

Abstract
The homomeric GABA‐ρ ligand‐gated ion channels (also known as GABAC or GABAA‐ρ receptors) are similar to heteromeric GABAA receptors in structure, function and mechanism of action. However, their distinctive pharmacological properties and distribution make them of special interest. This review focuses on GABA‐ρ ion channel structure, ligand selectivity toward ρ receptors over heteromeric GABAA receptor sub‐types and selectivity between different homomeric ρ sub‐type receptors. Several GABA analogues show selectivity at homomeric GABA‐ρ receptors over heteromeric GABAA receptors. More recently, some synthetic ligands have been found to show selectivity at receptors formed from one ρ subtype over others. The unique pharmacological profiles of these agents are discussed in this review. The classical binding site of GABA within the orthosteric site of GABA‐ρ homomeric receptors is discussed in detail regarding the loops and residues that constitute the binding site. The ligand‐residue interactions in this classical binding and those of mutant receptors are discussed. The structure and conformations of GABA are discussed in regard to its flexibility and molecular properties. Although the binding mode of GABA is difficult to predict, several interactions between GABA and the receptor assist in predicting its potential conformation and mode of action. The structure–activity relationships of GABA and structurally key ligands at ρ receptors are described and discussed.
Abbreviations
- 2‐MeGABA
4‐amino‐2‐methylbutanoic acid
- 2‐MeTACA
trans‐2‐methyl‐4‐aminocrotonic acid
- 3‐AMOHP
3‐(aminomethyl)‐1‐oxo‐1‐hydroxy‐phospholane
- 3‐AOHP
3‐(amino)‐1‐oxo‐1‐hydroxy‐phospholane
- 3‐GOHP
3‐(guanidino)‐1‐oxo‐1‐hydroxy‐phospholane
- 4‐ACPAM
4‐aminocyclopent‐1‐enecarboxamide
- 4‐ACPHA
4‐amino‐N‐hydroxycyclopent‐1‐enecarboxamide
- ACPBPA
3‐aminocyclopentenyl‐butylphosphinic acid
- (±)‐ACPECA
(±)‐4‐aminocyclopent‐2‐ene‐1‐carboxylic acid
- CACA
cis‐4‐aminocrotonic acid
- CAMP
cis‐2‐(aminomethyl)cyclopropane carboxylic acid
- I4AA
imidazole‐4‐acetic acid
- LGIC
ligand‐gated ion channel
- TACA
trans‐4‐aminocrotonic acid
- TAMP
trans‐2‐(aminomethyl) cyclopropane carboxylic acid
- THIP
4,5,6,7‐tetrahydroisoxazolo[5,4‐c] pyridin‐3‐ol
- TM
transmembrane
- TPMPA
(1,2,5,6‐tetrahydropyridin‐4‐yl) methylphosphinic acid
Cys‐loop pentameric ligand‐gated ion channels
The GABAA‐ρ receptors as designated by the International Union of Pharmacology (IUPHAR) (Alexander et al., 2015) and also known as GABA‐ρ or GABAC receptors, are homopentameric ligand‐gated ion channels (LGIC) composed of ρ subunits. They are members of the pentameric or Cys‐loop LGIC superfamily comprising excitatory cation selective receptors such as nicotinic acetylcholine receptors, 5‐HT3 receptors and zinc‐activated channels, and inhibitory anion‐selective receptors such as GABAA receptors, strychnine‐sensitive glycine receptors and invertebrate glutamate‐gated chloride channels (Thompson et al., 2010; Baenziger and Corringer, 2011). Receptors of this superfamily require five subunits to assemble a single ion channel. The ion channel may be homomeric formed by five identical subunits as is the case of GABA‐ρ receptors or heteromeric, consisting of a combination of at least two different subunits, such as GABAA receptors (Olsen and Sieghart, 2009).
The Cys‐loop receptors are analogous to each other in their structure, and they consist of three domains. The N‐terminal extracellular domain is generally formed by 10 β‐strands in two sheets that form a sandwich and two α‐helices (Figure 1). This domain contains the orthosteric binding site and also the Cys–Cys disulfide bond forming the characteristic Cys‐loop of 13 residues (also called β6–β7 loop). This loop is conserved across subunits belonging to this superfamily and is the basis of the name “Cys‐loop receptors” (Miller and Smart, 2010). This structure is believed to be important for both cell surface expression and cooperative interaction between the agonist binding sites and the channel gate (Wong et al., 2014). The second domain consists of the four transmembrane α‐helices (TM1–TM4). TM2 forms the pore of the ion channel, whereas the remaining three TM helices form a hydrophobic environment to incorporate the pore into the plasma membrane (Figure 1) (Corringer et al., 2000). The third domain is an intracellular loop between TM3 and TM4 that is variable and of unknown structure. This loop has little residue conservation between different subunits or subunits of different subtypes. There is evidence that the intracellular domain is involved in modulating the receptor by phosphorylation and binding to other intracellular molecules. There is a short extracellular C terminus after M4 (Filippova et al., 1999).
Figure 1.

Pentameric ligand‐gated ion channel receptor of GABA‐ρ homology model based on GluCl structure prepared has been previously described (Naffaa et al., 2015). A single ion channel showing the main domains with five identical subunits, coloured differently to show the intersubunit interfaces located between the principal (+) and complementary (−) sides. (A) Bottom view and (B) side view. As the structure of the intracellular domain has not yet been determined by crystallography, it is not included.
Ionotropic GABA receptor subunit composition
In humans, there are 19 isoforms of GABAA subunits, that is, six α, three β, three γ and one of δ, ε, π, θ, known to form heteromeric GABAA receptors, and three ρ subunits that were reclassified by the Nomenclature Committee of IUPHAR (Olsen and Sieghart, 2009) to GABAA from a distinct class of receptors known as GABAC. This reclassification is controversial due to the differing pharmacology, physiology and molecular biology of GABA receptors containing ρ‐subunits from those containing non ρ‐subunits. Most GABAA receptors are heteromeric and require at least two different GABAA subunits (Olsen and Sieghart, 2009). The predominant GABAA receptors in brain tissues are formed from two α, two β and one γ subunit, for example, GABAA α1β2γ. The formation of active GABA‐gated ion channels requires the presence of at least α and β subunits. However, GABA‐ρ receptors express as homomeric ion channels, and some studies suggest pseudoheteromeric channels consisting of different ρ subunits (Connolly et al., 1996). There is some limited in vitro evidence to support co‐assembly of ρ subunits with other GABAA subunits, particularly with α1 and γ2 (Milligan et al., 2004; Pétriz et al., 2014), and also with glycine receptor α1 and α2 subunits (Pan et al., 2000).
Significant differences have been identified between various ionotropic GABA receptors based on physiological, pharmacological and biochemical properties (Johnston, 2002). The amino acid sequence identity between various GABAA subunits and the ρ subunits ranges between 35 and 45% but is as high as 75% in the TM region (Le Novere and Changeux, 1999). GABA‐ρ ion channels expressed in Xenopus oocytes have different properties to other GABAA receptors in terms of potency, channel opening time and receptor desensitization. In general, GABA is between 10‐ and 100‐fold more potent at GABA‐ρ receptors than heteromeric GABAA receptors, with slow activation and deactivation and less readily desensitized (Feigenspan et al., 1993; Amin and Weiss, 1994; Feigenspan and Bormann, 1994; Bormann, 2000). Heteromeric GABAA receptors are also well known to be modulated by agents such as benzodiazepines, barbiturates and neurosteroids (Johnston, 1996; Bormann, 2000). GABA‐ρ receptors are insensitive to these GABAA receptor modulators. However, some GABAA modulators can also modulate GABA‐ρ receptors, such as zinc, lanthanides (Wu et al., 1993; Calvo et al., 1994; Chang et al., 1995) and some synthetic neurosteroids (Morris et al., 1999).
GABA‐ρ receptors
The concept of a third sub‐type of GABA receptor arose in 1984 from the studies demonstrating the lack of inhibition by cis‐4‐aminocrotonic acid (CACA) (Figure 2), a GABA analogue that has a bicuculline‐insensitive depressant action on the firing of cat spinal neurones, on the binding of [3H]‐baclofen to the rat cerebellar membranes and thus unlikely to act as an agonist on bicuculline‐sensitive GABAA or baclofen‐sensitive GABAB receptors (Drew et al., 1984). Later, bovine retinal mRNA expressed in Xenopus oocytes was found to result in receptors sensitive to GABA but insensitive to both bicuculline and baclofen (Polenzani et al., 1991), and the novel ρ1 subunit from a human retina cDNA library was cloned in the early 1990s (Cutting et al., 1991). Originally known as (and often still referred to as) GABAC receptors, the second member of this subfamily, ρ2 subunits were cloned from human retina a year later (Cutting et al., 1992) and is also found in brain tissue such as hippocampus, cerebellum and pituitary, with significant abundance (Lopez‐Chavez et al., 2005). A third member of this subfamily, ρ3, has been detected in retina and at lower expression levels in higher brain regions (Boue‐Grabot et al., 1998; Bailey et al., 1999). In the retina, ρ3 subunits are expressed in ganglion neurons, while ρ1 and ρ2 subunits are specifically expressed in bipolar and horizontal cells (Qian and Dowling, 1993; Fletcher et al., 1998; Lopez‐Chavez et al., 2005). ρ1 knockout mice studies have indicated that ρ receptors are present in the superior colliculus (Schlicker et al., 2009). The presence of GABA ρ1 and ρ2 subunits, either homomerically or combined with other GABAA (α1 and γ2) subunits, has also been identified in cultured cerebellar astrocytes. It is proposed that these GABA‐ρ subunits may contribute to the regulation of glial development in the cerebellum (Pétriz et al., 2014). Although GABA‐ρ receptors are mainly expressed in the CNS, they are also found in the peripheral nervous system, for example, in the gastrointestinal tract (Jansen et al., 2000), and sperm cells (Li et al., 2008). Members of this ρ subfamily share more than 70 and 95% amino acid identity and similarity respectively (Figure 3) (Zhang et al., 2001).
Figure 2.
