake MD, Rothberg BS, Soboloff J, Gill DL. Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat Commun. 2014;5:3183. doi: 10.1038/ncomms4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.McAndrew D, Grice DM, Peters AA, Davis FM, Stewart T, Rice M, Smart CE, Brown MA, Kenny PA, Roberts-Thomson SJ, Monteith GR. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol Cancer Ther. 2011;10:448–460. doi: 10.1158/1535-7163.MCT-10-0923. [DOI] [PubMed] [Google Scholar]
- 212.Aytes A, Mollevi DG, Martinez-Iniesta M, Nadal M, Vidal A, Morales A, Salazar R, Capella G, Villanueva A. Stromal interaction molecule 2 (STIM2) is frequently overexpressed in colorectal tumors and confers a tumor cell growth suppressor phenotype. Mol Carcinog. 2012;51:746–753. doi: 10.1002/mc.20843. [DOI] [PubMed] [Google Scholar]
- 213.Stanisz H, Saul S, Muller CS, Kappl R, Niemeyer BA, Vogt T, Hoth M, Roesch A, Bogeski I. Inverse regulation of melanoma growth and migration by Orai1/STIM2-dependent calcium entry. Pigment Cell Melanoma Res. 2014;27:442–453. doi: 10.1111/pcmr.12222. [DOI] [PubMed] [Google Scholar]
- 214.Scrimgeour NR, Wilson DP, Rychkov GY. Glu(1)(0)(6) in the Orai1 pore contributes to fast Ca(2)(+)-dependent inactivation and pH dependence of Ca(2)(+) release-activated Ca(2)(+) (CRAC) current. The Biochemical journal. 2012;441:743–753. doi: 10.1042/BJ20110558. [DOI] [PubMed] [Google Scholar]
- 215.Beck A, Fleig A, Penner R, Peinelt C. Regulation of endogenous and heterologous Ca(2)(+) release-activated Ca(2)(+) currents by pH. Cell calcium. 2014;56:235–243. doi: 10.1016/j.ceca.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Koh MY, Lemos R, Jr, Liu X, Powis G. The hypoxia-associated factor switches cells from HIF-1alpha- to HIF-2alpha-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer research. 2011;71:4015–4027. doi: 10.1158/0008-5472.CAN-10-4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Brock SE, Rendon BE, Yaddanapudi K, Mitchell RA. Negative regulation of AMP-activated protein kinase (AMPK) activity by macrophage migration inhibitory factor (MIF) family members in non-small cell lung carcinomas. The Journal of biological chemistry. 2012;287:37917–37925. doi: 10.1074/jbc.M112.378299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Auciello FR, Ross FA, Ikematsu N, Hardie DG. Oxidative stress activates AMPK in cultured cells primarily by increasing cellular AMP and/or ADP. FEBS letters. 2014;588:3361–3366. doi: 10.1016/j.febslet.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Nurbaeva MK, Schmid E, Szteyn K, Yang W, Viollet B, Shumilina E, Lang F. Enhanced Ca(2)(+) entry and Na+/Ca(2)(+) exchanger activity in dendritic cells from AMP-activated protein kinase-deficient mice. FASEB J. 2012;26:3049–3058. doi: 10.1096/fj.12-204024. [DOI] [PubMed] [Google Scholar]
- 220.Faubert B, Vincent EE, Poffenberger MC, Jones RG. The AMP-activated protein kinase (AMPK) and cancer: many faces of a metabolic regulator. Cancer Lett. 2015;356:165–170. doi: 10.1016/j.canlet.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 221.Bonini MG, Gantner BN. The multifaceted activities of AMPK in tumor progression--why the “one size fits all” definition does not fit at all? IUBMB Life. 2013;65:889–896. doi: 10.1002/iub.1213. [DOI] [PubMed] [Google Scholar]
- 222.Hardie DG. Molecular Pathways: Is AMPK a Friend or a Foe in Cancer? Clin Cancer Res. 2015;21:3836–3840. doi: 10.1158/1078-0432.CCR-14-3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, Brugge JS. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Sundararaman A, Amirtham U, Rangarajan A. Calcium-Oxidant Signaling Network Regulates AMP-activated Protein Kinase (AMPK) Activation upon Matrix Deprivation. The Journal of biological chemistry. 2016;291:14410–14429. doi: 10.1074/jbc.M116.731257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Alansary D, Schmidt B, Dorr K, Bogeski I, Rieger H, Kless A, Niemeyer BA. Thiol dependent intramolecular locking of Orai1 channels. Sci Rep. 2016;6:33347. doi: 10.1038/srep33347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Shin DH, Nam JH, Lee ES, Zhang Y, Kim SJ. Inhibition of Ca(2+) release-activated Ca(2+) channel (CRAC) by curcumin and caffeic acid phenethyl ester (CAPE) via electrophilic addition to a cysteine residue of Orai1. Biochem Biophys Res Commun. 2012;428:56–61. doi: 10.1016/j.bbrc.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 228.Saul S, Gibhardt CS, Schmidt B, Lis A, Pasieka B, Conrad D, Jung P, Gaupp R, Wonnenberg B, Diler E, Stanisz H, Vogt T, Schwarz EC, Bischoff M, Herrmann M, Tschernig T, Kappl R, Rieger H, Niemeyer BA, Bogeski I. A calcium-redox feedback loop controls human monocyte immune responses: The role of ORAI Ca2+ channels. Science signaling. 2016;9:ra26. doi: 10.1126/scisignal.aaf1639. [DOI] [PubMed] [Google Scholar]
