

Abstract
Although traditional cardiovascular risk factors ‘prime the soil’ for atherogenesis systemically, atherosclerosis primarily occurs in a site-specific manner with a predilection towards the inner wall of curvatures and outer wall of bifurcations with sparing of flow-dividers. Wall shear stress is a frictional force exerted parallel to the vessel wall that leads to alteration of the endothelial phenotype, endothelial cell signaling, gene and protein expression leading to a proinflammatory phenotype, reduced nitric oxide availability and disruption of the extracellular matrix, which in turn leads to plaque development. Clinical and experimental data are emerging that suggest the pathobiology associated with abnormal wall shear stress results in atherosclerotic plaque development and progression.
Keywords: atherosclerosis, endothelium, inflammation, nitric oxide, oxidative stress, plaque, wall shear stress
Although cardiovascular risk factors such as age, hypertension, diabetes, hyperlipidemia and smoking are associated with systemic atherogenesis, atherosclerosis primarily affects large and medium-sized elastic and muscular arteries in a site-specific manner, with a predilection towards the inner wall of curvatures and outer wall of bifurcations with sparing of flow-dividers, with the exception of the pulmonary circulation, which is devoid of atherosclerosis [1,2]. Local hemodynamic forces have been proposed to regulate the site-specific predilection of atherosclerosis and are primarily of two types. One is an outward deforming force that is exerted perpendicular to the vessel wall by the blood pressure and is called ‘tensile stress’. The other is a frictional force that is exerted parallel to the vessel wall by virtue of the viscosity of blood and is called ‘wall shear stress’ (WSS) [3]. It should be noted that the tensile stress is several orders of magnitude larger than the WSS.
Mechanical stress is defined as force per unit area (1 Pa = 1 N/m2 = 10 dynes/cm2). WSS is defined as the tangential stress on the endothelial surface of the arterial wall derived from the friction of the flowing blood [4]. WSS (τ) is directly proportional to the velocity of blood flow, and inversely proportional to the cube of the arterial radius (R), where flow rate is Q and fluid viscosity is μ: τ = 4 μ Q/R3. Shear rate at the arterial wall is the spatial gradient of blood flow velocity (dv/dy). Blood viscosity is a factor of internal friction that causes blood to resist flow and primarily depends on hematocrit and fibrinogen content. WSS (τ) can also be calculated as the product of blood viscosity (μ) and shear rate: τ = μ (dv/dy) [4,5]. Fluid mechanics models have shown that WSS values of ± 4 Pa are seen in areas susceptible to atherogenesis and are greater than 1.2 Pa in areas protected from atherogenesis. While the 0.5–1.2 Pa range is considered physiological WSS, values below and above this range are considered low and high WSS, respectively [6].
Two contradicting explanations were advanced in the 1960s to explain the effects of WSS in influencing atherogenesis. The first hypothesis was that high shear stress induced endothelial injury and increased permeability to lipids that favored atherogenesis [7,8]. The second hypo thesis was that low shear stress promoted modification of mass transport of atherogenic substances between the lumen and the vessel wall to cause atherosclerosis [9]. Over the years, low WSS has been shown to be predictive of plaque development.
Fluid dynamics & WSS
The nature of the flow of a fluid through a tube is dependent on the velocity of flow and the geometry of the tube. Flow is described as laminar if viscous forces prevail over inertial forces, resulting in the fluid flowing in concentric laminae parallel to the course of the artery, with the highest flow velocity in the center of the tube and the lowest in the periphery. Disturbed laminar flow is characterized by departure of flow from the streamline pattern such that there is formation of flow separation, recirculation and reattachment to forward flow of the laminae. Laminar flow occurs in relatively straight arterial segments and disturbed laminar flow in arterial segments with geometric irregularities, such as curvatures, branches, bifurcations and upstream or downstream from stenoses (FIGURE 1). Laminar flow occurs up to a certain velocity, called the ‘critical velocity’, above which turbulence occurs because inertial forces dominate over viscous forces. Turbulence is governed by the Reynold’s number (Re), which is proportional to the density of fluid (ρ), the diameter of the tube (D) under consideration and the flow velocity (V) while being inversely proportional to the viscosity (μ): Re = ρDV/μ. The higher the value of Re, the greater the probability of turbulence, such that flow is not turbulent if Re is less than 2000 and definitely turbulent if Re is greater than 3000 [4,10].
Figure 1. Schematic diagram of the carotid bifurcation demonstrating different types of flow.
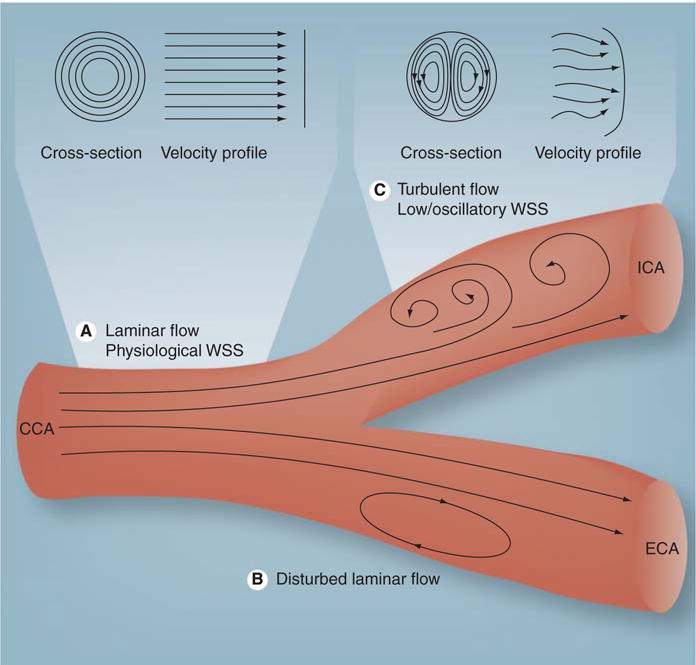
(A) Laminar flow in which the layers of fluid move in a streamlined and concentric fashion to produce a conical velocity profile. (B) Disturbed laminar flow in which layers of fluid separate, flow in the reverse direction and then reattach to maintain forward flow. (C) Turbulent flow in which velocity of fluid varies continually, but the overall flow is in the forward direction albeit with a flatter velocity profile.
CCA: Common carotid artery; ECA: External carotid artery; ICA: Internal carotid artery; WSS: Wall shear stress.
Adapted from [100].
The varying geometry of blood vessels and pulsatility of blood flow adds complexity to the patterns of blood flow and, as a result, the WSS vector in different areas of the body. In relatively straight arterial segments where laminar flow occurs, WSS is pulsatile and unidirectional, yielding a positive time-average, varying within the range of 1.5–7.0 Pa over the cardiac cycle [11,12]. In geometrically irregular sections, such as inner curves of vessels and upstream from stenoses, WSS maintains its predominantly unidirectional direction, but flow yields a relatively low positive time-average of less than 1.0–1.2 Pa [11,12]. In outer walls of bifurcations and downstream from stenoses, cyclic departure of the WSS vector from the predominant axial alignment occurs bi directionally during systole, accounting for oscillations in blood flow, and thereby creating oscillatory shear stress that yields a very low time-average (zero or even negative) [11,12].
Mechanisms relating WSS to atherosclerosis development
Effects on endothelial phenotype
The direct effects of WSS on endothelial cells have been elucidated primarily by in vitro experiments that investigated the effects of different types of shear flow on cultured endothelial cells lining the flow channel. One of the earliest observations was that sustained laminar flow and consequently physiological WSS induced an ellipsoidal endothelial cell morphology and alignment in the axial direction of flow in a time-dependent manner, whereas low WSS induced polygonal endothelial cell morphology and loss of alignment, findings that were subsequently confirmed in vivo [13–15]. Furthermore, laminar WSS was found to arrest endothelial cells in G0 or G1 phase of the cell cycle, preventing cell turnover, whereas low WSS accelerated cell turnover that subsequently leads to increases in lipid uptake in regions of disturbed flow through widening of gap junctions between polygonal endothelial cells [16–18].