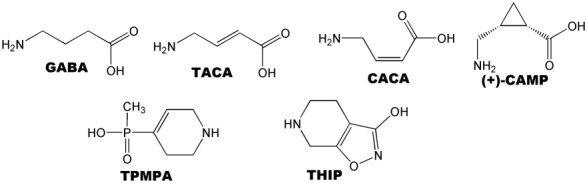
Chemical structures of ligands that selectively distinguish GABA‐ρ1 receptors from GABAA receptors.
Figure 3.
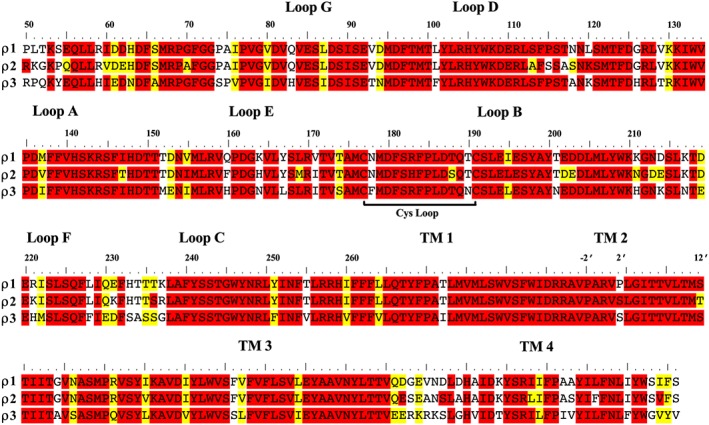
The amino acid sequence alignment of GABA‐ρ1, GABA‐ρ2 and GABA‐ρ3 subunits. Alignments were prepared as previously described (Naffaa et al., 2015). The amino acid sequence of various human GABA‐ρ receptors was obtained from the Universal Protein Resources (http://www.uniprot.org/) (UniProt Consortium, 2013). The UniProt IDs for GABA‐ρ1, GABA‐ρ2 and GABA‐ρ3 subunits are P24046, P28476 and A8MPY1 respectively.
The involvement of GABA‐ρ receptors in a range of physiological processes has been suggested, including the inhibition of ammonia‐induced apoptosis in hippocampal neurons (Yang et al., 2003) and hormone release in the pituitary (Boue‐Grabot et al., 2000). The GABA‐ρ antagonist (1,2,5,6‐tetrahydropyridin‐4‐yl) methylphosphinic acid; TPMPA) (Figure 2) was shown to improve the symptoms of retinitis pigmentosa in rats (Jensen, 2012) and inhibit the development of myopia in chicks (Chebib et al., 2009b). Both GABA‐ρ1 and ‐ρ2 receptors are found in the hippocampus as extrasynaptic receptors activated by GABA through spillover (Alakuijala et al., 2006) and are believed to be involved in paired‐pulse depression of inhibitory postsynaptic currents (Xu et al., 2009). More recently GABA‐ρ2 receptors expressed pre‐synaptically in the spinal dorsal horn have been implicated in pain perception and identified as a novel target for analgesia (Tadavartya et al., 2015). Behavioural pharmacological studies have shown an important role for ρ1 receptors in the sleep‐waking behaviour of rats (Arnaud et al., 2001), learning and memory in chicks and rats (Chebib et al., 2009b; Gibbs and Johnston, 2005), the inhibitory modulation of the olfactory bulb (Chen et al., 2007) and evidence that ρ1 and ρ2 receptors may be important for some specific, in vivo, effects of ethanol (Blednov et al., 2014).
Structure and function of GABA‐ρ1 receptors
Homology models of GABA‐ρ receptors (Harrison and Lummis, 2006b; Abdel‐Halim et al., 2008; Osolodkin et al., 2009; Tai et al., 2009; Naffaa et al., 2015) have been based on the five different LGIC crystal structures, AChBP, ELIC, GLIC, GABAA β3 and GluCl (Brejc et al., 2001; Hilf and Dutzler, 2008; Chen et al., 2010; Hibbs and Gouaux, 2011; Miller and Aricescu, 2014). With the exception of AChBP, these crystal structures are of full ion channels, with varying percentages of residues conserved with GABA‐ρ. AChBP, ELIC and GLIC have limited amino acid conservation with ρ1 (~20% amino acid identity), while for GABAA β3 and GluCl receptors, this is ~40% (Figure 4) (Naffaa et al., 2015).
Figure 4.
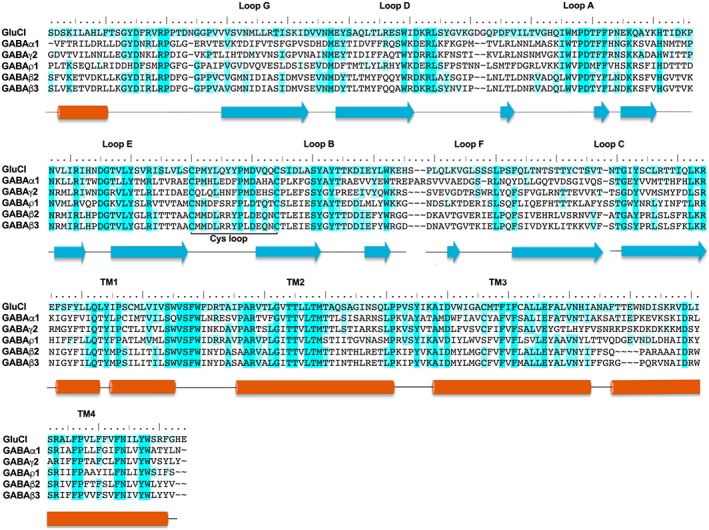
The amino acids sequence alignment of GluCl, GABA α1, GABAγ2, GABA ρ1, GABA β3 and GABA β2 subunits. The different shades of blue highlights show the conserved amino acids between the template ‘GluCl’ and the studied GABA subunits in this review. Dark blue indicates that the residue is conserved between all or most of the subunits, while light blue indicates that residue is conserved or related to another residue in two or three subunits only. Alignments were prepared as previously described (Naffaa et al., 2015). The amino acid sequence of human GABA and GluCl subunits was obtained from the Universal Protein Resources (http://www.uniprot.org/) (UniProt Consortium, 2013) and Protein Data Bank (PDB). UniProtKT ID for GABA α1, GABAγ2, GABAρ1 and GABA β2 subunits are P14867, P18507, P24046 and P47870–1 respectively. PDB ID for GluCl and GABA β3 are 3RIF and 4COF respectively.
The human homopentameric GABAA β3 receptor was the first of the GABAA receptor structures to be resolved (Miller and Aricescu, 2014). The β3 subunit has a relatively high sequence identity to the ρ1 subunit, and it is unlikely to form functional homomeric receptors in vivo (Tretter and Moss, 2008). In GABAA heteromeric receptors, the orthosteric binding site is between the β subunit on the principal (+) side (comprising loops A–C) and an α subunit on the complementary (−) side (comprising loops D–G), as defined in the same manner as shown for GABA‐ρ receptors (Figure 1). The (−) side of the GABA β3 homomeric receptor lacks the key arginine residue that forms a critical salt‐bridge between all α and ρ subunits and the carboxylate of GABA (Naffaa et al., 2015). Therefore, the (+) side of the interface may provide accurate predictions, but the (−) side is lacking residues that form essential interactions with GABA. The homology model based on the GABAA β3 template is not able to fully predict the critical interactions of GABA and its binding mode (Figure 4) (Baumann et al., 2002; Naffaa et al., 2015). In contrast, the ρ1 homology model based on GluCl is in excellent agreement with most previous ρ1 receptor experimental findings. Additionally, GluCl has been co‐crystalized with its endogenous ligand, L‐glutamate, which is similar to GABA in structure and flexibility that also helps to predict GABA interactions (Naffaa et al., 2015).
GABA‐ρ1 agonist binding site
Mutational studies on a number of ρ1 residues equivalent to some previously studied in the GABAA β2 subunit (Amin and Weiss, 1993) led to significant decreases in GABA sensitivity, revealing the importance of residues such as Tyr198, Tyr200, Tyr241, Tyr247 and Thr244 (Figure 5) (Amin and Weiss, 1994; Bormann and Feigenspan, 1995). Mutation of Tyr102 to serine was also found to produce spontaneously active receptors (Torres and Weiss, 2002), and structurally diverse antagonists were found to exhibit different effects at these spontaneously active receptors suggesting differing affinities for the open and closed state of the receptor (Yamamoto et al., 2012b). Other mutational studies have identified Tyr102, Tyr106, Phe138 and Phe240 in GABA‐ρ1 receptors to be major determinants for antagonist selectivity at ρ1 receptors when compared with the GABAA α1β2γ receptors (Figure 4) (Zhang et al., 2008). Mutation of the Tyr102 residue leads to significant changes in GABA activity but does not appear to be involved in stabilizing GABA in the binding site (Figure 5B). Therefore, Tyr102 may play a role in the conformational changes that lead to gating, as has been proposed for the equivalent residue (Phe64) in the GABAA α1 subunit (Szczot et al., 2014), or it may also have a critical role in stabilizing the protein by forming inter‐subunit interactions (Figure 5B) (Miller and Aricescu, 2014; Naffaa et al., 2015).
Figure 5.
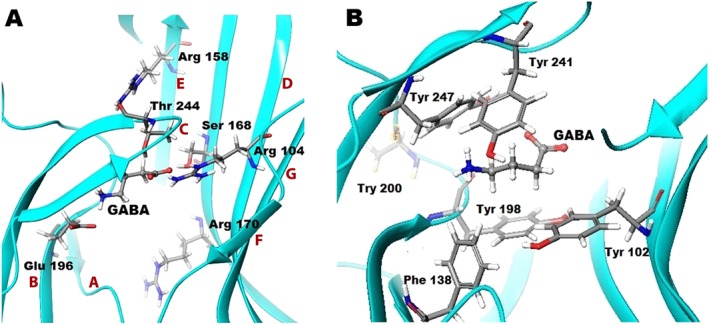
Molecular basis of GABA bound in the orthosteric binding site of GABA ρ1 homology model based on GluCl. (A) GABA bound GABA‐ρ1 homology model based on GluCl, showing loops A–G (labelled in red), and H‐bond interactions formed between GABA and Thr244 and Ser168, and salt bridges with Arg104 and Glu196. Arg158 and Arg170 are two important residues for protein stability either by forming interaction within the same subunit or between neighbouring subunits. (B) GABA and aromatic residues (Tyr102, Tyr198, Tyr241 and Tyr247) forming the aromatic box that stabilizes GABA in the binding site during the channel gating. The Phe138 residue may form important interactions as it has a benzene ring system that partially oriented to both the GABA binding site and the adjacent subunit.