- 229.Alansary D, Bogeski I, Niemeyer BA. Facilitation of Orai3 targeting and store-operated function by Orai1. Biochimica et biophysica acta. 2015;1853:1541–1550. doi: 10.1016/j.bbamcr.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 230.Holzmann C, Kilch T, Kappel S, Armbruster A, Jung V, Stockle M, Bogeski I, Schwarz EC, Peinelt C. ICRAC controls the rapid androgen response in human primary prostate epithelial cells and is altered in prostate cancer. Oncotarget. 2013;4:2096–2107. doi: 10.18632/oncotarget.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Mignen O, Thompson JL, Shuttleworth TJ. Both Orai1 and Orai3 are essential components of the arachidonate-regulated Ca2+-selective (ARC) channels. J Physiol. 2008;586:185–195. doi: 10.1113/jphysiol.2007.146258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Mignen O, Thompson JL, Shuttleworth TJ. The molecular architecture of the arachidonate-regulated Ca2+-selective ARC channel is a pentameric assembly of Orai1 and Orai3 subunits. J Physiol. 2009;587:4181–4197. doi: 10.1113/jphysiol.2009.174193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Gonzalez-Cobos JC, Zhang X, Zhang W, Ruhle B, Motiani RK, Schindl R, Muik M, Spinelli AM, Bisaillon JM, Shinde AV, Fahrner M, Singer HA, Matrougui K, Barroso M, Romanin C, Trebak M. Store-independent Orai1/3 channels activated by intracrine leukotriene C4: role in neointimal hyperplasia. Circ Res. 2013;112:1013–1025. doi: 10.1161/CIRCRESAHA.111.300220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Zhang X, Gonzalez-Cobos JC, Schindl R, Muik M, Ruhle B, Motiani RK, Bisaillon JM, Zhang W, Fahrner M, Barroso M, Matrougui K, Romanin C, Trebak M. Mechanisms of STIM1 activation of store-independent leukotriene C4-regulated Ca2+ channels. Mol Cell Biol. 2013;33:3715–3723. doi: 10.1128/MCB.00554-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Zhang X, Zhang W, Gonzalez-Cobos JC, Jardin I, Romanin C, Matrougui K, Trebak M. Complex role of STIM1 in the activation of store-independent Orai1/3 channels. J Gen Physiol. 2014;143:345–359. doi: 10.1085/jgp.201311084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Dubois C, Vanden Abeele F, Lehen'kyi V, Gkika D, Guarmit B, Lepage G, Slomianny C, Borowiec AS, Bidaux G, Benahmed M, Shuba Y, Prevarskaya N. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell. 2014;26:19–32. doi: 10.1016/j.ccr.2014.04.025. [DOI] [PubMed] [Google Scholar]
- 237.Faouzi M, Hague F, Potier M, Ahidouch A, Sevestre H, Ouadid-Ahidouch H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. Journal of cellular physiology. 2011;226:542–551. doi: 10.1002/jcp.22363. [DOI] [PubMed] [Google Scholar]
- 238.Jenkins J, Papkovsky DB, Dmitriev RI. The Ca2+/Mn2+-transporting SPCA2 pump is regulated by oxygen and cell density in colon cancer cells. The Biochemical journal. 2016;473:2507–2518. doi: 10.1042/BCJ20160477. [DOI] [PubMed] [Google Scholar]
- 239.Grupe M, Myers G, Penner R, Fleig A. Activation of store-operated I(CRAC) by hydrogen peroxide. Cell calcium. 2010;48:1–9. doi: 10.1016/j.ceca.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Zaidi A, Michaelis ML. Effects of reactive oxygen species on brain synaptic plasma membrane Ca(2+)-ATPase. Free radical biology & medicine. 1999;27:810–821. doi: 10.1016/s0891-5849(99)00128-8. [DOI] [PubMed] [Google Scholar]
- 241.Zaidi A, Fernandes D, Bean JL, Michaelis ML. Effects of paraquat-induced oxidative stress on the neuronal plasma membrane Ca(2+)-ATPase. Free radical biology & medicine. 2009;47:1507–1514. doi: 10.1016/j.freeradbiomed.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Muscella A, Calabriso N, Vetrugno C, Fanizzi FP, De Pascali SA, Storelli C, Marsigliante S. The platinum (II) complex [Pt(O,O′-acac)(gamma-acac)(DMS)] alters the intracellular calcium homeostasis in MCF-7 breast cancer cells. Biochem Pharmacol. 2011;81:91–103. doi: 10.1016/j.bcp.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 243.Bartlett RK, Bieber Urbauer RJ, Anbanandam A, Smallwood HS, Urbauer JL, Squier TC. Oxidation of Met144 and Met145 in calmodulin blocks calmodulin dependent activation of the plasma membrane Ca-ATPase. Biochemistry. 2003;42:3231–3238. doi: 10.1021/bi026956z. [DOI] [PubMed] [Google Scholar]
