

ABSTRACT
Hepatitis C virus (HCV) requires multiple receptors for its attachment to and entry into cells. Our previous studies found that human syndecan-1 (SDC-1), SDC-2, and T cell immunoglobulin and mucin domain-containing protein 1 (TIM-1) are HCV attachment receptors. Other cell surface molecules, such as CD81, Claudin-1 (CLDN1), Occludin (OCLN), SR-BI, and low-density lipoprotein receptor (LDLR), function mainly at postattachment steps and are considered postattachment receptors. The underlying molecular mechanisms of different receptors in HCV cell-free and cell-to-cell transmission remain elusive. In the present study, we used a clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 technology, gene-specific small interfering RNAs, and a newly developed luciferase-based reporter system to quantitatively determine the importance of individual receptors in HCV cell-free and cell-to-cell transmission. Knockouts of SDC-1 and SDC-2 resulted in remarkable reductions of HCV infection and cell attachment, whereas SDC-3 and SDC-4 knockouts did not affect HCV infection. Defective HCV attachment to SDC-1 and/or SDC-2 knockout cells was completely restored by SDC-1 and SDC-2 but not SDC-4 expression. Knockout of the attachment receptors SDC-1, SDC-2, and TIM-1 also modestly decreased HCV cell-to-cell transmission. In contrast, silencing and knockout of the postattachment receptors CD81, CLDN1, OCLN, SR-BI, and LDLR greatly impaired both HCV cell-free and cell-to-cell transmission. Additionally, apolipoprotein E was found to be important for HCV cell-to-cell spread, but very-low-density lipoprotein (VLDL)-containing mouse serum did not affect HCV cell-to-cell transmission, although it inhibited cell-free infection. These findings demonstrate that attachment receptors are essential for initial HCV binding and that postattachment receptors are important for both HCV cell-free and cell-to-cell transmission.
IMPORTANCE The importance and underlying molecular mechanisms of cell surface receptors in HCV cell-free and cell-to-cell transmission are poorly understood. The role of some of the HCV attachment and postattachment receptors in HCV infection and cell-to-cell spread remains controversial. Using CRISPR-Cas9-mediated knockouts of specific cellular genes, we demonstrate that both SDC-1 and SDC-2, but not SDC-3 or SDC-4, are bona fide HCV attachment receptors. We also used a newly developed luciferase-based reporter system to quantitatively determine the importance of attachment and postattachment receptors in HCV cell-to-cell transmission. SDC-1, SDC-2, TIM-1, and SR-BI were found to modestly promote HCV cell-to-cell spread. CD81, CLDN1, OCLN, and LDLR play more important roles in HCV cell-to-cell transmission. Likewise, apolipoprotein E (apoE) is critically important for HCV cell-to-cell spread, unlike VLDL-containing mouse serum, which did not affect HCV cell-to-cell spread. These findings suggest that the mechanism(s) of HCV cell-to-cell spread differs from that of cell-free infection.
KEYWORDS: hepatitis C virus, attachment receptor, postattachment receptor, infection, cell-to-cell transmission, cell-free infection
INTRODUCTION
Hepatitis C virus (HCV) is a hepatotropic virus, resulting in acute and chronic hepatitis, cirrhosis, and hepatocellular carcinoma (HCC) in humans. It contains a single-stranded and positive-sense RNA genome and a cellular membrane-derived envelope. The viral RNA genome is approximately 9.6 kb long and is composed of the 5′ untranslated region (UTR), a large open reading frame (ORF) encoding a polyprotein precursor of about 3,000 amino acids, and the 3′ UTR (1). The viral polyprotein is co- and/or posttranslationally cleaved by cellular peptidases and viral proteases into structural (core, E1, E2, and p7) and nonstructural (NS) viral proteins (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (2). Based on the similarity of the HCV genome organization to those of Flavivirus, Pestivirus, and Pegivirus, it is classified as a separate genus, Hepacivirus, in the Flaviviridae family (3, 4).
HCV enters cells via receptor-mediated endocytosis (5). A number of cell surface molecules have been identified as HCV receptors and/or coreceptors. Based on their distinct functions, they can be divided into two different groups, attachment receptors and postattachment receptors. Several previous studies have shown that heparan sulfate (HS) proteoglycans (HSPGs) play an important role in HCV infection (6–9). HSPGs are composed of a core protein such as syndecans (SDCs) (SDC-1 to -4), glypicans (glypican-1 [GPC1] to GPC6), perlecan (HSPG2), or agrin and one or more HS glycosaminoglycan (GAG) chains (10). Our previous work demonstrated that SDC-1, SDC-2, and T cell immunoglobulin and mucin domain-containing protein 1 (TIM-1) are major receptors for HCV attachment to the cell surface (11, 12). HCV attachment to cells is mediated primarily by the binding of cellular apolipoprotein E (apoE) and phosphatidylserine (PS) incorporated on the viral envelope to SDC-1/SDC-2-containing HSPGs and TIM-1 on the surface of hepatocytes, respectively (12–15). Postattachment receptors include CD81, Claudin-1 (CLDN1), Occludin (OCLN), SR-BI, and low-density lipoprotein receptor (LDLR), which specifically interact with the viral envelope glycoproteins E1 and E2 (16–18). Postattachment receptors are important for HCV cell entry and uncoating but do not play any role in cell attachment (13). Other cellular factors were also found to enhance HCV infection, including phosphatidylinositol 3-kinase (PI3K)–Akt (19), cell death-inducing DFFA-like effector b (CIDEB) (20), Niemann-Pick C1 (NPC1L1) (21), transferrin receptor 1 (TfR1) (22), epidermal growth factor receptor (EGFR), and ephrin receptor A2 (EphA2) (23). However, the precise functions and underlying molecular mechanisms of so many different postattachment receptors and other cellular factors in HCV infection remain unknown.
HCV infection occurs in two different forms, cell-free and cell-to-cell transmission. Cell-free transmission is the major route (>90%) of HCV infection, which can be blocked by E1/E2-specific monoclonal antibodies. Cell-cell transmission is responsible for the spread of HCV between neighboring cells and is not affected by HCV-neutralizing antibodies (24, 25). Thus, it is thought that cell-to-cell transmission may contribute to the escape of the host immune response against HCV, resulting in persistent infection. Recently, several studies suggested that some of the postattachment receptors are important for HCV cell-to-cell transmission, including CD81, CLDN1, OCLN, and SR-BI (26–29). Additionally, apoE is implicated in HCV cell-to-cell transmission (30, 31). Whether attachment receptors play a role in HCV cell-to-cell spread has not been experimentally examined.
In the present study, we used clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 gene-specific editing technology and small interfering RNAs (siRNAs) to interrogate the importance of each HCV attachment and postattachment receptor in HCV cell-free and cell-to-cell transmission. The results obtained from our present study demonstrate that both SDC-1 and SDC-2, but not SDC-3 or SDC-4, are HCV attachment receptors, unlike a previous report indicating that SDC-4 is an HCV attachment receptor (32). More significantly, we found that all three attachment receptors (SDC-1, SDC-2, and TIM-1) modestly enhanced HCV cell-to-cell spread. All postattachment receptors (CD81, CLDN1, OCLN, SR-BI, and LDLR) tested here were confirmed to play important roles not only in cell-free but also in cell-to-cell transmission. Similarly, apoE is a cellular factor important for HCV cell-to-cell transmission, but very-low-density lipoprotein (VLDL)-containing mouse serum had no effect on HCV cell-to-cell spread. These findings suggest that a different form(s) of HCV particles is likely responsible for cell-to-cell spread in vitro and in vivo.
RESULTS
Knockout of the SDC-1 and SDC-2 genes but not the SDC-3 or SDC-4 gene reduces HCV cell attachment and infection.
We previously found that SDC-1 served as a major HCV attachment receptor and that SDC-2 also affected HCV entry to a much lesser extent, as determined by siRNA-mediated silencing of syndecan expression (11). In contrast, another research group later argued that SDC-4 principally mediates HCV entry by interacting with apoE (32). To clarify the role of SDCs in HCV infection, we used the CRISPR-Cas9 gene-editing system to specifically knock out SDC-1, SDC-2, SDC-3, and SDC-4 individually as well as both SDC-1 and SDC-2 (33). Individual SDC-specific single-guard RNAs (sgRNAs) were expressed from lentiCRISPRv2 lentiviruses upon transduction into Huh-7.5 cells. A number of Huh-7.5 cell clones were selected with puromycin. Knockouts of SDC-1, SDC-2, SDC-3, and SDC-4 in puromycin-resistant Huh-7.5 cell clones were initially determined by DNA sequence analysis. The results showed that a 7-nucleotide (nt) deletion (nt 211 to 217) of the ORF was found in the SDC-1 knockout Huh-7.5 cells. SDC-2, SDC-3, and SDC-4 gene knockout Huh-7.5 cells contain a 76-nt deletion (nt 194 to 69), a 10-nt deletion (nt 622 to 631), and a 16-nt deletion (nt 574 to 589), respectively (Fig. 1A). To create SDC-1 and SDC-2 double-knockout cell lines, the SDC-2 knockout cell line was used for transduction with the lentiCRISPRv2-blasticidin/SDC-1 lentivirus. A Huh-7.5 cell clone contained a single nucleotide insertion in the sgRNA-targeting region (between nt 215 and 216) of the SDC-1 ORF, besides a 76-nt deletion in the SDC-2 ORF, as shown by DNA sequencing results (Fig. 1A). sgRNA-mediated nucleotide deletion and insertion could result in new ORFs by retaining the N-terminal regions prior to the deletion. To confirm the knockout of SDC-1, SDC-2, SDC-3, and SDC-4, Western blot (WB) analysis was carried out (Fig. 1B). SDC-1, SDC-2, SDC-3, and SDC-4 were not detected in their corresponding knockout cell lines by WB analysis. Additionally, we determined the levels of expression of the attachment receptor TIM-1 and the postattachment receptors CD81, CLDN1, OCLN, and SR-BI in the SDC knockout cell lines. Clearly, the levels of CD81, CLDN1, OCLN, SR-BI, and TIM-1 were similar among parent and SDC knockout Huh-7.5 cells (Fig. 1B).
FIG 1.
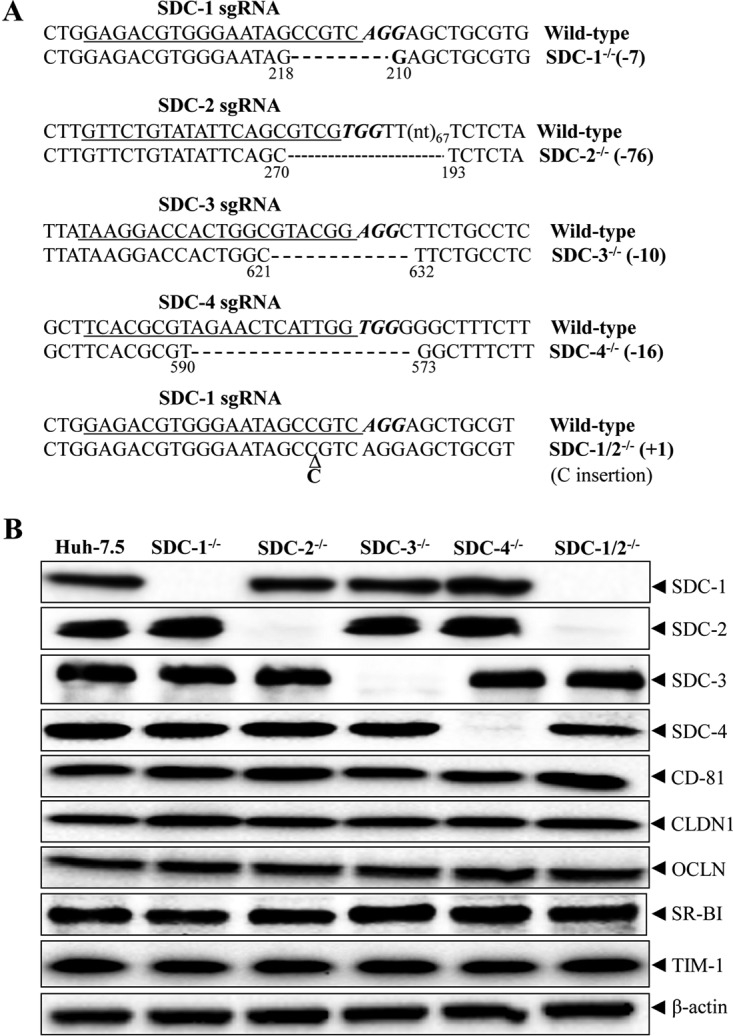
CRISPR-Cas9-induced knockout of SDC genes in Huh-7.5 cell lines. (A) Summary of target gene sequences in SDC knockout Huh-7.5 cell lines. DNAs extracted from SDC gene knockout cells were used for PCR amplification of the target gene using the primers described in Table 1. PCR DNA fragments were subject to DNA sequence analysis. DNA sequences of wild-type and gene knockout Huh-7.5 cells were compared. The SDC-1 gene knockout (SDC-1−/−), SDC-2−/−, SDC-3−/−, and SDC-4−/− contain a 7-nt deletion (−7), a 76-nt deletion (−76), a 10-nt deletion (−10), and a 16-nt deletion (−16), respectively. The positions flanking the nucleotide deletion are highlighted by numbers below the nucleotides beginning from the first nucleotide of the ORF. SDC-1−/− SDC-2−/− has a 1-bp insertion (+1) between nucleotides 215 and 216 of the SDC-1 ORF besides a 76-nt deletion in the SDC-2 gene. (B) Detection of expression of SDC-1 to -4, TIM-1, and postattachment receptors (CD81, CLDN1, OCLN, and SR-BI) by WB in SDC-1 to -4 knockout Huh-7.5 cells. β-Actin was used as an internal control. The knockout of SDC-1 to -4 is highlighted at the top, and individual proteins are indicated on the right.
