

ABSTRACT
There are marked differences in the spread and prevalence of HIV-1 subtypes worldwide, and differences in clinical progression have been reported. However, the biological reasons underlying these differences are unknown. Gag-protease is essential for HIV-1 replication, and Gag-protease-driven replication capacity has previously been correlated with disease progression. We show that Gag-protease replication capacity correlates significantly with that of whole isolates (r = 0.51; P = 0.04), indicating that Gag-protease is a significant contributor to viral replication capacity. Furthermore, we investigated subtype-specific differences in Gag-protease-driven replication capacity using large well-characterized cohorts in Africa and the Americas. Patient-derived Gag-protease sequences were inserted into an HIV-1 NL4-3 backbone, and the replication capacities of the resulting recombinant viruses were measured in an HIV-1-inducible reporter T cell line by flow cytometry. Recombinant viruses expressing subtype C Gag-proteases exhibited substantially lower replication capacities than those expressing subtype B Gag-proteases (P < 0.0001); this observation remained consistent when representative Gag-protease sequences were engineered into an HIV-1 subtype C backbone. We identified Gag residues 483 and 484, located within the Alix-binding motif involved in virus budding, as major contributors to subtype-specific replicative differences. In East African cohorts, we observed a hierarchy of Gag-protease-driven replication capacities, i.e., subtypes A/C < D < intersubtype recombinants (P < 0.0029), which is consistent with reported intersubtype differences in disease progression. We thus hypothesize that the lower Gag-protease-driven replication capacity of subtypes A and C slows disease progression in individuals infected with these subtypes, which in turn leads to greater opportunity for transmission and thus increased prevalence of these subtypes.
IMPORTANCE HIV-1 subtypes are unevenly distributed globally, and there are reported differences in their rates of disease progression and epidemic spread. The biological determinants underlying these differences have not been fully elucidated. Here, we show that HIV-1 Gag-protease-driven replication capacity correlates with the replication capacity of whole virus isolates. We further show that subtype B displays a significantly higher Gag-protease-mediated replication capacity than does subtype C, and we identify a major genetic determinant of these differences. Moreover, in two independent East African cohorts we demonstrate a reproducible hierarchy of Gag-protease-driven replicative capacity, whereby recombinants exhibit the greatest replication, followed by subtype D, followed by subtypes A and C. Our data identify Gag-protease as a major determinant of subtype differences in disease progression among HIV-1 subtypes; furthermore, we propose that the poorer viral replicative capacity of subtypes A and C may paradoxically contribute to their more efficient spread in sub-Saharan Africa.
KEYWORDS: HIV-1 subtype, Gag-protease, viral replication capacity
INTRODUCTION
HIV-1 group M accounts for greater than 95% of HIV infections worldwide (1, 2). It is considerably genetically diverse, consisting of 9 subtypes (A to D, F to H, J, and K) and over 70 circulating recombinant forms (CRFs), which are unevenly distributed globally (2–5). Subtype B, which predominates in the Americas, Europe, and Australia (3), is the best-characterized subtype; however, it accounts for only approximately 11% of infections (6). On the other hand, subtypes A, C, and D and intersubtype recombinants predominate in sub-Saharan Africa (2), which carries about 70% of the global burden of HIV-1 disease (7). In particular, HIV-1 subtype C, which is the most prevalent subtype globally (∼50% of infections), is responsible for nearly all HIV-1 infections in Southern Africa and the Ethiopia/Somalia region; it has also gained dominance in several east Asian countries and is the fastest-emerging subtype in South America (2, 8). Moreover, subtypes A and D and intersubtype recombinants predominate in East and Central Africa (2, 3); subtype A in particular has significantly increased in frequency in Uganda (9).
Differences in the rates of disease progression and transmission between HIV-1 subtypes could contribute to their differing prevalence and expansion. Studies in East Africa, where subtypes A and D cocirculate, have established that subtype D is associated with faster disease progression than subtype A (10–13). There is also evidence that recombinant forms have a higher rate of progression to AIDS than does subtype A (10, 14), although this was not observed in another study (12). There are conflicting reports regarding the rate of disease progression of subtype C relative to that of other subtypes (12, 15–18). While there is general agreement that subtype C has a transmissibility rate at least equal to those of other M group subtypes (19, 20), some studies have reported increased transmissibility of subtype C compared to subtypes A and D (21–23). Furthermore, the higher heterosexual transmission of subtype A than of subtype D (24) may have contributed to the expansion of subtype A in Uganda (9).
Differential prevalence and expansion of subtypes have important implications for future projections of the pandemic and for the development of vaccines and other preventative or therapeutic strategies. However, the biological reasons underlying these differences remain unclear. Ex vivo competition assays of limited numbers of viral isolates have indicated that recombinant viruses are fitter than their parental forms (25, 26) and that subtype C (from Africa) is substantially less fit than subtypes A, B, and D (19, 20, 27), although more isolates are required to delineate differences between subtypes A, B, and D (20). Although in vitro viral replication has been correlated with disease progression in some studies (28), the relationship between these factors remains incompletely characterized. Furthermore, biological differences between subtype C and subtype B and/or other subtypes in the Env (29), reverse transcriptase (30), protease (31), Vif (32), and long terminal repeat (LTR) regions (33, 34) have been reported; however, the implications of these factors for disease progression and epidemic spread remain unclear. More recently, a study of >100 clinical isolates showed that subtype D had higher Pol-driven replication capacity than subtype A and directly linked these differences to faster disease progression in the former than in the latter subtype (35). Furthermore, we recently demonstrated subtype-specific differences in Nef function (36) and showed that Nef functional differences in acute infection correlate with markers of disease progression (37). Taken together, these results support subtype-specific differences in HIV-1 protein function as determinants of disease progression.
We and others have demonstrated that Gag-protease (Gag-Pro) or Gag-driven replication capacity correlates with markers of disease progression (38–41). Given the evidence of Gag as a determinant of disease progression and the important role of Gag in HIV-1 replication (reviewed in reference 42), we sought to determine whether there are subtype-specific differences in Gag-protease-driven replication capacity using well-pedigreed clinical cohorts from Africa and the Americas. We first assessed the contribution of Gag-protease to the overall viral replicative capacity and demonstrated that the former reflects the latter. We next compared the Gag-protease-driven replication capacities of subtype B and C isolates derived from different geographical locations and determined that subtype C isolates have substantially lower replication capacity than subtype B isolates. Furthermore, we identified the genetic determinants of these differences. To further elucidate subtype-specific Gag-protease biological differences and whether they contribute to intersubtype differences in disease progression, we measured Gag-protease-driven replication capacity in a population where subtypes A, C, and D and intersubtype recombinants cocirculate. A hierarchy of Gag-protease-driven replication capacities that was consistent with reported intersubtype differences in disease progression was found. We discuss possible implications of these results in light of reported subtype differences in disease progression and epidemic spread.