- 244.Yao Y, Yin D, Jas GS, Kuczer K, Williams TD, Schoneich C, Squier TC. Oxidative modification of a carboxyl-terminal vicinal methionine in calmodulin by hydrogen peroxide inhibits calmodulin-dependent activation of the plasma membrane Ca-ATPase. Biochemistry. 1996;35:2767–2787. doi: 10.1021/bi951712i. [DOI] [PubMed] [Google Scholar]
- 245.Padanyi R, Paszty K, Hegedus L, Varga K, Papp B, Penniston JT, Enyedi A. Multifaceted plasma membrane Ca(2+) pumps: From structure to intracellular Ca(2+) handling and cancer. Biochimica et biophysica acta. 2016;1863:1351–1363. doi: 10.1016/j.bbamcr.2015.12.011. [DOI] [PubMed] [Google Scholar]
- 246.Aung CS, Ye W, Plowman G, Peters AA, Monteith GR, Roberts-Thomson SJ. Plasma membrane calcium ATPase 4 and the remodeling of calcium homeostasis in human colon cancer cells. Carcinogenesis. 2009;30:1962–1969. doi: 10.1093/carcin/bgp223. [DOI] [PubMed] [Google Scholar]
- 247.Ribiczey P, Tordai A, Andrikovics H, Filoteo AG, Penniston JT, Enouf J, Enyedi A, Papp B, Kovacs T. Isoform-specific up-regulation of plasma membrane Ca2+ATPase expression during colon and gastric cancer cell differentiation. Cell calcium. 2007;42:590–605. doi: 10.1016/j.ceca.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.VanHouten J, Sullivan C, Bazinet C, Ryoo T, Camp R, Rimm DL, Chung G, Wysolmerski J. PMCA2 regulates apoptosis during mammary gland involution and predicts outcome in breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11405–11410. doi: 10.1073/pnas.0911186107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Lee WJ, Roberts-Thomson SJ, Monteith GR. Plasma membrane calcium-ATPase 2 and 4 in human breast cancer cell lines. Biochem Biophys Res Commun. 2005;337:779–783. doi: 10.1016/j.bbrc.2005.09.119. [DOI] [PubMed] [Google Scholar]
- 250.Peters AA, Milevskiy MJ, Lee WC, Curry MC, Smart CE, Saunus JM, Reid L, da Silva L, Marcial DL, Dray E, Brown MA, Lakhani SR, Roberts-Thomson SJ, Monteith GR. The calcium pump plasma membrane Ca(2+)-ATPase 2 (PMCA2) regulates breast cancer cell proliferation and sensitivity to doxorubicin. Sci Rep. 2016;6:25505. doi: 10.1038/srep25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Curry M, Roberts-Thomson SJ, Monteith GR. PMCA2 silencing potentiates MDA-MB-231 breast cancer cell death initiated with the Bcl-2 inhibitor ABT-263. Biochem Biophys Res Commun. 2016;478:1792–1797. doi: 10.1016/j.bbrc.2016.09.030. [DOI] [PubMed] [Google Scholar]
- 252.Kuster GM, Lancel S, Zhang J, Communal C, Trucillo MP, Lim CC, Pfister O, Weinberg EO, Cohen RA, Liao R, Siwik DA, Colucci WS. Redox-mediated reciprocal regulation of SERCA and Na+-Ca2+ exchanger contributes to sarcoplasmic reticulum Ca2+ depletion in cardiac myocytes. Free radical biology & medicine. 2010;48:1182–1187. doi: 10.1016/j.freeradbiomed.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Glancy B, Balaban RS. Role of mitochondrial Ca2+ in the regulation of cellular energetics. Biochemistry. 2012;51:2959–2973. doi: 10.1021/bi2018909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Hunter DR, Haworth RA, Southard JH. Relationship between configuration, function, and permeability in calcium-treated mitochondria. The Journal of biological chemistry. 1976;251:5069–5077. [PubMed] [Google Scholar]
- 255.Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovascular research. 2004;61:372–385. doi: 10.1016/S0008-6363(03)00533-9. [DOI] [PubMed] [Google Scholar]
- 256.Quintana A, Schwarz EC, Schwindling C, Lipp P, Kaestner L, Hoth M. Sustained activity of calcium release-activated calcium channels requires translocation of mitochondria to the plasma membrane. The Journal of biological chemistry. 2006;281:40302–40309. doi: 10.1074/jbc.M607896200. [DOI] [PubMed] [Google Scholar]
- 257.Parekh AB. Mitochondrial regulation of store-operated CRAC channels. Cell calcium. 2008;44:6–13. doi: 10.1016/j.ceca.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 258.Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. The Journal of cell biology. 1997;137:633–648. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, Hajnoczky G. Structural and functional features and significance of the physical linkage between ER and mitochondria. The Journal of cell biology. 2006;174:915–921. doi: 10.1083/jcb.200604016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 261.Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:436–441. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Bernardi P, von Stockum S. The permeability transition pore as a Ca(2+) release channel: new answers to an old question. Cell calcium. 