To determine the importance of SDCs in HCV infection, a single-cycle HCV growth assay was initially used to compare the efficiencies of HCV infection among parent and individual SDC knockout Huh-7.5 cell lines. Huh-7.5 cells were infected with cell culture-grown HCV (HCVcc) at a multiplicity of infection (MOI) of approximately 10. At 24 h postinfection (p.i.), HCV-infected cells were lysed in radioimmunoprecipitation assay (RIPA) buffer. The levels of NS5A were determined by WB analysis. Strikingly, the levels of NS5A were reduced in SDC-1 knockout and SDC-1/SDC-2 double knockout Huh-7.5 cells by >90% (Fig. 2A), consistent with our previous findings with SDC-1-silencing Huh-7.5 cells (11). The SDC-2 knockout also resulted in a reduction of the NS5A level by 75% (Fig. 2A), suggesting its importance for HCV infection. However, SDC-3 and SDC-4 knockouts did not significantly affect HCV infection (Fig. 2A), in contrast to findings reported by others that SDC-4 is a principal mediator of HCV entry (32). To further confirm the role of SDC-1 and SDC-2 in HCV infection, we used Renilla luciferase-expressing HCV for infection, which is more sensitive and more quantitative than WB analysis. In a single-cycle HCV growth assay (Fig. 2B), the SDC-1 knockout reduced Renilla luciferase expression by more than 80%. The SDC-2 knockout decreased HCV infection by about 50%, similar to the knockout of TIM-1, which is another HCV attachment receptor, as shown by our recent work (12). Again, SDC-3 and SDC-4 knockouts did not have a significant effect on infection by Renilla luciferase-expressing HCV (Fig. 2B). Additionally, we used previously described CLDN1 and OCLN knockout Huh-7.5 cells as controls. Similar to previous findings (34), CLDN1 and OCLN knockouts in Huh-7.5 cells ablated HCV infection (Fig. 2B). These results were further validated by experiments using a multiple-cycle HCV growth assay (Fig. 2C). At each time point (24 h, 48 h, and 72 h p.i.), SDC-1, SDC-2, and SDC-1/SDC-2 knockouts resulted in reductions of luciferase activity by 85%, 72%, and 95%, respectively (Fig. 2C). Taken together, these results demonstrate that both SDC-1 and SDC-2 are important for HCV infection.
FIG 2.
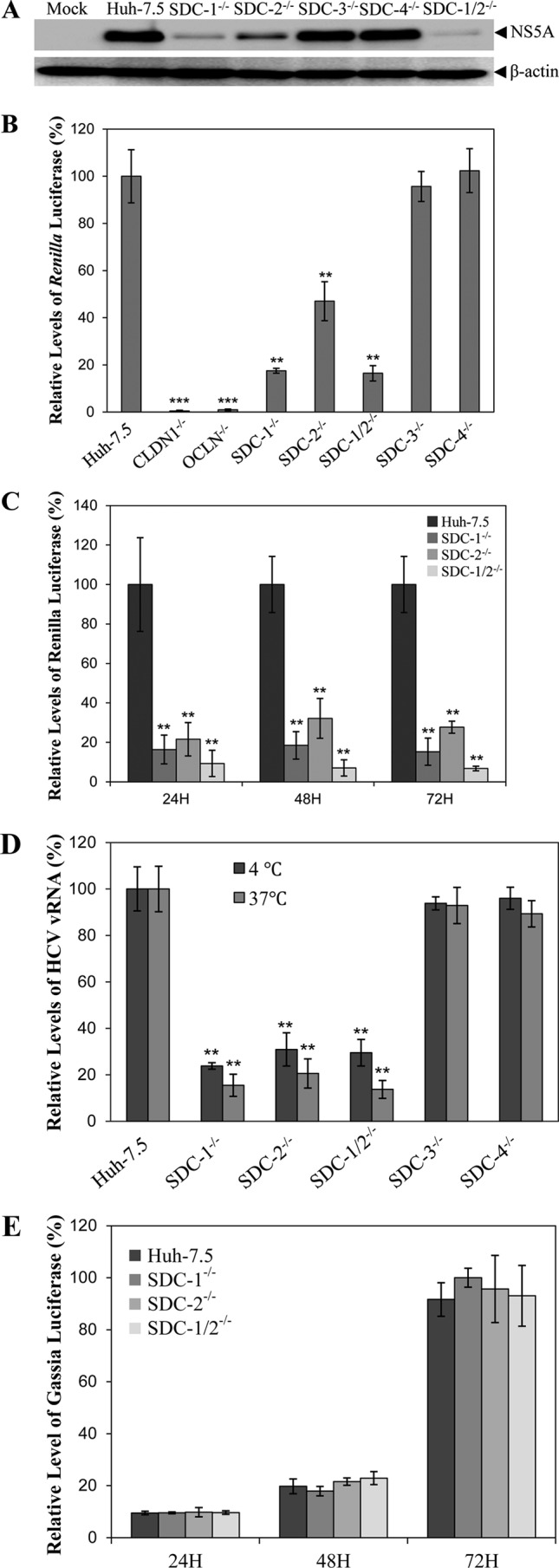
Effects of specific gene knockouts on HCV cell-free infection and cell attachment. (A) Effects of individual knockouts of SDC-1 to -4 on HCV cell-free infection. Parent and individual SDC-1 to -4 knockout Huh-7.5 cells in 12-well cell culture plates were infected with HCV at an MOI of 10 at 37°C for 2 h. Unbound HCV was removed by washing with 1× PBS three times. The HCV-infected cells were incubated at 37°C for 24 h. Cell lysates were collected for the detection of NS5A by WB analysis using monoclonal antibody 9E10. (B) Effects of individual knockouts of SDC-1 to -4 on infection by Renilla luciferase-expressing HCV (HCV-Rluc). Parent Huh-7.5, CLDN1 knockout (CLDN1−/−), OCLN knockout (OCLN−/−), and individual SDC-1 to -4 knockout (SDC-1−/−, SDC-2−/−, SDC-3−/−, and SDC-4−/−) Huh-7.5 cells in 48-well cell culture plates were infected with HCV-Rluc at 37°C for 2 h. Unbound HCV was removed by washing with 1× PBS. HCV-infected cells were incubated at 37°C for 24 h. Cell lysates were collected for the detection of Renilla luciferase. Average values from triplicate samples were used for the calculation of relative levels of luciferase as a percentage of the control, considering the level of luciferase in parent Huh-7.5 cells as 100%. (C) Determination of HCV-Rluc infection by measuring luciferase activity at different time points p.i. HCV infection was done in the same way as described above for panel B. The levels of Renilla luciferase expression were determined at 24 h, 48 h, and 72 h p.i. and were converted to a percentage of the control, with the level of luciferase expression in parent Huh-7.5 cells at each time point as 100%. (D) Effects of individual SDC1-4 gene knockouts on HCV attachment. An HCV attachment assay was carried out by incubation of HCV with parent Huh-7.5 or individual SDC-1 to -4 knockout Huh-7.5 cells at an MOI of 10 at 37°C for 1 h or on ice for 2 h. Unbound HCV was removed by washing with 1× PBS three times. Total RNAs were extracted with TRIzol reagent. The vRNA levels of cell-bound HCV particles were quantified by a real-time qRT-PCR method. Mean values from three different experiments were converted to a percentage of the control, considering the HCV vRNA level in parent Huh-7.5 cells as 100%. (E) Effects of individual SDC1-4 gene knockouts on HCV RNA replication. Gaussia luciferase-expressing subgenomic HCV RNA of HCV JHF1 was transfected into parent Huh-7.5, SDC-1−/−, SDC-2−/−, and SDC-1−/−/SDC-2−/− Huh-7.5 cells in 48-well cell culture plates using DMRIE-C reagent (Invitrogen). At 24, 48, and 72 h p.t., HCV RNA-transfected cells were lysed for the measurement of Gaussia luciferase activity. The relative levels of luciferase expression were converted to a percentage of the control, as described above for panels B and C. *, P < 0.05; **, P < 0.01.
Our previous study demonstrated that SDC-1 is a major HCV attachment receptor (11). Thus, we sought to directly determine the effects of SDC-1 and SDC-2 knockouts on HCV attachment. Parent and SDC-1 and SDC-2 knockout Huh-7.5 cells were incubated with HCV at 37°C for 1 h or on ice for 2 h. HCV can bind but is unable to enter cells when incubated on ice. Unbound virus was removed by washing with phosphate-buffered saline (PBS) three times. The virion RNA (vRNA) of HCVcc attached to cells was extracted and quantified by using a real-time reverse transcription-quantitative PCR (qRT-PCR) method (12). Similar to the above-described results for HCV infection, knockouts of the SDC-1 and SDC-2 genes in Huh-7.5 cells decreased the levels of HCV vRNA by 70% to 85% when assayed at 37°C or on ice (Fig. 2D). On the contrary, knockout of the SDC-3 gene or the SDC-4 gene did not significantly affect HCV attachment based on similar levels of HCV vRNA in SDC-3 and SDC-4 knockout Huh-7.5 cells compared to those in parent Huh-7.5 cells (Fig. 2D). Collectively, these findings demonstrate that both SDC-1 and SDC-2, but not SDC-3 and SDC-4, are HCV attachment receptors.
To exclude a possible effect of SDC-1 and SDC-2 gene knockouts on HCV RNA replication, a subgenomic HCV RNA replicon expressing a Gaussia luciferase reporter was transfected into parent and SDC-1/SDC-2 knockout Huh-7.5 cells. At 24 h, 48 h, and 72 h posttransfection (p.t.), the levels of Gaussia luciferase expression were determined by using a Gaussia luciferase assay kit. The results show that the levels of Gaussia luciferase expression at each time point are similar among parent and SDC-1 and SDC-2 knockout cells (Fig. 2E), suggesting that SDC-1 and SDC-2 knockouts did not affect viral RNA replication.
Restoration of HCV attachment and infection in SDC-1 and SDC-2 knockout cells by ectopic expression of SDC-1 and SDC-2 but not SDC-4.
To exclude possible off-target effects associated with CRISPR-Cas9-mediated gene knockouts, we sought to determine whether the ectopic expression of SDC-1 and SDC-2 would restore HCV infection and attachment in SDC-1 and SDC-2 knockout Huh-7.5 cells. Recombinant adenoviruses expressing SDC-1 and SDC-2 were previously constructed and used for their ectopic expression (11). A recombinant adenovirus expressing lacZ was used as a negative control. SDC-1 or SDC-2 knockout Huh-7.5 cells were infected with recombinant adenoviruses, which was followed by HCV infection. At 24 h p.i., the levels of the HCV NS5A, SDC-1, and SDC-2 proteins were determined by WB analysis. Both SDC-1 and SDC-2 were expressed in SDC-1- and SDC-2-deficient cells to levels comparable to those in parent Huh-7.5 cells (Fig. 3A and B). As a result, HCV infection was fully restored to that observed in parent Huh-7.5 cells by the ectopic expression of SDC-1 and SDC-2, respectively, as shown by the level of NS5A expression (Fig. 3A and B). In contrast, the negative-control adenovirus expressing LacZ did not rescue HCV infection. Similarly, the ectopic expression of SDC-1 and SDC-2 rescued defective HCV cell attachment in SDC-1 and SDC-2 knockout Huh-7.5 cells when an HCV attachment assay was carried out either at 37°C or on ice (Fig. 3C and D), as the nucleotide deletion in the SDC-4 ORF could result in a new ORF potentially encoding most of the SDC-4 protein with an additional 21 random amino acids at the C terminus. To further validate the possible role of SDC-4 in HCV infection, SDC-1 knockout and SDC-1/SDC-2 double knockout cells were used for the ectopic expression of SDC-1 and SDC-4, respectively. Similar to the above-described results (Fig. 3A and C), ectopically expressed SDC-1 rescued HCV infection in SDC-1 knockout or SDC-1/SDC-2 double knockout cells. However, the ectopic expression of SDC-4 did not restore HCV infection, similar to the negative LacZ control (Fig. 3E). Taken together, these findings demonstrate that both SDC-1 and SDC-2, but not SDC-4, are bona fide HCV attachment receptors.