RESULTS
Gag-protease-driven replication capacity correlates with replication capacity of whole virus isolates.
Previous studies have demonstrated that Gag-protease-driven replication capacity correlates with markers of disease progression, namely, plasma viral load, CD4+ T cell counts, and the rate of CD4+ T cell decline (38–41). To determine the contribution of Gag-protease to the overall HIV-1 replication capacity, we compared the in vitro replication of whole patient-derived HIV-1 isolates with that of their respective Gag-protease NL4-3 recombinant viruses for 16 chronic subtype C-infected individuals from the South African Sinikithemba (SK) cohort. There was a tendency for the recombinant viruses to have higher replication capacity than the isolates; nevertheless, a statistically significant correlation was found between them (Pearson's correlation, r = 0.51 and P = 0.04) (Fig. 1). This confirms that Gag-protease contributes significantly to overall viral replication capacity.
FIG 1.
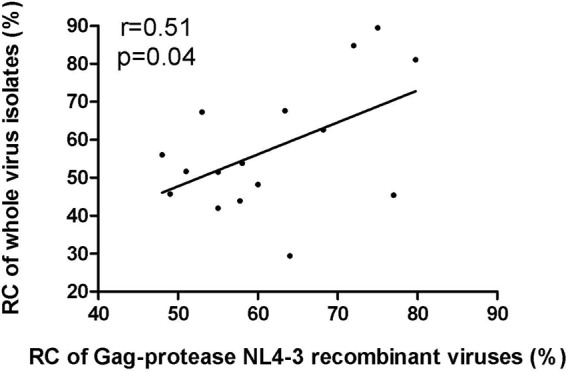
Contribution of Gag-protease to overall HIV-1 replication capacity. The graph shows a significant positive correlation (Pearson's correlation) between the replication capacities (RC) of HIV-1 subtype C isolated from patients and the corresponding NL4-3 recombinant viruses encoding Gag-protease derived from the same patients. Replication capacity was assayed in GXR cells using flow cytometry and normalized to the growth of the wild-type NL4-3 virus (replication capacity of 100%).
Subtype C Gag-protease NL4-3 recombinant viruses display significantly lower replication capacity than subtype B Gag-protease NL4-3 recombinant viruses.
To investigate Gag-protease-mediated differences in replication capacity between HIV-1 subtypes, the in vitro replication capacities of 803 and 406 NL4-3 recombinant viruses encoding Gag-protease from antiretroviral therapy-naive patients chronically infected with HIV-1 subtype B (41) and subtype C (39), respectively, were compared. Subtype B Gag-protease NL4-3 recombinant viruses exhibited significantly higher replication capacities (mean = 1; standard deviation [SD] = 0.15) than the subtype C Gag-protease recombinant viruses (mean = 0.62; SD = 0.1) (Mann-Whitney U test; P < 0.0001) (Fig. 2A). To exclude the possibility that this difference was due to interlaboratory variation (the subtype B and C recombinants were generated and assayed in different laboratories), a representative subset of the subtype B Gag-protease NL4-3 recombinants (n = 25) were generated and assayed in the same laboratory as the subtype C Gag-protease NL4-3 recombinants, with consistent results (Mann-Whitney U test; P < 0.0001) (Fig. 2B). Since viral replication capacity may be related to stage of disease (41, 43), a subset (n = 25) of HIV-1 subtype C-infected patients were matched with a subset (n = 25) of subtype B-infected patients in terms of CD4+ T cell counts and viral loads (medians [interquartile ranges], 140 [60 to 320] cells/mm3 and 5.4 [5.2 to 5.6] log10 copies/ml for subtype B, and 137 [76 to 341] cells/mm3 and 5.3 [4.9 to 5.7] log10 copies/ml for subtype C), and the analysis was repeated. The replication capacities of this subtype C subset (mean = 0.65; SD = 0.08) were still significantly lower than the replication capacities of the subtype B Gag-protease NL4-3 recombinants (mean = 0.95; SD = 0.13) (Mann-Whitney U-test; P < 0.0001) (Fig. 2C). These results suggest that subtype C Gag-protease is less functional than that of subtype B and further suggest that subtype-specific differences in this region could contribute to the lower in vitro replication capacity reported for HIV-1 subtype C isolates than for those of subtype B (20).
FIG 2.
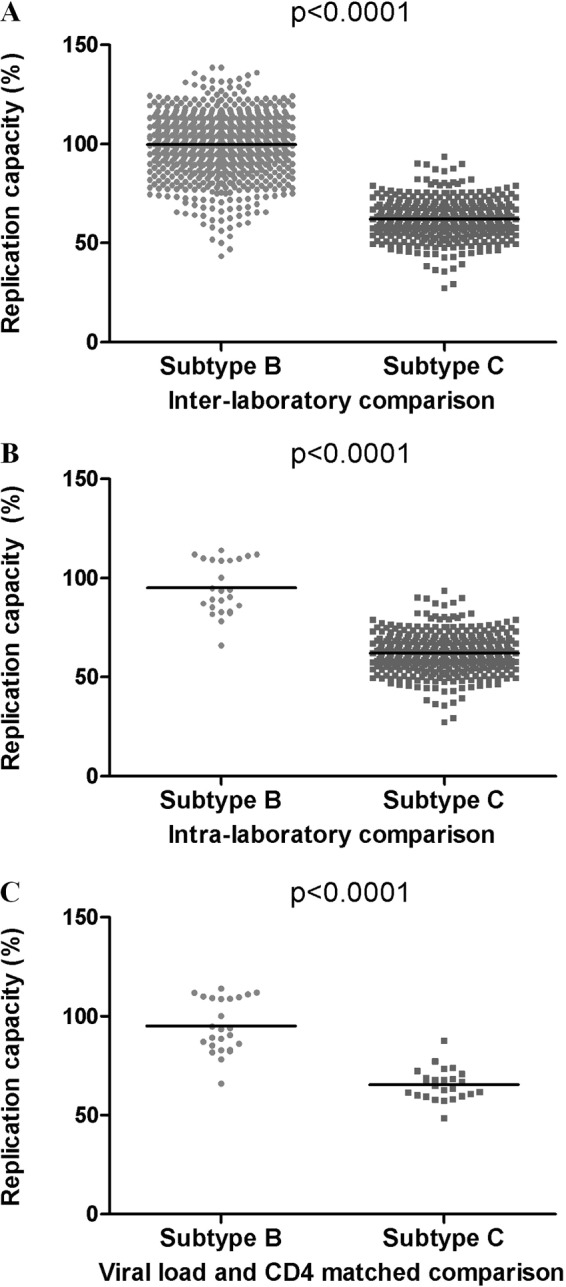
HIV-1 subtype B versus subtype C Gag-protease-mediated replication capacity. (A) Significant differences between the replication capacities of NL4-3 recombinant viruses encoding subtype B (n = 803) (41) and subtype C (n = 406) (39) Gag-proteases from patients chronically infected with HIV-1. (B) A comparison of subtype B (n = 25) and subtype C (n = 406) Gag-protease-mediated replication capacities performed in the same laboratory. (C) Significant differences between subtype B (n = 25) and subtype C (n = 25) Gag-protease-mediated replication capacity remain when samples matched for viral loads and CD4 counts are compared. Replication capacity was assayed in GXR cells using flow cytometry and normalized to the growth of the wild-type NL4-3 virus (replication capacity of 100%). The Mann-Whitney U test was used to test for significance, and lines represent the means.