Effects on endothelial cell signaling
Endothelial cells sense WSS on their luminal side through primary cilia that function as sensors and signal amplifiers of hydrodynamic forces [19]. Integrins, VEGFR-2, ion channels, G-protein-coupled receptors and trimeric G proteins, adhesion molecules (e.g., platelet endothelial cell adhesion molecule-1), gap junctions and membrane lipids have been found to modulate sensing of WSS by endothelial cells [20]. In addition to different mechano-sensing mechanisms interacting with one another to carry out cell signaling, the endothelial cyto skeleton has been found to play a central role in mediating mechano-transduction through conformational change [21,22]. The activation of these mechanosensors leads to triggering of phosphorylation cascades of signaling molecules and subsequent expression of proatherosclerotic genes, such as procoagulant, proapoptotic and proinflammatory genes that promote the atherosclerotic process [23,24].
Effects on low-density lipoprotein uptake
Sterol regulatory element-binding proteins (SREBPs) are key transcription factors that upregulate the expression of genes that encode the low-density lipoprotein (LDL) receptor and fatty acid synthase [25]. Laminar flow results in transient activation of SREBP, whereas disturbed flow results in sustained activation of SREBP [26], which results in subendothelial accumulation of LDL by enhanced permeability through endothelial junctions and LDL receptor-ediated endocytosis [27,28]. This LDL is subsequently oxidized to produce oxidized LDL, which is proatherogenic.
Effects on redox state
Reactive oxygen species (ROS) are a family of molecules including molecular oxygen and its derivatives, such as superoxide anion (O2−), hydroxyl radical (HO−) and lipid radicals. The three main sources of ROS are xanthine oxidase, NADH/NADPH oxidase and nitric oxide (NO) synthase [29].
Although, laminar WSS initially activates NADPH oxidase, the major source of O2− in vascular cells, this effects wanes after 24 h [30]. Laminar WSS also stimulates production of copper zinc-containing superoxide dismutase (SOD) [31] and vascular extracellular SOD [32], responsible for O2− scavenging, as well as glutathione (GSH) peroxidase, which is responsible for H2O2 scavenging [33], thereby having a predominant antioxidant effect.
Low and oscillatory WSS increases production of ROS and enhances gene expression of major oxidative enzymes, such as NADPH and xanthine oxidases at endothelial cell membranes, thereby stimulating not only sustained but also higher production of O2− [34], without an effect on Cu/Zn SOD [30]. Low and oscillatory WSS also decouple endothelial NO synthase (eNOS), which results in the production of ROS [34]. Therefore, low and oscillatory WSS have a discrete pro-oxidant effect, resulting in the induction of oxidative stress that refers to the excessive production of ROS such they overwhelm the endogenous antioxidant defense mechanisms to oxidize biological macromolecules (e.g., DNA, proteins and lipids), subsequently leading to altered endothelial function.
Effects on endothelial NO production & eNOS expression
The endothelium releases NO, which plays a central role in vascular homeostasis, not only by modulating vasomotor tone but also by inhibiting the expression of monocyte chemotactic protein-1 and VCAM-1, preventing propagation of lipid oxidation, inhibiting smooth muscle proliferation and decreasing platelet aggregation, all of which represent the earliest steps in atherosclerosis [35].
Laminar WSS affects endothelial function, which is measured in vitro by NO production primarily by two mechanisms. The first mechanism is acute and occurs within seconds of induction of laminar WSS, which induces an acute increase in intracellular calcium that enhances the binding of calmodulin to eNOS to increase eNOS activity and subsequent production of NO [36]. The second mechanism is chronic and occurs after several hours of laminar WSS, which stimulates an increase in eNOS mRNA transcription via the MAPK pathway, and sustained stability of this mRNA via tyrosine kinases. Impaired endothelial function in regions of low WSS has been recently been confirmed by our group in a mouse model (FIGURES 2C & 2D) [37].
Figure 2. Effects of wall shear stress on inflammation and endothelial function.
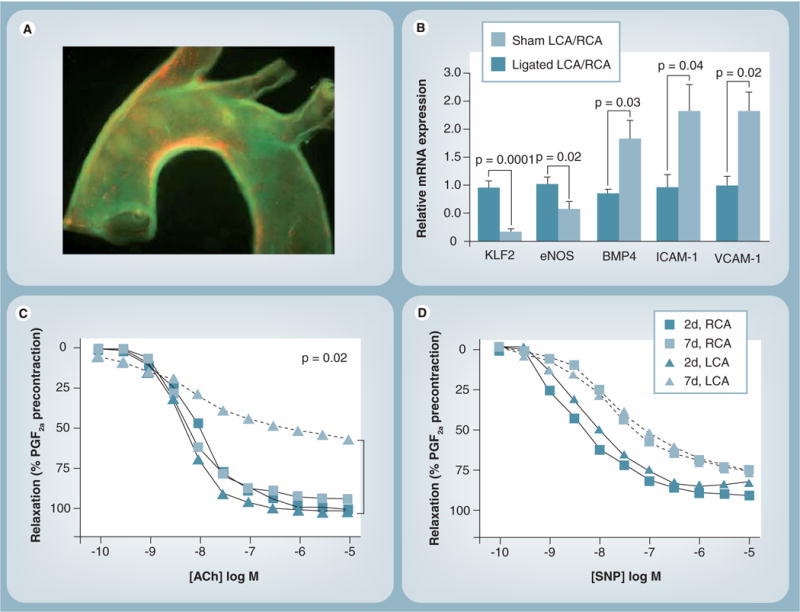
(A) Epifluorescence image of a whole mouse aortic mount showing the expression of ICAM-1 and VCAM-1 along the inner curvature of the arch and in the orifices of the arch vessels, areas that correspond to relatively lower values of mean wall shear stress (WSS). (B) Bar graphs depicting that partial ligation results in decreases in KLF-2 and eNOS, while increasing BMP4, ICAM-1 and VCAM-1 expression. (C) Impaired endothelium-dependent vasodilation as evident by lack of acetycholine-induced relaxation of arterial rings obtained from the left carotid artery of ApoE-knockout mice that underwent partial ligation and were fed a high-fat diet. (D) No impairment of endothelium-independent vasodilation as evident by complete relaxation induced by sodium nitroprusside.
ACh: Acetylcholine; BMP: Bone morphogenic protein; eNOS: Endothelial nitric oxide synthase; KLF: Krüppel-like factor; LCA: Left carotid artery; PGF: Prostaglandin F; RCA: Right carotid artery; SNP: Sodium nitroprusside.
Low and oscillatory WSS also induce NO production and eNOS expression [38], but through a different pathway and in addition, decouple eNOS to induce production of ROS such as O2− and H2O2, which are proinflammatory [34]. In vivo human studies using brachial artery reactivity have confirmed the direct relationship between WSS and endothelial function [39,40].
Effects on proinflammatory pathways
Laminar WSS produces anti-inflammatory effects through regulation of endothelial gene expression by transcription factors such as NF-κB and Krüppel-like factor (KLF)-2, thereby controlling inflammation, thrombosis/hemostasis, vascular tone and blood vessel development to effect atheroprotection [41–44].
By contrast, low WSS induces expression of inflammatory molecules such as VCAM-1 and ICAM-1, C-reactive protein, VEGF, IL-6 and upregulation of several broad-acting inflammatory cytokines and receptors, consistent with a proinflammatory phenotype (FIGURE 3) [45–77].
Figure 3. Schematic diagram broadly showing the regulation of transcription in the endothelial cells by wall shear stress.
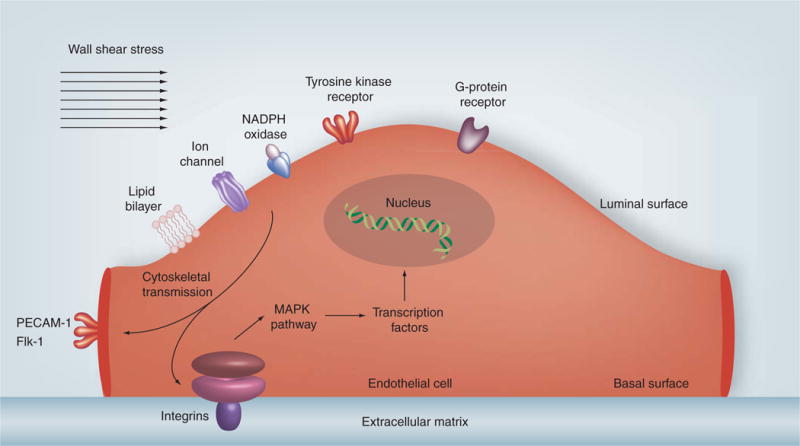
Wall shear stress is sensed on the luminal surface of endothelial cells by ionic channels, lipid layer, tyrosine kinase, G protein, NADPH oxidase and xanthine oxidase receptors that activate integrins on the basal surface of the endothelial cell or PECAM-1 and Flk-1 on the endothelial cell junctional surface via cytoskeletal transmission of shear forces. This initiates a downstream signaling cascade that activate MAPKs and other pathways to phosphorylate transcription factors, such as NF-κB and Krüppel-like factor 2, which regulate expression of atherogenic or atheroprotective genes.