2012;52:22–27. doi: 10.1016/j.ceca.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.De Marchi U, Santo-Domingo J, Castelbou C, Sekler I, Wiederkehr A, Demaurex N. NCLX protein, but not LETM1, mediates mitochondrial Ca2+ extrusion, thereby limiting Ca2+-induced NAD(P)H production and modulating matrix redox state. The Journal of biological chemistry. 2014;289:20377–20385. doi: 10.1074/jbc.M113.540898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Csordas G, Varnai P, Golenar T, Roy S, Purkins G, Schneider TG, Balla T, Hajnoczky G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Molecular cell. 2010;39:121–132. doi: 10.1016/j.molcel.2010.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Anelli T, Bergamelli L, Margittai E, Rimessi A, Fagioli C, Malgaroli A, Pinton P, Ripamonti M, Rizzuto R, Sitia R. Ero1alpha regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (MAM) Antioxidants & redox signaling. 2012;16:1077–1087. doi: 10.1089/ars.2011.4004. [DOI] [PubMed] [Google Scholar]
- 266.Gilady SY, Bui M, Lynes EM, Benson MD, Watts R, Vance JE, Simmen T. Ero1alpha requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM) Cell stress & chaperones. 2010;15:619–629. doi: 10.1007/s12192-010-0174-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Li G, Mongillo M, Chin KT, Harding H, Ron D, Marks AR, Tabas I. Role of ERO1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. The Journal of cell biology. 2009;186:783–792. doi: 10.1083/jcb.200904060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Verfaillie T, Rubio N, Garg AD, Bultynck G, Rizzuto R, Decuypere JP, Piette J, Linehan C, Gupta S, Samali A, Agostinis P. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell death and differentiation. 2012;19:1880–1891. doi: 10.1038/cdd.2012.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.May D, Itin A, Gal O, Kalinski H, Feinstein E, Keshet E. Ero1-L alpha plays a key role in a HIF-1-mediated pathway to improve disulfide bond formation and VEGF secretion under hypoxia: implication for cancer. Oncogene. 2005;24:1011–1020. doi: 10.1038/sj.onc.1208325. [DOI] [PubMed] [Google Scholar]
- 270.Kutomi G, Tamura Y, Tanaka T, Kajiwara T, Kukita K, Ohmura T, Shima H, Takamaru T, Satomi F, Suzuki Y, Torigoe T, Sato N, Hirata K. Human endoplasmic reticulum oxidoreductin 1-alpha is a novel predictor for poor prognosis of breast cancer. Cancer Sci. 2013;104:1091–1096. doi: 10.1111/cas.12177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Bonora M, Pinton P. The mitochondrial permeability transition pore and cancer: molecular mechanisms involved in cell death. Front Oncol. 2014;4:302. doi: 10.3389/fonc.2014.00302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Takeyama N, Matsuo N, Tanaka T. Oxidative damage to mitochondria is mediated by the Ca(2+)-dependent inner-membrane permeability transition. The Biochemical journal. 1993;294(Pt 3):719–725. doi: 10.1042/bj2940719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Mendes CC, Gomes DA, Thompson M, Souto NC, Goes TS, Goes AM, Rodrigues MA, Gomez MV, Nathanson MH, Leite MF. The type III inositol 1,4,5-trisphosphate receptor preferentially transmits apoptotic Ca2+ signals into mitochondria. The Journal of biological chemistry. 2005;280:40892–40900. doi: 10.1074/jbc.M506623200. [DOI] [PubMed] [Google Scholar]
- 274.De Stefani D, Bononi A, Romagnoli A, Messina A, De Pinto V, Pinton P, Rizzuto R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell death and differentiation. 2012;19:267–273. doi: 10.1038/cdd.2011.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Giorgi C, Ito K, Lin HK, Santangelo C, Wieckowski MR, Lebiedzinska M, Bononi A, Bonora M, Duszynski J, Bernardi R, Rizzuto R, Tacchetti C, Pinton P, Pandolfi PP. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science. 2010;330:1247–1251. doi: 10.1126/science.1189157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Bononi A, Bonora M, Marchi S, Missiroli S, Poletti F, Giorgi C, Pandolfi PP, Pinton P. Identification of PTEN at the ER and MAMs and its regulation of Ca(2+) signaling and apoptosis in a protein phosphatase-dependent manner. Cell death and differentiation. 2013;20:1631–1643. doi: 10.1038/cdd.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ. BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science. 2003;300:135–139. doi: 10.1126/science.1081208. [DOI] [PubMed] [Google Scholar]