Subtype C consensus Gag pZM246-F10 recombinant virus displays lower replication capacity than NL4-3 Gag pZM246-F10 recombinant virus.
It is conceivable that the engineering of subtype C Gag-protease into the subtype B NL4-3 viral backbone may have disrupted the natural context, resulting in suboptimal replication of the subtype C Gag-protease NL4-3 recombinant viruses. However, two observations argue against this possibility. First, greater similarity of the subtype C Gag sequences to the subtype B consensus did not correlate with higher subtype C Gag-protease NL4-3 recombinant virus replication capacity (39). Second, we engineered the subtype C consensus and subtype B NL4-3 Gag sequences into a subtype C viral backbone, pZM246-F10, and observed that the former replicated substantially slower than the latter (Fig. 3). It should be noted that the replication of wild-type pZM246-F10 was very poor in our experiments. This may be attributed to the several B*57/B*58:01-associated mutations, previously reported to reduce replication, encoded by pZM246-F10 Gag (44). Thus, the consensus C Gag was engineered into the pZM246-F10 backbone to represent subtype C. Together, these results suggest that subtype C Gag-protease is inherently less functional than subtype B Gag-protease.
FIG 3.
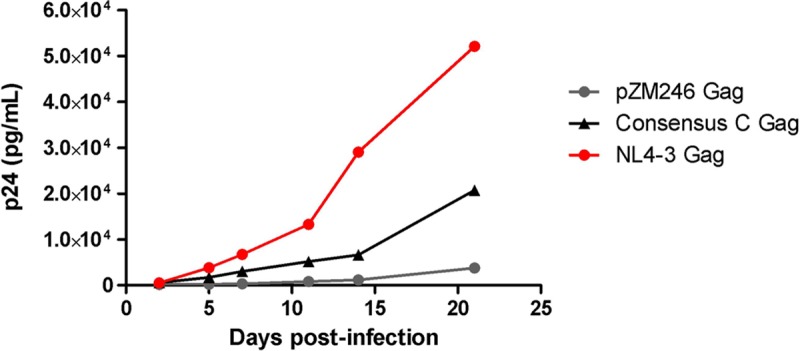
Replication kinetics of the subtype C consensus Gag pZM246-F10 and NL4-3 Gag pZM246-F10 recombinant viruses. Comparison of the replication kinetics of pZM246-F10 (subtype C) viral constructs carrying the gag genes from subtype C consensus and NL4-3. The wild-type pZM246-F10 virus was included as a control. The replication assay was carried out in activated pooled PBMCs from four donors and monitored using p24 ELISA.
Gag codons 483 and 484 are major determinants of subtype-specific replication capacity.
We next investigated the genetic determinants of the functional differences between subtypes. Since previous studies showed no difference (or increased efficiency) of subtype C compared to subtype B protease (30, 31), we hypothesized that Gag largely mediates the differences observed. HIV gag encodes four posttranslationally cleaved proteins: p17 (matrix), p24 (capsid), p7 (nucleocapsid), and p6. Given previous data underscoring the structural Gag p24 protein as a key determinant of HIV-1 replication capacity (45, 46), we initially compared the replication capacities of HIV-1 wild-type NL4-3 and of NL4-3 encoding the consensus C Gag p24. However, we observed that these viruses replicated similarly (Fig. 4), suggesting that the determinants underlying the functional difference between subtype B and subtype C Gag-proteases are outside p24. Codon function analysis of our subtype B and C Gag sequences identified subtype-specific polymorphisms at codons 67 (matrix), 473 (p6), and 483 (p6) that were strongly linked to altered viral replication capacities in our previous studies (39, 41) (Table 1). Specifically, 67A is the consensus amino acid in subtype C and is strongly associated with decreased replication capacity in subtype B (41), where 67S is the consensus. In both subtypes B and C, 473P is associated with increased replication capacity (39, 41), and the frequencies of this polymorphism are 39% and 1.7%, respectively, in the present subtype B- and subtype C-infected cohorts. In subtype B, the consensus 483L is associated with significantly increased replication capacity (41), while there is a deletion at positions 483 and 484 (indicated as 483−/484− here) in subtype C.
FIG 4.

Replication kinetics of consensus C Gag p24 NL4-3 recombinant virus and NL4-3 wild-type virus. The replication of NL4-3 encoding consensus C Gag p24 is similar to that of wild-type NL4-3. The replication assay was carried out in GXR cells, and viral replication was monitored using flow cytometry to measure GFP expression.
TABLE 1.
Subtype B and C consensus residues at Gag codons associated with altered HIV-1 replication capacitya
| Codon(s) | Residue(s) for subtype: |
Associations with RC | |
|---|---|---|---|
| B | C | ||
| 67 | S | A | 67A with decreased RC in subtype B (P < 0.0001) (41). |
| 473 | S | A | 473Pb with increased RC in subtypes B (P < 0.0001) and C (P = 0.01) (39, 41). |
| 483/484 | L/Y | −/−c | 483L with increased RC in subtype B (P < 0.0001) (41). |
RC, replication capacity.
473P is more frequent in subtype B (39%) than subtype C (1.7%).
−/−, deletion of residues.
To further investigate whether these differences contribute to subtype differences in Gag-protease-mediated replication capacity, subtype B- and C-specific Gag residues at these sites were introduced into subtype C (SK-254) and subtype B (NL4-3) Gag-proteases, respectively, by site-directed mutagenesis, and replication was measured in an NL4-3 backbone. While alterations at residues 67 and 473 did not appreciably alter replication capacity in either backbone (Fig. 5), deletion of Gag residues 483 and 484 from the subtype B Gag-protease decreased replication capacity to 81% of wild-type levels (Fig. 5A). Correspondingly, insertion of the subtype B amino acids at these codons into the subtype C Gag-protease increased replication capacity to 120% of wild-type levels (Fig. 5B). We further confirmed that insertion of 483L/484Y into subtype C increased replication capacity irrespective of the backbone subtype, by cloning the SK-254 Gag into a subtype C MJ4 backbone and measuring the replication capacity in GXR cells (an HIV-inducible GFP-reporter GXR T cell line) by detection of green fluorescent protein (GFP)-positive cells as well as reverse transcriptase activity as previously described (47). In the MJ4 backbone, insertion of 483L/484Y similarly increased replication capacity to 121% of wild-type levels (Fig. 5C). These results suggest that the higher Gag-protease-mediated replication capacity of subtype B versus C sequences may be mediated, at least in part, by the presence versus absence of Gag 483L/484Y in these subtypes, respectively.