PECAM-1: Platelet endothelial cell adhesion molecule-1.
Effects on vascular smooth muscle cells & extracellular matrix
Low WSS can stimulate vascular smooth muscle cell migration from the media to the intima, followed by proliferation that results in formation of a fibrous cap, neointimal formation and arterial wall thickening [48,49]. High WSS produces the opposite effect [50–54].
The extracellular matrix (ECM) consists of collagen and elastin fibers within a ground substance composed of proteoglycans and glycosaminoglycans. The ECM surrounds endothelial cells and provides scaffolding for the vascular wall and fibroatheroma. Matrix metalloproteinases (MMPs) are enzymes that degrade the ECM. Low WSS induces production of MMPs through upregulation of gene expression and stimulation by proinflammatory cytokine release from endothelial cells, macrophages and vascular smooth muscle cells that results in ECM degradation, and subsequent loss of this support outside the vascular wall to effect vascular remodeling [55–57].
High WSS can also stimulate production of MMPs in endothelial and smooth muscle cells that can degrade the internal elastic lamina of vessel and produce arterial enlargement. In addition, high WSS also stimulates endothelial cells to produce plasmin that dissolves the proteoglycan matrix [58,59] as well as NO and TGF-β that might suppress matrix production by smooth-muscle cells and induce apoptosis of smooth-muscle cells [60,61]. It is thought that if these molecular events occur after the formation of thin cap fibroatheromas in an area of low WSS, thinning of the fibrous cap can occur that makes plaque more vulnerable to fissuring and rupture [62].
Experimental models of WSS & atherosclerosis development
The cause–effect relationship between altered WSS patterns and plaque development was first studied in the carotid arteries of ApoE-knockout mice that were fed a high-fat diet. A conical perivascular shear stress modifier was applied around the carotid artery to create regions of low WSS, high WSS and oscillatory WSS, while maintaining undisturbed flow in the contralateral carotid artery that served as the control arm. Atherosclerotic lesions developed in the regions with low WSS and oscillatory WSS, whereas regions with high WSS were protected. The low WSS regions developed larger atherosclerotic lesions with greater lipid content, fewer smooth muscle cells, lesser collagen and greater outward remodeling compared with their counterparts in the oscillatory WSS region. In addition, regions with low WSS had higher expression of proatherogenic inflammatory mediators (e.g., VCAM-1, ICAM-1, C-reactive protein, VEGF and IL-6, as well as higher MMP activity) and intraplaque hemorrhages. In conclusion, the study showed that low WSS produced larger lesions that were of vulnerable phenotype, whereas oscillatory WSS produced smaller lesions that were of stable phenotype [63].
We have also observed a colocalization of proinflammatory adhesion molecules such as VCAM-1 and ICAM-1 with low WSS in with the inner curvature of the ApoE mouse (FIGURE 2A) [46]. In a subsequent study in ApoE-knockout mice using a novel partial carotid ligation model, we demonstrated that in zones of low WSS, the intimal RNA developed upregulation of pro atherogenic genes (such as those governing VCAM-1 and ICAM-1 expression), as well as downregulation of antiatherogenic genes (e.g., those governing eNOS and KLF-2 expression) (FIGURE 2B) [37]. In addition, the low WSS regions exhibited endothelial dysfunction and developed rapidly progressive atherosclerosis to advanced lesions within 4 weeks (FIGURES 2C & D). This model was unique in that it allows the study of atherosclerosis development and progression in a considerably shorter duration of time and allows for intimal RNA isolation in order to study alterations in genes induced by atherosclerosis-promoting environments.
In a swine diabetic model of atherosclerosis, Chatzizisis et al.demonstrated that low WSS promoted plaque development and progression to high-risk atherosclerotic plaques that were characterized by a large lipid core, inflammation, thin fibrous cap, severe internal elastic lamina degradation and excessive expansive remodeling [64].
Clinical studies of WSS & atherosclerosis development
WSS & early atheroma development
The role of WSS in plaque development was first suggested in ex vivo studies in human aortic specimens where fatty streaks and atheromatous plaque were found to be distributed in regions of low WSS [9,65]. Subsequent studies confirmed these findings in human carotid bifurcations using laser Doppler velocimetry [6,11], coronary vasculature using cinemicrographic techniques [66] and abdominal aorta using MRI velocimetry [67]. These studies described oscillations in the WSS vector during systole and formation of slow recirculatory secondary flows within areas of low WSS as promoters of atherogenesis and exercise as its inhibitor [6,11,66,67].
Cross-sectional studies of the carotid tree in humans using Doppler ultrasound demonstrated that, in patients with unilateral carotid plaques, the common carotid artery with plaque had a lower mean peak WSS compared with the contralateral side devoid of plaque [68]. Similar carotid ultrasonic studies demonstrated that patients with diabetes [69], hypertension [70] and left ventricular hypertrophy [71] had lower WSS compared with controls, and that accumulation of traditional cardiovascular risk factors, such as age and BMI, hypertension, hyperlipidemia, diabetes and smoking, was associated with lower WSS independent of carotid intima-media thickness [72]. In addition, studies using MRI have shown an inverse correlation between wall thickness and WSS [73].
In the coronary circulation, studies using quantitative coronary angiography have shown that low WSS correlates with atherosclerosis progression (FIGURE 4) [74]. Higher resolution evaluation of coronary WSS can be performed using intravascular ultrasound (IVUS), biplane angiography, Doppler flow velocity measurement and computational fluid dynamic models. These more detailed evaluations have shown that vessel wall thickness (an index of atherosclerosis) is inversely correlated with WSS [75]. Subsequently, prospective studies using these techniques demonstrated that low WSS is associated with development of atherosclerosis and outward remodeling [12], as well as neointimal thickness after stent implantation [76].
Figure 4.
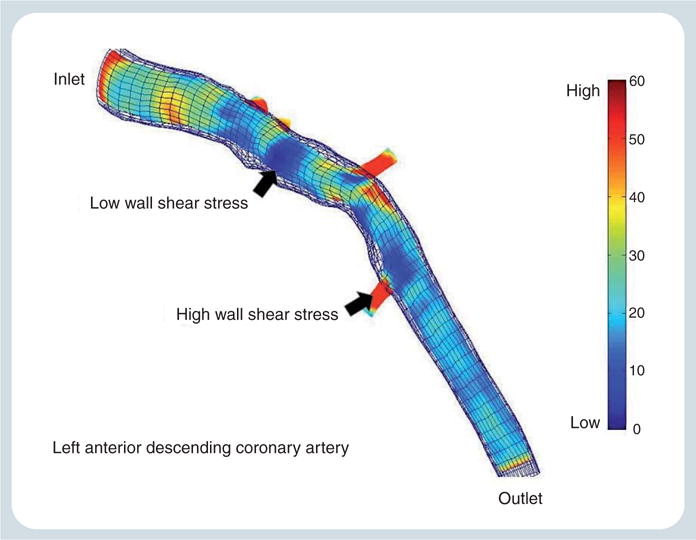
Wall shear stress profile of the left anterior descending coronary artery from a patient with nonobstructive coronary artery disease performed in our laboratory, outlining areas of low and high wall shear stress.
WSS & vascular remodeling
Vascular remodeling was first described in human autopsy studies as a compensatory change in vessel size as a result of increasing plaque burden and changes in local WSS in an effort to preserve the cross-sectional luminal area. Indeed, it was found that the development of luminal stenoses was delayed until the coronary plaque began to occupy 40% of the internal elastic lamina area [77]. This was later corroborated in vivo using IVUS and correlated with clinical presentations such that positive (outward) remodeling and larger plaque areas were associated with an unstable clinical presentation, and negative remodeling with a stable clinical presentation [78].
