

Summary
The Clp protease complex in Mycobacterium tuberculosis is unusual in its composition, functional importance, and activation mechanism. While most bacterial species contain a single ClpP protein that is dispensable for normal growth, mycobacteria have two ClpPs, ClpP1 and ClpP2, which are essential for viability and together form the ClpP1P2 tetradecamer. Acyldepsipeptide antibiotics of the ADEP class inhibit the growth of Gram-positive firmicutes by activating ClpP and causing unregulated protein degradation. Here we show that, in contrast, mycobacteria are killed by ADEP through inhibition of ClpP function. Although ADEPs can stimulate purified M. tuberculosis ClpP1P2 to degrade larger peptides and unstructured proteins, this effect is weaker than for ClpP from other bacteria and depends on the presence of an additional activating factor (e.g. the dipeptide benzyloxycarbonyl-leucyl-leucine in vitro) to form the active ClpP1P2 tetradecamer. The cell division protein FtsZ, which is a particularly sensitive target for ADEP-activated ClpP in firmicutes, is not degraded in mycobacteria. Depletion of the ClpP1P2 level in a conditional Mycobacterium bovis BCG mutant enhanced killing by ADEP unlike in other bacteria. In summary, ADEPs kill mycobacteria by preventing interaction of ClpP1P2 with the regulatory ATPases, ClpX or ClpC1, thus inhibiting essential ATP-dependent protein degradation.
Keywords: antitubercular agent, ADEP, Mycobacterium tuberculosis, ClpP peptidase, ClpC1
Introduction
Mycobacterium tuberculosis (MTB), the causative infectious agent of tuberculosis, is a major threat in hospital and community settings worldwide. Mycobacteria are intrinsically resistant to most antimicrobial agents essentially due to their thick, hydrophobic cell wall, with mycolic acids and phthiocerole-lipids, diverse ABC drug exporters and the expression of enzymes that modify antibiotics or their targets (Daffe and Draper, 1998; Buriankova et al., 2004; Louw et al., 2009). Multidrug resistant (MDR) strains of M. tuberculosis are already widespread and extensively drug resistant (XDR) as well as totally drug resistant (TDR) strains have emerged (Goldman et al., 2007; Calligaro et al., 2014). Moreover, clinically applied antibiotics only act against actively growing mycobacteria, but not persisters (Robertson et al., 2012; Fattorini et al., 2013), further emphasizing the need for new treatment strategies to target this pathogen (Balganesh et al., 2008).
The M. tuberculosis Clp protease complex is an attractive novel target for antitubercular drugs because it is essential for growth and virulence (Sassetti et al., 2003; Schmitt et al., 2011; Griffin et al., 2011; Ollinger et al., 2012; Raju et al., 2012a; Raju et al., 2012b; Vasudevan et al., 2013; Gavrish et al., 2014; Raju et al., 2014). ClpP forms the proteolytic core of the Clp protease complex, with fourteen subunits assembled into two heptameric rings around a spacious chamber that encloses the 14 catalytic triads. Small apical and distal entrance pores of the ClpP tetradecamer restrict access of substrates to the active sites (Alexopoulos et al., 2013; Brötz-Oesterhelt and Sass, 2014; Liu et al., 2014). Thus, ClpP alone is either dormant or limited to the degradation of small peptides (Yu and Houry, 2007; Kress et al., 2009; Molière and Turgay, 2009). For efficient protein degradation, ClpP strictly depends on the assistance of cognate Clp-ATPases, which widen the entrance pores for substrate passage, unfold the proteins and thread them through the pores into the degradation chamber (Lee et al., 2010; Li et al., 2010; Baker and Sauer, 2012).
While most bacterial species possess a single clpP gene, mycobacteria encode two copies, clpP1 and clpP2, which are organized in a single operon and are co-transcribed (Cole et al., 1998; Personne et al., 2013). Initial attempts to characterize the MTB ClpP proteins in vitro yielded inactive ClpP1 and ClpP2 oligomers of heptameric or lower order (Ingvarsson et al., 2007; Benaroudj et al., 2011) and even when homo-tetradecamers were obtained (Ingvarsson et al., 2007; Akopian et al., 2012), they did not exhibit any peptidase activity (Akopian et al., 2012). The active form of the enzyme was characterized only after the discovery of particular N-terminally blocked dipeptide activators such as benzyloxycarbonyl-leucyl-leucine (Z-LL) that promote the dissociation of the homo-tetradecamers into heptamers in vitro and their re-association into the active mixed ClpP1P2 tetradecamer, which is composed of one ClpP1 and one ClpP2 ring (Akopian et al., 2012). These rings influence each other’s conformations, and their interaction is indispensable for both peptidase activity and ATP-dependent degradation of proteins in collaboration with an AAA+-ATPase (Akopian et al., 2012; Schmitz and Sauer, 2014; Schmitz et al., 2014). In vivo, ClpP1P2 functions together with Clp-ATPases ClpC1 or ClpX, both of which are also essential for viability in mycobacteria (Sassetti et al., 2003; Griffin et al., 2011; Gavrish et al., 2014).
Here, we set out to investigate the effects of the novel class of acyldepsipeptide antibiotics called ADEP against mycobacteria in order to evaluate their potential use against this major pathogen. In previous studies, ADEPs have shown substantial antibacterial activity against several Gram-positive pathogens including multidrug-resistant Staphylococcus aureus in vitro and in rodent infection models (Brötz-Oesterhelt et al., 2005; Hinzen et al., 2006; Conlon et al., 2013), and even persisters were eradicated by ADEP treatment (Conlon et al., 2013). The mode of action of ADEP is distinct from all other antibiotics and it is important to note that it is based on a dual molecular mechanism (Brötz-Oesterhelt et al., 2005): 1) The binding of ADEP to hydrophobic pockets at the ClpP surface induces a conformational change that widens the gated pores for substrate entry (Kirstein et al., 2009; Lee et al., 2010; Li et al., 2010), thereby allowing uncontrolled ATP-independent degradation of nascent polypeptides and unstructured proteins in the absence of regulatory Clp-ATPases (Kirstein et al., 2009; Conlon et al., 2013); 2) in addition, by binding to these hydrophobic pockets, ADEPs prevent the interaction of ClpP with its regulatory ATPases (Kirstein et al., 2009; Lee et al., 2010), thereby precluding the selective degradation by the Clp protease machinery of its physiological substrates. In firmicutes, where ClpP is not essential for growth under moderate conditions, ADEP-mediated cell death is primarily a consequence of the nonselective degradation of indispensable proteins by activated ClpP alone. One such substrate is the essential cell division protein FtsZ, which is particularly sensitive to proteolysis by ADEP-activated ClpP in S. aureus and Bacillus subtilis (Sass et al., 2011). Although inhibition of ClpP’s physiological functions probably contributes somewhat to ADEP efficacy against pathogenic firmicutes, (e.g. Staphylococcus and Streptococcus) in which this protease contributes to virulence (Frees et al., 2014), this mechanism is not the primary cause of cell death and has received little attention.
ADEPs were shown to possess antibacterial activity against M. tuberculosis (Ollinger et al., 2012), although the mechanism of ADEP’s antitubercular activity was not investigated. We therefore set out to characterize the effects of ADEP on the function of purified M. tuberculosis ClpP1 and ClpP2 in the absence and presence of Z-LL using both peptide and protein substrates. In addition, we tackled the question of the primary killing event in mycobacteria. Because a functional Clp protease is essential for viability of M. tuberculosis under all conditions, it is a priori unclear whether ADEP kills primarily by activating nonselective proteolysis or by blocking the physiological functions of ClpP1P2 and its associated ATPases. To this end, we constructed a conditional clpP1P2 knock-down strain of Mycobacterium bovis BCG Pasteur and determined the growth inhibitory activity of ADEP with different cellular levels of ClpP.
Recently, an independent study was published describing the effects of ADEP on the activity of purified MTB ClpP1P2 as well as the crystal structure of ADEP bound to the active ClpP1P2 tetradecamer (Schmitz et al., 2014). Those observations on the structure of ClpP1P2/ADEP and our present results from substrate degradation assays confirm that ADEP opens the pore for substrate entry. Nonetheless, our findings also indicate that it is not excessive nonspecific protein degradation that kills mycobacteria, but the inability of the Clp protease complex to perform its essential physiological functions in eliminating potentially toxic proteins. A coherent picture of the antibacterial mechanism of ADEPs in mycobacteria has now emerged.
Results
Antibacterial activity of ADEP against mycobacteria
Moderate activity against M. tuberculosis was reported for ADEPs, with ADEP2 being the most active (minimal inhibitory concentration, MIC of 25 μg ml−1) among a small series of congeners tested (Ollinger et al., 2012). We corroborate this result for ADEP2 using a BSA free minimal medium and determined a slightly higher susceptibility for the closely related slow growing M. bovis BCG as well as slightly lower susceptibility for the fast growing Mycobacterium smegmatis (Table 1). Broadening the range of ADEP congeners to the ones depicted in figure 1, ADEP2 remained the most active. This finding is notable because ADEP4, which is particularly potent against S. aureus and other Gram-positive bacteria (Brötz-Oesterhelt et al., 2005; Conlon et al., 2013), was less effective than ADEP2 against mycobacteria. All ADEP congeners tested so far are less active against mycobacteria than against other Gram-positive bacteria, where MICs were generally in the nanomolar range (Brötz-Oesterhelt et al., 2005; Carney et al., 2014). One reason could be a lower uptake of ADEPs into the mycobacterial cell, as the computer model mycpermcheck (Merget et al., 2013) predicts 0% probability of ADEP passage across the mycobacterial cell wall. In addition, the activity of efflux pumps in M. tuberculosis was shown to reduce ADEP2 activity 2 to 4-fold (Ollinger et al., 2012). Alternatively, the cause for the comparably low MIC values of mycobacteria could be a technical problem rather than a physiological difference. While MIC determinations for most bacteria are based on an overnight incubation of cells with the antibiotic, the corresponding assay for slow growing M. tuberculosis and M. bovis takes 10 days and even for the faster growing M. smegmatis still takes 2 days, which demands a certain stability of the antibiotic over time in the medium. In B. subtilis, we observed that while freshly dissolved ADEP2 yielded an MIC of 0.06 μg ml−1 in the regular overnight assay, pre-incubation of ADEP2 in medium for 9 days prior to performing the same procedure resulted in a MIC of 8 μg ml−1 (Table 1). Following the time course of ADEP2 degradation under MIC assay conditions by HPLC revealed the degradation of 75–90% ADEP2 within a single day (Fig. S1).