FIG 5.
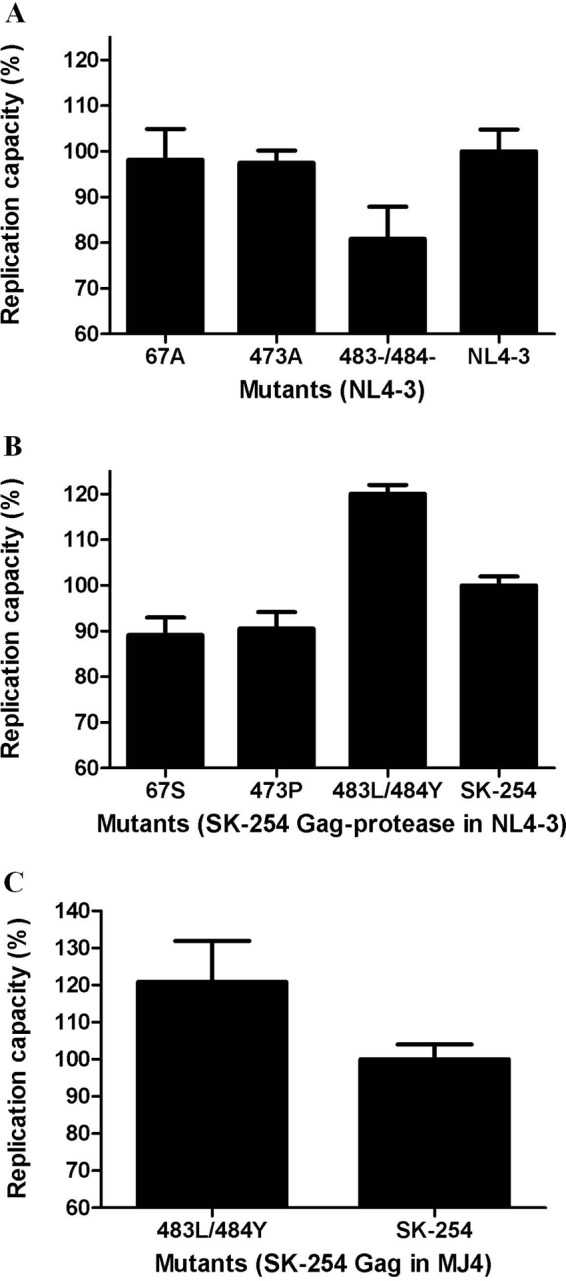
Replication capacities of HIV-1 subtype B and C viruses encoding subtype-specific mutations in Gag. (A) Replication capacities of subtype B NL4-3 wild-type virus and NL4-3 viruses encoding subtype C-specific Gag residues 67S, 473A, and 483−/484−. (B) Replication capacities of NL4-3 recombinant viruses encoding wild-type subtype C Gag derived from patient SK-254 and SK-254 Gag with introduced subtype B-specific Gag residues 67S, 473P, and 483L/484Y. (C) Replication capacities of MJ4 recombinant viruses encoding wild-type subtype C Gag derived from patient SK-254 and SK-254 Gag with introduced subtype B-specific Gag residues 483L/484Y. Replication capacities were assayed in GXR cells using flow cytometry and are expressed relative to the capacity of the respective wild-type virus, which represents 100% replication. Bars represent the means from at least 3 independent experiments, and error bars represent standard deviations from the means.
Gag-protease-mediated replication capacity differs significantly between subtypes A, C, and D and intersubtype recombinants.
The subtype B and C sequences studied here were derived from different populations that differ substantially with respect to sociodemographic, clinical, and host genetic characteristics (including HLA class I distributions, which are a major driver of Gag-protease-driven virus replication capacity [39, 41, 48]), rendering it difficult to tease apart the relative contributions of viral subtype versus other factors on HIV-1 disease progression (49–51). We therefore sought to further explore intersubtype differences in Gag-protease-driven replication capacity and their relationship to disease progression in a single population in which multiple subtypes cocirculate in order to mitigate such confounding factors.
The Gag-protease region was amplified and sequenced from 103 treatment-naive, chronically infected individuals from a Kenyan cohort. Of these, 57 (55.4%) were subtype A, 16 (15.5%) were subtype C, 13 (12.6%) were subtype D, and 17 (16.5%) were intersubtype recombinants (Fig. 6). NL4-3 viruses encoding the patient-derived Gag-proteases were constructed, and replication capacities were compared. Marked differences in replication capacities were observed between subtypes (Fig. 7A). Gag-protease intersubtype recombinants had the highest mean replication capacity, 113% (P < 0.001 compared to subtypes A, C and D), followed by subtype D with a mean replication capacity of 94% (P < 0.001 compared to subtype A, P < 0.01 compared to subtype C), followed by subtypes C and A with mean replication capacities of 78% and 69%, respectively, where 100% represents the replication capacity of wild-type NL4-3. Although we were limited by sample size, we did not observe significant differences in replication capacity between different intersubtype recombinants or those with different common (n ≥ 5) patterns of recombination (data not shown).
FIG 6.

Gag-protease subtype composition of the Kenyan Majengo cohort. (A) Maximum likelihood phylogenetic tree shows clustering of patient-derived Gag-protease gene sequences into distinct subtypes. Subtype reference A (A1, n = 37; A2, n = 20), C (n = 16), and D (n = 13) and intersubtype recombinant (n = 17) sequences are represented by black, red, purple, blue, and green, respectively. The scale bar indicates 2% nucleotide sequence divergence. (B) Illustration of Gag-protease intersubtype recombinants. The subtypes A, C, and D are represented by red, purple, and blue, respectively. Numbering is according to the HXB2 reference strain. Residues (150, 410, and 435) at which recombination breakpoints were common (observed 5 or more times) are highlighted in bold.
FIG 7.

Intersubtype comparison of Gag-protease-mediated replication capacity in East African cohorts. (A) Significant differences in Gag-protease-mediated replication capacities of NL4-3 recombinant viruses encoding patient-derived Gag-proteases of subtypes A, C, and D and intersubtype recombinants from a Kenyan cohort. (B) Significant differences in replication capacities between subtypes are reproducible in a Ugandan cohort. Replication capacity was assayed in GXR cells using flow cytometry and normalized to the growth of the wild-type NL4-3 virus (replication capacity of 100%). Subtypes A, C, and D and intersubtype recombinants are represented in red, purple, blue, and green, respectively. The bars and whiskers represent the means and interquartile ranges, respectively. ANOVA (P value shown) with Tukey post hoc tests was used to test for significant differences between subtypes. The number of asterisks denotes the level of significance: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
CD4+ T cell counts and viral loads were comparable across subtypes (Table 2), suggesting that our observations are not driven by differences in clinical parameters. Furthermore, subtype D is phylogenetically closer to subtype B (52, 53) than subtypes A or C, raising the possibility that subtype D Gag-protease sequences would be inherently more compatible in an NL4-3 backbone than subtypes A or C. Two lines of evidence argue against such effects. First, there was no significant difference in replication capacity between the subtype D-containing intersubtype recombinants and nonsubtype D intersubtype recombinants (data not shown). Second, the similarity of each Gag sequence to the consensus B Gag sequence did not correlate with replication capacity (data not shown), supporting that the subtype B backbone was not driving the differences in Gag-protease-driven replication capacity observed between viruses encoding different subtypes of Gag-protease in the Kenyan Majengo cohort.