Positively remodeled vessels often contain eccentric plaque, resulting in a portion of the cross-sectional area of the vessel covered with plaque (the plaque-rich wall) and the remaining portion free of plaque (the plaque-free wall). Two explanations for positive remodeling have evolved. The first suggests that the plaque-free wall expands in response to increased WSS, resulting in positive remodeling and expansion of the lumen, thereby maintaining a low WSS environment on the plaque-rich wall, further facilitating plaque development [77,79,80]. The second explanation is that an increase in vascular inflammation and MMP production results in fragmentation of the internal elastic lamina, extension of inflammation into the underlying media that causes atrophy of the media and digestion of the surrounding ECM, resulting in positive remodeling [77,81]. Evidence in favor of the former explanation includes studies that have shown that vessels with smaller the plaque-free circumferential wall area have lesser compensatory enlargement [82,83]. Interestingly, progression of coronary atherosclerosis can also be accompanied by an over-compensatory increase in luminal cross-sectional area; on the other hand, regression may be associated with no change in luminal cross-sectional area [84].
Negative (inward) remodeling refers to a local shrinkage of vessel size and, consequently, reduction in luminal area beyond that is explained by increasing plaque burden [78]. As eccentric plaques grow in size and encroach into the plaque-free wall, the ability of the vessel to undergo compensatory enlargement decreases and the plaque assumes a more concentric shape. Such plaques can rupture and heal by fibrosis, further constricting the vessel, resulting in negative remodeling [85,86].
WSS & plaque vulnerability & rupture
The term ‘vulnerable plaque’ is used to describe plaques that are prone to thrombosis and those that can undergo rapid progression and thus have a high probability of precipitating an acute coronary event. Several cardiovascular pathology studies have helped to characterize such plaques to create major and minor criteria for defining vulnerable plaques, as well as a histopathological classification system. Major criteria include the presence of active inflammation (monocyte, macrophage or T-cell infiltration), a thin cap (<65 μm) with a large lipid core (>40% of the total volume of the plaque), the presence of endothelial denudation with superficial platelet aggregation, fissuring of the surface of the plaque to promote platelet aggregation and the presence of hemodynamically significant stenosis (>90%). Minor criteria include the presence of superficial calcified nodules, intraplaque hemorrhage, endothelial dysfunction and expansive remodeling [87,88].
Current in vivo imaging techniques, such as IVUS and optical coherence tomography, allow us to clinically define coronary plaques with vulnerable features and prospectively evaluate their prognostic value. One such study is Providing Regional Observations to Study Predictors of Events in the Coronary Tree (PROSPECT), which investigated 3-year clinical outcomes in patients with acute coronary syndromes who underwent three vessel radiofrequency backscatter or ‘virtual histology’ (VH)-IVUS to assess plaque burden and content. The preliminary results of PROSPECT suggest that nonculprit lesions with high-risk IVUS features (plaque burden at least 70%, large necrotic core without a visible cap and minimum luminal area up to 4 mm2) are far more likely to be associated with future major cardiac events compared with nonculprit lesions without these high-risk IVUS features (17 vs 2%; p < 0.05) [89]. Experimental studies suggest that low WSS produces plaque with a vulnerable phenotype, whereas oscillatory WSS results in the development of a more stable phenotype [90]. In human carotid arteries, endarterectomy specimens have demonstrated higher macrophage and lipid concentrations in areas of low and oscillatory WSS [91]. Coronary circulation studies using VH-IVUS have shown that the plaque necrotic core content is higher in segments with low WSS [92,93]. It is possible, however, that a stable plaque phenotype in humans is produced in low WSS coronary segments by recurrent subclinical rupture vulnerable-type plaques progressively resulting in fibrotic lesions [94]. At some point in this process, the WSS increases into the physiologic range. Exactly what drives further plaque formation in segments with physiologic WSS is not known and may involve systemic risk factors, inflammation and endothelial dysfunction. However, as plaque continues to encroach into the lumen, the WSS becomes supraphysiologic or high due to the acceleration of blood flow through the constricted vessel. The ensuing high WSS is thought to induce several biological changes within the fibrous cap that can weaken it and make the plaque more vulnerable to fissuring or rupture [95–97]. As in early-stage vulnerable plaques, the fibrous cap of the more advanced plaque can rupture and heal by fibrosis, causing increasing luminal constriction, or develop overlying thrombus, occluding the vessel and manifesting as an acute coronary syndrome (FIGURE 5) [47,98]. Recent in vivo human studies have demonstrated plaque ulceration in areas of high WSS by IVUS of the human coronary arteries and MRI of human carotid arteries, respectively [95–97]. Furthermore, IVUS-derived palpography, a measure of plaque strain in human coronary arteries, has been associated with regions of high WSS [99]. Prospective studies are underway to evaluate the predictive value of WSS on regional plaque progression in human coronary atherosclerosis.
Figure 5. Development of atherosclerotic plaque and the role of wall shear stress in influencing plaque differentiation into quiescent, stenotic and vulnerable phenotypes.
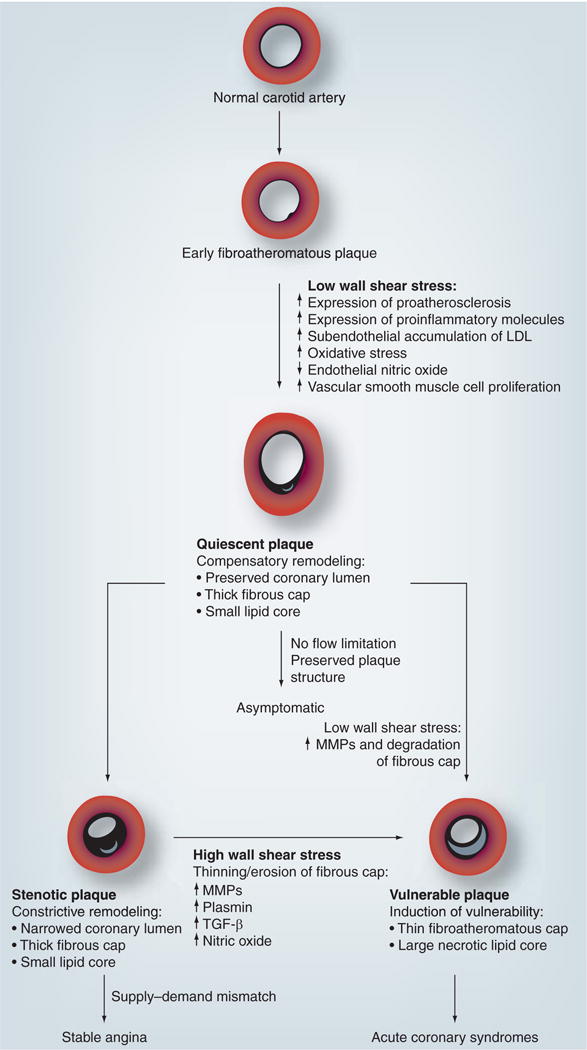
LDL: Low-density lipoprotein; MMP: Matrix metalloproteinase.
Expert commentary
Atherosclerosis begins with fatty streak formation in an atherogenic milieu that is created by the interplay of systemic cardiovascular risk factors and local hemodynamic forces, such as a low WSS. The precise interplay between WSS, vascular remodeling and further plaque development is not entirely clear and is currently under active investigation. However, it is thought that coronary segments that are geometrically susceptible to low WSS (i.e., outer hips of bifurcations, inner curvatures of vessels, immediately distal to bifurcations and distal to existing plaque) develop atherosclerosis. Within low-WSS segments, the developing plaque could take one of two courses: evolve into a vulnerable phenotype, such as a thin-cap fibroatheroma; or progress into a more stable phenotype, with gradual luminal narrowing that eventually results in an increase in WSS into the physiologic range. It can be speculated that the antiatherogenic effects of physiologic WSS might inhibit further plaque formation in some cases; however, continued exposure to atherogenic risk factors, inflammation and/or endothelial dysfunction can result in plaque progression despite physiologic WSS. However, as plaque continues to encroach into the lumen, the WSS becomes high due to acceleration of blood flow. The ensuing high WSS is thought to facilitate biological changes within the fibrous cap and the plaque itself, making it more vulnerable to fissuring or rupture. The plaque rupture can either heal by fibrosis, causing gradual luminal constriction, or be associated with thrombus, resulting in vessel occlusion and an acute coronary syndrome. Nevertheless, it should be emphasized that the pathobiology of human plaque progression and its translation into clinical events are still uncertain and remain to be fully elucidated by ongoing mechanistic and natural history studies.