In a mapping study on loops A, E and F (Figure 5A) (Sedelnikova et al., 2005), only residues Asp136 in loop A, Leu166, Ser168 and Arg170 in loop E and Gln226 in loop F were noted to significantly decrease GABA sensitivity when cysteine was introduced at these sites. Interestingly, cysteine mutation of Leu169, located in loop E but orientated away from the binding site and not thought to be involved in GABA binding, resulted in a several‐fold increase in the potency of GABA. Residues at the 169 position may be involved in the conformational changes of channel gating or form interactions with residues in the same subunit, with the interactions formed by the cysteine leading to greater stability. Trp133, Pro135 and Phe139 in loop A and Gln160 in loop E, when individually mutated to cysteine resulted in non‐functional receptors. As these residues are also some distance away from the GABA binding site, these residues may have critical roles in protein stability through intra‐subunit interactions rather than a direct effect on GABA binding. The same study showed that when cysteine replaces Gln160 in loop E, the receptors become spontaneously active, suggesting that this residue is involved in the conformational changes that lead to gating (Sedelnikova et al., 2005).
Mutations of residues in the binding site within a distance of 7 Å of the carboxylate group of GABA identified Arg104 and Ser168 (Figure 5A) (Harrison and Lummis, 2006a) to be essential for GABA activity and have been predicted by modelling studies to form a salt bridge and a H‐bond interaction, respectively, with GABA (Harrison and Lummis, 2006b; Abdel‐Halim et al., 2008; Melis et al., 2008; Osolodkin et al., 2009; Naffaa et al., 2015). However, Arg158 and Arg170 residues (Figure 5A), which are also in close proximity to the carboxylate group of GABA in the binding site and have been found to be critical for receptor activation by GABA, are predicted by modelling not to be involved in GABA binding but important for protein stability (Harrison and Lummis, 2006b; Abdel‐Halim et al., 2008; Osolodkin et al., 2009; Naffaa et al., 2015).
Similar to other LGIC receptors, the orthosteric binding site of GABA‐ρ1 receptors contains many aromatic residues. Mutational studies have demonstrated the significance of the aromatic residues Tyr198, Tyr241 and Tyr247 (Figure 5B). Mutation of these tyrosine residues to the aromatic amino acid phenylalanine leads to only a minor effect on GABA response whereas mutation to the aliphatic serine leads to a significant decrease (Lummis et al., 2012). These residues are predicted by modelling studies to have their aromatic group oriented toward the binding site and to form an aromatic box surrounding the ammonium group of GABA (Harrison and Lummis, 2006b; Abdel‐Halim et al., 2008; Osolodkin et al., 2009; Naffaa et al., 2015). Tyr200 (Figure 5B) was also found to be important for GABA activity. However, according to modelling studies, this residue has its functional group oriented away from the orthosteric binding site, indicating that it is unlikely to interact directly with GABA (Lummis et al., 2012; Naffaa et al., 2015).
Mutation of the ρ1 Thr244 residue in loop C, which has its hydroxyl group oriented toward the GABA binding site (Figure 5A), found that only the T244S mutation resulted in functional receptors, which were 35‐fold less sensitive to GABA (Amin and Weiss, 1994). A range of agonists studied at the T244S mutant receptor demonstrated many‐fold decreases in potency, while antagonist activity remained unaffected by the mutation (Yamamoto et al., 2012a). Thr244 is proposed to be essential for the formation of an H‐bond with agonists initiating conformational changes through movement of Loop C to open the channel (Naffaa et al., 2016).
GABA‐ρ1 channel
The structure of the TM2 domains of most LGIC receptors is highly conserved across species and subunits, with variation at only a few sites in the pore. The structures of the ρ1 and ρ2 subunits differ at two sites within TM2 regions only, Pro294 (−2′) and Ser304 (12′) in ρ1 are Ser290 (2′) and Thr300 (12′), respectively, in ρ2 subunits (Figure 6). Picrotoxinin is a pore‐blocker at many ligand‐gated ion channels (Akaike et al., 1985; Jarboe et al., 1968; Pribilla et al., 1992). It binds to a unique site in TM2 (Gurley et al., 1995; Wang et al., 1995), inhibiting the chloride flux through the anionic ligand‐gated channels (Inoue and Akaike, 1988; Etter et al., 1999).
Figure 6.
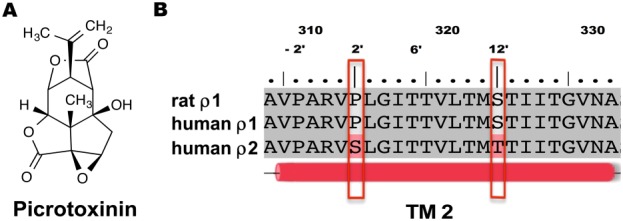
(A) Chemical structure of picrotoxinin. (B) The amino acids sequence alignment of the second TM domain of human GABA ρ1 and ρ2 subunits against the GABA rat ρ1 subunit.
Picrotoxinin has different effects at ρ1 and ρ2 homomeric receptors and also at native GABA‐ρ receptors from different species (Qian and Dowling, 1993; Lukasiewicz and Werblin, 1994; Zhang et al., 1995). In GABA and glycine channels, the residues located at the 2′ positions play significant roles in the picrotoxinin activity (Lynch et al., 1995; Shan et al., 2001; Sedelnikova et al., 2006). Introduction of mutations at Pro294 (−2′) and Pro298 (2′) (Figure 6) of human ρ1 homomeric receptors expressed in Xenopus oocytes changed not only picrotoxinin sensitivity but also the response of the mutant receptors to the agonists and partial agonists (Carland et al., 2004). Rat GABA‐ρ2 receptors are relatively insensitive to picrotoxinin (Feigenspan et al., 1993). Mutation of Thr314 (6′) in the TM2 of the rat ρ1 subunit to methionine (Met299), the equivalent residue in the rat ρ2 subunit, resulted in mutant receptors with smaller chloride currents (Figure 6). Co‐expression of rat WT ρ1 and rat T314 M ρ1 mutant subunits resulted in receptors with picrotoxinin sensitivity that is similar to co‐expressed rat WT ρ1 and ρ2 subunits, which led to the suggestion of native pseudo‐heteromeric GABAA ρ receptors composed of ρ1 and ρ2 subunits in rat (Zhang et al., 1995). In the ρ2 channel, the serine residue at the 2′ position is predicted by homology models to form multiple hydrogen bonds and hydrophobic interactions with picrotoxinin, whereas the homologous proline residues of ρ1 channels are predicted to form only hydrophobic contacts with picrotoxinin (Naffaa and Samad, 2016).
The Trp328 in TM3 of ρ1 receptors is important for sensitivity to pentobarbital. Although ρ1 wild‐type receptors are generally insensitive to pentobarbital, when Trp328 is mutated to various hydrophobic residues, these ρ1 mutant receptors become sensitive to pentobarbital. This is possibly due to a change in structure that results from substitution of Trp328 by smaller, hydrophobic residues exposing the gate of the channels to the extracellular membrane components or affording a binding cavity for pentobarbital (Amin, 1999). Moreover, mutation of Ile323 in TM2 to serine also renders human homomeric ρ1 mutant receptors sensitive to barbiturates, which is believed to be due to allosteric rather than direct effects (Belelli et al., 1999). Wild‐type GABA‐ρ receptors are also insensitive to modulation by benzodiazepines. However, the double mutation of I307S/W328 M in ρ1 subunits renders homomeric mutant receptors sensitive to micromolar concentrations of diazepam (Walters et al., 2000). These ρ1 I307S/W328 M mutant GABA receptors still resemble ρ1 WT receptors in terms of the pharmacological properties of agonists and antagonists, demonstrating that the receptors retain GABA‐ρ character (Hall et al., 2014).
Many divalent cations such as Zn2+, Ni2+ and Cu2+ have modulatory actions at ρ1 receptors (Calvo et al., 1994; Dong and Werblin, 1995). The zinc cation, which is present in the synaptic terminal of photoreceptors (Wu et al., 1993), is able to reduce GABA responses at ρ1 receptors with an effect that depends on the extracellular pH. Because of this pH effect, the His156 residue in the orthosteric binding site was suggested to have a direct role in zinc modulation. Introduction of a tyrosine at this site leads to receptors insensitive to divalent cations such as Zn2+. However, other pharmacological and electrophysiological properties were the consistent with those of wild‐type receptors, supporting the involvement of this residue in the modulation by Zn2+ (Calvo et al., 1994; Wang et al., 1995).
Ligand selectivity for GABA‐ρ receptors and structure activity relationships
Structure and conformations of GABA
GABA is a low molecular weight, zwitterionic ligand that has three rotatable carbon–carbon bonds, which afford flexibility and the ability to adopt many different conformations. The flexible rotation about C2–C3 and C3–C4 bonds allows GABA to exist in a range of low‐energy conformations. Because of the variation in binding residues and sensitivities of specific subunit combinations to GABA, the conformational flexibility is critical for its biological activity at different receptors sub‐types (Figure 2, Table 1). It is believed that GABA adopts different conformations at the orthosteric binding sites of different GABA receptor sub‐types (Crittenden et al., 2005a,c; Majumdar and Guha, 1988; Ottosson et al., 2014). However, the question of the conformation(s) of GABA in the binding site has not yet been fully answered, and there is currently no evidence to support whether GABA is able to activate a receptor subtype in only one or more than one conformation. Indeed, this conformational flexibility may be an important factor in the ability of GABA to activate receptors.
Table 1.