TABLE 2.
Characteristics of Majengo study subjects infected with HIV-1 subtypes A, C, and D and intersubtype recombinantsa
| Characteristic | Subtype A (n = 57) | Subtype C (n = 16) | Subtype D (n = 13) | Recombinants (n = 17) | P value |
|---|---|---|---|---|---|
| Age (yr) | 35 (31–40) | 37 (33–42) | 31 (25–35) | 35 (27–40) | 0.39 |
| CD4 count (cells/mm3) | 421 (342–590) | 376 (307–471) | 427 (356–509) | 358 (351–400) | 0.14 |
| Plasma HIV-1 RNA (log10 copies/ml) | 4.20 (3.71–5.18) | 4.04 (3.81–4.95) | 5.00 (4.21–5.73) | 5.23 (4.65–5.32) | 0.16 |
Values for subtypes and recombinants are medians, with interquartile ranges in parentheses. P values were calculated using the Kruskal-Wallis test.
Subtype differences in Gag-protease-mediated replication capacity are reproducible across cohorts and geographical regions.
To confirm that intersubtype differences in viral replication capacity are consistent across cohorts and regions, we generated and assessed the replication capacities of 30 Gag-protease recombinant viruses (10 samples each from subtypes A and D and AD recombinants) from the UARTO cohort in Uganda. To reduce confounding by clinical factors, selected samples were matched for CD4+ T cell counts and viral loads (medians [interquartile ranges], 171 [134.25 to 241.50] cells/mm3 and 5.2 [4.56 to 5.70] log10 copies/ml for subtype A, 154 [119.25 to 199] cells/mm3 and 5.0 [4.69 to 5.64] log10 copies/ml for subtype D, and 147 [107 to 204.75] cells/mm3 and 5.0 [4.28 to 5.65] log10 copies/ml for AD recombinants). Consistent with our previous observations, replication capacity differed markedly between subtypes (Fig. 7B). AD recombinants had the greatest replication capacity, 142% (P < 0.01 compared to subtype A), followed by subtype D with a mean of 107% (P < 0.05 compared to subtype A) and lastly subtype A with a mean of 76%, where 100% represents the replication capacity of wild-type NL4-3.
Subtype-specific differences in Gag-driven replication capacity are consistent using a subtype C backbone.
To more fully address the potential that subtype-specific differences in replication capacity simply reflect incompatibility with the NL4-3 subtype B backbone, additional experiments were performed using a pZM246-F10 subtype C backbone. A total of 5 patients (one each for A, B, C, and D subtypes and one for AD recombinants) for which their Gag-protease NL4-3 recombinant viruses represented the average replication capacity for each subtype were selected for the generation of patient-derived Gag pZM246-F10 recombinant viruses; the NL43 Gag-protease sequence was also inserted into pZM246-F10 as the normalization control (Table 3). It should be noted that wild-type pZM246-F10 was not included as a control, as it encodes B*57/B*58:01-associated mutations and replicates poorly (Fig. 3). Two independent attempts to generate the subtype A Gag pZM246-F10 recombinant virus failed, consistent with this subtype having the lowest replication capacity, although incompatibility with the subtype C backbone cannot be ruled out. Thus, conclusive data regarding the position of subtype A in the replication hierarchy using the pZM246-F10 backbone could not be obtained. The pZM246-F10 recombinant viruses encoding the other Gags of different subtypes were successfully generated; however, the virus growth using this backbone was particularly slow, which is consistent with previous reports of subtype C isolates having lower replication ability than other M group subtypes (20) and reports of poor growth of subtype C isolates in monocyte-macrophage cultures and T cell lines (54). A similar replication hierarchy of these viruses was observed in both GXR cells (Table 3; C < D < AD recombinant < B) and peripheral blood mononuclear cells (PBMCs) (Fig. 8; C < D < AD recombinant/B). Thus, Gag-protease replication is broadly consistent in both pZM246-F10 and NL4-3 backbones, viz., lower replication capacity of subtype C versus subtype B Gag-proteases and the replication hierarchy of A < D < intersubtype Gag-protease recombinants observed in East Africa. More data may be required to discriminate between Gag/Gag-protease-driven replication capacities in subtypes A versus C and subtypes B versus intersubtype recombinants.
TABLE 3.
Replication capacities of recombinant viruses encoding patient-derived Gag/Gag-proteases of different subtypes in GXR cells
| Subtype of Gag/Gag-proteasea | RC in NL4-3 backboneb | RC in pZM246-F10 backbonec |
|---|---|---|
| A | 0.66 | NDd |
| B | 0.99 | 1.05 |
| B (NL4-3) | 1 | 1 |
| C | 0.62 | 0.51 |
| D | 0.97 | 0.60 |
| AD recombinant | 1.08 | 0.90 |
Patient-derived Gag proteins and Gag-proteases of different subtypes as well as the laboratory strain NL4-3.
RCs of NL4-3 (subtype B) recombinant viruses encoding patient-derived Gag-proteases of different subtypes were normalized to wild-type NL4-3.
RCs of pZM246-F10 (subtype C) recombinant viruses encoding patient-derived Gags of different subtypes were normalized to NL4-3 Gag pZM246-F10.
ND, not determined: two independent attempts to generate pZM246-F10 recombinant viruses encoding the patient-derived A Gag failed, and thus RC could not be assessed.
FIG 8.
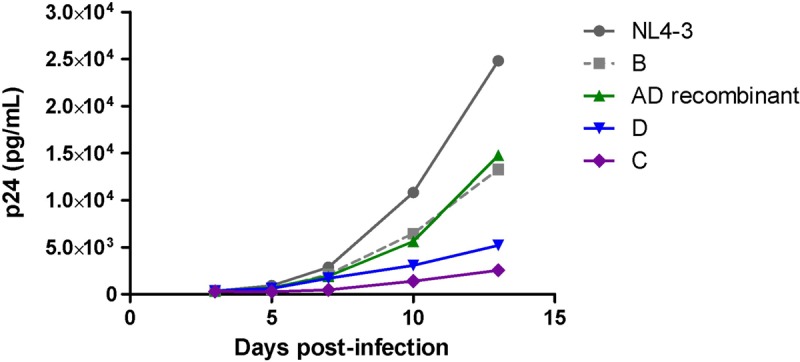
Intersubtype comparison of Gag-driven replication capacity in PBMCs using the subtype C pZM246-F10 backbone. Comparison of the replication kinetics of pZM246-F10 (subtype C) viral constructs carrying patient-derived gag genes of different subtypes as well as NL4-3 gag. The patients for whom the derived Gag-protease NL4-3 recombinant viruses represented the average replication capacity for each subtype were selected for generation of patient-derived Gag pZM246-F10 recombinant viruses. Subtypes B, C, and D and intersubtype recombinants are represented in gray, purple, blue, and green, respectively. The replication assay was carried out in activated pooled PBMCs from two donors and monitored using p24 ELISA.