Five-year view
With increased understanding of systemic and local factors governing plaque development and the advent of novel imaging techniques, such as optical coherence tomography, endothelial function testing and WSS measurement, the identification of coronary segments that are vulnerable to rapid progression or rupture can be anticipated. Very high-risk segments likely identified with such multimodality imaging could be preemptively treated to prevent acute coronary syndromes and improve clinical outcomes. A combination of focal therapy for targeted vulnerable coronary segments and systemic therapy would represent a paradigm shift in our treatment of chronic coronary artery disease, which is currently focused on relieving symptoms of ischemia but not improving outcomes.
Key issues.
Although cardiovascular risk factors such as age, hypertension, diabetes, hyperlipidemia and smoking ‘prime the soil’ for atherogenesis systemically, atherosclerosis primarily affects large and medium-sized elastic and muscular arteries in a site-specific manner, with a predilection towards the inner wall of curvatures and outer wall of bifurcations, with sparing of flow dividers.
Wall shear stress (WSS) is thought to determine the site-specific predilection of atherosclerosis.
At the cellular level, low WSS leads to alteration of the endothelial phenotype, endothelial cell signaling, and gene and protein expression, leading to a proinflammatory phenotype, reduced nitric oxide availability and disruption of the extracellular matrix leading to plaque development.
These concepts have also been shown in experimental models of atherosclerosis. The cause–effect relationship between altered WSS patterns and plaque development has been demonstrated in a number of experimental models of atherosclerosis.
The relationship between altered WSS and plaque development in human coronary atherosclerosis is not fully understood. However, it is thought that low WSS promotes early plaque development. Blood accelerates with luminal narrowing, thereby initially increasing WSS into the physiologic range. What governs further plaque development in the physiologic WSS environment is unknown, but probably involves systemic risk factors and inflammation. With further luminal encroachment, WSS approaches the supraphysiologic or high range. The ensuing high-WSS environment is associated with certain biological processes that promote plaque vulnerability and destabilization.
Natural history studies are required to fully elucidate the relationship between WSS, plaque progression and plaque rupture, as well as their translation into clinical events.
Footnotes
Financial & competing interests disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
No writing assistance was utilized in the production of this manuscript.
Contributor Information
Saurabh S Dhawan, Division of Cardiology, Department of Medicine, Emory University, Atlanta, GA, USA.
Ravi P Avati Nanjundappa, Division of Hospital Medicine, Department of Medicine, Emory University, Atlanta, GA, USA.
Jonathan R Branch, Division of Cardiology, Department of Medicine, Emory University, Atlanta, GA, USA.
W Robert Taylor, Division of Cardiology, Department of Medicine, Emory University, Atlanta, GA, USA.
Arshed A Quyyumi, Division of Cardiology, Department of Medicine, Emory University, Atlanta, GA, USA.
Hanjoong Jo, Division of Cardiology, Department of Medicine, Emory University, Atlanta, GA, USA.
Michael C McDaniel, Division of Cardiology, Department of Medicine, Emory University, Atlanta, GA, USA.
Jin Suo, Wallace H Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA, USA.
Don Giddens, Wallace H Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta, GA, USA.
Habib Samady, Professor of Medicine, Division of Cardiology, Department of Medicine, Emory University, 1364 Clifton Rd NE, Suite F606, Atlanta, GA 30322, USA, Tel.: +1 404 778 5299, Fax: +1 404 778 5278.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340:11–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Van der Laan P, Reardon C, Getz G. Site specificity of atherosclerosis site-selective responses to atherosclerotic modulators. Arterioscler Thromb Vasc Biol. 2004:12–22. doi: 10.1161/01.ATV.0000105054.43931.f0. [DOI] [PubMed] [Google Scholar]
- 3.Davies P. Flow-mediated endothelial mechanotransduction. Am Physiol Soc. 1995:51–60. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nichols W, O’Rourke M. McDonald’s Blood Flow in Arteries: Theoretical, Experimental and Clinical Principles. 5th. Hodder Arnold Publications; London, UK: 2005. [Google Scholar]
- 5••.Slager CJ, Wentzel JJ, Gijsen FJH, et al. The role of shear stress in the generation of rupture-prone vulnerable plaques. Nat Clin Pract Cardiovasc Med. 2005;2:401–407. doi: 10.1038/ncpcardio0274. Excellent review of the pathobiology of plaque development and the generation of rupture-prone vulnerable plaques. [DOI] [PubMed] [Google Scholar]
- 6.Zarins C, Giddens D, Bharadvaj B, Sottiurai V, Mabon R, Glagov S. Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circ Res. 1983;53:502–514. doi: 10.1161/01.res.53.4.502. [DOI] [PubMed] [Google Scholar]
- 7.Fry DL. Acute vascular endothelial changes associated with increased blood velocity gradients. Circ Res. 1968;22:165–197. doi: 10.1161/01.res.22.2.165. [DOI] [PubMed] [Google Scholar]
- 8.Fry DL. Certain histological and chemical responses of the vascular interface to acutely induced mechanical stress in the aorta of the dog. Circ Res. 1969;24:93–108. doi: 10.1161/01.res.24.1.93. [DOI] [PubMed] [Google Scholar]
- 9•.Caro C, Fitz-Gerald J, Schroter R. Arterial wall shear and distribution of early atheroma in man. Nature. 1969;223:115–160. doi: 10.1038/2231159a0. The first study to show that low wall shear stress predisposes to atheroma formation. [DOI] [PubMed] [Google Scholar]
- 10.Ganong W. Review of Medical Physiology. 23rd. McGraw-Hill; NY, USA: 2009. [Google Scholar]
- 11.Ku D, Giddens D, Zarins C, Glagov S. Pulsatile flow and atherosclerosis in the human carotid bifurcation. Positive correlation between plaque location and low oscillating shear stress. Arterioscler Thromb Vasc Biol. 1985;5:293–302. doi: 10.1161/01.atv.5.3.293. [DOI] [PubMed] [Google Scholar]
- 12•.Stone PH, Coskun AU, Kinlay S, et al. Effect of endothelial shear stress on the progression of coronary artery disease, vascular remodeling, and in-stent restenosis in humans: in vivo 6-month follow-up study. Circulation. 2003;108:43–44. doi: 10.1161/01.CIR.0000080882.35274.AD. The first study to describe the role of endothelial wall shear stress in human coronary arteries. [DOI] [PubMed] [Google Scholar]
- 13.Levesque MJ, Nerem RM. The elongation and orientation of cultured endothelial cells in response to shear stress. J Biomech Eng. 1985;107:341–347. doi: 10.1115/1.3138567. [DOI] [PubMed] [Google Scholar]
- 14.Dewey CF, Jr, Bussolari SR, Gimbrone MA, Jr, Davies PF. The dynamic response of vascular endothelial cells to fluid shear stress. J Biomech Eng. 1981;103:177–185. doi: 10.1115/1.3138276. [DOI] [PubMed] [Google Scholar]
- 15.Levesque MJ, Liepsch D, Moravec S, Nerem RM. Correlation of endothelial cell shape and wall shear stress in a stenosed dog aorta. Arterioscler Thromb Vasc Biol. 1986;6:22–29. doi: 10.1161/01.atv.6.2.220. [DOI] [PubMed] [Google Scholar]