FIG 3.
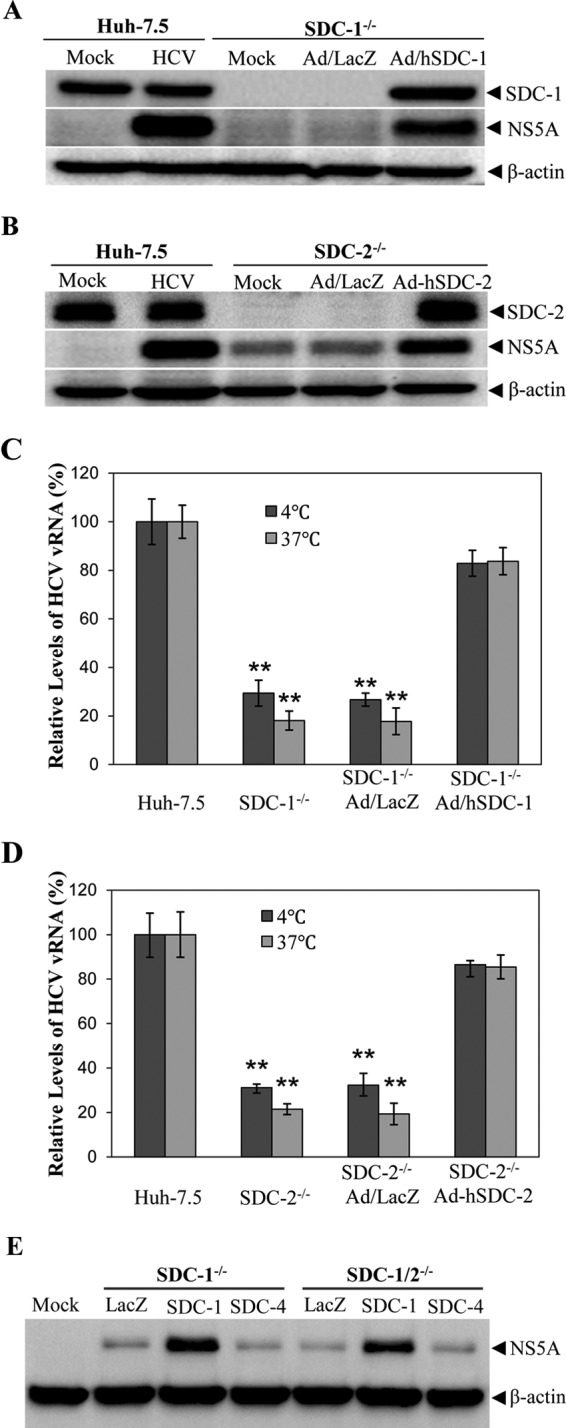
Restoration of HCV attachment and infection in SDC-1−/− and/or SDC-2−/− Huh-7.5 cells by ectopic expression of SDC-1 and SDC-2 but not SDC-4. (A) Restoration of HCV infection in SDC-1−/− Huh-7.5 cells by ectopic SDC-1 expression. SDC-1−/− Huh-7.5 cells in 12-well cell culture plates were infected with either a recombinant adenovirus expressing a lacZ gene (Ad/LacZ) or human SDC-1 (Ad-hSDC-1). At 48 h p.i., Ad/LacZ- or Ad-hSDC1-infected SDC-1−/− Huh-7.5 cells were infected with HCV at an MOI of 10 at 37°C for 2 h, with parent Huh-7.5 cells as a positive control. Twenty-four hours after HCV infection, cell lysates were collected for the detection of NS5A by WB analysis. (B) Rescue of HCV infection in SDC-2−/− cells by ectopic SDC-2 expression. The experiments were done in the same way as described above for panel A except that SDC-2−/− cells and a recombinant adenovirus expressing human SDC-2 (Ad-hSDC-2) were used. (C and D) Restoration of HCV cell attachment by ectopic SDC-1 (C) and SDC-2 (D) expression. SDC-1−/− and SDC-2−/− Huh-7.5 cells were infected with Ad/LacZ and Ad-hSDC-1 or Ad-hSDC-2, respectively. After 48 h p.i., adenovirus-infected SDC-1−/− and SDC-2−/− cells were incubated with HCV at an MOI of 10 at 37°C for 1 h or on ice for 2 h. Unbound HCV was removed by washing with 1× PBS three times. Total RNAs were extracted with TRIzol reagent. The vRNA levels of cell-bound HCVs were quantified by using a real-time qRT-PCR method. Average values from three independent experiments were calculated as a percentage of the control. The level of HCV vRNA in parent Huh-7.5 cells was considered 100%. (E) Determination of restoration of HCV infection by ectopic expression of SDC-1 or SDC-4 in SDC-1−/− and SDC-1−/−/SDC-2−/− Huh-7.5 cells. SDC-1−/− and SDC-1−/−/SDC-2−/− Huh-7.5 cells in 12-well cell culture plates were infected with Ad/LacZ, Ad/hSDC-1, and Ad/hSDC-4, similarly to the experiments described above for panels A and B. At 48 h p.i., cells were infected with HCV at an MOI of 1. Forty-eight hours after HCV infection, cells were lysed, and the levels of NS5A were determined by WB analysis in the same way as described above for panels A and B. Mock, uninfected cells. *, P < 0.05; **, P < 0.01.
Development of a luciferase-based reporter system for quantification of HCV cell-to-cell spread.
We previously developed an mCherry-based dual-reporter system for real-time monitoring of HCV cell-free and cell-to-cell transmission (34). In the dual-reporter system, a reversible tetracycline-controlled transcriptional activator (rtTA) is expressed as a fusion protein with mitochondrial antiviral signaling protein (MAVS). An HCV NS3/4A protease recognition peptide was inserted between the rtTA and MAVS. The rtTA is transported to the nucleus after cleavage by the HCV NS3/4A protease and then activates mCherry gene transcription in the presence of doxycycline (Dox). Thus, mCherry expression could be visualized in HCV-infected cells (34). To precisely quantify HCV cell-free and cell-to-cell spread, we modified the dual-reporter system by replacing the mCherry gene with a firefly luciferase gene. The firefly luciferase-based reporter was introduced into Huh-7.5 cells by lentiviral transduction, resulting in a luciferase reporter cell line designated NIrD-fluc-sc4. This cell line expresses a high level of firefly luciferase only when it is infected with HCV in the presence of doxycycline. Firefly luciferase activity was undetectable in the absence of either HCV infection or doxycycline (Fig. 4A). The NIrD-fluc-sc4 cell line was then used as recipient cells to determine the optimal conditions for the quantification of HCV cell-to-cell spread. The parameters examined included coculture time (Fig. 4A), MOIs used for infection of donor cells (Fig. 4B), the ratio between donor and recipient cells (Fig. 4C), and methods for blocking HCV cell-free infection (Fig. 4D). As described in Materials and Methods, parent Huh-7.5 cells infected with HCV at 24 h p.i. were cocultured with NIrD-fluc-sc4 cells. The level of firefly luciferase activity was highest 48 h after coculture of donor and recipient cells (Fig. 4A). The level of firefly luciferase activity was lower at 72 h p.i. because of the HCV-induced cytopathic effect (cell death). The level of firefly luciferase activity was also closely correlated with the increasing MOIs used for infection of donor cells (Fig. 4B). The ratios between donor and recipient cells were also critical for HCV cell-to-cell spread. A 1:4 ratio of donor to recipient cells was optimal for HCV cell-to-cell spread (Fig. 4C). Several previous studies suggested that HCV-specific neutralizing antibodies could efficiently block HCV cell-free infection but not cell-to-cell transmission (24, 25). It was also found that different cell culture-adapted HCV strains have different sensitivities to neutralizing antibodies (35–37). Therefore, we compared 2% methylcellulose in the medium with various concentrations of an E2 monoclonal antibody (CBH5) for blocking cell-free HCV infection. It turned out that 2% methylcellulose blocked HCV cell-free infection as efficiently as did 10 μg/ml of CBH5, resulting in a >90% reduction of HCV infectivity (Fig. 4D). Therefore, a coculture time of 48 h, an MOI of 10 for HCV infection of donor cells, a 1:4 ratio of donor to recipient cells, and culture medium containing 2% methylcellulose were subsequently used as the optimal conditions to determine the effects of cell surface receptors on HCV cell-to-cell spread.
FIG 4.
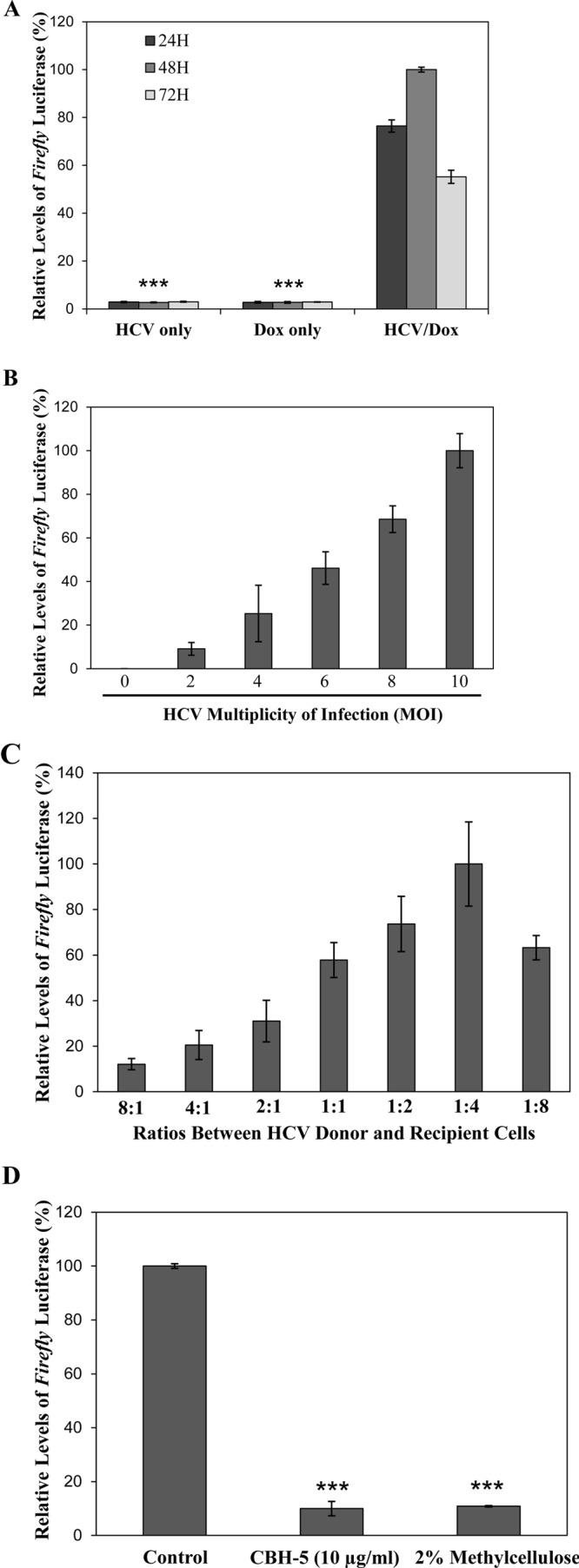
Optimization of a luciferase-based reporter system for quantification of HCV cell-to-cell spread. Our previously described dual-reporter system for monitoring HCV infection (34) was modified by replacing the mCherry gene with a firefly luciferase gene. A stable Huh-7.5 cell line, designated NIrD-fluc-sc4, was constructed by using the newly modified reporter plasmid. This novel reporter cell line was used to optimize the conditions for the quantification of HCV cell-to-cell spread. The parameters for optimization included the coculture time of HCV donor and recipient cells, the MOI used for HCV infection of donor cells, the ratio between HCV donor and recipient cells, and restriction of HCV cell-free dissemination. NIrD-fluc-sc4 cells, serving as HCV recipient cells, were transfected with a DNA vector expressing Renilla luciferase as a control for normalization, whereas Huh-7.5 cells were infected with HCV as donor cells. At 24 h p.t. and at 24 h p.i., respectively, HCV donor and recipient cells were mixed and cocultured in the presence or absence of 2 μg/ml of Dox and a 2% methylcellulose overlay. (A) HCV cell-to-cell spread at different time points (24 h, 48 h, and 72 h) of coculture of HCV donor and recipient cells. Cell lysates collected at 24 h, 48 h, and 72 h p.i. were used for measurement of both Renilla and firefly luciferase activities with the Dual-Glo luciferase assay system. Uninfected cells (Dox only) and coculture of HCV donor and recipient cells without Dox (HCV only) served as negative controls. (B) Dependence of HCV cell-to-cell spread on HCV infection of donor cells. Huh-7.5 cells were infected with HCV at different MOIs (0 to 10) (x axis) and then cocultured with pcDNA6/TR-Rluc-transfected NIrD-fluc-sc4 recipient cells for 48 h in the presence of 2 μg/ml of Dox. The levels of Renilla luciferase and firefly luciferase expression were determined as described above for panel A. (C) Determination of the optimal ratio between HCV donor and recipient cells. Huh-7.5 cells were infected with HCV at an MOI of 10 for 24 h. HCV-infected Huh-7.5 cells were then cocultured with pcDNA6/TR-Rluc-transfected NIrD-fluc-sc4 cells at different ratios (8:1, 4:1, 2:1, 1:1, 1:2, 1:4, and 1:8) of donor to recipient cells in the presence of 2 μg/ml of Dox and a 2% methylcellulose overlay. After 48 h of coculture, cells were harvested for the measurement of Renilla and firefly luciferase activities. (D) Blockade of HCV cell-free dissemination by E2-specific monoclonal antibody CBH5 (60) or 2% methylcellulose. HCV-infected Huh-7.5 cells (MOI of 10) were cocultured with pcDNA6/TR-Rluc-transfected NIrD-fluc-sc4 cells in the presence of Dox (2 μg/ml) and CBH5 (10 μg/ml) or a 2% methylcellulose overlay. After 48 h of coculture, cells were lysed for the quantification of Renilla and firefly luciferase expression. The relative levels of firefly luciferase (average values derived from three experiments) were normalized to the levels of Renilla luciferase (average values derived from three experiments) and converted to a percentage of the control, considering the conditions with the highest level of firefly luciferase expression as 100%. *, P < 0.05; **, P < 0.01.