Genetic correlates of Gag-protease-mediated replication capacity in East Africa. (i) Overrepresentation of the 483L/484Y motif in intersubtype recombinant sequences.
The Gag 483L/484Y motif, which likely explains in part the higher replication of subtype B than C sequences, is present in the subtype D consensus sequence but absent in the consensus A sequence (which has residues QDP instead of LY at this position). Of interest, of the 14 recombinants in the Majengo cohort that featured a subtype D component, 11 (79%) were subtype D at the 3′ end of Gag (Fig. 6) (two-tailed binomial test; P = 0.057). Moreover, the 483L/484Y motif was present in 10 of the 17 recombinants (59%) compared to only 8 of the 86 (9%) pure subtype A, C, or D sequences assessed (Fisher's exact test; P < 0.0001). Taken together, these results suggest that the Gag 483L/484Y motif is overrepresented among intersubtype recombinants in East Africa, which may in turn contribute to the enhanced replication capacity of these sequences.
(ii) Single-amino-acid variants associated with altered replication capacity.
We have previously identified single-amino-acid variants in subtype B and subtype C Gag-proteases that are associated with altered replication capacity (39, 41). Although we were limited by sample size, we performed an exploratory codon-by-codon analysis in our subtype A sequences to identify Gag-protease residues associated with altered replication capacity (our subtype D data set was not large enough to perform such an analysis). In doing so, we identified six amino acids significantly (P < 0.05 and q < 0.2) associated with reduced or increased replication capacity (Table 4). We next compared the frequency of these residues in 400 subtype A and 400 subtype D sequences retrieved from the Los Alamos HIV sequence database (http://www.hiv.lanl.gov). Four of the six residues differed significantly in frequency between the subtypes. Of note, 107L and 315N, both associated with reduced replication capacity, were significantly more frequent in subtype A than in subtype D (8% versus 0% and 66% versus 57%, respectively). However, these frequency differences are moderate, and therefore, while it is possible that these differences contribute to the lower replication capacity of subtype A sequences, further studies are required to investigate the genetic determinants of Gag-protease functional differences between subtypes A, C, and D and intersubtype recombinants.
TABLE 4.
Amino acids (observed 5 or more times) in Gag associated with altered Gag-protease-mediated replication capacity in subtype A
| Codon no.a | AAb | Consensus | Function (%)c |
No. of samplesd |
P value | q value | ||
|---|---|---|---|---|---|---|---|---|
| +AA | −AA | +AA | −AA | |||||
| 75 | I | L | 65 | 70 | 13 | 40 | 0.07 | 0.04 |
| 107 | L | I | 64 | 69 | 9 | 43 | 0.007 | 0.03 |
| 125 | S | S | 70 | 65 | 37 | 15 | 0.02 | 0.03 |
| 126 | S | S | 69 | 62 | 47 | 8 | 0.01 | 0.03 |
| 315 | N | N | 67 | 71 | 18 | 36 | 0.05 | 0.04 |
| 499 | S | S | 70 | 62 | 48 | 6 | 0.03 | 0.04 |
According to HXB2 numbering.
AA, amino acid variant associated with differences in replication capacity.
Median replication capacity (expressed as a percentage of wild-type NL4-3) of viruses with (+AA) and without (−AA) the amino acid variant.
Number of sequences with (+AA) and without (−AA) the amino acid variant. Amino acid totals vary, as gaps in the alignment are considered missing data.
DISCUSSION
There are striking differences in the spread and prevalence of different HIV-1 subtypes; however, the intrinsic biological properties that may contribute to these differences are not completely known (2, 4). In this study, we compared the functions of Gag-protease, which is essential for HIV-1 replication (42) and has previously been correlated with disease progression (39, 41), in different subtypes in large well-characterized cohorts. We demonstrate here that there are significant intersubtype differences in Gag-protease function: patient-derived subtype C Gag-protease recombinant viruses replicate significantly slower than their subtype B counterparts, and a hierarchy of Gag-protease-driven replication capacity—subtypes A/C < D < intersubtype recombinants—was observed in two independent East African cohorts.
The intersubtype differences in Gag-protease function are largely consistent with those observed for the replication capacity of whole virus isolates in studies where competition assays were performed in PBMCs for limited numbers of isolates of different subtypes (19, 20, 27). In those studies, subtype C isolates from Africa were shown to have lower replication capacity than other M group isolates, including those of subtype A, while we show that subtype C Gag-protease-driven replication capacity is significantly lower than that of subtypes B and D and intersubtype recombinants but similar to or lower than that of subtype A (indicating that genetic determinants other than Gag-protease may distinguish the replication capacities of subtype A and C isolates). Using an ex vivo competition assay in PBMCs, Abraha et al. described a replication capacity hierarchy of B ≥ D ≥ A > C (subtype A isolates replicated significantly slower than subtype B isolates; however, more isolates are required to confirm significant differences between each subtype) (20), which is consistent with our finding that subtype A Gag-proteases were less functional than those of subtype D (and by inference less functional than subtype B Gag-proteases, since they were on par with subtype C Gag-proteases). Furthermore, intersubtype recombinant isolates have been shown to outcompete isolates of their parental subtypes (25, 26), and we demonstrate here that viruses encoding intersubtype recombinant Gag-protease have a greater replication capacity than those with subtypes A, C, and D. These observations, taken together with our finding that the replication capacities of subtype C whole virus isolates correlated with that of recombinant viruses encoding the Gag-proteases derived from these isolates, confirm that Gag-protease is a significant determinant of overall viral replication capacity, in addition to Env (19) and protease-reverse transcriptase (55), which have also been shown to correlate with whole-isolate replication capacity. In further support of Gag as an important contributor to replication capacity, a recent study described a strong correlation between the replication capacities of 6 subtype C full-length infectious molecular clones and MJ4 recombinant viruses encoding their respective Gag proteins (56).