Table 1.
MIC determinations (μg ml−1) of ADEP derivatives against mycobacteria and B. subtilis.
| Strain / Medium | ADEP2 | ADEP4 | ADEP7 | ADEP8 | apramycin | isoniazid |
|---|---|---|---|---|---|---|
| Minimal medium | ||||||
| M. smegmatis mc2155 | 64 | >64 | >64 | >64 | nd | 4 |
| M. bovis BCG Pasteur | 16 (8) | >64 (16) | >64 (16) | 32 | 1 | 0.06 |
| M. tuberculosis H37Rv | 32 (16) | >64 | >64 | 32 | 1 | nd |
|
| ||||||
| 7H9 medium | ||||||
| M. bovis BCG Pasteur | 64 | >64 | >64 | 32 | nd | 0.06 |
|
| ||||||
| Mueller-Hinton broth | ||||||
| B. subtilis 168 fresh ADEP a | 0.06 | 0.13 | 0.25 | 0.03 | nd | nd |
| B. subtilis 168 preincubated ADEP b | 8 | 1 | 2 | 1 | nd | nd |
Data in brackets represent the concentration where partial growth inhibition was observed. nd, not determined.
MIC determinations using freshly diluted ADEP2.
MIC determinations using ADEP2, which had been pre-incubated for 9 days in Mueller-Hinton broth.
Figure 1. Structure of the natural product ADEP1 and its synthetic congeners used in this study.
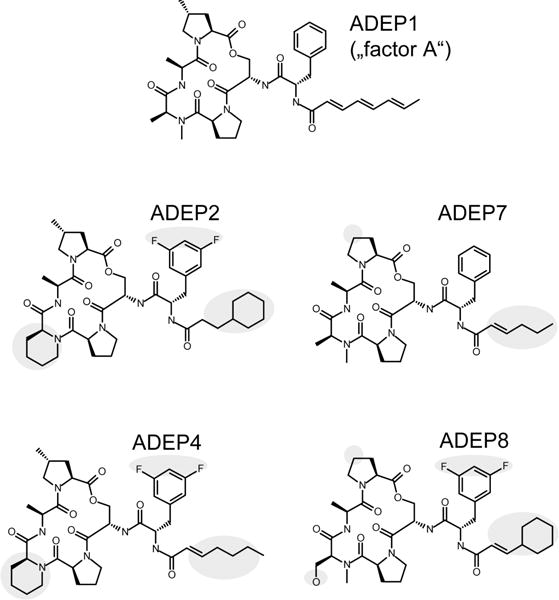
ADEP1 was originally isolated from a fermentation broth of Streptomyces hawaiiensis NRRL 15010 and briefly described as “factor A” in a patent (Michel and Kastner, 1982). The synthetic congeners ADEP2, 4 and 7 have been reported previously (Brötz-Oesterhelt et al., 2005; Hinzen et al., 2006), whereas ADEP8 represents an additional derivative that was synthesized by a previously described procedure (Hinzen et al., 2006). Regions where the synthetic congeners deviate from the natural product ADEP1 are highlighted.
ADEP alone fails to activate purified CIpP1, CIpP2 or mixed ClpP1P2
Assuming that the antibacterial activity of ADEP against mycobacteria is based on ClpP as the target, we performed a series of experiments with purified ClpP1 and ClpP2 of M. tuberculosis. First we investigated, whether ADEP might produce active tetradecamers from purified ClpP1, ClpP2 or a combination of both in the absence of the activating dipeptide Z-LL. Using the fluorogenic peptide substrate benzyloxycarbonyl-Gly-Gly-Leu-7-amino-4-methylcoumarin (Z-GGL-amc), no peptidase activity could be detected during 40 minutes incubation in the presence of ADEP up to 100 μg ml−1. The experiment was repeated using fluorescein isothiocyanate (FITC)-casein as a model protein substrate, but still no protease activity was observed with ADEP alone, in clear contrast to the strong stimulation of casein degradation observed with ADEP-activated homo-tetradecameric ClpP proteins from B. subtilis and E. coli (Kirstein et al., 2009). These differences highlight the unique nature and activation process of MTB ClpP.
ADEP and Z-LL synergistically stimulate peptidase activity of ClpP1P2
We next determined the effect of ADEPs on the peptidase activity of the mixed MTB ClpP1P2 tetradecamer, formed and pre-activated by Z-LL. ClpP1P2 activated by Z-LL alone was able to degrade the fluorogenic peptide substrate Z-GGL-amc (Fig. 2A). Pre-formation and pre-activation of ClpP1P2 by Z-LL was indispensable for ClpP1P2 to show activity in our assays. Consequently, Z-LL pretreated ClpP1P2 was used in all experiments described below. Nonetheless, several of our collection of ADEP derivatives (Fig. 1) caused a slight but reproducible increase in peptidase activity of Z-LL pre-activated ClpP1P2, with ADEP4, 7, and 8 being the most effective (Fig. 2B).
Figure 2. Peptide degradation assays using purified MTB ClpP1P2.
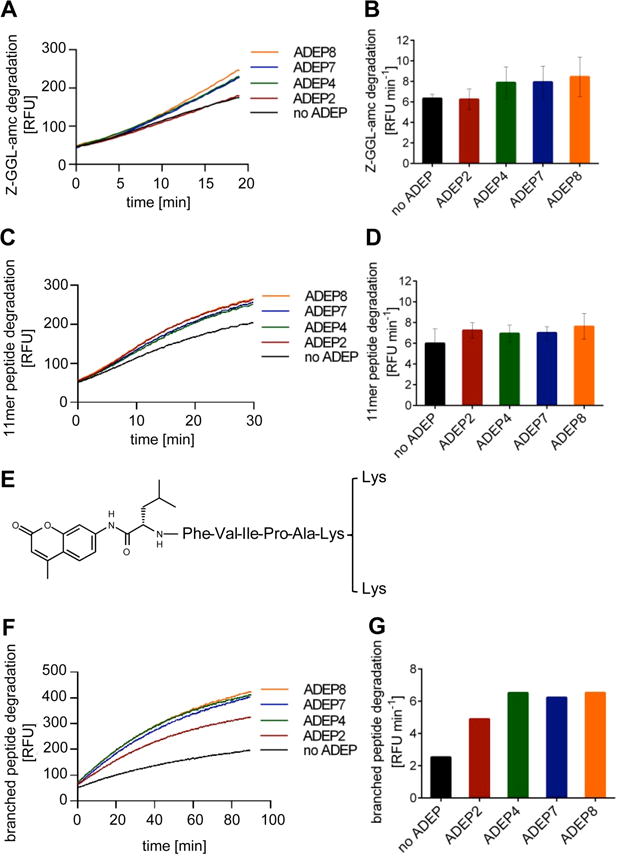
A. Time course of degradation of the tripeptide substrate Z-GGL-amc in the presence of different ADEP derivatives. B. Reaction rates (increase in relative fluorescence units, RFU, per minute) during the initial linear 5 min of the Z-GGL-amc degradation reaction in A. C. Time course of degradation of 11-mer peptides from the “FRET 25 Xaa peptide library” in the presence of different ADEP derivatives. D. Reaction rates (increase in RFU min−1) during the initial linear 5 min of the 11-mer peptide library degradation reaction in C. E. Chemical structure of the branched peptide used in this study. F. Time course of degradation of the branched peptide in the presence of different ADEP derivatives. G. Reaction rates (increase in RFU min−1) during the initial linear period (5 min) of enzyme activity in F. All data sets were reproduced in three independent experiments. In A, C, F and G, one representative experiment is shown. B and D show mean values of at least three independent experiments and error bars indicate standard deviations.
Next, we tested the degradation of longer peptides, using the “FRETs 25 Xaa peptide library” (Peptides International), which contains a collection of diverse quenched peptides containing 11 amino acids. These fluorogenic peptides were also cleaved by ClpP1P2 and again ADEPs stimulated this process weakly (Fig. 2C and D). We also studied a branched peptide (Buckley et al., 2011) consisting of a hexapeptide core with an N-terminally linked amc moiety and a C-terminal lysine branch (Fig. 2E). ADEPs markedly stimulated degradation of the branched peptide and led to an up to threefold higher degradation rate compared to the situation without antibiotic (Fig. 2F and G) in accord with the established ADEP mechanism of increasing the diameter of ClpP entrance pores.
Effects of ADEP on protein degradation by ClpP1P2 and its interaction with ClpC1
We next investigated, whether ADEP can also stimulate degradation of the model protein substrate FITC-casein. Addition of ADEPs significantly enhanced this process, with ADEP4, 7 and 8 increasing the degradation rate approximately five-fold and ADEP2 about two-fold (Fig. 3A). The effect of ADEPs was concentration-dependent as exemplified here by ADEP8, which showed half-maximal activation at 25 μM (Fig. 3B). Monitoring the degradation of unlabeled casein by SDS-PAGE confirmed the results of the fluorogenic assay (Fig. 3C). To test whether ADEP-activated ClpP1P2 is also able to degrade other unstructured proteins, we employed the microtubule-associated protein Tau, which is natively unfolded. ADEPs also significantly increased the digestion of Tau by ClpP1P2 (Fig. 3D), which, however, caused also some degradation of this substrate alone (Fig. 3D).
Figure 3. Degradation of unfolded proteins using purified MTB ClpP1P2.