Enhancement of HCV cell-to-cell spread by attachment receptors.
The role of the attachment receptors SDC-1, SDC-2, and TIM-1 in HCV cell-to-cell spread has not been experimentally examined, although they are important for HCV infection (11, 12). To determine the importance of attachment receptors in HCV cell-to-cell spread, two different cell culture model systems were used. Initially, firefly luciferase-expressing NIrD-fluc-sc4 cells were used as recipient cells, which were transfected with pcDNA6/Rluc (as an internal control) and SDC-1-, SDC-2-, SDC-3-, SDC-4-, and TIM-1-specific SmartPool siRNAs. HCV-infected Huh-7.5 cells were used as donor cells. At 24 h p.t. and at 24 h p.i., respectively, donor and recipient cells at a ratio of 1:4 were cocultured in the presence of 2 μg/ml of doxycycline and Dulbecco's modified Eagle's medium (DMEM) containing 2% methylcellulose for 48 h. The levels of Renilla and firefly luciferase activities were determined by using a Dual-Glo luciferase assay system. As expected, the levels of SDC-1, SDC-2, and TIM-1 were significantly reduced by their corresponding SmartPool siRNAs (Fig. 5A). Likewise, the firefly luciferase activity was reduced by the knockdown of SDC-1, SDC-2, and TIM-1 expression by about 24%, suggesting that attachment receptors modestly enhance HCV cell-to-cell spread (Fig. 5B). However, the knockdown of SDC-3 and SDC-4 expression did not affect HCV cell-to-cell spread (Fig. 5B), consistent with the findings derived from HCV attachment and infection assays (Fig. 2 and 3). Similar results were observed with SDC-1, SDC-2, SDC-3, SDC-4, and TIM-1 knockout Huh-7.5 cells that were transduced with the lenti-NIrD-fluc lentivirus as recipient cells (data not shown).
FIG 5.
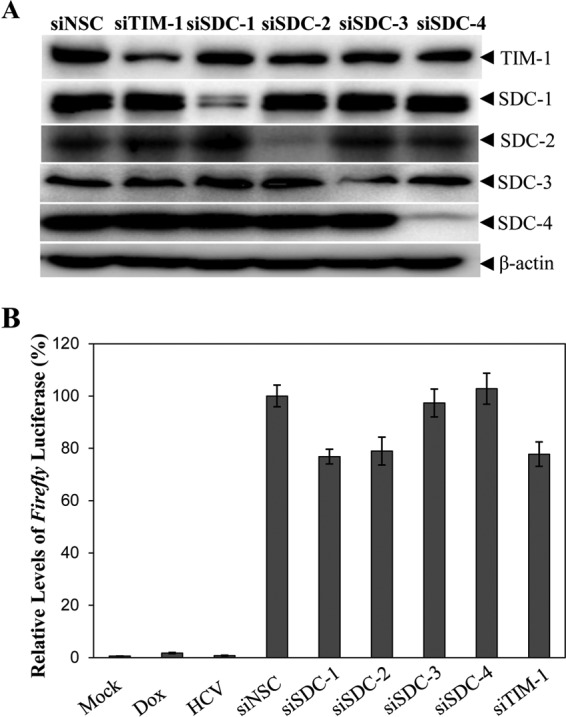
Enhancement of HCV cell-to-cell spread by attachment receptors. NIrD-fluc-sc4 cells were transfected with pcDNA6/TR-Rluc (as an internal control) and SDC-1, SDC-2, SDC-3, SDC-4, or TIM-1 gene-specific SmartPool siRNAs (50 nM). Huh-7.5 cells were infected with HCV at an MOI of 10. At 24 h p.t. and at 24 h p.i., respectively, HCV-infected Huh-7.5 donor cells and NIrD-fluc-sc4 recipient cells at a ratio of 1:4 were cocultured in the presence of 2 μg/ml of Dox and a 2% methylcellulose overlay for 48 h. (A) Determination of SDC-1, SDC-2, SDC-3, SDC-4, and TIM-1 expression by WB analysis. β-Actin was used as an internal control. (B) Effects of siRNA-mediated silencing of SDC-1, SDC-2, SDC-3, SDC-4, and TIM-1 on HCV cell-to-cell spread. Renilla and firefly luciferase activities were determined by using a Dual-Glo luciferase assay system. Relative firefly luciferase expression was calculated as a percentage of the control after normalization to Renilla luciferase expression, considering the level of firefly luciferase expression in cells transfected with a nonspecific control siRNA (siNSC) as 100%. Average values from three different experiments are plotted.
Importance of postattachment receptors for HCV cell-to-cell spread.
It was previously reported that the postattachment receptors CD81, CLDN1, OCLN, and SR-BI are all involved in HCV cell-to-cell transmission (24–26, 29, 38, 39). However, their importance in HCV cell-to-cell spread was somewhat controversial based on findings from different studies, probably due to the lack of a more quantitative assay system (24–26, 28, 29, 38, 39). Therefore, we used the above-described luciferase-based cell culture system to reevaluate the role of key postattachment receptors in HCV cell-to-cell spread. All five known postattachment receptors, CD81, CLDN1, OCLN, SR-BI, and LDLR, were downregulated by using gene-specific siRNAs in the NIrD-fluc-sc4 cell line prior to coculture with HCV-infected Huh-7.5 donor cells. SmartPool siRNAs specific for CLDN1, OCLN, CD81, SR-BI, and LDLR significantly knocked down the expression of their corresponding gene, resulting in a reduction of protein expression by up to 75% (Fig. 6A). As a result, the firefly luciferase activity was reduced to various degrees by siRNAs specific for different postattachment receptors. The silencing of CLDN1 and OCLN decreased HCV cell-to-cell spread by 55% and 65%, respectively. The knockdown of the expression of both the CD81 and LDLR genes decreased HCV cell-to-cell spread by nearly 50%. It appears that SR-BI is the least important postattachment receptor for HCV cell-to-cell spread, considering a 20% reduction of HCV cell-to-cell spread by the downregulation of its expression (Fig. 6B). These results suggest that there are different mechanisms of HCV cell-free and cell-to-cell transmission. HCV receptors at the tight junction play an important role in the mediation of HCV cell-to-cell spread.
FIG 6.
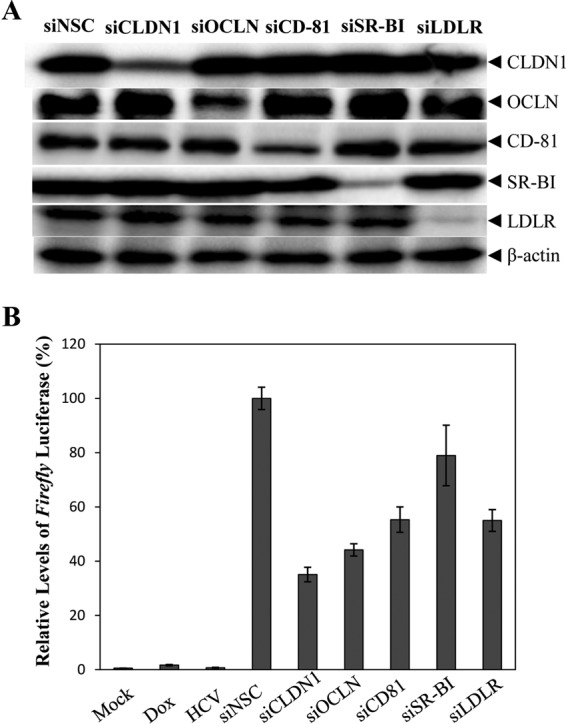
Effects of siRNA-induced knockdown of postattachment receptors on HCV cell-to-cell spread. NIrD-fluc-sc4 cells were transfected with pcDNA6/TR-Rluc (as an internal control) and CD81-, CLDN1-, OCLN-, SR-BI-, and LDLR-specific SmartPool siRNAs at a 50 nM concentration. Huh-7.5 cells were infected with HCV at an MOI of 10. At 24 h p.t. and at 24 h p.i., respectively, HCV-infected Huh-7.5 cells were cocultured with siRNA-transfected NIrD-fluc-sc4 cells in the presence of 2 μg/ml of Dox and a 2% methylcellulose overlay for 48 h. (A) Determination of CD81, CLDN1, OCLN, SR-BI, and LDLR expression by WB analysis using specific antibodies. β-Actin was used as a control. (B) Influence of postattachment receptors on HCV cell-to-cell spread. After coculture of HCV donor and recipient cells, the levels of Renilla and firefly luciferases were determined by using a Dual-Glo luciferase assay system. Relative levels of firefly luciferase expression in HCV recipient cells were calculated after normalization to the Renilla luciferase control and were converted to a percentage of the control, considering the level of firefly luciferase expression in NSC siRNA-transfected cells as 100%.
Effects of apoE and VLDL-containing mouse serum on HCV cell-to-cell spread.
Previous studies suggested that apoE is important for HCV cell-to-cell spread (30, 31, 40). We confirmed the role of apoE in HCV cell-to-cell spread using the newly developed NIrD-fluc-sc4 cell line (Fig. 4). Previous studies with short hairpin RNA (shRNA)-mediated apoE silencing suggested that apoE is involved in HCV cell-to-cell transmission (30, 31, 40), as apoE also plays an important role in HCV assembly. It was not clear whether the effect of shRNA-mediated apoE silencing on HCV cell-to-cell spread was due to a reduction of HCV production or apoE-mediated infection. Therefore, we tested an apoE-blocking monoclonal antibody in the HCV cell-to-cell transmission assay, which inhibits only HCV attachment, as demonstrated by our previous studies (13, 15). The results show that the apoE-blocking monoclonal antibody suppressed HCV cell-to-cell spread in a dose-dependent manner, resulting in up to an 80% reduction of firefly luciferase expression (Fig. 7A). apoE is known to mediate HCV attachment by binding to HSPGs on the cell surface of hepatocytes, similar to the mechanism of action of VLDL-containing serum in the inhibition of HCV infection (41). Thus, we sought to determine the effect of mouse serum, which potently blocked HCV infection (41), on HCV cell-to-cell spread. Surprisingly, the mouse serum did not significantly affect the levels of firefly luciferase expression (Fig. 7B), suggesting that it does not restrict HCV cell-to-cell transmission, even though it effectively inhibited HCV cell-free infection (data not shown) (41).
FIG 7.