It is likely that Gag, rather than protease, is the major contributor to the results observed here (at least for the comparison of subtypes B and C), since protease has previously been shown to have similar activity in subtypes B and C (30) or greater catalytic efficiency in subtype C than B (31), which is not consistent with the overall lower replication capacity of subtype C isolates. Furthermore, the replication hierarchies of NL4-3 recombinant viruses encoding Gag-protease of different subtypes and pZM246-F10 recombinant viruses encoding Gag of different subtypes were consistent, indicating that Gag and not protease was the main driver of these results. Consistent with our observations for Gag, the Env (29) and reverse transcriptase (30) (which are also significant determinants of overall replication capacity) as well as Nef proteins (36) have been shown to have lower functionality in subtype C than in subtype B. In contrast, protease (31), Vif (32), and LTR (34, 57) are more active in subtype C than in other subtypes, which might reflect compensatory mechanisms for reduced functionality of other proteins, although the overall replication capacity of subtype C isolates remains compromised relative to other subtypes.
The intersubtype differences in Gag-protease-driven replication capacity could influence the spread and consequently the prevalence of different subtypes. This could occur through the influence of the disease progression rate, which then affects the opportunity for transmission (17). Gag-protease function could significantly affect disease progression rate: Gag-protease function reflects the overall replication capacity of isolates, for which distinct differences have been shown between long-term nonprogressors and progressors, indicating that this parameter significantly influences the rate of disease progression (28, 45, 58), and furthermore, Gag-protease- and Gag-driven replication capacities have been independently correlated with markers of disease progression (38–41). Moreover, the hierarchy of Gag-protease-driven replication capacity—subtypes A < D < intersubtype recombinants—that we observed in the East African cohorts is overall consistent with intersubtype differences in the disease progression reported in this region: subtype A resulted in a slower disease progression than subtype D in cohorts from Uganda (10, 11), Kenya (13), and Tanzania (12), and in Uganda, intersubtype recombinants resulted in faster progression to AIDS/death than subtype A and had a slightly higher hazard ratio for death than did subtype D (10) (yet increased virulence of intersubtype recombinants was not observed in Tanzania [12]). In addition, in Guinea-Bissau, infection with recombinant A3/02 was associated with increased risk of AIDS or death compared with subtype A3 (14). There is also evidence for reduced subtype C Gag-protease function relative to subtype B, corresponding with slower disease progression in subtype C-infected individuals; in Brazil, where subtype C cocirculates with subtypes B and F1 and intersubtype recombinants, subtype C was associated with higher CD4 counts (18). However, there are conflicting reports of the disease progression rate of subtype C relative to that of subtypes A and D (12, 15–17, 59). Nevertheless, overall, our data suggest that the lower Gag-protease-driven replication capacity of subtypes A and C (in addition to the lower Pol-driven replication capacity for subtype A [35]) may contribute to slower disease progression in individuals infected with these subtypes. The associated increased longevity is expected to result in greater opportunity for transmission (17) and hence in an increased prevalence of these subtypes. Accordingly, subtype C is the most prevalent subtype worldwide and subtype A is the predominant subtype in East Africa (2, 3). Furthermore, reports indicate that these subtypes are increasing in prevalence (2, 8, 9, 17).
Aside from the hypothesis that Gag-protease-mediated replication capacity influences a differential spread of subtypes through affecting disease progression, viral replication capacity has been directly linked to transmissibility. Several studies on RNA viruses have shown that genetic bottlenecks (such as occur during transmission) reduce viral replication capacity (60–62). More recently, it was described that viruses transmitted heterosexually did not have an enhanced viral replication capacity (63). Furthermore, we have observed in mother-to-child transmission pairs that recombinant viruses encoding Gag-protease from the infants had significantly lower replication capacities than those from their mothers (Vanessa L. Naidoo, Jaclyn K. Mann, Christie Noble, Emily Adland, Jonathan M. Carlson, Jake Thomas, Chanson J. Brumme, Christina F. Thobakgale-Tshabalala, Zabrina L. Brumme, Mark A. Brockman, Philip J. R. Goulder, Thumbi Ndung’u, submitted for publication). Taken together, these data lead to the hypothesis that HIV-1 variants with a lower viral replication capacity or lower Gag-protease-mediated replication capacity may be more transmissible, and this may be a factor underlying the differential spread of subtypes. Indeed, subtypes A and C, which have the lowest Gag-protease-driven replication capacity in the present study, may have increased transmissibility relative to other subtypes (21, 22, 24). The mechanism underlying the potentially increased transmissibility of viruses with low replication capacity remains unknown. We speculate that the mechanism may relate to the half-life of productively infected cells in genital fluids that are transmitted. Free infectious virions have a short half-life; however, transmitted productively infected cells may continue to release virions after the sexual encounter (thus, productively infected cells in the transmitted genital fluid would increase transmission risk as opposed to free virions alone). A virus with a high replication capacity may result in a shortened half-life of productively infected cells due to a higher budding rate, thereby paradoxically decreasing transmission risk, while a virus with low replication capacity may result in a longer half-life of productively infected cells and therefore increased transmission risk. Another possible factor contributing to increased transmissibility of subtype C is the low frequency of switch from CCR5-tropic virus to CXCR4-tropic virus relative to other subtypes, resulting in subtype C having a larger proportion of viruses that use the CCR5 receptor (4), which is required for cell entry of the transmitted virus (64).
In this study, we investigated the genetic determinants of intersubtype differences in Gag-protease-driven replication capacity. We observed that recombinant viruses encoding subtype B and subtype C Gag p24 did not differ in replication capacity, indicating that the genetic determinants of differences in Gag-protease function were outside this region. An investigation of residues that differed in frequency between subtypes B and C and were also statistically linked to altered Gag-protease-mediated replication capacities in our previous studies (39, 41) revealed that subtype-specific differences at residues 483 and 484 in Gag p6 are a significant determinant of the difference in Gag-protease function between subtypes B and C. Specifically, deletion of these residues, which are not present in the vast majority of subtype C Gag sequences, from subtype B Gag-protease reduced viral replication capacity, and correspondingly, introduction of these residues (483L/484Y) into a subtype C Gag-protease increased replication capacity. Interestingly, the 483L/484Y motif (consensus for subtypes B and D) was present in 59% of the intersubtype recombinant Gag sequences yet present in only 9% of the other Gag sequences in the Majengo cohort. The overrepresentation of the 483L/484Y motif in the intersubtype recombinants suggests that it is advantageous and may partially contribute to the enhanced replication capacity of these viruses. Residues 483 and 484 are essential residues in a late domain for binding the Alix host protein (65), which acts in concert with the primary budding factor Tsg101 to mediate viral budding (66). Mutants with disrupted Alix binding have significantly reduced particle production and infectivity, demonstrating an important role for Alix in HIV-1 replication (66). Recently, it was shown that the subtype C Gag p6 cannot bind Alix and that replacement of NL4-3 Gag p6 with subtype C Gag p6 reduced viral replication, although this could not conclusively be attributed to the 483L/484Y deletion (67). Here, we directly demonstrate that the 483L/484Y deletion, present almost universally in subtype C sequences, reduces viral replication and is likely a major contributor to the lower replication capacity of viruses encoding subtype C Gag-proteases. It is also of interest that an unusual mutant with deletion at residues 482 and 483, shown to disrupt Alix binding (68), was previously associated with nonprogressive HIV-1 infection (69), indicating that the ability to bind Alix may influence disease progression. Furthermore, it was recently described that a PYxE insertion (at the same position as the LY motif) in some East African subtype C isolates enhanced the replication capacity and virulence of these isolates compared to subtype C isolates without the insertion (70).