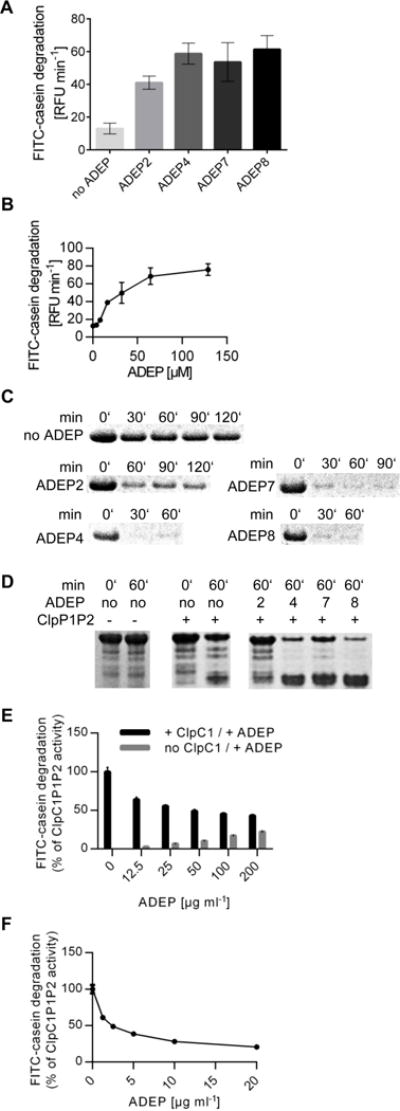
A. FITC-casein degradation rates (increase in RFU min−1) in the presence of ADEP calculated from the initial linear period of enzyme activity (5 min). B. Reaction rates of FITC-casein degradation at increasing ADEP8 concentrations. C. Time course of proteolysis of unlabeled casein analyzed by SDS-PAGE. D. Degradation of the eukaryotic model protein substrate Tau analyzed by SDS-PAGE. E. FITC-casein degradation by ClpP1P2 in the presence of competing amounts of ClpC1 versus ADEP2. Black bars indicate combined activation of ClpP1P2 by ClpC1 and increasing amounts of ADEP. Two-fold molar excess of ClpC1 over ClpP1P2 was kept constant, which in the absence of ADEP ensures efficient ClpC1P1P2 tetradecamer formation. ClpC1P1P2 activity in the absence of ADEP was taken as 100%. Grey bars indicate activating activity of ADEP in the absence of ClpC1 (as determined in parallel reactions). F. Inhibition of ClpC1P1P2 proteolytic activity by ADEP. Reaction rates for ClpC1P1P2 calculated from the values presented in E. The contribution of ADEP (grey bars in panel E) was subtracted from the combined reaction rates (black bars in panel E). In A, B, E, and F data represents the mean values of three experiments and error bars indicate the respective standard deviations.
The hexameric ATPase ClpC1 in M. tuberculosis catalyzes ATP-dependent degradation of casein by ClpP1P2 (Akopian et al., 2012). Although ADEP activated casein hydrolysis by ClpP1P2, it was not as efficient as ClpC1. The degradation rate of casein in the presence of 200 μg ml−1 ADEP2 did not exceed 20% of the rate in the presence of ClpC1 (Fig. 3E). This large difference in their stimulatory activities allowed us to monitor the effect of ADEP on the interaction of ClpC1 with ClpP1P2. Adding ADEP to the ClpC1P1P2 complex in the presence of ATP substantially reduced the degradation of casein. Thus, ADEP competed with ClpC1 for ClpP1P2 binding and blocked ATP-dependent proteolysis (Fig. 3F).
Binding mode of Z-LL to ClpP
Even though we could measure activation of MTB ClpP1P2 by ADEP, the magnitude of stimulation was much lower than with B. subtilis ClpP (BS ClpP) (Fig. S2) or as reported for E. coli ClpP (Kirstein et al., 2009; Li et al., 2010; Leung et al., 2011). One obvious difference is the presence of Z-LL in the mycobacterial system, which was indispensable for forming the active conformation of the ClpP1P2 tetradecamer. The mechanism of activation of Z-LL and related dipeptides is unclear. As Z-LL is a hydrophobic dipeptide, it could potentially bind to the active sites and interfere with the catalytic activity of MTB ClpP1P2. Alternatively, Z-LL could bind to the hydrophobic ATPase-binding pockets and stimulate in a similar way as ADEP. To distinguish these possibilities directly with MTB ClpP1P2 was not possible in vitro, as we did not obtain catalytically active protein in the absence of Z-LL. However, using BS ClpP as a model, we were able to observe an inhibitory effect of Z-LL on catalytic activity. In order to focus on catalysis and to minimize effects of ADEP-mediated pore opening, we measured the hydrolysis of the small fluorogenic peptide substrate N-succinyl-Lys-Tyr-amc (suc-LY-amc) by BS ClpP in the presence and absence of either Z-LL or ADEP2, as well as both compounds together. While ADEP2 and Z-LL were non-competitive (Fig. 4A and B), suc-LY-amc and Z-LL showed typical competitive behavior (i.e. nearly constant VMax values and increasing KM values with increasing Z-LL concentrations) (Fig. 4C and D). Thus, Z-LL binds to the active sites and decreases hydrolysis of the fluorogenic substrate. We further tested whether Z-LL does not only bind, but also may be hydrolyzed by BS ClpP. Our HPLC analyses showed that it is not a substrate, as the amount of Z-LL was not reduced after 3 hours incubation with BS ClpP, under typical assay conditions (Fig. S3). Thus, at concentrations used typically, Z-LL has the potential to interact with the active site but not with the hydrophobic pocket, where ADEPs and Clp-ATPases bind. The question, whether the presence of Z-LL in addition to its special activating function also affects hydrolysis by MTB ClpP1P2 remains uncertain. These results are in line with the published ClpP1P2-ADEP Z-Ile-Leu crystal structure (Schmitz et al., 2014), and our recent X-ray analysis of MTB ClpP1P2-CBZ-LL (Li et al., 2016), which shows the activating peptide benzoyl-Leu-Leu in all 14 active sites and not in the hydrophobic pockets.
Figure 4. Inhibition of BS ClpP by Z-LL.
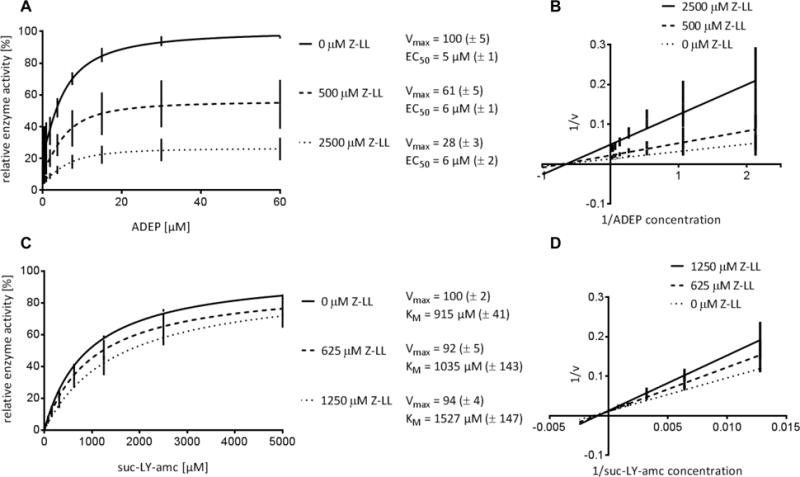
Z-LL is non-competitive with ADEP (A and B) but competitive with suc-LY-amc (C and D). Mean values of at least three independent experiments are presented. Standard deviations are indicated in brackets. Reaction rates are given in % of the maximum value in the absence of Z-LL. EC50 = ADEP concentration where half of the maximum activation is reached. The activating effect of ADEP on peptide hydrolysis by BS ClpP is based on stabilization of the active tetradecameric conformation as reported previously (Lee et al., 2010).
In mycobacteria FtsZ is not degraded in the presence of ADEP
We had previously observed that ADEPs inhibit bacterial cell division in B. subtilis and S. aureus by stimulating the degradation of the essential cell division protein FtsZ by ClpP (Sass et al., 2011). Therefore, we tested whether ADEPs can activate MTB ClpP1P2 to digest mycobacterial FtsZ (MTB FtsZ). MTB ClpP1P2 alone degraded a small fraction of our MTB FtsZ preparation (Fig. 5A), which might represent a residual amount of insufficiently folded FtsZ protein. However, the presence of ADEPs did not increase degradation further (Fig. 5A, panel 1). By contrast, MTB FtsZ was rapidly and completely hydrolyzed by ADEP-activated BS ClpP in a control sample (Fig. 5A, panel 2), whereas ADEPs could not activate MTB ClpP1P2 to degrade B. subtilis FtsZ (BS FtsZ) (Fig. 5A, panel 3). Thus, these results reflect the unique properties of MTB ClpP1P2 rather than structural differences between the FtsZ proteins from the different species.
Figure 5. Growth of M. bovis BCG is inhibited by ADEP2, but FtsZ is not degraded.
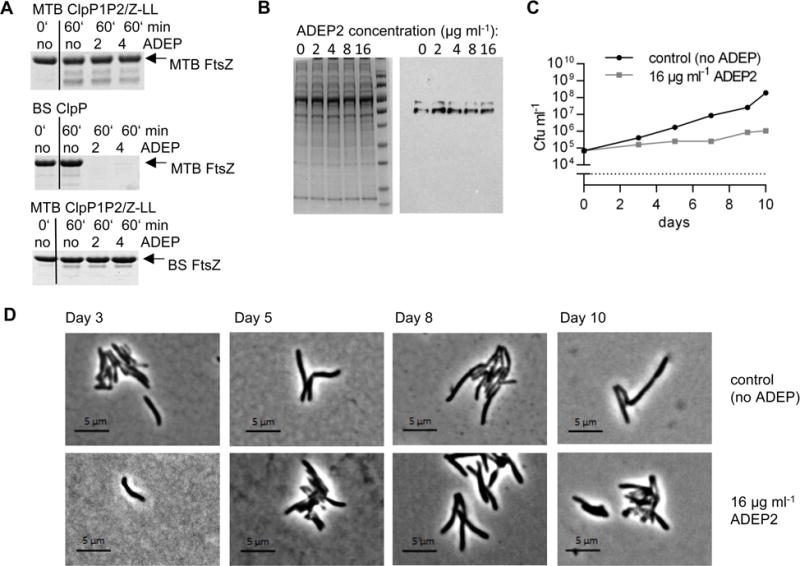
A. In vitro degradation of FtsZ from M. bovis BCG (MTB FtsZ) or B. subtilis 168 (BS FtsZ) with ClpP proteins from M. tuberculosis or B. subtilis in the absence or presence of ADEP2 and ADEP4. B. Lysates of M. bovis BCG wildtype (wt), grown for 10–12 days in the presence of rising ADEP2 concentrations. SDS page (left), Western blot using an anti-MTB FtsZ antibody (right). C. Growth curve of M. bovis BCG wt in the absence or presence of ADEP2. D. Phase contrast images of M. bovis BCG wt without ADEP (upper lane) or with 16 μg ml−1 ADEP2 (lower lane) at distinct points in time.