The activities of the ligands at various GABA‐ρ, GABAA and GABAB receptors.
| GABAA‐ρ1 | GABAA‐ρ2 | GABAA‐ρ3 | GABAA (α1β2γ2L) | GABAB | |
|---|---|---|---|---|---|
| GABA | EC50 = 2.5μMa | EC50 = 2.5μMb | EC50 = 2.5μMb | EC50 = 21.1μMc | EC50 = 1.7 μMc |
| TACA | EC50 = 0.44μMd | EC50 = 0.3μMa | EC50 = 3.8 μMe | EC50 = 133 μMf | Inactive |
| CACA |
EC50 = 74 μMg
(70% efficacy) |
EC50 = 70 μMg | EC50 = 139 μMe | Inactive | Inactive |
| (+)‐CAMP | EC50 = 40 μMg | EC50 = 17 μMg | EC50 = 28 μMg | Weak antagonist ≥ 1mMg | Inactive |
| TPMPA | KB = 2.1 μMh | KB = 15.6 μMi | KB = 10.2 μMe | Weak Antagonist KB = 320 μMh | Weak agonist EC50 ~ 500 μMh |
| THIP | Ki = 25 μMk | ‐ | IC50 = 10.2 μMe | EC50 = 355 μMl | Inactive |
| Picrotoxinin | IC50 = 48μMm | IC50 = 4.8μMm | ‐ | IC50 = 5 μMn | Inactive |
| I4AA | IC50 = 1.45 μMi | IC50 = 3.18 μMj | IC50 = 12.6 μMi | EC50 = 138 μMa | ‐ |
| 2‐MeTACA | Kb = 45 μMi | Partial agonist KB = 101 μM, IMAX = 34%)i | Inactivee | ||
| (‐)‐TAMP | EC50 = 9 μM, IMAX = 39%g | EC50 = 55 μM, IMAX = 55%g | Antagonistg | EC50 = 50 μM, IMAX = 51%g | Inactive |
| (R)‐ACPBPA | KB = 60 μMc | KB = 6 μMo | ‐ | Inactivec | Inactivec |
| (S)‐ACPBPA | KB = 5 μMc | KB = 11 μMo | ‐ | Inactivec | Inactivec |
| (S)‐2MeGABA | EC50 = 65μMp | EC50 = 20μMp | EC50 = 25μMq | ‐ | ‐ |
| (R)‐2MeGABA | IC50 = 16μMp | IC50 = 36μMp | ‐ | ‐ | ‐ |
| (+)‐ACPECA | EC50 = 135μMp | EC50 = 60μMp | EC50 = 110μMp | ‐ | ‐ |
| (−)‐ACPECA | IC50 = 300 μM Inhibit 25% of GABA EC50 p | ‐ | IC50 = 300 μM Inhibit 25% of GABA (3μM)p | ‐ | ‐ |
| (+)‐TACP | EC50 = 2.7 μM, IMAX = 83%q | EC50 = 1.5 μM, IMAX = 85%q | EC50 = 2.7 μM, IMAX = 27%q | ‐ | ‐ |
| (−)‐TACP(100 μM) | 2% activation (56% blockade of 1 μM GABA)q | 4.5% activation (62% blockade of 1 μM GABA)q | ‐ | ‐ | ‐ |
| Aza‐THIP | Ki = 31 μMk | ‐ | ‐ | Ki > 100 μMk | ‐ |
| Thio‐THIP | Ki = 91 μMk | ‐ | ‐ | Ki = 52 μMk | ‐ |
| 3‐AMOHP | IC50 = 20 μMr | IC50 = 60 μMr | ‐ | Inactiveq | Weak agonistr |
| 3‐GOHP | IC50 = 30 μMr | IC50 = 50 μMq | ‐ | Inactiver | Inactiver |
| 3‐AOHP | Inactiver | ‐ | ‐ | Inactiver | Inactiver |
| 4‐ACPAM | IC50 = 10 μMs | ‐ | ‐ | IC50 = 23 μMs | Inactives |
| 4‐ACPHA | IC50 = 13 μMs | ‐ | ‐ | Inactives | Inactives |
Note: KB = the molar concentration of a competitive antagonist that would occupy 50% of the receptors at equilibrium; Ki = the molar concentration of a competing ligand that would occupy 50% of the receptors if no radioligand was present; IC50 = the molar concentration of an agonist or antagonist which produces 50% of its maximum possible inhibition in a functional assay. 2‐MeTACA, trans‐2‐methyl‐4‐aminocrotonic acid; 3‐AMOHP, 3‐(aminomethyl)‐1‐oxo‐1‐hydroxy‐phospholane; 3‐AOHP, 3‐(amino)‐1‐oxo‐1‐hydroxy‐phospholane; 3‐GOHP, 3‐(guanidino)‐1‐oxo‐1‐hydroxy‐phospholane; 4‐ACPAM, 4‐aminocyclopent‐1‐enecarboxamide; 4‐ACPHA, 4‐amino‐N‐hydroxycyclopent‐1‐enecarboxamide.
Data from Kusama et al. (1993a).
Data from Kusama et al. (1993b).
Data from Kumar et al. (2008).
Data from Chebib et al. (1997).
Data from Vien et al. (2002).
Data from Ragozzino (1996).
Data from Duke et al. (2000).
Data from Murata (1996).
Data from Chebib et al. (1998).
Unpublished data Nafaa et al
Data from Krehan et al. (2003a).
Data from Ebert (1997).
Data from Wang et al. (1995).
Data from Xu (1995).
Data from Kim et al. (2008).
Data from Crittenden et al. (2006).
Data from Chebib et al. (2001).
Data from Gavande et al. (2011).
Data from Locock et al. (2013).
Structure activity relationships of GABA‐ρ receptors
The synthesis of conformationally restricted GABA analogues led to the initial identification of GABA‐ρ receptors using ligands that are selective at this subfamily, relative to heteromeric GABAA receptors (Johnston et al., 1975). The conformationally restricted GABA analogues, CACA and trans‐3‐aminocrotonic acid (TACA) possess an unsaturated bond at C2–C3 and are therefore only freely rotatable at the C3‐C4 bond. The cis‐isomer (CACA, Figure 2, Table 1) offers a folded conformation and is a potent and selective GABA receptor partial agonist. CACA depressed firing of cat spinal neurons with bicuculline‐insensitive properties, and this depressant effect, not reproduced by baclofen, suggested pharmacologically distinct GABA receptors in the mammalian CNS, which were not GABAA or GABAB (Johnston, 1996). CACA is inactive at heteromeric GABAA receptors but is a partial agonist (70% efficacy) at GABA‐ρ1 recombinant homomeric receptors. The trans‐isomer (TACA, Figure 2, Table 1) exhibits an extended conformation and is a potent agonist. TACA has greater potency than GABA at ρ receptors but is non‐selective, also acting as an agonist at heteromeric GABAA receptors (Woodward et al., 1993; Kusama et al., 1993a; Kerr and Ong, 1995).
The restricted cyclic analogue, (+)‐cis‐2‐(aminomethyl)cyclopropane carboxylic acid (CAMP) (Figure 2), demonstrates greater selectivity for GABA‐ρ receptors than CACA, which also inhibits GABA reuptake transporters. Interestingly, (+)‐ and (−)‐CAMP show opposite pharmacological actions, with (+)‐CAMP being a full agonist and (−)‐CAMP being a weak antagonist on both ρ1 and ρ2 receptors (Duke et al., 2000). Both (±)‐CAMP and (±)‐trans‐2‐(aminomethyl) cyclopropane carboxylic acid (TAMP) (Figure 7, Table 1) have been shown to differentiate between subtypes of GABA‐ρ receptors (Table 1). (±)‐CAMP is a partial agonist at ρ3 receptors while (±)‐TAMP is an antagonist (Vien et al., 2002). The area of steric interaction encountered by the antagonists (−)‐TACP (Figure 8, Table 1) and (−)‐CAMP (Figure 2, Table 1) is thought to be the same area of the binding site (Chebib et al., 2001) and may be responsible for the antagonist action of these ligands.
Figure 7.
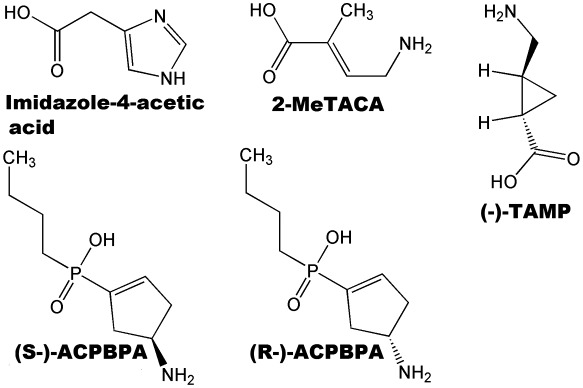
Chemical structures of ligands demonstrate subunit selectivity between homomeric receptors composed of different GABA ρ subunits.
Figure 8.
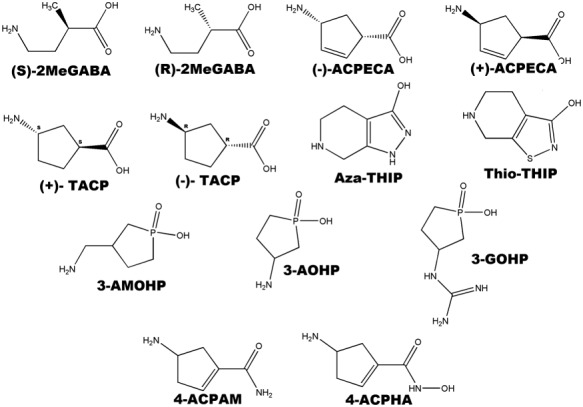
Chemical structures of ligands used to study structure activity relationships at GABA‐ρ receptors.
(S)‐2MeGABA (Figure 7) and (+)‐4‐aminocyclopent‐2‐ene‐1‐carboxylic acid [(+)‐ACPECA] (Figure 8, Table 1) are full agonists at ρ1 and ρ2 receptors with preferred orientations that place the bulk of the molecule behind the plane. Their enantiomeric pairs, (R)‐2MeGABA and (−)‐ACPECA, are antagonists and orientate such that the bulk of the molecule is in front of the plane (Figure 8) (Crittenden et al., 2006). This difference in orientation is thought to contribute to their opposing pharmacological activities. This finding is in accordance with the pharmacological profile of GABA analogues at the ρ1 receptor where substitution behind the double bond leads to agonist activity while substitution in front of the double bond results in antagonist activity (Crittenden et al., 2005b). These results are also consistent with the observed antagonist activity of (−)‐CAMP. Substitution in the front of the plane may lead to steric interactions that prevent the movement of loop C required for receptor activation, thus resulting in antagonist activity (Abdel‐Halim et al., 2008; Naffaa and Samad, 2016).