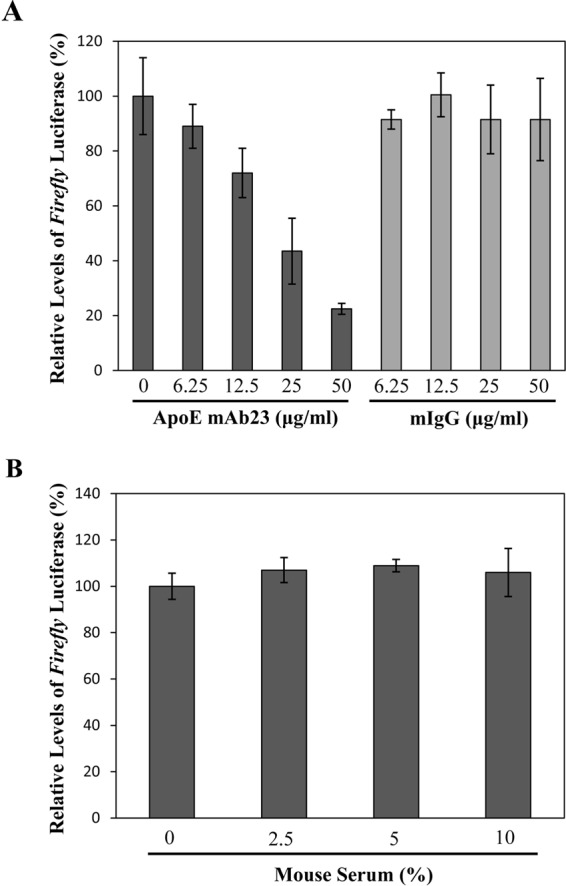
Effects of apoE and VLDL-containing mouse serum on HCV cell-to-cell spread. HCV-infected Huh-7.5 cells were cocultured with pcDA6/TR-Rluc-transfected NIrD-fluc-sc4 cells at a ratio of 1:4 in the presence of different concentrations (0, 6.25, 12.5, 25, and 50 μg/ml) of apoE-blocking monoclonal antibody mAb23 (A) or various percentages (0%, 2.5%, 5%, and 10%) of mouse serum (B). Normal mouse IgG (mIgG) was used as a control for the apoE monoclonal antibody. After 48 h of coculture, the levels of Renilla and firefly luciferase expression were determined and converted to a percentage of the control in the same way as described in the legends of Fig. 5 and 6.
DISCUSSION
Virus infection is ubiquitously mediated by initial binding (attachment) to cell surface receptors and subsequent receptor-mediated endocytic internalization, vesicular tracking, membrane fusion, and uncoating (42). Most viruses infect cells through viral protein-mediated binding to cell surface receptors. In most cases, the same receptors are used for both virus attachment and cell entry. In contrast, HCV uses cellular components instead of viral envelope proteins for its initial cell attachment, including apoE and PS incorporated on the viral envelope (12, 13, 15, 43). apoE binds to cell surface HSPGs (13, 15, 43), whereas PS interacts with TIM-1 (12), both of which mediate HCV attachment. Thus, HSPGs and TIM-1 are considered HCV attachment receptors (9, 12, 44). The HCV envelope proteins E1 and E2 are responsible for interacting with other cell surface receptors such as CD81, CLDN1, OCLN, SR-BI, and LDLR (16, 45–49), which function mainly at postattachment steps such as internalization, membrane fusion, and uncoating prior to the release of the viral RNA genome to the cytosol (9, 16, 50–53). Accordingly, HCV receptors can be divided into attachment and postattachment receptors based on their different ligand-binding properties and distinct functions during HCV infection.
Our previous studies suggested that SDC-1, one of the HSPG core proteins, serves as the major HCV attachment receptor (11). Consistent with the siRNA-mediated silencing of SDC-1 expression (11), SDC-1 gene knockout in Huh-7.5 cells reduced HCV cell attachment and infection by 80% (Fig. 1 and 2), which could be fully restored by ectopic SDC-1 expression (Fig. 3). Additionally, results from the present study demonstrate that SDC-2 also plays an important role in HCV attachment and infection (Fig. 1 and 2), which was neglected in our previous report based on the modest reduction of HCV cell attachment in SDC-2-silencing cells (11). In the present study, SDC-2 gene knockout was shown to cause a more detrimental effect on HCV infection, resulting in a decrease of HCV cell attachment by about 70% (Fig. 2). Ectopic SDC-2 expression completely rescued HCV cell attachment and infection in SDC-2 knockout Huh-7.5 cells (Fig. 3). However, we did not reproduce those observations suggesting that SDC-4 principally mediated HCV entry, as previously reported by others (32), nor did SDC-3 or SDC-4 gene knockout affect HCV cell attachment or infection (Fig. 1 and 2), suggesting that SDC-3 and SDC-4 do not play any role in HCV infection. These results are in line with our previous findings that SDC-1 and SDC-2, but not SDC-3 and SDC-4, are important for HCV attachment and infection (11). The ectopic expression of SDC-4 also failed to restore the defect of HCV infection in SDC-1 knockout and SDC-1/SDC-2 double knockout cells (Fig. 3E). The importance of SDC-1 and SDC-2 for HCV attachment and infection was not due to their higher levels of expression. We quantified the mRNA levels of SDC-1, SDC-2, SDC-3, and SDC-4 using a real-time qRT-PCR method. The relative levels of SDC-1, SDC-2, SDC-3, and SDC-4 mRNAs in Huh-7.5 cells were 1.7-, 10-, 1-, and 21-fold, respectively (results not shown). Strong support for the importance of SDC-1 and SDC-2 in HCV cell entry also comes from two elegant studies recently reported by others (54, 55). Zhang et al. demonstrated that the expression of both SDC-1 and SDC-2 is transcriptionally regulated by SMAD6 and SMAD7 (55). The siRNA-mediated silencing of the SMAD6 gene resulted in the downregulation of SDC-1 and SDC-2 expression and, consequently, reduced HCV cell entry and infection. Conversely, the overexpression of SMAD6 increased SDC-1 and SDC-2 expression and therefore enhanced HCV infection. The expressions of SMAD6, SMAD7, and SDC-1 were also upregulated in liver biopsy samples of chronic hepatitis C patients, suggesting a physiological relevance of SDC-1 for HCV infection (55). Interestingly, SDC-1 was found to interact with CD81 in primary and transformed human hepatocytes, providing a direct link between attachment and postattachment receptors (54). It was previously shown that CD81 could form a complex with another postattachment receptor, CLDN1 (56, 57). Thus, attachment receptors may co-opt postattachment receptors by forming functional complexes that govern HCV cell entry. Interestingly, SDC-4 is a cytoplasmic protein and does not localize to the cell membrane of primary human hepatocytes, unlike SDC-1, CD-81, and CLDN-1, as demonstrated by confocal microscopy (54). Collectively, these findings suggest that both SDC-1 and SDC-2, but not SDC-3 or SDC-4, are bona fide HCV attachment receptors in vitro and in vivo.
Like many other viruses (58), HCV can spread to neighboring cells via direct cell-to-cell contact besides cell-free dissemination. All known attachment and postattachment receptors are required for efficient HCV cell-free transmission (Fig. 2 and data not shown) (59). HCV cell-free infection can be effectively blocked by HCV E1/E2-specific neutralizing antibodies (60, 61). In contrast, HCV cell-to-cell spread is resistant to HCV E1/E2 antibody-mediated neutralization (24, 25), suggesting that cell-to-cell spread is mechanistically different from cell-free infection. However, the importance and underlying molecular mechanisms of cell surface receptors in HCV cell-to-cell spread remain elusive, with controversial observations even among different studies (24–26, 28, 29, 38, 39). Previous studies on HCV cell-to-cell spread relied mainly on the visualization of HCV-infected cells by immunofluorescence microscopy. The discrepancy in different results on the requirement of cellular factors for HCV cell-to-cell spread was likely due to the lack of a sensitive and quantitative assay system. With this in mind, we developed and optimized a luciferase-based reporter system to quantify HCV cell-to-cell spread. Firefly luciferase was expressed only in recipient cells (NIrD-fluc-4sc) when HCV spread from HCV-infected Huh-7.5 donor cells (Fig. 4A and B). HCV cell-free transmission was completely restricted by either a methylcellulose overlay or an E2-specific monoclonal antibody, CBH5 (Fig. 4D). Therefore, the level of firefly luciferase expression directly reflects the magnitude of HCV cell-to-cell spread. This reporter system can accurately and quantitatively measure HCV cell-to-cell spread. Using this reporter system, we found that the siRNA-mediated knockdown or CRISPR-Cas9-induced gene knockout of SDC-1, SDC-2, or TIM-1 resulted in about a 20% reduction of HCV cell-to-cell spread (Fig. 5 and data not shown). Again, SDC-3 and SDC-4 gene knockdowns or knockouts did not affect HCV cell-to-cell spread, similar to their ineffectiveness in HCV attachment and cell-free infection (Fig. 2). These findings suggest that the attachment receptors SDC-1, SDC-2, and TIM-1 can modestly promote HCV cell-to-cell spread. Similarly, silencing of the SR-BI gene and an SR-BI-specific inhibitor, BLT-1, decreased HCV cell-to-cell spread by about 20% (Fig. 6 and data not shown). SR-BI was implicated in HCV cell-to-cell spread (25, 26, 39, 62), although its importance in HCV cell-free and cell-to-cell spread also depends on the virus strain (26, 63). On the contrary, other postattachment receptors, such as CLDN1, OCLN, CD81, and LDLR, appeared to be more important for HCV cell-to-cell spread. The siRNA-mediated silencing of CLDN1, OCLN, CD81, and LDLR resulted in a >50% decrease in HCV cell-to-cell spread (Fig. 6). The knockout of the CLDN1 and OCLN genes reduced HCV cell-to-cell spread by 60% to 80% (data not shown), suggesting that these two postattachment receptors at the tight junction are the most important receptors for HCV cell-to-cell spread among all attachment and postattachment receptors tested in this study. The importance of CD81 and LDLR in HCV cell-to-cell spread falls between SR-BI and the tight-junction receptors CLDN1 and OCLN (Fig. 6). Even though postattachment receptors are important for efficient HCV cell-to-cell spread, the lack of an individual receptor did not ablate it. The occurrence of a significant portion (20% to 50%) of HCV cell-to-cell spread in the absence of an individual postattachment receptor suggests a distinct mechanism independent of cell-free infection, which strictly depends on the postattachment receptors.
Several previous studies suggested that apoE is involved in HCV cell-to-cell spread (30, 31, 40). We confirmed the importance of apoE in HCV cell-to-cell spread using the newly developed luciferase reporter system in conjunction with an apoE-blocking monoclonal antibody. The apoE-blocking antibody mAb23 blocked HCV cell-to-cell spread by up to 80% (Fig. 7A), similar to its inhibition of HCV cell-free infection (13, 14, 64). apoE is known to mediate HCV attachment by binding to cell surface HSPGs, with the core proteins SDC-1 and SDC-2 serving as the major HCV attachment receptors (11, 13, 15, 43, 65). Surprisingly, HCV cell-to-cell spread was not affected by VLDL-containing mouse serum (Fig. 7B), which was previously shown to potently inhibit HCV attachment and cell-free infection similarly to human serum (41). VLDL in mouse and human sera would compete with apoE incorporated on the HCV envelope for HSPG binding. The failure of VLDL-containing serum to block HCV cell-to-cell spread may explain the pathological characteristics observed in liver biopsy samples of chronic hepatitis C patients. About 7% to 20% of hepatocytes were detected for HCV infection and replication in the liver of hepatitis C patients. HCV-infected hepatocytes were found in clusters, suggesting the spread of the virus from cell to cell (66). We speculated that plasma VLDL serves as a restriction factor for HCV cell-free dissemination in vivo (41). The resistance of HCV cell-to-cell spread to VLDL may also contribute to persistent HCV infection in vivo. Additionally, the opposing effects of apoE and VLDL-containing mouse serum on HCV cell-to-cell spread suggest that different mechanisms are involved in HCV cell-free and cell-to-cell spread.
The underlying molecular mechanism of HCV cell-free and cell-to-cell spread remains an area of intense investigation in HCV research. HCV cell-to-cell spread accounts for less than 10% of total HCV infection, at least in vitro (Fig. 4). It may play a more prominent role in HCV persistence in vivo (39). The above-described discrepancy in receptor dependence between HCV cell-free and cell-to-cell transmission suggests that there are different virus particles involved in different routes of virus spread. HCV particles produced in cell culture are highly heterogeneous in size (ranging from 40 nm to 100 nm in diameter) and pleomorphic, with vesicular particles resembling exosomes, as revealed by cryo-electron microscopy (67, 68). A number of studies demonstrated that exosomes produced from HCV-infected hepatocytes in cell culture and in hepatitis C patients were infectious (69–73). More significantly, exosome-associated HCV particles contain apoE, are resistant to HCV-neutralizing antibodies, and are less dependent on HCV receptors (69, 71, 73). Therefore, it is possible that exosome-associated HCV particles are responsible for HCV cell-to-cell spread, which is also dependent on apoE and less dependent on HCV attachment and postattachment receptors, unlike HCV cell-free infection. It was reported previously that an intracellular cholesterol transport inhibitor and knockdown of autophagy could inhibit the release of exosome-dependent HCV particles (74, 75). It will be interesting to determine whether inhibitors of the exosomal pathway will have therapeutic value for hepatitis C by blocking HCV cell-to-cell transmission.
MATERIALS AND METHODS
Viruses and cell culture.