To make sure that we did not overlook potential effects on MTB FtsZ due to limitations of the in vitro assay, we also monitored the FtsZ concentration in ADEP-treated M. bovis BCG by Western blotting cell extracts and probing with polyclonal antiserum against MTB FtsZ. At an ADEP2 concentration corresponding to the MIC (16 μg ml−1), FtsZ was present in the extract at the same levels as in untreated controls (Fig. 5B), even though bacterial growth was strongly impaired (Fig. 5C). Because degradation of FtsZ in B. subtilis causes filamentation (Sass et al., 2011), we recorded the shape of M. bovis BCG in the presence of ADEP2 at different points of the growth curve, but never observed filamented mycobacteria (Fig. 5D). These data further confirm that in mycobacteria, FtsZ is not degraded by ADEP-activated ClpP1P2.
Down-regulation of clpP1P2 expression increases ADEP susceptibility of M. bovis
To focus on the question of the primary killing event in mycobacteria, we constructed the conditional clpP1P2 knock-down strain M. bovis BCG clpP1-tetoff (Fig. 6A, S4 and S5) that allows for clpP1 gene silencing in the presence of anhydrotetracycline (ATc). As shown for M. tuberculosis, clpP1 and clpP2 are co-transcribed (Personne et al., 2013) and ATc-induced down-regulation of clpP1 concomitantly silences gene expression of clpP2 (Raju et al., 2014). The use of M. bovis BCG allowed us to avoid the safety containment required for M. tuberculosis, while profiting from the particularly high homology between the two species (overall homology 95–99%, (Behr et al., 1999)). Of note, clpP1, clpP2, and ftsZ genes as well as the promoter region and operon structure of the bicistronic clpP1P2 operon show 100% sequence identity between the two species and genes of the AAA+-ATPases clpC1 and clpX 99%. A concentration as low as 0.1 ng ml−1 ATc slowed down the growth rate of the clpP1-tetoff strain (Fig. 6B) and at 0.5 ng ml−1 ATc the cells were dying (Fig. 6C), confirming that ClpP is also essential in M. bovis. In contrast, the growth curve of M. bovis wildtype (wt) was not affected at 2 ng ml−1 ATc (Fig. 6D) and even 25 ng ml−1 did not impede growth of the wildtype strain as indicated by the MIC growth controls (Table 2). Western blot analyses using an anti-ClpP2 antiserum proved that protein levels of ClpP2 were indeed decreased upon addition of ATc in a concentration-dependent manner in the conditional mutant and demonstrated that the ClpP1P2 protein level could be efficiently regulated from a wildtype-like level in the absence of ATc to a considerably reduced level at 0.1 μg ml−1 ATc (Fig. 6E). MIC determinations of the clpP1-tetoff strain showed that the presence of ATc caused a strong increase in ADEP2 sensitivity (Fig. 6F, 6G and Table 2). This ATc-effect was restricted to ADEP and did not occur with other antibiotics like apramycin or isoniazid, the mechanisms of which are unrelated to ClpP (Table 2). The synergy between ADEP2 and ATc was specific for the clpP1-tetoff mutant and was not observed with the wildtype and can, thus, be linked to the reduced ClpP1P2 protein level. Improved activity of an antibacterial agent upon target down-regulation is a widely accepted means of validating that the compound acts through inhibition of a particular target (Chen et al., 2000; Haas et al., 2001; Ji et al., 2004). Consequently, our data reveal 1) that ClpP is the target for ADEP also in mycobacteria and 2) that preventing the essential natural functions of the ClpP1P2/Clp-ATPase complex by ADEP is responsible for mycobacterial death.
Figure 6. Impact of clpP1P2 down-regulation on growth and ADEP sensitivity of M. bovis BCG.
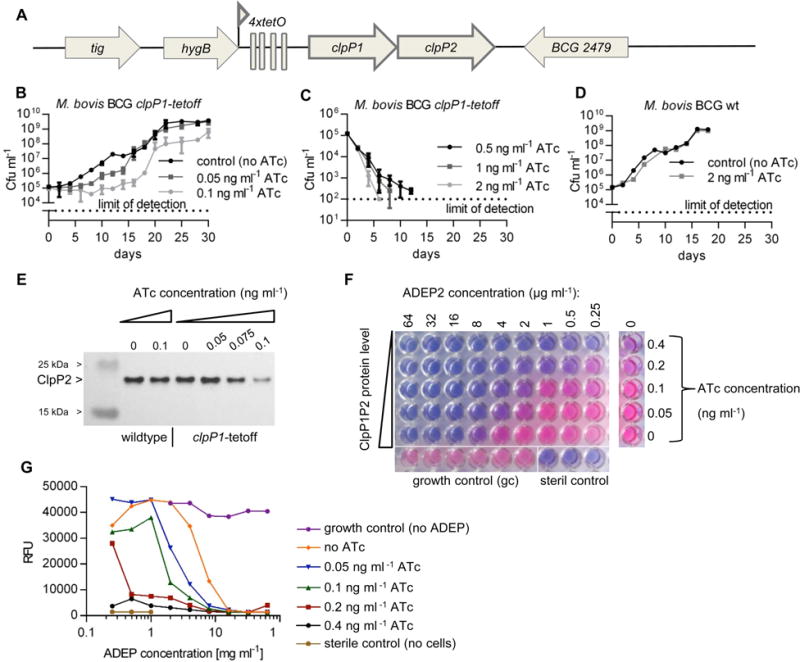
A. Genomic organization of the clpP region in M. bovis BCG clpP1-tetoff. The original promoter in front of clpP1 was replaced by the Pmyc1 promoter from M. smegmatis engineered to contain four tetO operator sites. In the presence of anhydrotetracycline (ATc) a Tet-repressor binds to TetO, thereby shutting down transcription of the bicistronic clpP1P2 operon. B. and C. Growth of M. bovis BCG clpP1-tetoff is inhibited with increasing ATc concentrations. D. Growth of M. bovis BCG wildtype (wt) is not affected by ATc. E. ClpP2 protein levels in M. bovis BCG clpP1-tetoff compared to wildtype in the absence and presence of increasing ATc concentrations. Immunodetection of ClpP2 with anti-MTB ClpP2 antiserum. F. Down-regulation of clpP1P2 sensitizes M. bovis BCG clpP1-tetoff to ADEP. Titration of ClpP1P2 protein levels against ADEP2 concentration. Photograph shows the microtiter plate after 10 days of incubation in the presence of ADEP. In living, metabolically active cells the blue, non-fluorescent resazurin dye is reduced to the pink, fluorescent resorufin. G. Graphical presentation of RFU values of all wells from F. The data show mean values and standard deviations of at least three independent experiments (B, C, D, and E) or one representative experiment of at least three independent biological replicates (F and G).
Table 2.
Down-regulation of clpP1P2 increases susceptibility of M. bovis BCG clpP1-tetoff to ADEP2.
| no ATc | 0.05 ng ml−1 ATc | 0.1 ng ml−1 ATc | 0.2 ng ml−1 ATc | 25 ng ml−1 ATc | |
|---|---|---|---|---|---|
| MIC (μg ml−1) against M. bovis BCG clpP1-tetoff | |||||
| ADEP2 | 16 (8) | 8 (4) | 4 (2) | 2 (0.5) | no growth |
| apramycin | 1 | 1 | nd | 1 | no growth |
| isoniazid | 0.03 | 0.03 | 0.03 | 0.03 | no growth |
|
| |||||
| MIC (μg ml−1) against M. bovis BCG wt | |||||
| ADEP2 | 16 | Nd | nd | nd | 16 |
| apramycin | 1 | Nd | nd | nd | 1 |
| isoniazid | 0.06 | Nd | nd | nd | 0.06 |
MIC values against M. bovis BCG Pasteur wildtype (wt) and M. bovis BCG Pasteur clpP1-tetoff in the presence of increasing ATc concentrations. In this conditional system clpP1P2 expression is suppressed by ATc resulting in decreased ClpP1P2 protein levels (for further information on strain regulation compare figure S5A). For ease of comparison with table 3, data are arranged with decreasing ClpP levels from left to right. Data in brackets represent the concentration where partial growth inhibition was observed. nd, not determined.
Our conclusions for mycobacteria became even clearer, when we compared the situation described above with that in B. subtilis. Using a conditional B. subtilis pX2-clpP strain, where clpP expression can be regulated by xylose (Gerth et al., 2004) (Fig. S5), we found that in B. subtilis — unlike in mycobacteria — down-regulation of clpP expression led to ADEP resistance (Table 3). In accordance with our previous finding that B. subtilis primarily suffers from cell division inhibition due to FtsZ degradation by ADEP-deregulated ClpP (Sass et al., 2011), our current results confirm that in B. subtilis, ADEP strictly depends on sufficiently high ClpP levels in order to exert its lethal action. In summary, while in Bacillus and other firmicutes (based on our previous data (Sass et al., 2011)), ADEP kills by causing non-specific proteolysis by ClpP, mycobacteria cannot cope with the consequences of ADEP’s preventing ClpP1P2 from binding its cognate ATPases, ClpX (Schmitz et al., 2014) and ClpC1 (Fig. 3F).
Table 3.
Down-regulation of clpP reduces susceptibility of B. subtilis 168-pX2-clpP to ADEP2.
| 10% | 8% | 5% | 4% | 3% | 2% | 1% | no xylose | |
|---|---|---|---|---|---|---|---|---|
| ADEP2 MIC (μg ml−1) in LB medium | ||||||||
| B. subtilis 168 wt | 0.5 | nd | nd | nd | nd | nd | nd | 0.5 |
| B. subtilis 168-pX2-clpP | 0.25 | 1 | 4 | 8 | 16 | 16 | >32 | >32 |
| B. subtilis 168ΔclpP | >32 | nd | nd | nd | nd | nd | nd | >32 |
|
| ||||||||
| apramycin MIC (μg ml−1) in LB medium | ||||||||
| B. subtilis 168 wt | 2 | nd | nd | nd | nd | nd | nd | 4 |
| B. subtilis 168-pX2-clpP | 4 | 4 | 2 | 4 | 2 | 2 | 1 | 2 |
| B. subtilis 168ΔclpP | 1 | nd | nd | nd | nd | nd | nd | 2 |
MIC values against B. subtilis 168 wildtype (wt), B. subtilis 168-pX2-clpP and B. subtilis 168ΔclpP in the presence of different xylose concentrations. In this conditional system clpP expression is induced by xylose (for further information on strain regulation compare figure S5B). For ease of comparison with table 2, data are arranged with decreasing ClpP levels from left to right. nd, not determined.