- 16.Davies PF, Remuzzi A, Gordon EJ, Dewey CF, Gimbrone MA. Turbulent fluid shear stress induces vascular endothelial cell turnover in vitro. Proc Natl Acad Sci. 1986;83:2114–2117. doi: 10.1073/pnas.83.7.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin K, Hsu PP, Chen BP, et al. Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc Natl Acad Sci USA. 2000;97(17):9385–9389. doi: 10.1073/pnas.170282597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chien S. Molecular and mechanical bases of focal lipid accumulation in arterial wall. Prog Biophys Mol Biol. 2003;83:13–51. doi: 10.1016/s0079-6107(03)00053-1. [DOI] [PubMed] [Google Scholar]
- 19.Van der Heiden K, Hierck B, Krams R, et al. Endothelial primary cilia in areas of disturbed flow are at the base of atherosclerosis. Atherosclerosis. 2008;196:542–550. doi: 10.1016/j.atherosclerosis.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 20.Li Y-SJ, Haga JH, Chien S. Molecular basis of the effects of shear stress on vascular endothelial cells. J Biochem. 2005;38:1949–1971. doi: 10.1016/j.jbiomech.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Miao H, Li S, et al. Interplay between integrins and FLK-1 in shear stress-induced signaling. Am J Physiol Cell Physiol. 2002;283:C1540–C1547. doi: 10.1152/ajpcell.00222.2002. [DOI] [PubMed] [Google Scholar]
- 22.Davies PF, Barbee KA, Volin MV, et al. Spatial relationships in early signaling events of flow-mediated endothelial mechanotransduction. Ann Rev Physiol. 1997;59:527–549. doi: 10.1146/annurev.physiol.59.1.527. [DOI] [PubMed] [Google Scholar]
- 23.Brooks AR, Lelkes PI, Rubanyi GM. Gene expression profiling of human aortic endothelial cells exposed to disturbed flow and steady laminar flow. Physiol Genom. 2002;9:27–41. doi: 10.1152/physiolgenomics.00075.2001. [DOI] [PubMed] [Google Scholar]
- 24.Dai G, Kaazempur-Mofrad MR, Natarajan S, et al. Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc Natl Acad Sci. 2004;101:14871–14876. doi: 10.1073/pnas.0406073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown M, Goldstein J. The SREBP pathway: regulation review of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 26.Liu Y, Chen B, Lu M, et al. Shear stress activation of SREBP1 in endothelial cells is mediated by integrins. Arterioscler Thromb Vasc Biol. 2002:76–81. doi: 10.1161/hq0102.101822. [DOI] [PubMed] [Google Scholar]
- 27.Weinbaum S, Chien S. Lipid transport aspects of atherogenesis. J Biomech Eng. 1993;115:602–610. doi: 10.1115/1.2895547. [DOI] [PubMed] [Google Scholar]
- 28.Goldstein J, Kita T, Brown M. Defective lipoprotein receptors and atherosclerosis. Lessons from an animal counterpart of familial hypercholesterolemia. N Engl J Med. 1983;309(5):288–296. doi: 10.1056/NEJM198308043090507. [DOI] [PubMed] [Google Scholar]
- 29.Cai H, Harrison D. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 30.De Keulenaer G, Chappell D, Ishizaka N, Nerem R, Alexander R, Griendling K. Oscillatory and steady laminar shear stress differentially affect human endothelial redox state role of a superoxide-producing NADH oxidase. Circulation. 1998:1094–1101. doi: 10.1161/01.res.82.10.1094. [DOI] [PubMed] [Google Scholar]
- 31.Inoue N, Ramasamy S, Fukai T, Nerem R, Harrison D. Shear stress modulates expression of Cu/Zn superoxide dismutase in human aortic endothelial cells. Circ Res. 1996;79:32–37. doi: 10.1161/01.res.79.1.32. [DOI] [PubMed] [Google Scholar]
- 32.Fukai T, Siegfried M, Ushio-Fukai M, Cheng Y, Kojda G, Harrison D. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J Clin Invest. 2000;105:1631–1639. doi: 10.1172/JCI9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeshita S, Inoue N, Ueyama T, Kawashima S, Yokoyama M. Shear stress enhances glutathione peroxidase expression in endothelial cells. Biochem Biophys Res Commun. 2000;273:66–71. doi: 10.1006/bbrc.2000.2898. [DOI] [PubMed] [Google Scholar]
- 34.McNally J, Davis M, Giddens D, et al. Role of xanthine oxidoreductase and NAD (P) H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am J Physiol Heart Circ Physiol. 2003;285(6):H2290–H2297. doi: 10.1152/ajpheart.00515.2003. [DOI] [PubMed] [Google Scholar]
- 35.Harrison D, Widder J, Grumbach I, Chen W, Weber M, Searles C. Endothelial mechanotransduction, nitric oxide and vascular inflammation. J Intern Med. 2006;259:351–363. doi: 10.1111/j.1365-2796.2006.01621.x. [DOI] [PubMed] [Google Scholar]
- 36.Boo Y, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am J Physiol Cell Physiol. 2003;285(3):C499–C508. doi: 10.1152/ajpcell.00122.2003. [DOI] [PubMed] [Google Scholar]
- 37•.Nam D, Ni C, Rezvan A, et al. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. Am J Physiol Heart Circ Physiol. 2009;297:H1535–H1543. doi: 10.1152/ajpheart.00510.2009. Used a unique mouse model that permits intimal RNA isolation to study genetic changes induced by atherosclerosis-promoting environments. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai H, McNally J, Weber M, Harrison D. Oscillatory shear stress upregulation of endothelial nitric oxide synthase requires intracellular hydrogen peroxide and CaMKII. J Mol Cell Cardiol. 2004;37:121–125. doi: 10.1016/j.yjmcc.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 39.Gnasso A, Carallo C, Irace C, et al. Association between wall shear stress and flow-mediated vasodilation in healthy men. Atherosclerosis. 2001;156:171–176. doi: 10.1016/s0021-9150(00)00617-1. [DOI] [PubMed] [Google Scholar]
- 40.Silber H, Bluemke D, Ouyang P, Du Y, Post W, Lima J. The relationship between vascular wall shear stress and flow-mediated dilation: endothelial function assessed by phase-contrast magnetic resonance angiography. J Am Coll Cardiol. 2001;38:1859–1865. doi: 10.1016/s0735-1097(01)01649-7. [DOI] [PubMed] [Google Scholar]
- 41.Chen B, Li Y, Zhao Y, et al. DNA microarray analysis of gene expression in endothelial cells in response to 24-h shear stress. Physiol Genom. 2001;7:55–63. doi: 10.1152/physiolgenomics.2001.7.1.55. [DOI] [PubMed] [Google Scholar]
- 42.Resnick N, Yahav H, Shay-Salit A, et al. Fluid shear stress and the vascular endothelium: for better and for worse. Prog Biophys Mol Biol. 2003;81:177–199. doi: 10.1016/s0079-6107(02)00052-4. [DOI] [PubMed] [Google Scholar]
- 43.Lehoux S, Tedgui A. Cellular mechanics and gene expression in blood vessels. J Biochem. 2003;36:631–643. doi: 10.1016/s0021-9290(02)00441-4. [DOI] [PubMed] [Google Scholar]
- 44.Dekker R, van Soest S, Fontijn R, et al. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel-like factor (KLF2) Blood. 2002;100:1689–1698. doi: 10.1182/blood-2002-01-0046. [DOI] [PubMed] [Google Scholar]
- 45.Walpola P, Gotlieb A, Cybulsky M, Langille B. Expression of ICAM-1 and VCAM-1 and monocyte adherence in arteries exposed to altered shear stress. Arterioscler Thromb Vasc Biol. 1995;15(1):2–10. doi: 10.1161/01.atv.15.1.2. [DOI] [PubMed] [Google Scholar]
- 46.Suo J, Ferrara DE, Sorescu D, Guldberg RE, Taylor WR, Giddens DP. Hemodynamic shear stresses in mouse aortas: implications for atherogenesis. Arterioscler Thromb Vasc Biol. 2007;27:346–351. doi: 10.1161/01.ATV.0000253492.45717.46. [DOI] [PubMed] [Google Scholar]