4,5,6,7‐Tetrahydroisoxazolo[5,4‐c] pyridin‐3‐ol (THIP) (Gaboxadol, Figure 2, Table 1), a relatively rigid analogue of GABA, is a potent antagonist at all GABA‐ρ receptor subtypes (Johnston et al., 2003) but acts as a partial agonist at synaptic GABAA receptors and as a potent super‐agonist at extra‐synaptic α4β3δ receptors (Krogsgaard‐Larsen et al., 2004). THIP has demonstrated therapeutic effects on the area of sleep and analgesia (Krogsgaard‐Larsen et al., 2004). The THIP derivative, aza‐THIP (Figure 8, Table 1), is approximately equipotent to THIP as an antagonist at GABA‐ρ1 receptors but is more selective for GABA‐ρ receptors than THIP, with negligible activity at heteromeric GABAA receptors. Thio‐THIP (Figure 8, Table 1) in which the isoxazole oxygen is replaced by a bulkier sulfur atom has threefold lower potency than THIP at GABA‐ρ receptors, which is likely due to increased steric bulk of the sulfur atom (Krehan et al., 2003a).
The interaction between the acidic moiety of ligands and the GABA‐ρ receptor proteins is a common interaction among many GABA ligands. Ligands containing different acidic groups including carboxylic, phosphinic, methylphosphinic, phosphonic and seleninic acids have been investigated for their activity at GABA‐ρ receptor (Chebib et al., 1997; Krehan et al., 2003b; Kumar et al., 2008; Chebib et al., 2009a,b; Gavande et al., 2011). Additionally, replacement of the carboxylic acid moiety with either a hydroxamic acid (4‐amino‐N‐hydroxycyclopent‐1‐enecarboxamide) or amide group (4‐aminocyclopent‐1‐enecarboxamide) (Figure 8, Table 1) results in antagonists of moderate potency with increased selectivity for GABA‐ρ receptors. This indicates that a zwitterionic structure is not essential for strong ligand–receptor interactions (Locock et al., 2013) but is required for activation of the receptor. The proposed antagonist binding site as determined by homology modelling has been predicted to be larger in the apo conformation of the GABA‐ρ receptor than in the open conformation of the receptor, allowing the antagonists to be substituted with larger acidic moieties (Abdel‐Halim et al., 2008). This has been confirmed through a number of studies where changing the carboxylic acid to a larger acid such as alkyl phosphinic, phosphonic or seleninic acid leads to the conversion of agonist to antagonist activity (Chebib et al., 1997; Krehan et al., 2003b; Kumar et al., 2008).
Modification of the amine moiety of amino‐substituted phospholanes has been used to investigate the role of the basic functional group (−NH2). 3‐(aminomethyl)‐1‐oxo‐1‐hydroxy‐phospholane (Figure 8, Table 1) is a potent antagonist at GABA‐ρ1 receptors but inactive at GABAA (Gavande et al., 2011). Shortening the carbon backbone by one carbon, 3‐(amino)‐1‐oxo‐1‐hydroxy‐phospholane (Table 1), led to loss of activity across all GABA receptors. This demonstrates the importance of the distance between the acid and amine groups in determining the ligand affinity, potency and selectivity (Chang et al., 2000; Chebib et al., 2009a; Gavande et al., 2011). 3‐(Guanidino)‐1‐oxo‐1‐hydroxy‐phospholane (Figure 8 Table 1), with a guanidino group replacing the amine shows greater selectivity for GABA‐ρ receptors with reduced inhibition at GABAA receptors (Gavande et al., 2011). Replacement of the amine group by a carboxylic acid or hydroxyl group led to total loss of activity at mM concentrations at all GABA receptors. This indicates that a basic moiety such as an amine or guanidine group is essential for the affinity and potency of GABA (Gavande et al., 2011).
Subunit‐selectivity of ligands at GABA‐ρ receptors
The GABA‐ρ1 receptors are the most studied of the GABA‐ρ subfamily, while ρ2 and ρ3 receptors have been much less studied. Although the three subunits are highly similar in their amino acid sequences across most regions, there are some important differences between them. However, a detailed study of these differences has not yet been undertaken.
Some ligands that bind in the orthosteric binding site can differentiate between different GABA‐ρ subunits. Imidazole‐4‐acetic acid (I4AA) (Figure 7, Table 1) is a partial agonist at both ρ1 and ρ2 homomeric receptors expressed in oocytes. This ligand is a weak, low‐efficacy partial agonist (EC50 = 60 μM, Im = 8%) at ρ1 receptors, while it is potent and moderately efficacious (EC50 = 3 μM, Im = 40%) at ρ2 receptors (Kusama et al., 1993b; Chebib et al., 1998; Madsen et al., 2007). As a partial agonist at GABA‐ρ receptors, I4AA also acts as an antagonist and shows a similar potency at both ρ subtypes (Table 1). Consistent with its weaker agonist potency and efficacy, I4AA is more efficacious as an antagonist at GABA‐ρ1 receptors, inhibiting 97 and 20% of GABA responses at GABA‐ρ1 and GABA‐ρ2 homomeric receptors respectively (MN Nafaa et al., unpublished data).
trans‐2‐Methyl‐4‐aminocrotonic acid (Figure 7, Table 1) is a moderate antagonist at ρ1 receptors, a partial agonist at ρ2 receptors (Chebib et al., 1998), but inactive at ρ3 receptors (Vien et al., 2002). However, trans‐2‐aminomethylcyclopropane carboxylic acid (−)‐TAMP (Table 1) is a partial agonist at both human ρ1 and ρ2 receptors but an antagonist at ρ3 receptors (Vien et al., 2002), although it should be noted that the only data available for (−)‐TAMP at ρ3 receptors is from rat subunits. (S)‐4‐Amino‐2‐methylbutanoic acid ((S)‐2MeGABA) (Figure 8) is an agonist at ρ1 and ρ2 receptors and a partial agonist (90%) at ρ3 receptors. The stereoisomer, (R)‐4‐amino‐2‐methylbutanoic acid ((R)‐2MeGABA) (Figure 8), is an antagonist at all three receptor subtypes. Furthermore, the stereoisomers showed selective potencies at ρ1 and ρ2 receptors as (S)‐2MeGABA is a more potent agonist at ρ2 whereas (R)‐2MeGABA is a more potent antagonist at ρ1 (Crittenden et al., 2006).
The achiral antagonist, TPMPA (Table 1), is approximately equipotent at all GABA‐ρ receptor subtypes. However, (S)‐ and (R)‐aminocyclopentenyl‐butylphosphinic acids ((S)‐/(R)‐ACPBPA) (Figure 8, Table 1) are potent stereoisomeric antagonists with differential selectivity at ρ1 and ρ2 receptors. (R)‐ACPBPA, the only antagonist known at this time to be selective for ρ2 over ρ1 receptors, shows 10‐fold selectivity at ρ2 receptors, while (S)‐ACPBPA is a weakly selective for ρ1 over ρ2 receptors showing twofold selectivity (Kim et al., 2008).
Conclusions
The testing of conformationally restricted GABA analogues led to the initial discovery of a distinct class of GABA receptors that did not fit the pharmacology of the sub‐types known at that time. Following the naming strategy, these new receptors were termed GABAC and shown later to be composed of ρ subunits that were first cloned from the retina (ρ indicating retina) and later shown to form homopentameric LGICs. Although ρ subunits showed clear sequence homology to GABAA subunits, both the structure and pharmacology of these GABAC receptors were found to be much simpler than the complex heteromeric GABAA receptors with multiple subunit combinations. Many initial studies on these receptors focused on their physiological role in the retina, and many in the ophthalmic physiology field refer to the receptors simply as GABA‐ρ, indicating that they are GABA‐sensitive receptors composed of ρ subunits. The potential confusion through the identification of this class of receptor as either GABAC or GABA‐ρ led the IUPHAR to review the nomenclature of GABA receptors (Olsen and Sieghart, 2008; Alexander et al., 2015). It was ‘especially recommended that the name GABAC receptor should not be used as the sole name for the ρ‐receptors in an article including, especially, the title and abstract’ (Olsen and Sieghart, 2008). Instead, GABAC/GABA‐ρ receptors should be referred to as a sub‐class of GABAA receptor ‘GABAA‐ρ’. Rather than removing the confusion around the identity of these homomeric GABA LGICs, the classification of GABAA‐ρ has introduced a third term for these receptors, one that is in fact used by few researchers active in the field. This has further increased the possibility for confusion, complicating literature searches, and suggests that it may be time for a further review of GABA receptor nomenclature.
Despite confusion regarding the nomenclature, it is the lack of a GABA‐ρ receptor crystal structure that has significantly hampered efforts to design selective ligands. However, the availability of X‐ray crystal structures of related LGIC receptors has led to the development of GABA‐ρ homology models that have been used to study interactions between the receptor and ligands. The development of ligands that are selective either for GABA‐ρ over GABAA/B receptors or between different ρ subunit homomeric receptors has led to some understanding of the different structural features required for activity at the different receptors. It is particularly difficult to determine the binding conformation adopted in the receptor, as many ligands are able to exist in a number of low‐energy conformations. The use of conformationally restricted GABA analogues and mutational studies combined with computational docking studies using homology models has facilitated understanding binding interactions and the conformational changes that may be required for receptors activation. However, there has not yet been any detailed study examining differences in interactions between conformationally restricted ligands at different ρ subunits.
Although the physiological function of GABA‐ρ receptors is not fully understood, the selective distribution of ρ subunits indicates that GABA‐ρ are specifically involved in visual image processing. The limited distribution and lower abundance of GABA‐ρ receptors relative to heteromeric GABAA receptors suggests that GABA‐ρ ligands are promising leads for developing agents with improved selectivity and therefore reduced unwanted effects. Further studies are encouraged to understand that the binding interactions and development of ligands with improved selectivity will facilitate greater knowledge of GABA‐ρ receptors at both the molecular and physiological levels.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
The authors would like to acknowledge their co‐workers and students who have contributed to this work.