Discussion
In this study, we demonstrate that ADEPs target ClpP1P2 of mycobacteria, and exert their antibacterial action by abrogating the interaction between ClpP1P2 and its cognate Clp-ATPases. There are two likely reasons for this unique effect on mycobacteria, one related to the unusual mechanism of activation of ClpP1P2 in mycobacteria and the second related to its essential role in eliminating certain proteins, whose accumulation is toxic.
The enzymatic activation of mycobacterial ClpP proteins is exceptional because it requires the association of a ClpP1 and a ClpP2 ring, while either ClpP variant alone is inactive, even when present as tetradecamers. Simply mixing ClpP1 and ClpP2 is not sufficient for significant peptidase activity, which in vitro was only demonstrable in the presence of N-terminally blocked hydrophobic peptides (such as Z-LL used in this study), related peptides or peptide derivatives (Akopian et al., 2012; Schmitz et al., 2014). It is presently unclear whether in vivo, a similar activating peptide, a non-peptidic low molecular weight compound, or an assembly chaperone, serves this essential activating function. In a recent study by Schmitz and Sauer ClpP1P2 activity was also observed when the cognate AAA+-partner ClpX translocated a folded protein substrate through the pores into the mixed tetradecamer (Schmitz and Sauer, 2014). They suggested that the ATP-driven substrate delivery into the degradation chamber by a bound AAA+-ATPase provides the necessary stimulus for ClpP1P2 activation in the mycobacterial cell, and that sub-stoichiometric active-site occupancy by substrate peptides stabilizes the active conformation (Schmitz and Sauer, 2014).
In vitro, these two roles, activating and stabilizing, are served by the hydrophobic dipeptides, such as Z-LL used here and previously (Akopian et al., 2012) or Z-IL (Schmitz et al., 2014), at rather high concentrations (1 to 5 mM). Our kinetic measurements demonstrating competitive inhibition of peptide hydrolysis by Z-LL are in accord with the crystal structure of ClpP1P2-ADEP displaying Z-IL within the active sites (Schmitz et al., 2014) and a structure of ClpP1P2 in the presence of benzoyl-LL but without ADEP (Li et al., 2016). It is impressive how occupancy of active sites by Z-LL or related compounds can induce these major conformational changes in ClpP1 and ClpP2. In the present study, the response to ADEP was completely dependent on Z-LL-mediated formation of active ClpP1P2, just as we previously observed with ClpC1, which only stimulated casein degradation in the presence of the dipeptide activator (Akopian et al., 2012). The inability of ADEP to substitute for Z-LL in activating ClpP1P2 is consistent with the fact that in crystal structures ADEPs were never seen within the proteolytic chamber of ClpP (Lee et al., 2010; Li et al., 2010; Schmitz et al., 2014). The antibiotics bind to the hydrophobic pockets at the ClpP periphery in a position, where the subunits join, and where under physiological conditions the I/LGF/L loops of the cognate Clp-ATPases dock. The additivity between activation by Z-LL and ADEP that we observed in our assays indicates that both types of activators serve a different function in MTB ClpP1P2.
While ADEP occupied all 14 hydrophobic pockets in the homo-tetradecameric ClpP from B. subtilis and E. coli (Lee et al., 2010; Li et al., 2010; Schmitz et al., 2014), the antibiotic was only found at ClpP2 in the structure of MTB ClpP1P2 (Lee et al., 2010; Li et al., 2010; Schmitz et al., 2014). Interestingly, even with this partial occupancy at ClpP2 the pores of both rings, ClpP1 and ClpP2, were open (Schmitz et al., 2014). Our findings that ADEP stimulated the degradation of longer and branched peptides as well as of the unstructured proteins casein and Tau by Z-LL activated ClpP1P2 more strongly than the degradation of small peptides is in accordance with such widened pores. However, in the mycobacterial system with its special activation requirements pore opening is not sufficient for ClpP deregulation. ADEPs neither hydrolyze ATP nor actively translocate proteins into the proteolytic chamber. If this ATP-dependent translocation process conducted by the Clp-ATPase partners contributes to activation of ClpP1P2, as proposed by Schmitz and Sauer (Schmitz and Sauer, 2014), then it may explain why ADEPs do not unleash non-specific protein degradation in mycobacteria as they do in firmicutes and proteobacteria. Our observation that ADEPs do not stimulate mycobacterial ClpP1P2 to degrade FtsZ, unlike they do with ClpP from other bacteria (Sass et al., 2011), supports the notion that ADEP does not over-activate mycobacterial ClpP as severely as it does with other ClpP homologs. Although, without proteome analyses, we cannot exclude that other proteins might become targeted and degraded in mycobacteria by deregulated ClpP1P2/ADEP, especially at elevated ADEP concentrations, our current results exclude this as the primary cause of mycobacterial death.
In mycobacteria ADEP toxicity results primarily from prevention of the physiological functions of the ClpP1P2/Clp-ATPase complex. When ADEP occupies the hydrophobic pockets of ClpP, the Clp-ATPases can no longer bind. Even pre-assembled ClpP/Clp-ATPase complexes were shown to disassemble upon ADEP addition (Kirstein et al., 2009), because of competition between ADEP and the I/LGF/L loops for the same ClpP binding sites. Consequently, ADEP prevents ClpP and Clp-ATPases from interacting to degrade their folded protein substrates. Decreased interaction was described for B. subtilis ClpCP and ClpXP, E. coli ClpAP and ClpXP (Kirstein et al., 2009; Lee et al., 2010; Leung et al., 2011) as well as for M. tuberculosis ClpXP1P2 (Schmitz et al., 2014). Here, we show that ADEPs also block the interaction between ClpC1 and ClpP1P2. While in all bacteria investigated thus far, ADEPs can prevent the Clp-ATPase/peptidase systems from performing their physiological functions, in most species, this does not cause growth defects. For instance, in B. subtilis clpP deletion has pleiotropic effects, including impaired sporulation, loss of genetic competence and motility, heat sensitivity and reduced survival in stationary phase (Msadek et al., 1998; Gerth et al., 1998; Gerth et al., 2004), while in S. aureus virulence is strongly reduced in the absence of functional ClpP (Frees et al., 2014). However, under moderate growth conditions in rich media, ClpP is dispensable in these firmicutes, although it might rapidly lead to suppressor mutations such as in spx, a toxic ClpP substrate in B. subtilis (Nakano et al., 2002a; Nakano et al., 2002b).
In contrast, in M. tuberculosis all components of the Clp protease machinery, i.e. ClpP1, ClpP2, ClpC1, and ClpX, are essential for growth (Sassetti et al., 2003; Griffin et al., 2011; Ollinger et al., 2012; Raju et al., 2012b) probably by proteolytically preventing the accumulation of global transcription factors and other toxic proteins (Raju et al., 2014). One such important ClpP substrate in mycobacteria is the transcription factor WhiB1, whose accumulation was shown to be lethal to M. tuberculosis (Raju et al., 2014). A second likely substrate for ClpP1P2 is CarD (Raju et al., 2014). Although CarD accumulation is not directly lethal, it was shown to play a role in the stringent response controlling rRNA transcription (Stallings et al., 2009) and could, thus, slow down vegetative growth. Evidence that disturbing the function of the Clp protease complex leads to cell death in mycobacteria comes also from the studies of three cyclic peptide antibiotics, all of which target ClpC1. Cyclomarin, by binding to the ATPase, increased hydrolysis of a model protein in M. smegmatis by a still unknown mechanism (Schmitt et al., 2011), while lassomycin (Gavrish et al., 2014) and ecumicin (Gao et al., 2014) were shown to stimulate ATP-hydrolysis by ClpC1, while uncoupling it from protein degradation by ClpP1P2.
In summary, by docking to the hydrophobic pockets of ClpP, the ADEPs 1) release ClpP from its regulatory constraints in e.g. firmicutes, setting ClpP free to work as an independent protease, but 2) they also prevent the physiological functions of the Clp protease system in protein homeostasis, which leads to cell death in species where Clp-mediated proteolysis is essential for viability. The present findings further validate prior indications that mycobacterial ClpP is a promising antitubercular drug target (Schmitt et al., 2011; Akopian et al., 2012; Raju et al., 2012b; Gavrish et al., 2014; Gao et al., 2014) and demonstrate that ClpP1P2 is druggable, i.e. that its function can be blocked by a chemical agent to prevent mycobacterial growth. Development of ADEPs as potential antitubercular agents will require compound optimization including improved stability, solubility, oral bioavailability, and reduced efflux. Our data indicate that among our small collection of congeners, the most potent derivatives against the isolated mycobacterial enzyme (ADEP4, 7 and 8) were inferior to ADEP2 against whole cells. As in most other drug optimization programs, target affinity is only one critical parameter. Reaching sufficient intracellular concentrations is equally important and ADEP2 might have an advantage here due to better uptake or lower efflux compared to the other congeners. In general, ADEPs will probably benefit from the combination approach that is widely used in tuberculosis therapy. Rifampicin, which was shown to act synergistically with ADEP against S. aureus (Conlon et al., 2013), is widely used against M. tuberculosis and another interesting option could be a combination of ADEP with streptomycin, as down-regulation of clpP1P2 expression in M. smegmatis increased susceptibility against aminoglycosides (Raju et al., 2012b).