- 278.Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- 279.Rasola A, Bernardi P. Reprint of “The mitochondrial permeability transition pore and its adaptive responses in tumor cells”. Cell calcium. 2015;58:18–26. doi: 10.1016/j.ceca.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 280.McStay GP, Clarke SJ, Halestrap AP. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. The Biochemical journal. 2002;367:541–548. doi: 10.1042/BJ20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. The Journal of biological chemistry. 2011;286:40184–40192. doi: 10.1074/jbc.M111.243469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Hurst S, Hoek J, Sheu SS. Mitochondrial Ca2+ and regulation of the permeability transition pore. J Bioenerg Biomembr. 2016 doi: 10.1007/s10863-016-9672-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I, Lippe G, Bernardi P. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:5887–5892. doi: 10.1073/pnas.1217823110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. The Journal of experimental medicine. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Vervliet T, Parys JB, Bultynck G. Bcl-2 proteins and calcium signaling: complexity beneath the surface. Oncogene. 2016 doi: 10.1038/onc.2016.31. [DOI] [PubMed] [Google Scholar]
- 286.Zhong F, Harr MW, Bultynck G, Monaco G, Parys JB, De Smedt H, Rong YP, Molitoris JK, Lam M, Ryder C, Matsuyama S, Distelhorst CW. Induction of Ca(2)+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood. 2011;117:2924–2934. doi: 10.1182/blood-2010-09-307405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Akl H, Monaco G, La Rovere R, Welkenhuyzen K, Kiviluoto S, Vervliet T, Molgo J, Distelhorst CW, Missiaen L, Mikoshiba K, Parys JB, De Smedt H, Bultynck G. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis. 2013;4:e632. doi: 10.1038/cddis.2013.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Greenberg EF, McColl KS, Zhong F, Wildey G, Dowlati A, Distelhorst CW. Synergistic killing of human small cell lung cancer cells by the Bcl-2-inositol 1,4,5-trisphosphate receptor disruptor BIRD-2 and the BH3-mimetic ABT-263. Cell Death Dis. 2015;6:e2034. doi: 10.1038/cddis.2015.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.McCormack JG, Denton RM. Mitochondrial Ca2+ transport and the role of intramitochondrial Ca2+ in the regulation of energy metabolism. Developmental neuroscience. 1993;15:165–173. doi: 10.1159/000111332. [DOI] [PubMed] [Google Scholar]
- 290.Hong JH, Moon SJ, Byun HM, Kim MS, Jo H, Bae YS, Lee SI, Bootman MD, Roderick HL, Shin DM, Seo JT. Critical role of phospholipase Cgamma1 in the generation of H2O2-evoked [Ca2+]i oscillations in cultured rat cortical astrocytes. The Journal of biological chemistry. 2006;281:13057–13067. doi: 10.1074/jbc.M601726200. [DOI] [PubMed] [Google Scholar]
- 291.Ishii K, Hirose K, Iino M. Ca2+ shuttling between endoplasmic reticulum and mitochondria underlying Ca2+ oscillations. EMBO Rep. 2006;7:390–396. doi: 10.1038/sj.embor.7400620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- 293.Pralong WF, Spat A, Wollheim CB. Dynamic pacing of cell metabolism by intracellular Ca2+ transients. The Journal of biological chemistry. 1994;269:27310–27314. [PubMed] [Google Scholar]
- 294.Satrustegui J, Pardo B, Del Arco A. Mitochondrial transporters as novel targets for intracellular calcium signaling. Physiol Rev. 2007;87:29–67. doi: 10.1152/physrev.00005.2006. [DOI] [PubMed] [Google Scholar]
- 295.Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochimica et biophysica acta. 2009;1787:1309–1316. doi: 10.1016/j.bbabio.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 296.Pettit FH, Roche TE, Reed LJ. Function of calcium ions in pyruvate dehydrogenase phosphatase activity. Biochem Biophys Res Commun. 1972;49:563–571. doi: 10.1016/0006-291x(72)90448-2. [DOI] [PubMed] [Google Scholar]
- 297.Vassylyev DG, Symersky J. Crystal structure of pyruvate dehydrogenase phosphatase 1 and its functional implications. J Mol Biol. 2007;370:417–426. doi: 10.1016/j.jmb.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, Thompson CB, Birnbaum MJ, Hallows KR, Foskett JK. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell. 2010;142:270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Cardenas C, Muller M, McNeal A, Lovy A, Jana F, Bustos G, Urra F, Smith N, Molgo J, Diehl JA, Ridky TW, Foskett JK. Selective Vulnerability of Cancer Cells by Inhibition of Ca(2+) Transfer from Endoplasmic Reticulum to Mitochondria. Cell Rep. 2016;14:2313–2324. doi: 10.1016/j.celrep.2016.