The cell culture-adapted HCV strain of genotype 2a (JFH1) was grown in Huh-7.5 cells, as described in our previous work (76, 77). This cell culture-grown HCV is designated HCVcc. Huh-7.5, the lentivirus packaging cell line HEK293T, and the adenovirus packaging cell line AD293 (Agilent Technologies) were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 0.1 mM nonessential amino acids (Sigma), 100 U/ml of penicillin, and 100 μg/ml of streptomycin at 37°C in a 5% CO2 incubator.
Antibodies and reagents.
HCV NS5A monoclonal antibody (9E10) was provided by Tim Tellinghuisen. β-Actin monoclonal antibody (AC15) and human SDC-2 monoclonal antibody (1B2) were obtained from Sigma-Aldrich. Human SDC-1 (DL101), SDC-4 (5G9), and CD81 (5A6) monoclonal antibodies were purchased from Santa Cruz. Human SDC-3 monoclonal antibody (AF3539) and TIM-1 monoclonal antibody (MAB1750) were obtained from R&D Systems. CLDN1 (catalog no. 71-7800)-, OCLN (catalog no. 71-1500)-, and SR-BI (catalog no. 486800)-specific rabbit polyclonal antibodies were obtained from Invitrogen. LDLR-specific monoclonal antibody (HL1) was provided by Jin Ye (78). HCV E2-specific neutralizing antibody (CBH5) was provided by Steven Foung (60). The Western blotting ECL substrate was obtained from Bio-Rad. Gene-specific siRNAs and nonspecific control (NSC) siRNA were synthesized by Dharmacon. The Dual-Luciferase Reporter Assay system was purchased from Promega. One-Step qRT-PCR kits were obtained from Thermo Fisher Scientific.
Plasmid construction.
The lentiCRISPRv2-blasticidin plasmid was made by replacing the puromycin-resistant gene in the lentiCRISPRv2 vector (33) with a blasticidin-resistant gene. sgRNAs specifically targeting the SDC-1, SDC-2, SDC-3, and SDC-4 genes were designed based on the protocols described at the website of Feng Zhang (http://crispr.mit.edu/). All sgRNAs were designed with minimal potential off-target effects according to bioinformatics analysis (33). A pair of oligonucleotides was used for the construction of the lentiCRISPRv2-puromycin and lentiCRISPRv2-blasticidin vectors expressing SDC-1, SDC-2, SDC-3, and SDC-4 sgRNAs (Table 1). Two oligonucleotides were initially treated with T4 polynucleotide kinase and then annealed in T4 DNA ligation buffer. The double-stranded oligonucleotides were cloned into BsmBI-digested lentiCRISPRv2-puromycin (Addgene) and the above-described lentiCRISPRv2-blasticidin plasmids by using T4 DNA ligase. The ligation mixture was transformed into Stbl3 competent cells (TransGen Biotech). Plasmid DNAs were prepared by using a miniprep DNA isolation kit (Qiagen). The resulting lentiCRISPRv2-SDC-1, lentiCRISPRv2-SDC-2, lentiCRISPRv2-SDC-3, and lentiCRISPRv2-SDC-4 plasmids were confirmed by DNA sequence analysis. To better quantify HCV infection, the previously described HCV dual-reporter system (pLenti-NIrD) (34) was modified by replacing the mCherry gene with a firefly luciferase gene. The firefly luciferase gene was amplified by PCR using oligonucleotide primers Fluc-F-XbaI (5′-TGCTCTAGAATGGAAGACGCC-3′) and Fluc-R-MluI (5′-CGACGCGTTTACACGGCGATCT-3′). The resulting PCR DNA fragment was digested with the restriction enzymes XbaI and MluI and inserted into the pLenti-NIrD plasmid between the XbaI and MluI sites. A Renilla luciferase gene was amplified by PCR and inserted into the pcDNA6-RT vector (Invitrogen) between the HindIII and EcoRI restriction enzyme sites, resulting in a Renilla luciferase-expressing vector designated pcDNA6/TR-Rluc. A subgenomic JFH1 replicon expressing a Gaussia luciferase reporter was made for the measurement of HCV RNA replication. The Gaussia luciferase gene was fused with a blasticidin resistance gene in subgenomic JFH1 replicon plasmid pSGR-JFH1-BLAST, as described previously (79). The Gaussia luciferase-expressing replicon plasmid is designated pSGR-JFH1-Blast/Gluc.
TABLE 1.
Oligonucleotide primers used for construction of SDC gene sgRNAs and sequencinga
| Gene | sgRNA forward primer sequence | sgRNA reverse primer sequence | Forward primer for PCR of target gene | Reverse primer for PCR of target gene |
|---|---|---|---|---|
| SDC-1 | CACCGGAGACGTGGGAATAGCCGTC | AAACGACGGCTATTCCCACGTCTCC | GACAGCTCTCCTTGTCCATTT | TAGGACCAACCCACTCAATTTC |
| SDC-2 | CACCGGTTCTGTATATTCAGCGTCG | AAACCGACGCTGAATATACAGAACC | AGACTACCCACAGACACC | AGACTAGGACCACCTCAA |
| SDC-3 | CACCGTAAGGACCACTGGCGTACGG | AAACCCGTACGCCAGTGGTCCTTAC | GCCTGGATTCAACCTCATAG | CCTTCTCAACACCCTCACC |
| SDC-4 | CACCGTCACGCGTAGAACTCATTGG | AAACCCAATGAGTTCTACGCGTGAC | CACAGGCAGTGGTGATGG | TCAGTATGGGCTTGAGGG |
Sequences are in the 5′-to-3′ direction.
Recombinant lentivirus production.
HEK293T cells in 6-well plates were transfected with 0.58 μg of pVSVg, 1.25 μg of psPAX2, and 0.83 μg of lentiCRISPR vectors expressing Cas9 and SDC-1, SDC-2, SDC-3, and SDC-4 sgRNAs by using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen). Similarly, the lentivirus expressing HCV-inducible firefly luciferase (lenti-NIrD-fluc) was constructed by transfecting 4 μg of the pLenti-NirD-fluc vector, 0.4 μg of pVSVg, and 4 μg of pR8.74 DNAs into HEK293T cells. At 72 h p.t., the supernatant was collected and filtered through a 0.45-μm low-protein-binding membrane unit (Millipore). The resulting recombinant lentiviruses were aliquoted and stored at −80°C in a freezer.
Construction of SDC knockout cell lines.
Huh7.5 cells in 100-mm cell culture dishes were transduced with lentiCRISPRv2-puromycin lentiviruses expressing SDC-1, SDC-2, SDC-3, and SDC-4 sgRNAs. Cell clones were selected with 2 μg/ml of puromycin for 2 to 3 weeks. Individual cell clones were transferred to 24-well cell culture plates for expansion. Genomic DNAs were extracted from each cell clone and used for PCR amplification of each specific gene using primers complementary to both sides of the sgRNA targeting region. PCR products were used for DNA sequence analysis. The knockout of specific genes was further validated by Western blotting. To make SDC-1 and SDC-2 double knockout cell lines, SDC-2 knockout Huh-7.5 cells were transduced with lentiCRISPRv2-blasticidin lentivirus expressing an SDC-1-specific sgRNA. The knockout of both SDC-1 and SDC-2 was validated by DNA sequence analysis and Western blotting.
Construction of a stable Huh-7.5 cell line with an HCV dual-reporter system expressing firefly luciferase.
Parent and gene-specific knockout Huh-7.5 cells were transduced with the above-described lenti-NIrD-fluc lentivirus. Parent Huh-7.5 cells transduced with lenti-NIrD-fluc were selected with 10 μg/ml of blasticidin. Individual Huh-7.5 cell clones were picked and screened for HCV-inducible firefly luciferase expression. Individual cell clones were infected with HCV and then incubated with medium in the presence or absence of 2 μg/ml of Dox. The levels of firefly luciferase in NIrD-fluc-containing Huh-7.5 cells with or without HCV infection in the presence or absence of 2 μg/ml of Dox were determined by using a luciferase assay kit from Promega. One Huh-7.5 cell clone, designated NIrD-fluc-sc4, was chosen based on its high level of firefly luciferase expression only when infected with HCV. This stable NIrD-fluc-sc4 Huh-7.5 cell line was used for the study of HCV cell-cell transmission. Gene-specific knockout Huh-7.5 cells were also transduced with lenti-NIrD-fluc and incubated with blasticidin for 8 days. The pool of lenti-NIrD-fluc-transduced Huh-7.5 cells with a specific gene knockout was used for determining the effects of cellular gene knockout on HCV cell-cell transmission.
siRNA-mediated silencing of cellular genes.
Parent and NIrD-fluc-sc4 Huh-7.5 cells were transfected with 50 nM gene-specific siRNAs or NSC siRNA by using the RNAiMax transfection reagent (Invitrogen), as previously described (11). At 48 h p.t., the levels of mRNA and protein of each targeted cellular gene were determined by a qRT-PCR method and Western blotting, respectively, using the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or β-actin as an internal control. The effects of siRNA-mediated gene silencing on HCV infection and cell-cell transmission were subsequently determined by using HCV infection and cell-cell transmission assays.
HCV attachment and infection assays.
Parent Huh-7.5 cells and SDC knockout Huh-7.5 cells were seeded into 12-well cell culture plates. An HCV attachment assay was carried out by incubating Huh-7.5 cells with HCVcc at an MOI of 10 on ice for 2 h or at 37°C for 1 h. Unbound HCV was removed by washing with 1× PBS three times. Total RNA was extracted with TRIzol reagent (Invitrogen) and used for the quantification of HCV vRNA by a real-time qRT-PCR method. HCV infection was conducted by incubation of HCVcc with Huh-7.5 cells at 37°C for 1.5 h. The HCV-infected cells were then washed with PBS and incubated with fresh DMEM at 37°C for 24 h (single-cycle growth). The HCV-infected Huh-7.5 cells were lysed in RIPA buffer (50 mM Tris-HCl [pH 7.5], 150 mM sodium chloride, 1% Nonidet P-40, 0.5% sodium deoxycholate) for the detection of HCV NS5A and cellular proteins by Western blotting using specific monoclonal antibodies. Total RNA was extracted from HCV-infected cells with TRIzol reagent and was used for the quantification of positive-stranded HCV RNA by qRT-PCR.
HCV cell-to-cell transmission.
Parent Huh-7.5 cells in 100-mm cell culture dishes were infected with HCV at an MOI of 10. After 24 h p.i., HCV-infected Huh-7.5 cells were trypsinized and used for HCV cell-cell transmission experiments. The HCV-infected Huh-7.5 cells are designated HCV donor cells. NIrD-fluc-sc4 Huh-7.5 cells and lenti-NIrD-fluc-transduced Huh-7.5 cells with specific gene silencing or knockout were used as recipient cells. In order to normalize parent and gene knockout cell lines, pcDNA6/TR-Rluc plasmid DNA was transfected into recipient cells as a control. At 24 h p.t., cells were trypsinized and collected. HCV cell-to-cell transmission was done by mixing a total of 5 × 104 cells/well of the above-described HCV donor and recipient cells at a predetermined optimal ratio in 48-well cell culture plates. Cell numbers were determined by using a TC20 automated cell counter (Bio-Rad). After 2 h of incubation at 37°C (cell attachment), cell culture medium was replaced with fresh DMEM containing 10% FBS, 2% methylcellulose, and 2 μg/ml of doxycycline. After 48 h of coculture, the levels of Renilla and firefly luciferase expression in the recipient cells were quantified by using the Dual-Glo luciferase assay system (Promega) according to the manufacturer's instructions.
Transfection of subgenomic HCV replicon RNA expressing Gaussia luciferase.
The pSGR-JFH1-Blast/Gluc plasmid was linearized by XbaI digestion and blunted by treatment with mung bean nuclease (NEB). The subgenomic HCV RNAs were transcribed in vitro by using a RiboMAX T7 RNA transcription kit (Promega) and purified with a Qiagen RNeasy RNA isolation kit. Parent and gene-specific knockout Huh-7.5 cells in 12-well tissue culture plates were transfected with 1 μg/well of SGR-Blast/Gluc RNA mixed with 2 μl DMRIE-C reagent in Opti-MEM (Invitrogen). At 24 h, 48 h, and 72 h p.t., RNA-transfected cells were lysed for the quantification of Gaussia luciferase expression using BioLux Gaussia luciferase assay kits (New England BioLabs) to determine any effect of the specific gene knockout on HCV RNA replication.
Ectopic expression of SDC-1, SDC-2, and SDC-4 with recombinant adenoviruses.
Recombinant adenoviruses expressing SDC-1, SDC-2, and SDC-4 were described in our previous work (11). To ectopically express SDC-1, SDC-2, or SDC-4, SDC-1 or SDC-2 knockout Huh-7.5 cells in 12-well plates were infected with adenoviruses at an MOI of 10 at 37°C for 6 h. The virus-infected cells were washed with 1× PBS and incubated with fresh DMEM. At 48 h p.i., the adenovirus-infected Huh-7.5 cells were subsequently used for HCV attachment and infection assays as described above.