Experimental procedures
Bacterial strains and growth conditions
B. subtilis 168 (Anagnostopoulos and Spizizen, 1961), B. subtilis ΔclpP (Msadek et al., 1998) and B. subtilis pX2-clpP (Gerth et al., 1998) were cultured in Mueller-Hinton broth or on Mueller-Hinton agar plates at 37 °C. Liquid cultures of M. smegmatis mc2155 (Snapper et al., 1990), M. tuberculosis H37Rv, M. bovis BCG Pasteur (Institute Pasteur) and M. bovis BCG clpP1-tetoff (this study) were cultured in 10 ml minimal medium (Yam et al., 2009) at 37 °C and 80 rpm. Selection pressure in M. bovis BCG clpP1-tetoff was applied by 50 μg ml−1 hygromycin B (Roth) and 20 μg ml−1 kanamycin (Sigma). Mycobacteria were also cultured on Middlebrook 7H10 (BD) agar plates containing ADS (50 mg ml−1 BSA, 8.1 mg ml−1 NaCl, 20 mg ml−1 glucose) and OADC Enrichment (BD). Agar plates streaked with M. smegmatis were incubated for 2 days and agar plates with M. bovis BCG and M. bovis BCG clpP1-tetoff were incubated for 3 weeks and 5 weeks, respectively, at 37 °C and 5% CO2.
Construction of the conditional M. bovis BCG clpP1-tetoff mutant
For establishing regulated expression of the clpP1 gene, a synthetic gene cassette (hyg-Pmyc1-4xtetO; M. Alber and R. Kalscheuer, unpublished results) comprising a hygromycin resistance gene and the Pmyc1 promoter from M. smegmatis engineered to contain four tetO operator sites (serving as the DNA binding sites for the cognate repressor protein TetR) was inserted immediately upstream of the clpP1 start codon in M. bovis BCG Pasteur. Targeted gene knock-in was achieved by specialized transduction employing temperature-sensitive mycobacteriophages essentially as described previously (Bardarov et al., 2002). Briefly, for generation of allelic exchange constructs for site-specific insertion in M. bovis BCG of the hyg-Pmyc1-4xtetO cassette, upstream- and downstream DNA regions flanking the clpP1 start codon were amplified by PCR employing the primer clpP1-F1-fwd and clpP1-F1-rev as well as clpP1-F2-fwd and clpP1-F2-rev (Table S1). Subsequently, the upstream and downstream flanks were digested with the indicated restriction enzymes, and ligated with Van91I-digested pcRv1327c-4xtetO vector arms (M. Alber and R. Kalscheuer, unpublished results). The resulting knock-in plasmid was then linearized with PacI and cloned and packaged into the temperature-sensitive phage phAE159 (J. Kriakov and W. R. Jacobs, Jr., unpublished results), yielding a knock-in phage which was propagated in M. smegmatis at 30 °C. Allelic exchange in M. bovis BCG using the knock-in phage at the non-permissive temperature of 37 °C was achieved by specialized transduction using hygromycin (50 μg ml−1) for selection, resulting in site-specific insertion of the hyg-Pmyc1-4xtetO cassette (Fig. S4). The obtained BCG knock-in mutant c-clpP1 was verified by PCR, using the primer pair c-clpP1-fwd and c-clpP1-rev followed by sequencing of the PCR product with the primer seq-clpP1-fwd and seq-clpP2-rev. For achieving controlled gene expression of the target gene clpP1, a synthetic gene (rev-tetR) derived from Tn10 tetR encoding a mutated TetR protein with reversed binding affinity to tetO sites upon binding of tetracycline (Klotzsche et al., 2009) was heterologously expressed in the knock-in mutant. For this, the rev-tetR gene was amplified by PCR employing the primer pair rev-tetR-fwd and rev-tetR-rev (Table S1) and using the plasmid pTC-28S15-0X (Addgene plasmid 20316) as a template and cloned using the restriction enzymes EcoRI and HindIII into the episomal E. coli-mycobacterium shuttle plasmid pMV261-RBS-D, which is a derivative of plasmid pMV261 (Stover et al., 1991) harbouring a mutated ribosome binding site (M. Alber and R. Kalscheuer, unpublished results). The resulting plasmid pMV261::rev-tetR-RBS-D providing constitutive gene expression from the HSP60 promoter in mycobacteria was transformed by electroporation into the M. bovis BCG c-clpP1 knock-in mutant using solid medium containing 50 μg ml−1 hygromycin and 20 μg ml−1 kanamycin for selection. This yielded the conditional mutant BCG c-clpP1 pMV261::rev-tetR-RBS-D (here referred to as M. bovis BCG clpP1-tetoff).
Growth analyses
M. bovis BCG wt and M. bovis BCG clpP1-tetoff were grown as described above with increasing ATc concentrations (0, 0.05, 0.075, 0.1, 0.2, 0.5, 1 and 2 ng ml−1). At distinct points in time 30 μl samples were retrieved and serial dilutions were plated on Middlebrook 7H10 plates, containing ADS and OADC. After 3–5 weeks incubation at 37 °C and 5% CO2, colony forming units were counted.
MIC determination
MICs were determined by broth microdilution in 96-well plates using minimal medium (Yam et al., 2009) for mycobacteria and Mueller-Hinton broth (BD) for B. subtilis. ADEPs were diluted in DMSO. Isoniazid or apramycin were diluted in H2O. ADEPs were synthesized as described previously (Hinzen et al., 2006). Colonies of B. subtilis 168 or M. smegmatis mc2155 were diluted in 0.9% NaCl and the optical density at 600 nm (OD600nm) was measured.
Pre-cultures of M. tuberculosis H37Rv, M. bovis BCG wt and M. bovis BCG clpP1-tetoff were grown as described above, containing either no ATc or 0.1 ng ml−1 ATc, until an OD600nm of 0.6–1 was reached. All bacteria were diluted in medium to 1×105 cfu ml−1 and 50 μl of the inoculum was added per well. The cells were incubated for 9 days (M. bovis and M. tuberculosis) or 2 days (M. smegmatis) at 37 °C and 5% CO2 followed by addition of resazurin (10 μl; 100 μg ml−1) (Applichem) and fluorescence measurement (560 nmex, 600 nmem) at day 10. B. subtilis was incubated for 16–18 h at 37 °C in ambient air and the MIC was determined as the absence of visible bacterial growth according to CLSI (Clinical and Laboratory Standards Institute) standards (CLSI M07-09, 2012).
Isolation of total RNA
Cultures (10 ml) of M. bovis BCG wt or clpP1-tetoff (OD600nm 0.7–1), both grown either in the absence or in the presence of 0.1 ng ml−1 ATc, were pelleted and incubated overnight in 1 ml RNA protect (Qiagen) at room temperature (RT). After storing the samples at −80 °C, RNA was isolated with the RNeasy Kit (Qiagen), including on column DNase I digestion. Further DNA digestion was performed with the Turno DNase (Life Technologies) followed by concentration of the samples with the NucleoSpin RNA Clean-up Kit XS (Macherey-Nagel). Quality and quantity of the RNA were controlled by gel electrophoresis (3% agarose) and by absorption spectra measurements using the Nanodrop spectrophotometer (Thermo Scientific).
Preparation of cell lysates and Western blotting
Cultures of M. bovis BCG (8 ml, OD600nm 0.7–1) was pelleted and lysed in 500 μl PBS buffer containing 0.05% Tween 80 and glass beads (150–212 μm, Sigma-Aldrich) using the Precellys 24 homogenizer (Belkin/Peqlab). Protein concentration of the lysate was determined by measuring the absorbance at 280 nm using the Nanodrop spectrophotometer. Lysates were diluted, mixed with 4x LDS sample buffer (Thermo Scientific) and incubated for 10 min at 99 °C. Equal protein concentrations of all lysates were confirmed by SDS-PAGE. Proteins were transferred to an Amersham Hybond-ECL membrane (GE Healthcare) via semi-dry blotting. The membrane was incubated with polyclonal antiserum against FtsZ (Dziadek et al., 2002) or ClpP2 of M. tuberculosis followed by incubation with anti-rabbit IgG horse radish peroxidase-linked antibody (Cell signaling) as secondary antibody. Detection was performed with Amersham ECL prime western blotting detection reagent (GE Healthcare).
Protein expression and purification
MTB ClpP1 and MTB ClpP2 were expressed separately in M. smegmatis mc2155 and purified as previously described (Akopian et al., 2012). BS ClpP and BS FtsZ were expressed in E. coli and were purified as described earlier (Sass et al., 2011). MTB FtsZ was cloned via NcoI and HindIII restriction sites into the IPTG-inducible expression vector pET22bΔpelB (Sass and Bierbaum, 2007) and was expressed in E. coli BL21 (DE3) with a C-terminal 6xHis tag. Genomic DNA from M. bovis BCG was used as PCR template, as the ftsZ gene of this non-pathogenic model organism shows 100% nucleotide sequence identity to ftsZ of M. tuberculosis. MTB ftsZ-fwd and MTB ftsZ-rev (Table S1) served as primers. Protein expression was induced at an OD600 of 0.6 with 1 mM IPTG for 5 h at 37 °C. Cells were lysed in 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8 using a French Press. FtsZ-His6 was purified by Ni-NTA column chromatography and eluted with 50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazol. For storage 5% glycerol was added to the eluate.
Formation of the MTB ClpP1P2 tetradecamer
For the formation of catalytically active MTB ClpP1P2, equal volumes of MTB ClpP1 and MTB ClpP2 were mixed with 5 mM Z-LL and were then incubated for 4 h at RT. After confirming peptidase activity the active enzyme was stored at 4 °C.
Degradation of fluorescent peptides and proteins
Degradation assays were performed in 96-well plates as previously described (Akopian et al., 2012). For short peptide substrates, 0.1 mM Z-GGL-amc or 0.1 mM suc-LY-amc (Enzo life science, USA) were used with 1.5 μg ml−1 MTB ClpP1P2. For more complex peptide model substrates, 10 μM of the three-generation peptides were used with 0.8 μg ml−1 MTB ClpP1P2. Fluorescence of amc was measured in a SpectraMax M5 plate reader (Molecular Devices) at 380 nmex and 460 nmem. As protein substrate, 4 μg ml−1 FITC-casein (Mobitec) was employed with either 6.25 μg ml−1 MTB ClpP1P2 or 2 μg ml−1 BS ClpP. Here, fluorescence was measured at 492 nmex and 518 nmem. All assays were performed in 80 μl buffer A (50 mM phosphate buffer, pH 7.6; 100 mM KCl, 5% glycerol). Z-LL was used at a concentration of 5 mM. If not indicated otherwise ADEP was applied at 100 μg ml−1.