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Monaco G, Decrock E, Arbel N, van Vliet AR, La Rovere RM, De Smedt H, Parys JB, Agostinis P, Leybaert L, Shoshan-Barmatz V, Bultynck G. The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. The Journal of biological chemistry. 2015;290:9150–9161. doi: 10.1074/jbc.M114.622514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Arbel N, Ben-Hail D, Shoshan-Barmatz V. Mediation of the antiapoptotic activity of Bcl-xL protein upon interaction with VDAC1 protein. The Journal of biological chemistry. 2012;287:23152–23161. doi: 10.1074/jbc.M112.345918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Arzoine L, Zilberberg N, Ben-Romano R, Shoshan-Barmatz V. Voltage-dependent anion channel 1-based peptides interact with hexokinase to prevent its anti-apoptotic activity. The Journal of biological chemistry. 2009;284:3946–3955. doi: 10.1074/jbc.M803614200. [DOI] [PubMed] [Google Scholar]
- 303.Galluzzi L, Kepp O, Tajeddine N, Kroemer G. Disruption of the hexokinase-VDAC complex for tumor therapy. Oncogene. 2008;27:4633–4635. doi: 10.1038/onc.2008.114. [DOI] [PubMed] [Google Scholar]
- 304.Huang H, Shah K, Bradbury NA, Li C, White C. Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death Dis. 2014;5:e1482. doi: 10.1038/cddis.2014.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Wiel C, Lallet-Daher H, Gitenay D, Gras B, Le Calve B, Augert A, Ferrand M, Prevarskaya N, Simonnet H, Vindrieux D, Bernard D. Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun. 2014;5:3792. doi: 10.1038/ncomms4792. [DOI] [PubMed] [Google Scholar]
- 306.Liu T, O'Rourke B. Regulation of mitochondrial Ca2+ and its effects on energetics and redox balance in normal and failing heart. J Bioenerg Biomembr. 2009;41:127–132. doi: 10.1007/s10863-009-9216-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Joseph SK. Role of thiols in the structure and function of inositol trisphosphate receptors. Curr Top Membr. 2010;66:299–322. doi: 10.1016/S1063-5823(10)66013-9. [DOI] [PubMed] [Google Scholar]
- 308.Niggli E, Ullrich ND, Gutierrez D, Kyrychenko S, Polakova E, Shirokova N. Posttranslational modifications of cardiac ryanodine receptors: Ca(2+) signaling and EC-coupling. Biochimica et biophysica acta. 2013;1833:866–875. doi: 10.1016/j.bbamcr.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Lock JT, Sinkins WG, Schilling WP. Protein S-glutathionylation enhances Ca2+-induced Ca2+ release via the IP3 receptor in cultured aortic endothelial cells. J Physiol. 2012;590:3431–3447. doi: 10.1113/jphysiol.2012.230656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Higo T, Hattori M, Nakamura T, Natsume T, Michikawa T, Mikoshiba K. Subtype-specific and ER lumenal environment-dependent regulation of inositol 1,4,5-trisphosphate receptor type 1 by ERp44. Cell. 2005;120:85–98. doi: 10.1016/j.cell.2004.11.048. [DOI] [PubMed] [Google Scholar]
- 311.Bansaghi S, Golenar T, Madesh M, Csordas G, Ramachandra Rao S, Sharma K, Yule DI, Joseph SK, Hajnoczky G. Isoform- and species-specific control of inositol 1,4,5-trisphosphate (IP3) receptors by reactive oxygen species. The Journal of biological chemistry. 2014;289:8170–8181. doi: 10.1074/jbc.M113.504159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, Cipolat S, Costa V, Casarin A, Gomes LC, Perales-Clemente E, Salviati L, Fernandez-Silva P, Enriquez JA, Scorrano L. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–171. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Pelicano H, Lu W, Zhou Y, Zhang W, Chen Z, Hu Y, Huang P. Mitochondrial dysfunction and reactive oxygen species imbalance promote breast cancer cell motility through a CXCL14-mediated mechanism. Cancer research. 2009;69:2375–2383. doi: 10.1158/0008-5472.CAN-08-3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Marchi S, Rimessi A, Giorgi C, Baldini C, Ferroni L, Rizzuto R, Pinton P. Akt kinase reducing endoplasmic reticulum Ca2+ release protects cells from Ca2+-dependent apoptotic stimuli. Biochem Biophys Res Commun. 2008;375:501–505. doi: 10.1016/j.bbrc.2008.07.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Szado T, Vanderheyden V, Parys JB, De Smedt H, Rietdorf K, Kotelevets L, Chastre E, Khan F, Landegren U, Soderberg O, Bootman MD, Roderick HL. Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2427–2432. doi: 10.1073/pnas.0711324105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Leslie NR, Batty IH, Maccario H, Davidson L, Downes CP. Understanding PTEN regulation: PIP2, polarity and protein stability. Oncogene. 2008;27:5464–5476. doi: 10.1038/onc.2008.243. [DOI] [PubMed] [Google Scholar]