Naffaa, M. M. , Hung, S. , Chebib, M. , Johnston, G. A. R. , and Hanrahan, J. R. (2017) GABA‐ρ receptors: distinctive functions and molecular pharmacology. British Journal of Pharmacology, 174: 1881–1894. doi: 10.1111/bph.13768.
References
- Abdel‐Halim H, Hanrahan JR, Hibbs DE, Johnston GAR, Chebib M (2008). A molecular basis for agonist and antagonist actions at GABAC receptors. Chem Biol Drug Des 71: 306–327. [DOI] [PubMed] [Google Scholar]
- Akaike N, Hattori K, Oomura Y, Carpenter DO (1985). Bicuculline and picrotoxin block γ‐aminobutyric acid‐gated Cl− conductance by different mechanisms. Experientia 41: 70–71. [DOI] [PubMed] [Google Scholar]
- Alakuijala A, Alakuijala J, Pasternack M (2006). Evidence for a functional role of GABA receptors in the rat mature hippocampus. Eur J Neurosci 23: 514–520. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Peters JA, Kelly E, Marrion N, Benson HE, Faccenda E et al. (2015). The concise guide to PHARMACOLOGY 2015/16: Ligand‐gated ion channels. Br J Pharmacol 172: 5870–5903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin J (1999). A single hydrophobic residue confers barbiturate sensitivity to γ‐aminobutyric acid type C receptor. Mol Pharmacol 55: 411–423. [PubMed] [Google Scholar]
- Amin J, Weiss DS (1993). GABAA receptor needs two homologous domains of the beta‐subunit for activation by GABA but not by pentobarbital. Nature 366: 565–569. [DOI] [PubMed] [Google Scholar]
- Amin J, Weiss DS (1994). Homomeric rho 1 GABA channels: activation properties and domains. Receptors Channels 2: 227–236. [PubMed] [Google Scholar]
- Arnaud C, Gauthier P, Gottesmann C (2001). Study of a GABAC receptor antagonist on sleep‐waking behavior in rats. Psychopharmacology (Berl) 154: 415–419. [DOI] [PubMed] [Google Scholar]
- Baenziger JE, Corringer PJ (2011). 3D structure and allosteric modulation of the transmembrane domain of pentameric ligand‐gated ion channels. Neuropharmacology 60: 116–125. [DOI] [PubMed] [Google Scholar]
- Bailey ME, Albrecht BE, Johnson KJ, Darlison MG (1999). Genetic linkage and radiation hybrid mapping of the three human GABAC receptor ρ subunit genes: GABRR1, GABRR2 and GABRR3. Biochim Biophys Acta 1447: 307–312. [DOI] [PubMed] [Google Scholar]
- Baumann SW, Baur R, Sigel E (2002). Forced subunit assembly in α1β2γ2 GABAA receptors. Insight into the absolute arrangement. J Biol Chem 277: 46020–46025. [DOI] [PubMed] [Google Scholar]
- Belelli D, Pau D, Cabras G, Peters JA, Lambert JJ (1999). A single amino acid confers barbiturate sensitivity upon the GABA ρ1 receptor. Br J Pharmacol 127: 601–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Benavidez JM, Black M, Leiter CR, Osterndorff‐Kahanek E, Johnson D et al. (2014). GABAA receptors containing ρ1 subunits contribute to in vivo effects of ethanol in mice. PLoS One 9: e85525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bormann J (2000). The 'ABC' of GABA receptors. Trends Pharmacol Sci 21: 16–19. [DOI] [PubMed] [Google Scholar]
- Bormann J, Feigenspan A (1995). GABAC receptors. Trends Neurosci 18: 515–519. [DOI] [PubMed] [Google Scholar]
- Boue‐Grabot E, Roudbaraki M, Bascles L, Tramu G, Bloch B, Garret M (1998). Expression of GABA receptor ρ subunits in rat brain. J Neurochem 70: 899–907. [DOI] [PubMed] [Google Scholar]
- Boue‐Grabot E, Taupignon A, Tramu G, Garret M (2000). Molecular and electrophysiological evidence for a GABAC receptor in thyrotropin‐secreting cells. Endocrinology 141: 1627–1632. [DOI] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB et al. (2001). Crystal structure of an ACh‐binding protein reveals the ligand‐binding domain of nicotinic receptors. Nature 411: 269–276. [DOI] [PubMed] [Google Scholar]
- Calvo DJ, Vazquez AE, Miledi R (1994). Cationic modulation of ρ1 type γ‐aminobutyrate receptors expressed in Xenopus oocytes. Proc Natl Acad Sci U S A 91: 12725–12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carland JE, Moore AM, Hanrahan JR, Mewett KN, Duke RK, Johnston GAR et al. (2004). Mutations of the 2' proline in the M2 domain of the human GABAC ρ1 subunit alter agonist responses. Neuropharmacology 46: 770–781. [DOI] [PubMed] [Google Scholar]
- Chang Y, Amin J, Weiss DS (1995). Zinc is a mixed antagonist of homomeric rho 1 gamma‐aminobutyric acid‐activated channels. Mol Pharmacol 47: 595–602. [PubMed] [Google Scholar]
- Chang Y, Covey DF, Weiss DS (2000). Correlation of the apparent affinities and efficacies of γ‐aminobutyric acidC receptor agonists. Mol Pharmacol 58: 1375–1380. [DOI] [PubMed] [Google Scholar]
- Chebib M, Duke RK, Allan RD, Johnston GAR (2001). The effects of cyclopentane and cyclopentene analogues of GABA at recombinant GABAC receptors. Eur J Pharmacol 430: 185–192. [DOI] [PubMed] [Google Scholar]
- Chebib M, Gavande N, Wong KY, Park A, Premoli I, Mewett KN et al. (2009a). Guanidino acids act as ρ1 GABAC receptor antagonists. Neurochem Res 34: 1704–1711. [DOI] [PubMed] [Google Scholar]
- Chebib M, Hinton T, Schmid KL, Brinkworth D, Qian H, Matos S et al. (2009b). Novel, potent, and selective GABAC antagonists inhibit myopia development and facilitate learning and memory. J Pharmacol Exp Ther 328: 448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chebib M, Mewett KN, Johnston GAR (1998). GABAC receptor antagonists differentiate between human ρ1 and ρ2 receptors expressed in Xenopus oocytes. Eur J Pharmacol 357: 227–234. [DOI] [PubMed] [Google Scholar]
- Chebib M, Vandenberg RJ, Froestl W, Johnston GAR (1997). Unsaturated phosphinic analogs of γ‐aminobutyric acid as GABAC receptor antagonists. Eur J Pharmacol 329: 223–229. [PubMed] [Google Scholar]
- Chen Q, Cheng MH, Xu Y, Tang P (2010). Anesthetic binding in a pentameric ligand‐gated ion channel: GLIC. Biophys J 99: 1801–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhou D, Zhou K, Ren Y, Dai W, Xu M et al. (2007). Study on olfactory function in GABAC receptor/channel ρ1 subunit knockout mice. Neurosci Lett 427: 10–15. [DOI] [PubMed] [Google Scholar]
- Connolly CN, Krishek BJ, McDonald BJ, Smart TG, Moss SJ (1996). Assembly and cell surface expression of heteromeric and homomeric γ‐aminobutyric acid type A receptors. J Biol Chem 271: 89–96. [DOI] [PubMed] [Google Scholar]
- Corringer PJ, Le Novere N, Changeux JP (2000). Nicotinic receptors at the amino acid level. Annu Rev Pharmacol Toxicol 40: 431–458. [DOI] [PubMed] [Google Scholar]
- Crittenden DL, Chebib M, Jordan MJ (2005a). Stabilization of zwitterions in solution: GABA analogues. J Phys Chem A 109: 4195–4201. [DOI] [PubMed] [Google Scholar]
- Crittenden DL, Chebib M, Jordan MJT (2005b). A quantitative structure‐activity relationship investigation into agonist binding at GABAC receptors. J Mol Struct (THEOCHEM) 755: 81–89. [Google Scholar]
- Crittenden DL, Kumar RJ, Hanrahan JR, Chebib M, Jordan MJT (2005c). Stabilization of zwitterions in solution: phosphinic and phosphonic acid GABA analogues. J Phys Chem A 109: 8398–8409. [DOI] [PubMed] [Google Scholar]
- Crittenden DL, Park A, Qiu J, Silverman RB, Duke RK, Johnston GAR et al. (2006). Enantiomers of cis‐constrained and flexible 2‐substituted GABA analogues exert opposite effects at recombinant GABA(C) receptors. Bioorg Med Chem 14: 447–455. [DOI] [PubMed] [Google Scholar]
- Cutting GR, Curristin S, Zoghbi H, O'Hara B, Seldin MF, Uhl GR (1992). Identification of a putative γ‐aminobutyric acid (GABA) receptor subunit rho2 cDNA and colocalization of the genes encoding rho2 (GABRR2) and rho1 (GABRR1) to human chromosome 6q14‐q21 and mouse chromosome 4. Genomics 12: 801–806. [DOI] [PubMed] [Google Scholar]
- Cutting GR, Lu L, O'Hara BF, Kasch LM, Montrose‐Rafizadeh C, Donovan DM et al. (1991). Cloning of the γ‐aminobutyric acid (GABA) ρ1 cDNA: a GABA receptor subunit highly expressed in the retina. Proc Natl Acad Sci U S A 88: 2673–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong CJ, Werblin FS (1995). Zinc downmodulates the GABAC receptor current in cone horizontal cells acutely isolated from the catfish retina. J Neurophysiol 73: 916–919. [DOI] [PubMed] [Google Scholar]
- Drew CA, Johnston GA, Weatherby RP (1984). Bicuculline‐insensitive GABA receptors: studies on the binding of (−)‐baclofen to rat cerebellar membranes. Neurosci Lett 52: 317–321. [DOI] [PubMed] [Google Scholar]
- Duke RK, Chebib M, Balcar VJ, Allan RD, Mewett KN, Johnston GAR (2000). (+)‐ and (−)‐cis‐2‐aminomethylcyclopropanecarboxylic acids show opposite pharmacology at ρ2 GABA(C) receptors. J Neurochem 75 26022610. [DOI] [PubMed] [Google Scholar]
- Ebert B, Thompson SA, Saounatsou K, McKernan R, Krogsgaard‐Larsen P, Wafford KA (1997). Differences in agonist/antagonist binding affinity and receptor transduction using recombinant human γ‐aminobutyric acid type A receptors. Mol Pharmacol 52: 1150–1156. [PubMed] [Google Scholar]