Competitive assays with BS ClpP
To investigate potential kinetic interactions between Z-LL and either peptide substrate or ADEP we used BS ClpP and suc-LY-amc in 96-well plates with 3 μM BS ClpP-His6 in a previously described activity buffer (50 mM TrisHCl, pH8; 100 mM KCl, 25 mM MgCl2, 2 mM DTT) (Turgay et al., 1998). To study Z-LL in competition with a substrate, mixtures were prepared in activity buffer containing 0, 500 or 2500 μM Z-LL, each supplemented with a constant amount of 3 μM BS ClpP. A serial dilution of suc-LY-amc with final concentrations ranging from 0–5000 μM was added to start the reaction. To study Z-LL together with ADEP2 a serial dilution of ADEP2 with final concentrations ranging from 0–60 μM was mixed with a constant amount of 3 μM BS ClpP in activity buffer. Premixes of 0, 500 or 2500 μM Z-LL and a constant amount of 30 μM of suc-LY-amc were added to start the reaction. Fluorescence was measured using the Infinite M200pro plate reader (Tecan) with 380 nmex and 430 nmem. The data was analyzed via Michaelis Menten fittings using Graph Pad Prism software.
Degradation of unlabeled protein substrates
Degradation assays with unlabeled proteins were performed using either 6.25 μg ml−1 MTB ClpP1P2 or 2 μg ml−1 BS ClpP in 30 μl of buffer A as described above. As substrates, 8 μg ml−1 Tau, 7.5 μg ml−1 FtsZ, or 7.5 μg ml−1 casein were used. Samples were incubated for 30–120 min at 37 °C in the absence or presence of 50 μg ml−1 ADEP before stopping the reaction with 4x LDS sample buffer 5.0. The degradation of substrates was analyzed by SDS-PAGE.
HPLC analyses
To determine Z-LL stability in the presence of ClpP, Z-LL (1 mM) was incubated with 3 μM BS ClpP in 1 ml activity buffer at 37 °C. Immediately and after 3 h aliquots of 50 μl were analyzed using a 1100 series HPLC (Agilent) with an EC 250/3 Nucleodur C18 HTec column of 5 μm diameter (Macherey-Nagel) and the following gradient of methanol (solvent A) and water (solvent B): 0–10 min 10% solvent A; 10–40 min 10 to 100% solvent A; 40–50 min 100% solvent A. To determine ADEP stability in aqueous culture broth over time, 16 μg ml−1 ADEP2 was incubated in 2 ml minimal medium (Yam et al., 2009) for 10 days at 37 °C. At distinct points in time 100 μl aliquots were analyzed via HPLC using the following gradient of methanol (solvent A) and water (solvent B): 0–5 min 0% solvent A; 5–10 min 0 to 60% solvent A; 10–40 min 60 to 100% solvent A; 40–50 min 100% solvent A. The data was analyzed via ChemStation software (Agilent).
Supplementary Material
Acknowledgments
We thank Anne Wochele for expert technical assistance. We are grateful to Holger Paulsen and Siegfried Raddatz (Bayer Pharma AG, Wuppertal) for ADEP synthesis, to Craig M. Crews (Yale University, New Haven) for providing the branched peptide, to Eckhard Mandelkow (DESY, Hamburg) for sharing Tau protein, to Malini Rajagopalan (University of Texas Health Science Center) for providing the polyclonal antiserum against MTB FtsZ, to D. Schnappinger (Weill Cornell Medical College, NY) for sharing plasmid pTC-28S15-0X, and to U. Gerth (University of Greifswald) for providing pX2-clpP. This work was supported by a grant of the German Research Foundation (DFG; FOR854, BR 3783/1-2) to H.B.O. and P.S. and by a grant of the Jürgen Manchot Foundation to R.K. H.R.S. reports holding stock options in AiCuris;
Footnotes
all other authors declare no competing financial interests.
References
- Akopian T, Kandror O, Raju RM, Unnikrishnan M, Rubin EJ, Goldberg AL. The active ClpP protease from M. tuberculosis is a complex composed of a heptameric ClpP1 and a ClpP2 ring. EMBO J. 2012;31:1529–1541. doi: 10.1038/emboj.2012.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulos J, Ahsan B, Homchaudhuri L, Husain N, Cheng YQ, Ortega J. Structural determinants stabilizing the axial channel of ClpP for substrate translocation. Mol Microbiol. 2013;90:167–180. doi: 10.1111/mmi.12356. [DOI] [PubMed] [Google Scholar]
- Anagnostopoulos C, Spizizen J. Requirements for transformation in Bacillus subtilis. J Bacteriol. 1961;81:741–746. doi: 10.1128/jb.81.5.741-746.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker TA, Sauer RT. ClpXP, an ATP-powered unfolding and protein-degradation machine. Biochim Biophys Acta. 2012;1823:15–28. doi: 10.1016/j.bbamcr.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balganesh TS, Alzari PM, Cole ST. Rising standards for tuberculosis drug development. Trends Pharmacol Sci. 2008;29:576–581. doi: 10.1016/j.tips.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Bardarov S, Bardarov Jr S, Jr, Pavelka MSJ, Jr, Sambandamurthy V, Larsen M, Tufariello J, et al. Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology. 2002;148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- Behr MA, Wilson MA, Gill WP, Salamon H, Schoolnik GK, Rane S, Small PM. Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science. 1999;284:1520–1523. doi: 10.1126/science.284.5419.1520. [DOI] [PubMed] [Google Scholar]
- Benaroudj N, Raynal B, Miot M, Ortiz-Lombardia M. Assembly and proteolytic processing of mycobacterial ClpP1 and ClpP2. BMC Biochem. 2011;12:61. doi: 10.1186/1471-2091-12-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brötz-Oesterhelt H, Beyer D, Kroll HP, Endermann R, Ladel C, Schroeder W, et al. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat Med. 2005;11:1082–1087. doi: 10.1038/nm1306. [DOI] [PubMed] [Google Scholar]
- Brötz-Oesterhelt H, Sass P. Bacterial caseinolytic proteases as novel targets for antibacterial treatment. Int J Med Microbiol. 2014;304:23–30. doi: 10.1016/j.ijmm.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Buckley DL, Corson TW, Aberle N, Crews CM. HIV protease-mediated activation of sterically capped proteasome inhibitors and substrates. J Am Chem Soc. 2011;133:698–700. doi: 10.1021/ja109377p. [DOI] [PubMed] [Google Scholar]
- Buriankova K, Doucet-Populaire F, Dorson O, Gondran A, Ghnassia JC, Weiser J, Pernodet JL. Molecular basis of intrinsic macrolide resistance in the Mycobacterium tuberculosis complex. Antimicrob Agents Chemother. 2004;48:143–150. doi: 10.1128/AAC.48.1.143-150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calligaro GL, Moodley L, Symons G, Dheda K. The medical and surgical treatment of drug-resistant tuberculosis. J Thorac Dis. 2014;6:186–195. doi: 10.3978/j.issn.2072-1439.2013.11.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney DW, Schmitz KR, Truong JV, Sauer RT, Sello JK. Restriction of the conformational dynamics of the cyclic acyldepsipeptide antibiotics improves their antibacterial activity. J Am Chem Soc. 2014;136:1922–1929. doi: 10.1021/ja410385c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen DZ, Patel DV, Hackbarth CJ, Wang W, Dreyer G, Young DC, et al. Actinonin, a naturally occurring antibacterial agent, is a potent deformylase inhibitor. Biochemistry. 2000;39:1256–1262. doi: 10.1021/bi992245y. [DOI] [PubMed] [Google Scholar]
- CLSI M07-09. Methods for dilution antimicrobial susceptibility tests for bacteria that grow; approved standard. (ninth) 2012 [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- Conlon BP, Nakayasu ES, Fleck LE, LaFleur MD, Isabella VM, Coleman K, et al. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature. 2013;503:365–370. doi: 10.1038/nature12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daffe M, Draper P. The envelope layers of mycobacteria with reference to their pathogenicity. Adv Microb Physiol. 1998;39:131–203. doi: 10.1016/s0065-2911(08)60016-8. [DOI] [PubMed] [Google Scholar]
- Dziadek J, Madiraju MV, Rutherford SA, Atkinson MA, Rajagopalan M. Physiological consequences associated with overproduction of Mycobacterium tuberculosis FtsZ in mycobacterial hosts. Microbiology. 2002;148:961–971. doi: 10.1099/00221287-148-4-961. [DOI] [PubMed] [Google Scholar]
- Fattorini L, Piccaro G, Mustazzolu A, Giannoni F. Targeting dormant bacilli to fight tuberculosis. Mediterr J Hematol Infect Dis. 2013;5:e2013072. doi: 10.4084/MJHID.2013.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frees D, Gerth U, Ingmer H. Clp chaperones and proteases are central in stress survival, virulence and antibiotic resistance of Staphylococcus aureus. Int J Med Microbiol. 2014;304:142–149. doi: 10.1016/j.ijmm.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Gao W, Kim JY, Chen SN, Cho SH, Choi J, Jaki BU, et al. Discovery and characterization of the tuberculosis drug lead ecumicin. Org Lett. 2014;16(23):6044–6047. doi: 10.1021/ol5026603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrish E, Sit CS, Cao S, Kandror O, Spoering A, Peoples A, et al. Lassomycin, a ribosomally synthesized cyclic peptide, kills mycobacterium tuberculosis by targeting the ATP-dependent protease ClpC1P1P2. Chem Biol. 2014;21:509–518. doi: 10.1016/j.chembiol.2014.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth U, Kirstein J, Mostertz J, Waldminghaus T, Miethke M, Kock H, Hecker M. Fine-tuning in regulation of Clp protein content in Bacillus subtilis. J Bacteriol. 2004;186:179–191. doi: 10.1128/JB.186.1.179-191.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth U, Kruger E, Derre I, Msadek T, Hecker M. Stress induction of the Bacillus subtilis clpP gene encoding a homologue of the proteolytic component of the Clp protease and the involvement of ClpP and ClpX in stress tolerance. Mol Microbiol. 1998;28:787–802. doi: 10.1046/j.1365-2958.1998.00840.x. [DOI] [PubMed] [Google Scholar]
- Goldman RC, Plumley KV, Laughon BE. The evolution of extensively drug resistant tuberculosis (XDR-TB): history, status and issues for global control. Infect Disord Drug Targets. 2007;7:73–91. doi: 10.2174/187152607781001844. [DOI] [PubMed] [Google Scholar]
- Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLOS Pathog. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas M, Beyer D, Gahlmann R, Freiberg C. YkrB is the main peptide deformylase in Bacillus subtilis, a eubacterium containing two functional peptide deformylases. Microbiology. 2001;147:1783–1791. doi: 10.1099/00221287-147-7-1783. [DOI] [PubMed] [Google Scholar]
- Hinzen B, Raddatz S, Paulsen H, Lampe T, Schumacher A, Häbich D, et al. Medicinal chemistry optimization of acyldepsipeptides of the enopeptin class antibiotics. ChemMedChem. 2006;1:689–693. doi: 10.1002/cmdc.200600055. [DOI] [PubMed] [Google Scholar]
- Ingvarsson H, Mate MJ, Hogbom M, Portnoi D, Benaroudj N, Alzari PM, et al. Insights into the inter-ring plasticity of caseinolytic proteases from the X-ray structure of Mycobacterium tuberculosis ClpP1. Acta Crystallogr D Biol Crystallogr. 2007;63:249–259. doi: 10.1107/S0907444906050530. [DOI] [PubMed] [Google Scholar]
- Ji Y, Yin D, Fox B, Holmes DJ, Payne D, Rosenberg M. Validation of antibacterial mechanism of action using regulated antisense RNA expression in Staphylococcus aureus. FEMS Microbiol Lett. 2004;231:177–184. doi: 10.1016/S0378-1097(03)00931-5. [DOI] [PubMed] [Google Scholar]
- Kirstein J, Hoffmann A, Lilie H, Schmidt R, Rübsamen-Waigmann H, Brötz-Oesterhelt H, et al. The antibiotic ADEP reprogrammes ClpP, switching it from a regulated to an uncontrolled protease. EMBO Mol Med. 2009;1:37–49. doi: 10.1002/emmm.200900002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotzsche M, Ehrt S, Schnappinger D. Improved tetracycline repressors for gene silencing in mycobacteria. Nucleic Acids Res. 2009;37:1778–1788. doi: 10.1093/nar/gkp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress W, Maglica Z, Weber-Ban E. Clp chaperone-proteases: structure and function. Res Microbiol. 2009;160:618–628. doi: 10.1016/j.resmic.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Lee BG, Park EY, Lee KE, Jeon H, Sung KH, Paulsen H, et al. Structures of ClpP in complex with acyldepsipeptide antibiotics reveal its activation mechanism. Nat Struct Mol Biol. 2010;17:471–478. doi: 10.1038/nsmb.1787. [DOI] [PubMed] [Google Scholar]
- Leung E, Datti A, Cossette M, Goodreid J, McCaw SE, Mah M, et al. Activators of cylindrical proteases as antimicrobials: identification and development of small molecule activators of ClpP protease. Chem Biol. 2011;18:1167–1178. doi: 10.1016/j.chembiol.2011.07.023. [DOI] [PubMed] [Google Scholar]
- Li DH, Chung YS, Gloyd M, Joseph E, Ghirlando R, Wright GD, et al. Acyldepsipeptide antibiotics induce the formation of a structured axial channel in ClpP: A model for the ClpX/ClpA-bound state of ClpP. Chem Biol. 2010;17:959–969. doi: 10.1016/j.chembiol.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Kandror O, Akopian T, Dharkar P, Wlodawer A, Maurizi MR, Goldberg AL. Structure and functional properties of the active form of the proteolytic complex, ClpP1P2, from Mycobacterium tuberculosis. J Biol Chem. 2016 doi: 10.1074/jbc.M115.700344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Ologbenla A, Houry WA. Dynamics of the ClpP serine protease: A model for self-compartmentalized proteases. Crit Rev Biochem Mol Biol. 2014;49:400–412. doi: 10.3109/10409238.2014.925421. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Louw GE, Warren RM, Gey van Pittius NC, McEvoy CR, van Helden PD, Victor TC. A balancing act: efflux/influx in mycobacterial drug resistance. Antimicrob Agents Chemother. 2009;53:3181–3189. doi: 10.1128/AAC.01577-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merget B, Zilian D, Muller T, Sotriffer CA. MycPermCheck: the Mycobacterium tuberculosis permeability prediction tool for small molecules. Bioinformatics. 2013;29:62–68. doi: 10.1093/bioinformatics/bts641. [DOI] [PubMed] [Google Scholar]
- Michel KH, Kastner RE. A54556 antibiotics and process for production thereof. US patent 4492650 1982
- Molière N, Turgay K. Chaperone-protease systems in regulation and protein quality control in Bacillus subtilis. Res Microbiol. 2009;160:637–644. doi: 10.1016/j.resmic.2009.08.020. [DOI] [PubMed] [Google Scholar]
- Msadek T, Dartois V, Kunst F, Herbaud ML, Denizot F, Rapoport G. ClpP of Bacillus subtilis is required for competence development, motility, degradative enzyme synthesis, growth at high temperature and sporulation. Mol Microbiol. 1998;27:899–914. doi: 10.1046/j.1365-2958.1998.00735.x. [DOI] [PubMed] [Google Scholar]
- Nakano MM, Nakano S, Zuber P. Spx (YjbD), a negative effector of competence in Bacillus subtilis, enhances ClpC-MecA-ComK interaction. Mol Microbiol. 2002a;44:1341–1349. doi: 10.1046/j.1365-2958.2002.02963.x. [DOI] [PubMed] [Google Scholar]
- Nakano S, Zheng G, Nakano MM, Zuber P. Multiple pathways of Spx (YjbD) proteolysis in Bacillus subtilis. J Bacteriol. 2002b;184:3664–3670. doi: 10.1128/JB.184.13.3664-3670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nde CW, Toghrol F, Jang HJ, Bentley WE. Toxicogenomic response of Mycobacterium bovis BCG to peracetic acid and a comparative analysis of the M. bovis BCG response to three oxidative disinfectants. Appl Microbiol Biotechnol. 2011;90:277–304. doi: 10.1007/s00253-010-2931-6. [DOI] [PubMed] [Google Scholar]
- Ollinger J, O’Malley T, Kesicki EA, Odingo J, Parish T. Validation of the essential ClpP protease in Mycobacterium tuberculosis as a novel drug target. J Bacteriol. 2012;194:663–668. doi: 10.1128/JB.06142-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Personne Y, Brown AC, Schuessler DL, Parish T. Mycobacterium tuberculosis ClpP proteases are co-transcribed but exhibit different substrate specificities. PLoS One. 2013;8:e60228. doi: 10.1371/journal.pone.0060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju RM, Goldberg AL, Rubin EJ. Bacterial proteolytic complexes as therapeutic targets. Nat Rev Drug Discov. 2012a;11:777–789. doi: 10.1038/nrd3846. [DOI] [PubMed] [Google Scholar]
- Raju RM, Jedrychowski MP, Wei JR, Pinkham JT, Park AS, O’Brien K, et al. Post-translational regulation via Clp protease is critical for survival of Mycobacterium tuberculosis. PLOS Pathog. 2014;10:e1003994. doi: 10.1371/journal.ppat.1003994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raju RM, Unnikrishnan M, Rubin D, Krishnamoorthy V, Kandror O, Akopian T, et al. Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLOS Pathog. 2012b;8(2):e1002511. doi: 10.1371/journal.ppat.1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson BD, Altmann D, Barry C, Bishai B, Cole S, Dick T, et al. Detection and treatment of subclinical tuberculosis. Tuberculosis (Edinb) 2012;92:447–452. doi: 10.1016/j.tube.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Sass P, Bierbaum G. Lytic activity of recombinant bacteriophage phi11 and phi12 endolysins on whole cells and biofilms of Staphylococcus aureus. Appl Environ Microbiol. 2007;73:347–352. doi: 10.1128/AEM.01616-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sass P, Josten M, Famulla K, Schiffer G, Sahl HG, Hamoen L, Brötz-Oesterhelt H. Antibiotic acyldepsipeptides activate ClpP peptidase to degrade the cell division protein FtsZ. Proc Natl Acad Sci U S A. 2011;108:17474–17479. doi: 10.1073/pnas.1110385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH, Rubin EJ. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- Schmitt EK, Riwanto M, Sambandamurthy V, Roggo S, Miault C, Zwingelstein C, et al. The natural product cyclomarin kills Mycobacterium tuberculosis by targeting the ClpC1 subunit of the caseinolytic protease. Angew Chem Int Ed Engl. 2011;50:5889–5891. doi: 10.1002/anie.201101740. [DOI] [PubMed] [Google Scholar]
- Schmitz KR, Carney DW, Sello JK, Sauer RT. Crystal structure of Mycobacterium tuberculosis ClpP1P2 suggests a model for peptidase activation by AAA+ partner binding and substrate delivery. Proc Natl Acad Sci U S A. 2014;111:E4587–E4595. doi: 10.1073/pnas.1417120111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz KR, Sauer RT. Substrate delivery by the AAA+ ClpX and ClpC1 unfoldases activates the mycobacterial ClpP1P2 peptidase. Mol Microbiol. 2014;93:617–628. doi: 10.1111/mmi.12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR., Jr Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol. 1990;4:1911–1919. doi: 10.1111/j.1365-2958.1990.tb02040.x. [DOI] [PubMed] [Google Scholar]
- Stallings CL, Stephanou NC, Chu L, Hochschild A, Nickels BE, Glickman MS. CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell. 2009;138:146–159. doi: 10.1016/j.cell.2009.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, et al. New use of BCG for recombinant vaccines. Nature. 1991;351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- Turgay K, Hahn J, Burghoorn J, Dubnau D. Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J. 1998;17:6730–6738. doi: 10.1093/emboj/17.22.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan D, Rao SP, Noble CG. Structural basis of mycobacterial inhibition by cyclomarin A. J Biol Chem. 2013;288:30883–30891. doi: 10.1074/jbc.M113.493767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam KC, D’Angelo I, Kalscheuer R, Zhu H, Wang JX, Snieckus V, et al. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLOS Pathog. 2009;5:e1000344. doi: 10.1371/journal.ppat.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AY, Houry WA. ClpP: a distinctive family of cylindrical energy-dependent serine proteases. FEBS Lett. 2007;581:3749–3757. doi: 10.1016/j.febslet.2007.04.076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.