- 317.Leslie NR, Bennett D, Lindsay YE, Stewart H, Gray A, Downes CP. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. The EMBO journal. 2003;22:5501–5510. doi: 10.1093/emboj/cdg513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Han D, Antunes F, Canali R, Rettori D, Cadenas E. Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. The Journal of biological chemistry. 2003;278:5557–5563. doi: 10.1074/jbc.M210269200. [DOI] [PubMed] [Google Scholar]
- 319.De Pinto V, Reina S, Gupta A, Messina A, Mahalakshmi R. Role of cysteines in mammalian VDAC isoforms' function. Biochimica et biophysica acta. 2016;1857:1219–1227. doi: 10.1016/j.bbabio.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Prezma T, Shteinfer A, Admoni L, Raviv Z, Sela I, Levi I, Shoshan-Barmatz V. VDAC1-based peptides: novel pro-apoptotic agents and potential therapeutics for B-cell chronic lymphocytic leukemia. Cell Death Dis. 2013;4:e809. doi: 10.1038/cddis.2013.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Dinnen RD, Mao Y, Qiu W, Cassai N, Slavkovich VN, Nichols G, Su GH, Brandt-Rauf P, Fine RL. Redirecting apoptosis to aponecrosis induces selective cytotoxicity to pancreatic cancer cells through increased ROS, decline in ATP levels, and VDAC. Mol Cancer Ther. 2013;12:2792–2803. doi: 10.1158/1535-7163.MCT-13-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Sharaf el dein O, Gallerne C, Brenner C, Lemaire C. Increased expression of VDAC1 sensitizes carcinoma cells to apoptosis induced by DNA cross-linking agents. Biochem Pharmacol. 2012;83:1172–1182. doi: 10.1016/j.bcp.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 323.De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010;467:291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Jhun BS, Mishra J, Monaco S, Fu D, Jiang W, Sheu SS, OU J. The mitochondrial Ca2+ uniporter: regulation by auxiliary subunits and signal transduction pathways. Am J Physiol Cell Physiol. 2016;311:C67–80. doi: 10.1152/ajpcell.00319.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, Koch WJ, Cheung JY, Madesh M, Elrod JW. The Mitochondrial Calcium Uniporter Matches Energetic Supply with Cardiac Workload during Stress and Modulates Permeability Transition. Cell Rep. 2015;12:23–34. doi: 10.1016/j.celrep.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol. 2013;15:1464–1472. doi: 10.1038/ncb2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012;151:630–644. doi: 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL, Corbally DP, Nemani N, Fairweather JP, Cutri AR, Zhang X, Song J, Jana F, Huang J, Barrero C, Rabinowitz JE, Luongo TS, Schumacher SM, Rockman ME, Dietrich A, Merali S, Caplan J, Stathopulos P, Ahima RS, Cheung JY, Houser SR, Koch WJ, Patel V, Gohil VM, Elrod JW, Rajan S, Madesh M. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016;15:1673–1685. doi: 10.1016/j.celrep.2016.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Marchi S, Lupini L, Patergnani S, Rimessi A, Missiroli S, Bonora M, Bononi A, Corra F, Giorgi C, De Marchi E, Poletti F, Gafa R, Lanza G, Negrini M, Rizzuto R, Pinton P. Downregulation of the mitochondrial calcium uniporter by cancer-related miR-25. Curr Biol. 2013;23:58–63. doi: 10.1016/j.cub.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Hall DD, Wu Y, Domann FE, Spitz DR, Anderson ME. Mitochondrial calcium uniporter activity is dispensable for MDA-MB-231 breast carcinoma cell survival. PLoS One. 2014;9:e96866. doi: 10.1371/journal.pone.0096866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Curry MC, Peters AA, Kenny PA, Roberts-Thomson SJ, Monteith GR. Mitochondrial calcium uniporter silencing potentiates caspase-independent cell death in MDA-MB-231 breast cancer cells. Biochem Biophys Res Commun. 2013;434:695–700. doi: 10.1016/j.bbrc.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 333.Tang S, Wang X, Shen Q, Yang X, Yu C, Cai C, Cai G, Meng X, Zou F. Mitochondrial Ca(2)(+) uniporter is critical for store-operated Ca(2)(+) entry-dependent breast cancer cell migration. Biochem Biophys Res Commun. 2015;458:186–193. doi: 10.1016/j.bbrc.2015.01.092. [DOI] [PubMed] [Google Scholar]