Western blot analysis.
Protein concentrations of cell lysates were determined by using a protein assay reagent (Bio-Rad). Twenty micrograms of total protein for each sample was loaded onto 10% SDS-PAGE gels, followed by electrophoresis and protein transfer onto a polyvinylidene difluoride (PVDF) membrane. After blocking with 5% nonfat milk, the membrane was incubated with primary antibodies specific for viral NS5A or cellular genes as indicated. Proteins were visualized with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit secondary antibodies and a chemiluminescence (ECL) substrate (Bio-Rad).
Quantification of HCV RNAs by real-time qRT-PCR.
Total RNAs in HCV-infected or HCV-bound cells were extracted with TRIzol reagent. The levels of positive-strand RNA were quantified by a real-time qRT-PCR method using HCV-specific primers and a TaqMan probe as previously described (12). Cellular GAPDH mRNA was used as an internal control and was quantified by using a mixture of primers and a probe from Life Technology.
Statistical analysis.
All data were analyzed by using GraphPad Prism5 software (GraphPad Software Inc.). A P value of <0.05 was considered a statistically significant difference. Vector NTI was used to analyze the sequencing results.
ACKNOWLEDGMENTS
We thank Charlie Rice (Rockefeller University), Takaji Wakita (National Institute of Infectious Diseases, Japan), Timothy Tellinghuisen (The Scripps Research Institute, FL), and Jin Ye (UT Northwestern Medical Center) for providing the Huh-7.5 cell line, JFH1 cDNA, NS5A monoclonal antibody 9E10, and the LDLR monoclonal antibody, respectively.
This work was supported by the National Natural Science Foundation of China (NSFC) (grant 81130082), NIH grants AI097318 and AI091953, the University of Alabama at Birmingham (UAB) Center for AIDS Research (CFAR), an NIH-funded program (grant P30 AI027767), and the UAB Comprehensive Cancer Center (grant P30 CA013148). Huahao Fan and Junfen Fan are sponsored by the China Scholarship Council, China (CSC) (file no. 20143026).
REFERENCES
- 1.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. 1989. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 2.Lindenbach BD, Rice CM. 2005. Unravelling hepatitis C virus replication from genome to function. Nature 436:933–938. doi: 10.1038/nature04077. [DOI] [PubMed] [Google Scholar]
- 3.Pringle CR. 1999. Virus taxonomy—1999. The universal system of virus taxonomy, updated to include the new proposals ratified by the International Committee on Taxonomy of Viruses during 1998. Arch Virol 144:421–429. doi: 10.1007/s007050050515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robertson B, Myers G, Howard C, Brettin T, Bukh J, Gaschen B, Gojobori T, Maertens G, Mizokami M, Nainan O, Netesov S, Nishioka K, Shin i T, Simmonds P, Smith D, Stuyver L, Weiner A. 1998. Classification, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. International Committee on Virus Taxonomy. Arch Virol 143:2493–2503. doi: 10.1007/s007050050479. [DOI] [PubMed] [Google Scholar]
- 5.Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouille Y. 2006. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J Virol 80:6964–6972. doi: 10.1128/JVI.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basu A, Beyene A, Meyer K, Ray R. 2004. The hypervariable region 1 of the E2 glycoprotein of hepatitis C virus binds to glycosaminoglycans, but this binding does not lead to infection in a pseudotype system. J Virol 78:4478–4486. doi: 10.1128/JVI.78.9.4478-4486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basu A, Kanda T, Beyene A, Saito K, Meyer K, Ray R. 2007. Sulfated homologues of heparin inhibit hepatitis C virus entry into mammalian cells. J Virol 81:3933–3941. doi: 10.1128/JVI.02622-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koutsoudakis G, Kaul A, Steinmann E, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R. 2006. Characterization of the early steps of hepatitis C virus infection by using luciferase reporter viruses. J Virol 80:5308–5320. doi: 10.1128/JVI.02460-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morikawa K, Zhao Z, Date T, Miyamoto M, Murayama A, Akazawa D, Tanabe J, Sone S, Wakita T. 2007. The roles of CD81 and glycosaminoglycans in the adsorption and uptake of infectious HCV particles. J Med Virol 79:714–723. doi: 10.1002/jmv.20842. [DOI] [PubMed] [Google Scholar]
- 10.Bishop JR, Schuksz M, Esko JD. 2007. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 11.Shi Q, Jiang J, Luo G. 2013. Syndecan-1 serves as the major receptor for attachment of hepatitis C virus to the surfaces of hepatocytes. J Virol 87:6866–6875. doi: 10.1128/JVI.03475-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Qiao L, Hou Z, Luo G. 2017. TIM-1 promotes hepatitis C virus cell attachment and infection. J Virol 91:e01583-16. doi: 10.1128/JVI.01583-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang J, Cun W, Wu X, Shi Q, Tang H, Luo G. 2012. Hepatitis C virus attachment mediated by apolipoprotein E binding to cell surface heparan sulfate. J Virol 86:7256–7267. doi: 10.1128/JVI.07222-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang J, Luo G. 2009. Apolipoprotein E but not B is required for the formation of infectious hepatitis C virus particles. J Virol 83:12680–12691. doi: 10.1128/JVI.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiang J, Wu X, Tang H, Luo G. 2013. Apolipoprotein E mediates attachment of clinical hepatitis C virus to hepatocytes by binding to cell surface heparan sulfate proteoglycan receptors. PLoS One 8:e67982. doi: 10.1371/journal.pone.0067982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans MJ, von Hahn T, Tscherne DM, Syder AJ, Panis M, Wolk B, Hatziioannou T, McKeating JA, Bieniasz PD, Rice CM. 2007. Claudin-1 is a hepatitis C virus co-receptor required for a late step in entry. Nature 446:801–805. doi: 10.1038/nature05654. [DOI] [PubMed] [Google Scholar]
- 17.Helle F, Dubuisson J. 2008. Hepatitis C virus entry into host cells. Cell Mol Life Sci 65:100–112. doi: 10.1007/s00018-007-7291-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sourisseau M, Michta ML, Zony C, Israelow B, Hopcraft SE, Narbus CM, Parra Martin A, Evans MJ. 2013. Temporal analysis of hepatitis C virus cell entry with occludin directed blocking antibodies. PLoS Pathog 9:e1003244. doi: 10.1371/journal.ppat.1003244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Z, Tian Y, Machida K, Lai MM, Luo G, Foung SK, Ou JH. 2012. Transient activation of the PI3K-AKT pathway by hepatitis C virus to enhance viral entry. J Biol Chem 287:41922–41930. doi: 10.1074/jbc.M112.414789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu X, Lee EM, Hammack C, Robotham JM, Basu M, Lang J, Brinton MA, Tang H. 14 May 2014. Cell death-inducing DFFA-like effector b is required for hepatitis C virus entry into hepatocytes. J Virol doi: 10.1128/JVI.00081-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sainz B Jr, Barretto N, Martin DN, Hiraga N, Imamura M, Hussain S, Marsh KA, Yu X, Chayama K, Alrefai WA, Uprichard SL. 2012. Identification of the Niemann-Pick C1-like 1 cholesterol absorption receptor as a new hepatitis C virus entry factor. Nat Med 18:281–285. doi: 10.1038/nm.2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin DN, Uprichard SL. 2013. Identification of transferrin receptor 1 as a hepatitis C virus entry factor. Proc Natl Acad Sci U S A 110:10777–10782. doi: 10.1073/pnas.1301764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, Doffoel M, Raffelsberger W, Poch O, McKeating JA, Brino L, Baumert TF. 2011. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat Med 17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Timpe JM, Stamataki Z, Jennings A, Hu K, Farquhar MJ, Harris HJ, Schwarz A, Desombere I, Roels GL, Balfe P, McKeating JA. 2008. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 47:17–24. doi: 10.1002/hep.21959. [DOI] [PubMed] [Google Scholar]
- 25.Brimacombe CL, Grove J, Meredith LW, Hu K, Syder AJ, Flores MV, Timpe JM, Krieger SE, Baumert TF, Tellinghuisen TL, Wong-Staal F, Balfe P, McKeating JA. 2011. Neutralizing antibody-resistant hepatitis C virus cell-to-cell transmission. J Virol 85:596–605. doi: 10.1128/JVI.01592-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Catanese MT, Loureiro J, Jones CT, Dorner M, von Hahn T, Rice CM. 2013. Different requirements for scavenger receptor class B type I in hepatitis C virus cell-free versus cell-to-cell transmission. J Virol 87:8282–8293. doi: 10.1128/JVI.01102-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mailly L, Xiao F, Lupberger J, Wilson GK, Aubert P, Duong FH, Calabrese D, Leboeuf C, Fofana I, Thumann C, Bandiera S, Lutgehetmann M, Volz T, Davis C, Harris HJ, Mee CJ, Girardi E, Chane-Woon-Ming B, Ericsson M, Fletcher N, Bartenschlager R, Pessaux P, Vercauteren K, Meuleman P, Villa P, Kaderali L, Pfeffer S, Heim MH, Neunlist M, Zeisel MB, Dandri M, McKeating JA, Robinet E, Baumert TF. 2015. Clearance of persistent hepatitis C virus infection in humanized mice using a claudin-1-targeting monoclonal antibody. Nat Biotechnol 33:549–554. doi: 10.1038/nbt.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Witteveldt J, Evans MJ, Bitzegeio J, Koutsoudakis G, Owsianka AM, Angus AG, Keck ZY, Foung SK, Pietschmann T, Rice CM, Patel AH. 2009. CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J Gen Virol 90:48–58. doi: 10.1099/vir.0.006700-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fofana I, Xiao F, Thumann C, Turek M, Zona L, Tawar RG, Grunert F, Thompson J, Zeisel MB, Baumert TF. 2013. A novel monoclonal anti-CD81 antibody produced by genetic immunization efficiently inhibits hepatitis C virus cell-cell transmission. PLoS One 8:e64221. doi: 10.1371/journal.pone.0064221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gondar V, Molina-Jimenez F, Hishiki T, Garcia-Buey L, Koutsoudakis G, Shimotohno K, Benedicto I, Majano PL. 2015. Apolipoprotein E, but not apolipoprotein B, is essential for efficient cell-to-cell transmission of hepatitis C virus. J Virol 89:9962–9973. doi: 10.1128/JVI.00577-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hueging K, Doepke M, Vieyres G, Bankwitz D, Frentzen A, Doerrbecker J, Gumz F, Haid S, Wolk B, Kaderali L, Pietschmann T. 2014. Apolipoprotein E codetermines tissue tropism of hepatitis C virus and is crucial for viral cell-to-cell transmission by contributing to a postenvelopment step of assembly. J Virol 88:1433–1446. doi: 10.1128/JVI.01815-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lefevre M, Felmlee DJ, Parnot M, Baumert TF, Schuster C. 2014. Syndecan 4 is involved in mediating HCV entry through interaction with lipoviral particle-associated apolipoprotein E. PLoS One 9:e95550. doi: 10.1371/journal.pone.0095550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren Q, Li C, Yuan P, Cai C, Zhang L, Luo GG, Wei W. 2015. A dual-reporter system for real-time monitoring and high-throughput CRISPR/Cas9 library screening of the hepatitis C virus. Sci Rep 5:8865. doi: 10.1038/srep08865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anjum S, Wahid A, Afzal MS, Albecka A, Alsaleh K, Ahmad T, Baumert TF, Wychowski C, Qadri I, Penin F, Dubuisson J. 2013. Additional glycosylation within a specific hypervariable region of subtype 3a of hepatitis C virus protects against virus neutralization. J Infect Dis 208:1888–1897. doi: 10.1093/infdis/jit376. [DOI] [PubMed] [Google Scholar]