- 47••.Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling molecular, cellular, and vascular behavior. J Am Coll Cardiol. 2007;49:2379–2393. doi: 10.1016/j.jacc.2007.02.059. Excellent review that comprehensively describes the role of shear forces in the development and progression of atherosclerotic lesions at molecular, cellular and vascular levels. [DOI] [PubMed] [Google Scholar]
- 48.Bentzon JF, Sondergaard CS, Kassem M, Falk E. Smooth muscle cells healing atherosclerotic plaque disruptions are of local, not blood, origin in apolipoprotein E knockout mice. Circulation. 2007;116(18):2053–2061. doi: 10.1161/CIRCULATIONAHA.107.722355. [DOI] [PubMed] [Google Scholar]
- 49.Bentzon JF, Weile C, Sondergaard CS, Hindkjaer J, Kassem M, Falk E. Smooth muscle cells in atherosclerosis originate from the local vessel wall and not circulating progenitor cells in ApoE knockout mice. Arterioscler Thromb Vasc Biol. 2006;26(12):2696–2702. doi: 10.1161/01.ATV.0000247243.48542.9d. [DOI] [PubMed] [Google Scholar]
- 50•.Palumbo R, Gaetano C, Melillo G, Toschi E, Remuzzi A, Capogrossi MC. Shear stress downregulation of platelet-derived growth factor receptor-β and matrix metalloprotease-2 is associated with inhibition of smooth muscle cell invasion and migration. Circulation. 2000;102:225–230. doi: 10.1161/01.cir.102.2.225. Describes the role of shear stress in modulating neointimal proliferation after vascular injury. [DOI] [PubMed] [Google Scholar]
- 51.Liu S, Goldman J. Role of blood shear stress in the regulation of vascular smooth muscle cell migration. IEEE Trans Biomed Eng. 2001;48:474–483. doi: 10.1109/10.915714. [DOI] [PubMed] [Google Scholar]
- 52.Redmond E, Cullen J, Cahill P, et al. Endothelial cells inhibit flow-induced smooth muscle cell migration role of plasminogen activator inhibitor-1. Am Heart Assoc. 2001;103(4):597–603. doi: 10.1161/01.cir.103.4.597. [DOI] [PubMed] [Google Scholar]
- 53.Ueba H, Kawakami M, Yaginuma T. Shear stress as an inhibitor of vascular smooth muscle cell proliferation: role of transforming growth factor-fl1 and tissue-type plasminogen activator. Arterioscler Thromb Vasc Biol. 1997;17:1512–1516. doi: 10.1161/01.atv.17.8.1512. [DOI] [PubMed] [Google Scholar]
- 54.Palumbo R, Gaetano C, Antonini A, et al. Different effects of high and low shear stress on platelet-derived growth factor isoform release by endothelial cells consequences for smooth muscle cell migration. Arterioscler Thromb Vasc Biol. 2002:405–411. doi: 10.1161/hq0302.104528. [DOI] [PubMed] [Google Scholar]
- 55.Bassiouny H, Song R, Hong X, Singh A, Kocharyan H, Glagov S. Flow regulation of 72-kD collagenase IV (MMP-2) after experimental arterial injury. Circulation. 1998:157–163. doi: 10.1161/01.cir.98.2.157. [DOI] [PubMed] [Google Scholar]
- 56.Galis Z, Johnson C, Godin D, et al. Targeted disruption of the matrix metalloproteinase-9 gene impairs smooth muscle cell migration and geometrical arterial remodeling. Am Heart Assoc. 2002:852–859. doi: 10.1161/01.res.0000041036.86977.14. [DOI] [PubMed] [Google Scholar]
- 57.Braddock M, Schwachtgen JL, Houston P, Dickson MC, Lee MJ, Campbell CJ. Fluid shear stress modulation of gene expression in endothelial cells. News Physiol Sci. 1998;13:241–246. doi: 10.1152/physiologyonline.1998.13.5.241. [DOI] [PubMed] [Google Scholar]
- 58.Kenagy RD, Fischer JW, Davies MG, et al. Increased plasmin and serine proteinase activity during flow-induced intimal atrophy in baboon PTFE grafts. Arterioscler Thromb Vasc Biol. 2002;22:400–404. doi: 10.1161/hq0302.105376. [DOI] [PubMed] [Google Scholar]
- 59.Lijnen HR. Plasmin and matrix metalloproteinases in vascular remodeling. Thromb Haemost. 2001;86:324–333. [PubMed] [Google Scholar]
- 60.Casey PJ, Dattilo JB, Dai G, et al. The effect of combined arterial hemodynamics on saphenous venous endothelial nitric oxide production. J Vasc Surg. 2001;33:1199–1205. doi: 10.1067/mva.2001.115571. [DOI] [PubMed] [Google Scholar]
- 61.Kolpakov V, Gordon D, Kulik TJ. Nitric oxide–generating compounds inhibit total protein and collagen synthesis in cultured vascular smooth muscle cells. Circ Res. 1995;76:305–309. doi: 10.1161/01.res.76.2.305. [DOI] [PubMed] [Google Scholar]
- 62.Mattsson EJ, Kohler TR, Vergel SM, Clowes AW. Increased blood flow induces regression of intimal hyperplasia. Arterioscler Thromb Vasc Biol. 1997;17:2245–2249. doi: 10.1161/01.atv.17.10.2245. [DOI] [PubMed] [Google Scholar]
- 63.Cheng C, Tempel D, van Haperen R, et al. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation. 2006;113:2744–2753. doi: 10.1161/CIRCULATIONAHA.105.590018. [DOI] [PubMed] [Google Scholar]
- 64••.Chatzizisis YS, Jonas M, Coskun AU, et al. Prediction of the localization of high-risk coronary atherosclerotic plaques on the basis of low endothelial shear stress: an intravascular ultrasound and histopathology natural history study. Circulation. 2008;117:993–1002. doi: 10.1161/CIRCULATIONAHA.107.695254. Demonstrated an inverse relationship between the magnitude of low wall shear stress and the severity of high-risk features in atherosclerotic plaques. [DOI] [PubMed] [Google Scholar]
- 65.Mustard J, Rowsell H, Murphy E, Downie H, Jones RJ. Evolution of Atherosclerotic Plaque. University of Chicago Press; IL, USA: 1963. Intimal thrombosis in atherosclerosis; p. 183. [Google Scholar]
- 66.Asakura T. Flow patterns and spatial distribution of atherosclerotic lesions in human coronary arteries. Circ Res. 1990;66:1045–1066. doi: 10.1161/01.res.66.4.1045. [DOI] [PubMed] [Google Scholar]
- 67.Moore J, Jr, Xu C, Glagov S, Zarins C, Ku D. Fluid wall shear stress measurements in a model of the human abdominal aorta: oscillatory behavior and relationship to atherosclerosis. Atherosclerosis. 1994;110:225–240. doi: 10.1016/0021-9150(94)90207-0. [DOI] [PubMed] [Google Scholar]
- 68.Gnasso A, Irace C, Carallo C, et al. In vivo association between low wall shear stress and plaque in subjects with asymmetrical carotid atherosclerosis. Stroke. 1997;28:993–998. doi: 10.1161/01.str.28.5.993. [DOI] [PubMed] [Google Scholar]
- 69.Irace C. NIDDM is associated with lower wall shear stress of the common carotid artery. Diabetes. 1999;48:193–197. doi: 10.2337/diabetes.48.1.193. [DOI] [PubMed] [Google Scholar]
- 70.Jiang Y, Kohara K, Hiwada K. Low wall shear stress contributes to atherosclerosis of the carotid artery in hypertensive patients. Hypertens Res. 1999;22:203–207. doi: 10.1291/hypres.22.203. [DOI] [PubMed] [Google Scholar]
- 71.Jiang Y, Kohara K, Hiwada K. Low wall shear stress in carotid arteries in subjects with left ventricular hypertrophy. Am J Hypertens. 2000;13(8):892–898. doi: 10.1016/s0895-7061(00)00275-2. [DOI] [PubMed] [Google Scholar]
- 72.Jiang Y, Kohara K, Hiwada K. Association between risk factors for atherosclerosis and mechanical forces in carotid artery. Stroke. 2000:2319–2324. doi: 10.1161/01.str.31.10.2319. [DOI] [PubMed] [Google Scholar]
- 73.Wentzel JJ, Corti R, Fayad ZA, et al. Does shear stress modulate both plaque progression and regression in the thoracic aorta? Human study using serial magnetic resonance imaging. J Am Coll Cardiol. 2005;45:846–854. doi: 10.1016/j.jacc.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 74.Gibson C, Diaz L, Kandarpa K, et al. Relation of vessel wall shear stress to atherosclerosis progression in human coronary arteries. Arterioscler Thromb Vasc Biol. 1993;13:310–315. doi: 10.1161/01.atv.13.2.310. [DOI] [PubMed] [Google Scholar]