- Etter A, Cully DF, Liu KK, Reiss B, Vassilatis DK, Schaeffer JM et al. (1999). Picrotoxin blockade of invertebrate glutamate‐gated chloride channels: subunit dependence and evidence for binding within the pore. J Neurochem 72: 318–326. [PubMed] [Google Scholar]
- Feigenspan A, Bormann J (1994). Differential pharmacology of GABAA and GABAC receptors on rat retinal bipolar cells. Eur J Pharmacol 288: 97–104. [DOI] [PubMed] [Google Scholar]
- Feigenspan A, Wassle H, Bormann J (1993). Pharmacology of GABA receptor Cl‐ channels in rat retinal bipolar cells. Nature 361: 159–162. [DOI] [PubMed] [Google Scholar]
- Filippova N, Dudley R, Weiss DS (1999). Evidence for phosphorylation‐dependent internalization of recombinant human ρ1 GABAC receptors. J Physiol 518 (Pt 2): 385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher EL, Koulen P, Wassle H (1998). GABAA and GABAC receptors on mammalian rod bipolar cells. J Comp Neurol 396: 351–365. [DOI] [PubMed] [Google Scholar]
- Gavande N, Yamamoto I, Salam NK, Ai T‐H, Burden PM, Johnston GAR et al. (2011). Novel cyclic phosphinic acids as GABAC ρ receptor antagonists: design, synthesis, and pharmacology. ACS Med Chem Lett 2: 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs ME, Johnston GAR (2005). Opposing roles for GABAA and GABAC receptors in short‐term memory formation in young chicks. Neuroscience 131: 567–576. [DOI] [PubMed] [Google Scholar]
- Gurley D, Amin J, Ross PC, Weiss DS, White G (1995). Point mutations in the M2 region of the alpha, beta, or gamma subunit of the GABAA channel that abolish block by picrotoxin. Receptors Channels 3: 13–20. [PubMed] [Google Scholar]
- Hall BJ, Karim N, Chebib M, Johnston GAR, Hanrahan JR (2014). Modulation of ionotropic GABA receptors by 6‐methoxyflavanone and 6‐methoxyflavone. Neurochem Res 39: 1068–1078. [DOI] [PubMed] [Google Scholar]
- Harrison NJ, Lummis SC (2006a). Locating the carboxylate group of GABA in the homomeric rho GABAA receptor ligand‐binding pocket. J Biol Chem 281: 24455–24461. [DOI] [PubMed] [Google Scholar]
- Harrison NJ, Lummis SC (2006b). Molecular modeling of the GABA(C) receptor ligand‐binding domain. J Mol Model 12: 317324. [DOI] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E (2011). Principles of activation and permeation in an anion‐selective Cys‐loop receptor. Nature 474: 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilf RJ, Dutzler R (2008). X‐ray structure of a prokaryotic pentameric ligand‐gated ion channel. Nature 452: 37–79. [DOI] [PubMed] [Google Scholar]
- Inoue M, Akaike N (1988). Blockade of γ‐aminobutyric acid‐gated chloride current in frog sensory neurons by picrotoxin. Neurosci Res 5: 380–394. [DOI] [PubMed] [Google Scholar]
- Jansen A, Hoepfner M, Herzig KH, Riecken EO, Scherubl H (2000). GABA(C) receptors in neuroendocrine gut cells: a new GABA‐binding site in the gut. Pflugers Arch 441: 294–300. [DOI] [PubMed] [Google Scholar]
- Jarboe CH, Poerter LA, Buckler RT (1968). Structural aspects of picrotoxinin action. J Med Chem 11: 729–731. [DOI] [PubMed] [Google Scholar]
- Jensen RJ (2012). Blocking GABA(C) receptors increases light responsiveness of retinal ganglion cells in a rat model of retinitis pigmentosa. Exp Eye Res 105: 21–26. [DOI] [PubMed] [Google Scholar]
- Johnston GAR (1996). GABA(A) receptor pharmacology. Pharmacol Ther 69: 173–198. [DOI] [PubMed] [Google Scholar]
- Johnston GAR (2002). Medicinal chemistry and molecular pharmacology of GABAC receptors. Curr Top Med Chem 2: 903–913. [DOI] [PubMed] [Google Scholar]
- Johnston GAR, Chebib M, Hanrahan JR, Mewett KN (2003). GABA(C) receptors as drug targets. Curr Drug Targets CNS Neurol Disord 2: 260–268. [DOI] [PubMed] [Google Scholar]
- Johnston GAR, Curtis DR, Beart PM, Game CJA, Mcculloch RM, Twitchin B (1975). cis‐4‐Aminocrotonic and trans‐4‐aminocrotonic acid as GABA analogs of restricted conformation. J Neurochem 24: 157–160. [DOI] [PubMed] [Google Scholar]
- Kerr DI, Ong J (1995). GABAB receptors. Pharmacol Ther 67: 187–246. [DOI] [PubMed] [Google Scholar]
- Kim H‐L, Kumar RJ, Hanrahan JR, Fernandez SP, Johnston GAR, Chebib M (2008). GABAC antagonists as memory enhancers. Society for Neuroscience, Neuroscience, Washington DC, Abstract 531.21/D22.
- Krehan D, Frolund B, Ebert B, Nielsen B, Krogsgaard‐Larsen P, Johnston GAR et al. (2003a). Aza‐THIP and related analogues of THIP as GABAC antagonists. Bioorg Med Chem 11: 4891–4896. [DOI] [PubMed] [Google Scholar]
- Krehan D, Frølund B, Krogsgaard‐Larsen P, Kehler J, Johnston GA, Chebib M (2003b). Phosphinic, phosphonic and seleninic acid bioisosteres of isonipecotic acid as novel and selective GABA C receptor antagonists. Neurochem Int 42: 561–565. [DOI] [PubMed] [Google Scholar]
- Krogsgaard‐Larsen P, Frølund B, Liljefors T, Ebert B (2004). GABAA agonists and partial agonists: THIP (Gaboxadol) as a non‐opioid analgesic and a novel type of hypnotic. Biochem Pharmacol 68: 1573–1580. [DOI] [PubMed] [Google Scholar]
- Kumar RJ, Chebib M, Hibbs DE, Kim H‐L, Johnston GAR, Salam NK et al. (2008). Novel γ‐aminobutyric acid ρ1 receptor antagonists; synthesis, pharmacological activity and structure‐activity relationships. J Med Chem 51: 3825–3840. [DOI] [PubMed] [Google Scholar]
- Kusama T, Spivak CE, Whiting P, Dawson VL, Schaeffer JC, Uhl GR (1993a). Pharmacology of GABA ρ1 and GABA α/β receptors expressed in Xenopus oocytes and COS cells. Br J Pharmacol 109: 200–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusama T, Wang TL, Guggino WB, Cutting GR, Uhl GR (1993b). GABA rho 2 receptor pharmacological profile: GABA recognition site similarities to rho 1. Eur J Pharmacol 245: 83–84. [DOI] [PubMed] [Google Scholar]
- Le Novere N, Changeux JP (1999). The ligand gated ion channel database. Nucleic Acids Res 27: 340–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zhang Y, Liu H, Yan Y, Li Y (2008). Identification and expression of GABAC receptor in rat testis and spermatozoa. Acta Biochim Biophys Sin (Shanghai) 40: 761–767. [PubMed] [Google Scholar]
- Locock KES, Yamamoto I, Tran P, Hanrahan JR, Chebib M, Johnston GAR et al. (2013). γ‐Aminobutyric acid(C) (GABAC) selective antagonists derived from the bioisosteric modification of 4‐aminocyclopent‐1‐enecarboxylic acid: amides and hydroxamates. J Med Chem 56: 5626–5630. [DOI] [PubMed] [Google Scholar]
- Lopez‐Chavez A, Miledi R, Martinez‐Torres A (2005). Cloning and functional expression of the bovine GABAC ρ2 subunit. Molecular evidence of a widespread distribution in the CNS. Neurosci Res 53: 421–427. [DOI] [PubMed] [Google Scholar]
- Lukasiewicz PD, Werblin FS (1994). A novel GABA receptor modulates synaptic transmission from bipolar to ganglion and amacrine cells in the tiger salamander retina. J Neurosci 14: 1213–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lummis SC, Harrison NJ, Wang J, Ashby JA, Millen KS, Beene DL et al. (2012). Multiple tyrosine residues contribute to GABA binding in the GABA(C) receptor binding pocket. ACS Chem Nerosci 3: 186–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch JW, Rajendra S, Barry PH, Schofield PR (1995). Mutations affecting the glycine receptor agonist transduction mechanism convert the competitive antagonist, picrotoxin, into an allosteric potentiator. J Biol Chem 270: 13799–13806. [DOI] [PubMed] [Google Scholar]
- Madsen C, Jensen AA, Liljefors T, Kristiansen U, Nielsen B, Hansen CP et al. (2007). 5‐Substituted imidazole‐4‐acetic acid analogues: synthesis, modeling, and pharmacological characterization of a series of novel γ‐aminobutyric acidC receptor agonists. J Med Chem 50: 4147–4161. [DOI] [PubMed] [Google Scholar]
- Majumdar D, Guha S (1988). Conformation, electrostatic potential and pharmacophoric pattern of GABA (gamma‐aminobutyric acid) and several GABA inhibitors. J Mol Struct (THEOCHEM) 49: 125–140. [Google Scholar]
- Melis C, Lummis SCR, Molteni C (2008). Molecular dynamics simulations of GABA binding to the GABA(C) receptor: the role of Arg(104). Biophys J 95: 4115–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PS, Aricescu AR (2014). Crystal structure of a human GABA receptor. Nature 512: 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PS, Smart TG (2010). Binding, activation and modulation of Cys‐loop receptors. Trends Pharmacol Sci 31: 161–174. [DOI] [PubMed] [Google Scholar]
- Milligan CJ, Buckley NJ, Garret M, Deuchars J, Deuchars SA (2004). Evidence for inhibition mediated by coassembly of GABAA and GABAC receptor subunits in native central neurons. J Neurosci 24: 7241–7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris KD, Moorefield CN, Amin J (1999). Differential modulation of the γ‐aminobutyric acid type C receptor by neuroactive steroids. Mol Pharmacol 56: 752–759. [PubMed] [Google Scholar]