- 36.Keck ZY, Xia J, Wang Y, Wang W, Krey T, Prentoe J, Carlsen T, Li AY, Patel AH, Lemon SM, Bukh J, Rey FA, Foung SK. 2012. Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog 8:e1002653. doi: 10.1371/journal.ppat.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wasilewski LN, El-Diwany R, Munshaw S, Snider AE, Brady JK, Osburn WO, Ray SC, Bailey JR. 2016. A hepatitis C virus envelope polymorphism confers resistance to neutralization by polyclonal sera and broadly neutralizing monoclonal antibodies. J Virol 90:3773–3782. doi: 10.1128/JVI.02837-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barretto N, Sainz B Jr, Hussain S, Uprichard SL. 2014. Determining the involvement and therapeutic implications of host cellular factors in hepatitis C virus cell-to-cell spread. J Virol 88:5050–5061. doi: 10.1128/JVI.03241-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Z, He JJ. 2013. Cell-cell contact-mediated hepatitis C virus (HCV) transfer, productive infection, and replication and their requirement for HCV receptors. J Virol 87:8545–8558. doi: 10.1128/JVI.01062-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao F, Zhao T, Deng L, Lv D, Zhang X, Pan X, Xu J, Long G. 2017. Visualizing the essential role of complete virion assembly machinery in efficient hepatitis C virus cell-to-cell transmission by a viral infection-activated split-intein-mediated reporter system. J Virol 91:e01720-16. doi: 10.1128/JVI.01720-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tao J, Kang KD, Hall SD, Laube AH, Liu J, Renfrow MB, Novak J, Luo G. 2015. The serum very-low-density lipoprotein serves as a restriction factor against hepatitis C virus infection. J Virol 89:6782–6791. doi: 10.1128/JVI.00194-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helenius A. 2013. Virus entry and uncoating, p 87–104. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 43.Liu S, McCormick KD, Zhao W, Zhao T, Fan D, Wang T. 2012. Human apolipoprotein E peptides inhibit hepatitis C virus entry by blocking virus binding. Hepatology 56:484–491. doi: 10.1002/hep.25665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barth H, Schnober EK, Zhang F, Linhardt RJ, Depla E, Boson B, Cosset FL, Patel AH, Blum HE, Baumert TF. 2006. Viral and cellular determinants of the hepatitis C virus envelope-heparan sulfate interaction. J Virol 80:10579–10590. doi: 10.1128/JVI.00941-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agnello V, Abel G, Elfahal M, Knight GB, Zhang QX. 1999. Hepatitis C virus and other flaviviridae viruses enter cells via low density lipoprotein receptor. Proc Natl Acad Sci U S A 96:12766–12771. doi: 10.1073/pnas.96.22.12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu S, Yang W, Shen L, Turner JR, Coyne CB, Wang T. 2009. Tight junction proteins claudin-1 and occludin control hepatitis C virus entry and are downregulated during infection to prevent superinfection. J Virol 83:2011–2014. doi: 10.1128/JVI.01888-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R, Weiner AJ, Houghton M, Rosa D, Grandi G, Abrignani S. 1998. Binding of hepatitis C virus to CD81. Science 282:938–941. doi: 10.1126/science.282.5390.938. [DOI] [PubMed] [Google Scholar]
- 48.Ploss A, Evans MJ, Gaysinskaya VA, Panis M, You H, de Jong YP, Rice CM. 2009. Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457:882–886. doi: 10.1038/nature07684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scarselli E, Ansuini H, Cerino R, Roccasecca RM, Acali S, Filocamo G, Traboni C, Nicosia A, Cortese R, Vitelli A. 2002. The human scavenger receptor class B type I is a novel candidate receptor for the hepatitis C virus. EMBO J 21:5017–5025. doi: 10.1093/emboj/cdf529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benedicto I, Molina-Jimenez F, Bartosch B, Cosset FL, Lavillette D, Prieto J, Moreno-Otero R, Valenzuela-Fernandez A, Aldabe R, Lopez-Cabrera M, Majano PL. 2009. The tight junction-associated protein occludin is required for a postbinding step in hepatitis C virus entry and infection. J Virol 83:8012–8020. doi: 10.1128/JVI.00038-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fofana I, Krieger SE, Grunert F, Glauben S, Xiao F, Fafi-Kremer S, Soulier E, Royer C, Thumann C, Mee CJ, McKeating JA, Dragic T, Pessaux P, Stoll-Keller F, Schuster C, Thompson J, Baumert TF. 2010. Monoclonal anti-claudin 1 antibodies prevent hepatitis C virus infection of primary human hepatocytes. Gastroenterology 139:953.e4–964.e4. doi: 10.1053/j.gastro.2010.05.073. [DOI] [PubMed] [Google Scholar]
- 52.Haberstroh A, Schnober EK, Zeisel MB, Carolla P, Barth H, Blum HE, Cosset FL, Koutsoudakis G, Bartenschlager R, Union A, Depla E, Owsianka A, Patel AH, Schuster C, Stoll-Keller F, Doffoel M, Dreux M, Baumert TF. 2008. Neutralizing host responses in hepatitis C virus infection target viral entry at postbinding steps and membrane fusion. Gastroenterology 135:1719.e1–1728.e1. doi: 10.1053/j.gastro.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 53.Zahid MN, Turek M, Xiao F, Thi VL, Guerin M, Fofana I, Bachellier P, Thompson J, Delang L, Neyts J, Bankwitz D, Pietschmann T, Dreux M, Cosset FL, Grunert F, Baumert TF, Zeisel MB. 2013. The postbinding activity of scavenger receptor class B type I mediates initiation of hepatitis C virus infection and viral dissemination. Hepatology 57:492–504. doi: 10.1002/hep.26097. [DOI] [PubMed] [Google Scholar]
- 54.Grigorov B, Reungoat E, Gentil Dit Maurin A, Varbanov M, Blaising J, Michelet M, Manuel R, Parent R, Bartosch B, Zoulim F, Ruggiero F, Pecheur EI. 23 December 2016. Hepatitis C virus infection propagates through interactions between Syndecan-1 and CD81 and impacts the hepatocyte glycocalyx. Cell Microbiol doi: 10.1111/cmi.12711. [DOI] [PubMed] [Google Scholar]
- 55.Zhang F, Sodroski C, Cha H, Li Q, Liang TJ. 2017. Infection of hepatocytes with HCV increases cell surface levels of heparan sulfate proteoglycans, uptake of cholesterol and lipoprotein, and virus entry by up-regulating SMAD6 and SMAD7. Gastroenterology 152:257.e7–270.e7. doi: 10.1053/j.gastro.2016.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harris HJ, Davis C, Mullins JG, Hu K, Goodall M, Farquhar MJ, Mee CJ, McCaffrey K, Young S, Drummer H, Balfe P, McKeating JA. 2010. Claudin association with CD81 defines hepatitis C virus entry. J Biol Chem 285:21092–21102. doi: 10.1074/jbc.M110.104836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harris HJ, Farquhar MJ, Mee CJ, Davis C, Reynolds GM, Jennings A, Hu K, Yuan F, Deng H, Hubscher SG, Han JH, Balfe P, McKeating JA. 2008. CD81 and claudin 1 coreceptor association: role in hepatitis C virus entry. J Virol 82:5007–5020. doi: 10.1128/JVI.02286-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mothes W, Sherer NM, Jin J, Zhong P. 2010. Virus cell-to-cell transmission. J Virol 84:8360–8368. doi: 10.1128/JVI.00443-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dubuisson J, Helle F, Cocquerel L. 2008. Early steps of the hepatitis C virus life cycle. Cell Microbiol 10:821–827. doi: 10.1111/j.1462-5822.2007.01107.x. [DOI] [PubMed] [Google Scholar]
- 60.Keck ZY, Xia J, Cai Z, Li TK, Owsianka AM, Patel AH, Luo G, Foung SK. 2007. Immunogenic and functional organization of hepatitis C virus (HCV) glycoprotein E2 on infectious HCV virions. J Virol 81:1043–1047. doi: 10.1128/JVI.01710-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pierce BG, Keck ZY, Lau P, Fauvelle C, Gowthaman R, Baumert TF, Fuerst TR, Mariuzza RA, Foung SK. 26 October 2016. Global mapping of antibody recognition of the hepatitis C virus E2 glycoprotein: implications for vaccine design. Proc Natl Acad Sci U S A doi: 10.1073/pnas.1614942113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lacek K, Vercauteren K, Grzyb K, Naddeo M, Verhoye L, Slowikowski MP, Fafi-Kremer S, Patel AH, Baumert TF, Folgori A, Leroux-Roels G, Cortese R, Meuleman P, Nicosia A. 2012. Novel human SR-BI antibodies prevent infection and dissemination of HCV in vitro and in humanized mice. J Hepatol 57:17–23. doi: 10.1016/j.jhep.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 63.Grove J, Nielsen S, Zhong J, Bassendine MF, Drummer HE, Balfe P, McKeating JA. 2008. Identification of a residue in hepatitis C virus E2 glycoprotein that determines scavenger receptor BI and CD81 receptor dependency and sensitivity to neutralizing antibodies. J Virol 82:12020–12029. doi: 10.1128/JVI.01569-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chang K-S, Jiang J, Cai Z, Luo G. 2007. Human apolipoprotein E is required for infectivity and production of hepatitis C virus in cell culture. J Virol 81:13783–13793. doi: 10.1128/JVI.01091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xu Y, Martinez P, Seron K, Luo G, Allain F, Dubuisson J, Belouzard S. 2015. Characterization of hepatitis C virus interaction with heparan sulfate proteoglycans. J Virol 89:3846–3858. doi: 10.1128/JVI.03647-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liang Y, Shilagard T, Xiao SY, Snyder N, Lau D, Cicalese L, Weiss H, Vargas G, Lemon SM. 2009. Visualizing hepatitis C virus infections in human liver by two-photon microscopy. Gastroenterology 137:1448–1458. doi: 10.1053/j.gastro.2009.07.050. [DOI] [PubMed] [Google Scholar]
- 67.Catanese MT, Uryu K, Kopp M, Edwards TJ, Andrus L, Rice WJ, Silvestry M, Kuhn RJ, Rice CM. 2013. Ultrastructural analysis of hepatitis C virus particles. Proc Natl Acad Sci U S A 110:9505–9510. doi: 10.1073/pnas.1307527110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gastaminza P, Dryden KA, Boyd B, Wood MR, Law M, Yeager M, Chisari FV. 2010. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J Virol 84:10999–11009. doi: 10.1128/JVI.00526-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bukong TN, Momen-Heravi F, Kodys K, Bala S, Szabo G. 2014. Exosomes from hepatitis C infected patients transmit HCV infection and contain replication competent viral RNA in complex with Ago2-miR122-HSP90. PLoS Pathog 10:e1004424. doi: 10.1371/journal.ppat.1004424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Devhare PB, Sasaki R, Shrivastava S, Di Bisceglie AM, Ray R, Ray RB. 11 January 2017. Exosome mediated intercellular communication between hepatitis C virus infected hepatocytes and hepatic stellate cells. J Virol doi: 10.1128/JVI.02225-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu Z, Zhang X, Yu Q, He JJ. 2014. Exosome-associated hepatitis C virus in cell cultures and patient plasma. Biochem Biophys Res Commun 455:218–222. doi: 10.1016/j.bbrc.2014.10.146. [DOI] [PubMed] [Google Scholar]
- 72.Patman G. 2014. Hepatitis: exosomal route of HCV transmission exists in patients. Nat Rev Gastroenterol Hepatol 11:704. doi: 10.1038/nrgastro.2014.179. [DOI] [PubMed] [Google Scholar]
- 73.Ramakrishnaiah V, Thumann C, Fofana I, Habersetzer F, Pan Q, de Ruiter PE, Willemsen R, Demmers JA, Stalin Raj V, Jenster G, Kwekkeboom J, Tilanus HW, Haagmans BL, Baumert TF, van der Laan LJ. 2013. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. Proc Natl Acad Sci U S A 110:13109–13113. doi: 10.1073/pnas.1221899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Elgner F, Ren H, Medvedev R, Ploen D, Himmelsbach K, Boller K, Hildt E. 2016. The intracellular cholesterol transport inhibitor U18666A inhibits the exosome-dependent release of mature hepatitis C virus. J Virol 90:11181–11196. doi: 10.1128/JVI.01053-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shrivastava S, Devhare P, Sujijantarat N, Steele R, Kwon YC, Ray R, Ray RB. 2015. Knockdown of autophagy inhibits infectious hepatitis C virus release by the exosomal pathway. J Virol 90:1387–1396. doi: 10.1128/JVI.02383-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Carlsson P, Presto J, Spillmann D, Lindahl U, Kjellen L. 2008. Heparin/heparan sulfate biosynthesis: processive formation of N-sulfated domains. J Biol Chem 283:20008–20014. doi: 10.1074/jbc.M801652200. [DOI] [PubMed] [Google Scholar]
- 77.Jiang J, Luo G. 2012. Cell culture-adaptive mutations promote viral protein-protein interactions and morphogenesis of infectious hepatitis C virus. J Virol 86:8987–8997. doi: 10.1128/JVI.00004-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Owen DM, Huang H, Ye J, Gale M Jr. 2009. Apolipoprotein E on hepatitis C virion facilitates infection through interaction with low-density lipoprotein receptor. Virology 394:99–108. doi: 10.1016/j.virol.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chang KS, Cai Z, Zhang C, Sen GC, Williams BR, Luo G. 2006. Replication of hepatitis C virus (HCV) RNA in mouse embryonic fibroblasts: protein kinase R (PKR)-dependent and PKR-independent mechanisms for controlling HCV RNA replication and mediating interferon activities. J Virol 80:7364–7374. doi: 10.1128/JVI.00586-06. [DOI] [PMC free article] [PubMed] [Google Scholar]