- 334.Tosatto A, Sommaggio R, Kummerow C, Bentham RB, Blacker TS, Berecz T, Duchen MR, Rosato A, Bogeski I, Szabadkai G, Rizzuto R, Mammucari C. The mitochondrial calcium uniporter regulates breast cancer progression via HIF-1alpha. EMBO Mol Med. 2016;8:569–585. doi: 10.15252/emmm.201606255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Hou T, Zhang X, Xu J, Jian C, Huang Z, Ye T, Hu K, Zheng M, Gao F, Wang X, Cheng H. Synergistic triggering of superoxide flashes by mitochondrial Ca2+ uniport and basal reactive oxygen species elevation. The Journal of biological chemistry. 2013;288:4602–4612. doi: 10.1074/jbc.M112.398297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS, Anderson ME. CaMKII determines mitochondrial stress responses in heart. Nature. 2012;491:269–273. doi: 10.1038/nature11444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O'Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. 2006;99:172–182. doi: 10.1161/01.RES.0000232546.92777.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Feldman B, Fedida-Metula S, Nita J, Sekler I, Fishman D. Coupling of mitochondria to store-operated Ca(2+)-signaling sustains constitutive activation of protein kinase B/Akt and augments survival of malignant melanoma cells. Cell calcium. 2010;47:525–537. doi: 10.1016/j.ceca.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 339.Brostrom MA, Brostrom CO. Calcium dynamics and endoplasmic reticular function in the regulation of protein synthesis: implications for cell growth and adaptability. Cell calcium. 2003;34:345–363. doi: 10.1016/s0143-4160(03)00127-1. [DOI] [PubMed] [Google Scholar]
- 340.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 341.Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell death and differentiation. 2004;11:372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- 342.Sharov VS, Dremina ES, Galeva NA, Williams TD, Schoneich C. Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxidation during biological aging. The Biochemical journal. 2006;394:605–615. doi: 10.1042/BJ20051214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 344.Ye ZW, Zhang J, Ancrum T, Manevich Y, Townsend DM, Tew KD. Glutathione S-Transferase P-Mediated Protein S-Glutathionylation of Resident Endoplasmic Reticulum Proteins Influences Sensitivity to Drug-Induced Unfolded Protein Response. Antioxidants & redox signaling. 2016 doi: 10.1089/ars.2015.6486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Knyushko TV, Sharov VS, Williams TD, Schoneich C, Bigelow DJ. 3-Nitrotyrosine modification of SERCA2a in the aging heart: a distinct signature of the cellular redox environment. Biochemistry. 2005;44:13071–13081. doi: 10.1021/bi051226n. [DOI] [PubMed] [Google Scholar]
- 346.Xu S, Ying J, Jiang B, Guo W, Adachi T, Sharov V, Lazar H, Menzoian J, Knyushko TV, Bigelow D, Schoneich C, Cohen RA. Detection of sequence-specific tyrosine nitration of manganese SOD and SERCA in cardiovascular disease and aging. Am J Physiol Heart Circ Physiol. 2006;290:H2220–2227. doi: 10.1152/ajpheart.01293.2005. [DOI] [PubMed] [Google Scholar]
- 347.Blaskovic D, Drzik F, Viskupicova J, Zizkova P, Veverka M, Horakova L. Effects of novel quercetin derivatives on sarco/endoplasmic reticulum Ca2+-ATPase activity. Neuro Endocrinol Lett. 2012;33(3):190–197. [PubMed] [Google Scholar]
- 348.Dremina ES, Sharov VS, Kumar K, Zaidi A, Michaelis EK, Schoneich C. Anti-apoptotic protein Bcl-2 interacts with and destabilizes the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) The Biochemical journal. 2004;383:361–370. doi: 10.1042/BJ20040187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Dremina ES, Sharov VS, Schoneich C. Heat-shock proteins attenuate SERCA inactivation by the anti-apoptotic protein Bcl-2: possible implications for the ER Ca2+-mediated apoptosis. The Biochemical journal. 2012;444:127–139. doi: 10.1042/BJ20111114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Pierro C, Cook SJ, Foets TC, Bootman MD, Roderick HL. Oncogenic K-Ras suppresses IP(3)-dependent Ca(2)(+) release through remodelling of the isoform composition of IP(3)Rs and ER luminal Ca(2)(+) levels in colorectal cancer cell lines. Journal of cell science. 2014;127:1607–1619. doi: 10.1242/jcs.141408. [DOI] [PubMed] [Google Scholar]
- 351.Giorgi C, Bonora M, Missiroli S, Poletti F, Ramirez FG, Morciano G, Morganti C, Pandolfi PP, Mammano F, Pinton P. Intravital imaging reveals p53-dependent cancer cell death induced by phototherapy via calcium signaling. Oncotarget. 2015;6:1435–1445. doi: 10.18632/oncotarget.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Giorgi C, Bonora M, Sorrentino G, Missiroli S, Poletti F, Suski JM, Galindo Ramirez F, Rizzuto R, Di Virgilio F, Zito E, Pandolfi PP, Wieckowski MR, Mammano F, Del Sal G, Pinton P. p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:1779–1784. doi: 10.1073/pnas.1410723112. [DOI] [PMC free article] [PubMed] [Google Scholar]