- Murata Y, Woodward RM, Miledi R, Overman LE, (1996). The first selective antagonist for a GABAC receptor. Bioorg Med Chem Lett 6: 2073–2076. [Google Scholar]
- Naffaa MM, Absalom N, Solomon VR, Chebib M, Hibbs DE, Hanrahan JR (2016). Investigating the role of loop C hydrophilic residue 'T244' in the binding site of ρ1 GABAC receptors via site mutation and partial agonism. PLoS One 11: e0156618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naffaa MM, Chebib M, Hibbs DE, Hanrahan JR (2015). Comparison of templates for homology model of ρ1 GABAC receptors: more insights to the orthosteric binding site's structure and functionality. J Mol Graph Model 62: 43–55. [DOI] [PubMed] [Google Scholar]
- Naffaa MM, Samad A (2016). The binding mode of picrotoxinin in GABAA‐ρ receptors: insight into the subunit's selectivity in the transmembrane domain. Comput Biol Chem 64: 202–209. [DOI] [PubMed] [Google Scholar]
- Olsen RW, Sieghart W (2008). International union of pharmacology. LXX. Subtypes of γ‐aminobutyric acid A receptors: classification on the basis of subunit composition, pharmacology, and function. Update. Pharmacol Rev 60: 243–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen RW, Sieghart W (2009). GABA‐A receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology 56: 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osolodkin DI, Chupakhin VI, Palyulin VA, Zefirov NS (2009). Molecular modeling of ligand‐receptor interactions in GABA C receptor. J Mol Graph Model 27: 813–821. [DOI] [PubMed] [Google Scholar]
- Ottosson N, Pastorczak M, van der Post ST, Bakker HJ (2014). Conformation of the neurotransmitter γ‐aminobutyric acid in liquid water. Phys Chem Chem Phys 16: 10433–10437. [DOI] [PubMed] [Google Scholar]
- Pan ZH, Zhang D, Zhang X, Lipton SA (2000). Evidence for coassembly of mutant GABAC rho1 with GABAA gamma2S, glycine alpha1 and glycine alpha2 receptor subunits in vitro. Eur J Neurosci 12: 3137–3145. [DOI] [PubMed] [Google Scholar]
- Pétriz A, Reyes‐Haro D, González‐González M, Miledi R, Martínez‐Torres A (2014). GABAρ subunits confer a bicuculline‐insensitive component to GFAP+ cells of cerebellum. Proc Natl Acad Sci U S A 111: 17522–17527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polenzani L, Woodward RM, Miledi R (1991). Expression of mammalian γ‐aminobutyric acid receptors with distinct pharmacology in Xenopus oocytes. Proc Natl Acad Sci U S A 88: 4318–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribilla I, Takagi T, Langosch D, Bormann J, Betz H (1992). The atypical M2 segment of the beta subunit confers picrotoxinin resistance to inhibitory glycine receptor channels. EMBO J 11: 4305–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H, Dowling JE (1993). Novel GABA responses from rod‐driven retinal horizontal cells. Nature 361: 162–164. [DOI] [PubMed] [Google Scholar]
- Ragozzino D, Woodward RM, Murata Y, Eusebi F, Overman LE, Miledi R (1996). Design and in vitro pharmacology of a selective γ‐aminobutyric acid C receptor antagonist. Mol Pharmacol 50: 1024–1030. [PubMed] [Google Scholar]
- Schlicker K, McCall MA, Schmidt M (2009). GABAC receptor‐mediated inhibition is altered but not eliminated in the superior colliculus of GABAC ρ1 knockout mice. J Neurophysiol 101: 2974–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelnikova A, Erkkila BE, Harris H, Zakharkin SO, Weiss DS (2006). Stoichiometry of a pore mutation that abolishes picrotoxin‐mediated antagonism of the GABAA receptor. J Physiol 577: 569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedelnikova A, Smith CD, Zakharkin SO, Davis D, Weiss DS, Chang Y (2005). Mapping the ρ1 GABAC receptor agonist binding pocket. Constructing a complete model. J Biol Chem 280: 1535–1542. [DOI] [PubMed] [Google Scholar]
- Shan Q, Haddrill JL, Lynch JW (2001). A single beta subunit M2 domain residue controls the picrotoxin sensitivity of alphabeta heteromeric glycine receptor chloride channels. J Neurochem 76: 1109–1120. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucl Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczot M, Kisiel M, Czyzewska MM, Mozrzymas JW (2014). Alpha1F64 residue at GABA(A) receptor binding site is involved in gating by influencing the receptor flipping transitions. J Neurosci 34: 3193–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadavartya R, Hwanga J, Rajputb PS, Sojab PJ, Kumarb U, Sastry BR (2015). Are presynaptic GABA‐C ρ2 receptors involved in anti‐nociception? Neurosci Lett 606: 145–150. [DOI] [PubMed] [Google Scholar]
- Tai K, Fowler P, Mokrab Y, Stansfeld P, Sansom MS (2009). Molecular modeling and simulation studies of ion channel structures, dynamics and mechanisms. Methods Cell Biol 90: 233–265. [DOI] [PubMed] [Google Scholar]
- Thompson AJ, Lester HA, Lummis SC (2010). The structural basis of function in Cys‐loop receptors. Q Rev Biophys 43: 449–499. [DOI] [PubMed] [Google Scholar]
- Torres VI, Weiss DS (2002). Identification of a tyrosine in the agonist binding site of the homomeric ρ1 γ‐aminobutyric acid (GABA) receptor that, when mutated, produces spontaneous opening. J Biol Chem 277: 43741–43748. [DOI] [PubMed] [Google Scholar]
- Tretter V, Moss SJ (2008). GABA(A) receptor dynamics and constructing GABAergic synapses. Front Mol Neurosci 1: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium (2013). Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic Acids Res 41: D43–D47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vien J, Duke RK, Mewett KN, Johnston GAR, Shingai R, Chebib M (2002). trans‐4‐Amino‐2‐methylbut‐2‐enoic acid (2‐MeTACA) and (±)‐trans‐2‐aminomethylcyclopropanecarboxylic acid ((±)‐TAMP) can differentiate rat ρ3 from human ρ1 and ρ2 recombinant GABAC receptors. Br J Pharmacol 135: 883–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters RJ, Hadley SH, Kendall DWM, Amin J (2000). Benzodiazepines act on GABAA receptors via two distinct and separable mechanisms. Nat Neurosci 3: 1274–1281. [DOI] [PubMed] [Google Scholar]
- Wang TL, Hackam A, Guggino WB, Cutting GR (1995). A single histidine residue is essential for zinc inhibition of GABA ρ1 receptors. J Neurosci 15: 7684–7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LW, Tae HS, Cromer BA (2014). Role of the ρ1 GABAC receptor N‐terminus in assembly, trafficking and function. ACS Chem Nerosci 5: 1266–1277. [DOI] [PubMed] [Google Scholar]
- Woodward RM, Polenzani L, Miledi R (1993). Characterization of bicuculline/baclofen‐insensitive (ρ‐like) γ‐aminobutyric acid receptors expressed in Xenopus oocytes. II. Pharmacology of γ‐aminobutyric acidA and γ‐aminobutyric acidB receptor agonists and antagonists. Mol Pharmacol 43: 609–625. [PubMed] [Google Scholar]
- Wu SM, Qiao X, Noebels JL, Yang XL (1993). Localization and modulatory actions of zinc in vertebrate retina. Vision Res 33: 2611–2616. [DOI] [PubMed] [Google Scholar]
- Xu JY, Yang B, Sastry BR (2009). The involvement of GABA‐C receptors in paired‐pulse depression of inhibitory postsynaptic currents in rat hippocampal CA1 pyramidal neurons. Exp Neurol 216: 243–246. [DOI] [PubMed] [Google Scholar]
- Xu M, Covey DF, Akabas MH, (1995). Interaction of picrotoxin with GABAA receptor channel‐lining residues probed in cysteine mutants. Biophys J 69: 1858–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto I, Absalom N, Carland JE, Doddareddy MR, Gavande N, Johnston GAR et al. (2012a). Differentiating enantioselective actions of GABOB: a possible role for threonine 244 in the binding site of GABAC ρ1 receptors. ACS Chem Nerosci 3: 665–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto I, Carland JE, Locock K, Gavande N, Absalom N, Hanrahan JR et al. (2012b). Structurally diverse GABA antagonists interact differently with open and closed conformational states of the ρ1 receptor. ACS Chem Nerosci 3: 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Omori K, Omori K, Otani H, Suzukawa J, Inagaki C (2003). GABAC receptor agonist suppressed ammonia‐induced apoptosis in cultured rat hippocampal neurons by restoring phosphorylated BAD level. J Neurochem 87: 791–800. [DOI] [PubMed] [Google Scholar]
- Zhang D, Pan ZH, Awobuluyi M, Lipton SA (2001). Structure and function of GABA(C) receptors: a comparison of native versus recombinant receptors. Trends Pharmacol Sci 22: 121–132. [DOI] [PubMed] [Google Scholar]
- Zhang D, Pan ZH, Zhang X, Brideau AD, Lipton SA (1995). Cloning of a γ‐aminobutyric acid type C receptor subunit in rat retina with a methionine residue critical for picrotoxinin channel block. Proc Natl Acad Sci U S A 92: 11756–11760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Xue F, Chang Y (2008). Structural determinants for antagonist pharmacology that distinguish the ρ1 GABAC receptor from GABAA receptors. Mol Pharmacol 74: 941–951. [DOI] [PubMed] [Google Scholar]