- 75•.Krams R, Wentzel JJ, Oomen JAF, et al. Evaluation of endothelial shear stress and 3D geometry as factors determining the development of atherosclerosis and remodeling in human coronary arteries in vivo: combining 3D reconstruction from angiography and IVUS (ANGUS) with computational fluid dynamics. Arterioscler Thromb Vasc Biol. 1997;17:2061–2065. doi: 10.1161/01.atv.17.10.2061. Describes the use of 3D reconstruction to study development of atherosclerosis and remodeling in human coronary arteries. [DOI] [PubMed] [Google Scholar]
- 76.Wentzel JJ, Krams R, Schuurbiers JCH, et al. Relationship between neointimal thickness and shear stress after Wallstent implantation in human coronary arteries. Circulation. 2001:1740–1745. doi: 10.1161/01.cir.103.13.1740. [DOI] [PubMed] [Google Scholar]
- 77••.Glagov S, Weisenberg E, Zarins CK, Stankunavicius R, Kolettis GJ. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987:1371–1375. doi: 10.1056/NEJM198705283162204. One of the first studies to show compensatory enlargement and remodeling in human coronary arteries. [DOI] [PubMed] [Google Scholar]
- 78.Schoenhagen P, Ziada KM, Kapadia SR, Crowe TD, Nissen SE, Tuzcu EM. Extent and direction of arterial remodeling in stable versus unstable coronary syndromes an intravascular ultrasound study. Am Heart Assoc. 2000:598–603. doi: 10.1161/01.cir.101.6.598. [DOI] [PubMed] [Google Scholar]
- 79.Kubis N, Checoury A, Tedgui A, Levy B. Adaptive common carotid arteries remodeling after unilateral internal carotid artery occlusion in adult patients. Cardiovasc Res. 2001;50:597–602. doi: 10.1016/s0008-6363(01)00206-1. [DOI] [PubMed] [Google Scholar]
- 80.Zarins C, Zatina M, Giddens D, Ku D, Glagov S. Shear stress regulation of artery lumen diameter in experimental atherogenesis. J Vasc Surg. 1987;5:413–420. [PubMed] [Google Scholar]
- 81.Bentzon J, Pasterkamp G, Falk E. Expansive remodeling is a response of the plaque-related vessel wall in aortic roots of apoE-deficient mice: an experiment of nature. Arterioscler Thromb Vasc Biol. 2003:257–262. doi: 10.1161/01.atv.0000051387.70962.79. [DOI] [PubMed] [Google Scholar]
- 82.Von Birgelen C, Mintz G, De Vrey E, et al. Atherosclerotic coronary lesions with inadequate compensatory enlargement have smaller plaque and vessel volumes: observations with three dimensional intravascular ultrasound in vivo. BMJ. 1998;79:137–142. doi: 10.1136/hrt.79.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chatzizisis Y, Jonas M, Coskun A, et al. Prediction of the localization of high-risk coronary atherosclerotic plaques on the basis of low endothelial shear stress: an intravascular ultrasound and histopathology natural history study. Circulation. 2008;117:993–1002. doi: 10.1161/CIRCULATIONAHA.107.695254. [DOI] [PubMed] [Google Scholar]
- 84.Sipahi I, Tuzcu E, Schoenhagen P, et al. Paradoxical increase in lumen size during progression of coronary atherosclerosis: observations from the REVERSAL trial. Atherosclerosis. 2006;189:229–235. doi: 10.1016/j.atherosclerosis.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 85.Davies M, Thomas A. Thrombosis and acute coronary-artery lesions in sudden cardiac ischemic death. N Engl J Med. 1984;310:1137–1140. doi: 10.1056/NEJM198405033101801. [DOI] [PubMed] [Google Scholar]
- 86.Burke A, Kolodgie F, Farb A, et al. Healed plaque ruptures and sudden coronary death evidence that subclinical rupture has a role in plaque progression. Circulation. 2001:934–940. doi: 10.1161/01.cir.103.7.934. [DOI] [PubMed] [Google Scholar]
- 87••.Naghavi M, Libby P, Falk E, et al. Circulation. 2003;108:1664–1672. doi: 10.1161/01.CIR.0000087480.94275.97. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: part I. Important consensus document that discusses the clinicopathological classification of plaque and defining criteria for vulnerable plaque. [DOI] [PubMed] [Google Scholar]
- 88.Schaar J, Muller J, Falk E, et al. Terminology for high-risk and vulnerable coronary artery plaques. Eur Heart J. 2004;25:1077–1082. doi: 10.1016/j.ehj.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 89.Stone GW. Providing regional observations to study predictors of events in the coronary tree (PROSPECT) Transcath Cardiovasc Ther. 2009 Abstract. [Google Scholar]
- 90.Cheng C, Tempel D, van Haperen R, et al. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation. 2006;113:2744–2753. doi: 10.1161/CIRCULATIONAHA.105.590018. [DOI] [PubMed] [Google Scholar]
- 91.Kaazempur-Mofrad M, Isasi A, Younis H, et al. Characterization of the atherosclerotic carotid bifurcation using MRI, finite element modeling, and histology. Ann Biomed Eng. 2004;32:932–946. doi: 10.1023/b:abme.0000032456.16097.e0. [DOI] [PubMed] [Google Scholar]
- 92.Rodriguez-Granillo GA, GarcÌa-GarcÌa HM, Wentzel J, et al. Plaque composition and its relationship with acknowledged shear stress patterns in coronary arteries. J Am Coll Cardiol. 2006;47:884–885. doi: 10.1016/j.jacc.2005.11.027. [DOI] [PubMed] [Google Scholar]
- 93.McDaniel MC, Suo J, Oshinski JN, et al. Low endothelial wall shear stress is associated with higher necrotic core and calcium percentage in patients with coronary artery disease. Transcath Cardiovasc Ther. 2008 Abstract. [Google Scholar]
- 94.Kolodgie FD, Gold HK, Burke AP, et al. Intraplaque hemorrhage and progression of coronary atheroma. N Engl J Med. 2003;349:2316–2326. doi: 10.1056/NEJMoa035655. [DOI] [PubMed] [Google Scholar]
- 95.Tang D, Teng Z, Canton G, et al. Sites of rupture in human atherosclerotic carotid plaques are associated with high structural stresses: an in vivo MRI-based 3D fluid–structure interaction study. Stroke. 2009;40:3258–3263. doi: 10.1161/STROKEAHA.109.558676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fukumoto Y, Hiro T, Fujii T, et al. Localized elevation of shear stress is related to coronary plaque rupture: a 3-dimensional intravascular ultrasound study with in-vivo color mapping of shear stress distribution. J Am Coll Cardiol. 2008;51:645–650. doi: 10.1016/j.jacc.2007.10.030. [DOI] [PubMed] [Google Scholar]
- 97.Groen H, Gijsen F, van der Lugt A, et al. Plaque rupture in the carotid artery is localized at the high shear stress region: a case report. Stroke. 2007;38:2379–2381. doi: 10.1161/STROKEAHA.107.484766. [DOI] [PubMed] [Google Scholar]
- 98.Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Stone PH, Feldman CL. Risk stratification of individual coronary lesions using local endothelial shear stress: a new paradigm for managing coronary artery disease. Curr Opin Cardiol. 2007;22:552–564. doi: 10.1097/HCO.0b013e3282f07548. [DOI] [PubMed] [Google Scholar]
- 99.Gijsen F, Wentzel J, Thury A, et al. Strain distribution over plaques in human coronary arteries relates to shear stress. Am J Physiol Heart Circ Physiol. 2008;295:H1608–H1614. doi: 10.1152/ajpheart.01081.2007. [DOI] [PubMed] [Google Scholar]
- 100.Stone P, Coskun A, Yeghiazarians Y, et al. Prediction of sites of coronary atherosclerosis progression: in vivo profiling of endothelial shear stress, lumen, and outer vessel wall characteristics to predict vascular behavior. Curr Opin Cardiol. 2003;18:458–470. doi: 10.1097/00001573-200311000-00007. [DOI] [PubMed] [Google Scholar]