The assay employed in this study has several limitations; namely, it does not take into account interactions between different genes, the use of an NL4-3 backbone resulted in mixing of subtypes, and a cell line rather than primary cells was used to measure replication. Nevertheless, the assay yields results that correlate with the replication capacity of whole virus isolates and consistently correlate with markers of disease progression (38, 39, 41). Although we cannot conclusively rule out that the NL4-3 backbone may have partially influenced differences in Gag-protease-driven replication capacity between subtypes, the subtype-specific differences in Gag-driven replication capacity using a subtype C backbone were consistent with subtype-specific Gag-protease-driven replication capacity differences observed in the subtype B NL4-3 backbone.
In summary, we show that Gag-protease is an important determinant of viral replication capacity and that it differs substantially in functionality between HIV-1 subtypes, with subtype C showing lower functionality than subtype B and a functional hierarchy of subtypes (A/C < D < intersubtype recombinants) observed within East African populations. Since previous studies suggest an important influence of Gag-protease on disease progression, it is likely that the lower functionality of subtype A and C Gag-proteases slows disease progression in individuals infected with these subtypes, leading to a greater opportunity for transmission (as well as potential increased transmissibility) and a consequently increased prevalence of these subtypes. Importantly, we demonstrate that the hierarchy of Gag-protease-driven replication capacity in East Africa, where these subtypes cocirculate, is consistent with reported intersubtype differences in disease progression in this population, supporting that Gag-protease-driven replication capacity is a determinant of disease progression and differential spread of subtypes. Furthermore, our study sheds light on the genetic determinants of intersubtype differences in Gag-protease-mediated replication capacity, although studies to further investigate this are warranted. Future studies using full-length infectious molecular clones and whole-genome sequencing in large cohorts in which several subtypes cocirculate could further elucidate the biological determinants of subtype differences in disease and spread.
MATERIALS AND METHODS
Study subjects.
All study subjects were antiretroviral naive and chronically infected. For comparison of subtypes B and C, 803 individuals from the British Columbia HAART (highly active antiretroviral therapy) Observational Medical Evaluation and Research (BC HOMER) cohort (n = 762) and Boston chronic progressors cohort (n = 41) (41) and 406 individuals from the South African Sinikithemba (SK) cohort (39) were studied. To compare different subtypes in a population in which they cocirculate, we studied 103 individuals from the Kenyan Majengo sex worker cohort (71) who were infected with subtypes A (n = 57), C (n = 16), and D (n = 13) and intersubtype recombinants (n = 17). In addition, we investigated the reproducibility of subtype differences in 30 individuals from the Ugandan UARTO cohort (72) who were infected with subtypes A (n = 10) and D (n = 10) and AD recombinants (n = 10).
Specimens from the BC HOMER cohort comprised historic plasma samples that were anonymized according to Research Ethics Board (REB)-approved procedures prior to study. Approval was granted by the Research Ethics Board of Providence Health Care/University of British Columbia. Written informed consent was obtained from all other study participants, and ethical approval was granted from the following: Institutional Review Board at Massachusetts General Hospital (Boston chronic progressors cohort), Biomedical Research Ethics Committee of the University of KwaZulu-Natal (Sinikithemba cohort), Biomedical Research Ethics Committee of the University of KwaZulu-Natal and the Ethics Review Committee of the Kenyatta National Hospital Ethics Review Board (Majengo cohort), and institutional review boards at Mbarara University, Massachusetts General Hospital, and the University of California, San Francisco (UARTO cohort).
Whole-virus isolation.
For 16 subtype C-infected individuals from the SK cohort, HIV-1 was isolated from peripheral blood mononuclear cells (PBMCs) as previously described (73). Briefly, patient PBMCs were cocultured with prestimulated PBMCs from two anonymous HIV-1-negative donors, and the virus concentration in the supernatants was monitored by p24 enzyme-linked immunosorbent assay (ELISA; bioMérieux, Netherlands) every 3 days. Isolates were briefly cultured in a CEM-derived GFP reporter GXR T cell line (CEM-GXR25, here abbreviated to GXR) (74) to generate high titer virus stocks.
Amplification and sequencing of Gag-protease.
Gag-protease was amplified and sequenced as described previously (39). Sequence data were aligned to HIV-1 subtype B reference strain HXB2 (GenBank accession number K03455), and insertions with respect to HXB2 were stripped out. HIV-1 subtype was confirmed using the REGA subtyping tool (75). A maximum likelihood phylogenetic tree was drawn using PhyML, available at http://www.hiv.lanl.gov (76).
Generation of Gag-protease NL4-3 recombinant viruses.
Gag-protease NL4-3 recombinant viruses were constructed for all study subjects as described previously (39, 41, 77). Briefly, recombinant viruses encoding patient-derived Gag-protease were generated by electroporation of an HIV-inducible GFP-reporter GXR T cell line (GXR) with plasma-derived Gag-protease PCR products (amplified with primers complementary to NL4-3) and linearized Gag-protease-deleted HIV-1 subtype B NL4-3 plasmid. Protease was included together with Gag to maintain the important interaction between these proteins for each virus.
Generation of consensus C Gag pZM246-F10 and NL4-3 Gag pZM246-F10 recombinant viruses.
The pZM246-F10 HIV-1 subtype C infectious molecular clone, donated by Beatrice Hahn, was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH. The 2004 subtype C consensus Gag sequence (available at http://www.hiv.lanl.gov/content/sequence/NEWALIGN/align.html) was synthesized (Integrated DNA Technologies) and cloned into pZM246-F10, using BssHII and XhoI restriction enzymes as previously described (44). The NL4-3 Gag sequence was similarly cloned into pZM246_F10. Virus stocks were generated by transfection of HEK293T cells (obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH, from Andrew Rice [78]) with 7.5 μg of plasmid DNA, followed by harvesting of supernatants after 72 h (44). The capsid concentration of the viral stocks was quantified by p24 ELISA.
Generation of consensus C Gag p24 NL4-3 recombinant virus.
The consensus C Gag p24 NL4-3 recombinant virus was constructed as previously described (44). Briefly, the consensus C Gag p24 was amplified using 100-bp-long primers matching the NL4-3 sequence upstream and downstream of p24, and the virus stock was generated by electroporation of GXR cells with the amplicon as well as gag p24-deleted NL4-3 plasmid.
Generation of pZM246-F10 (subtype C) recombinant viruses encoding patient-derived Gags of different subtypes.
Gag pZM246-F10 recombinant viruses were constructed using methods similar to that described above. A total of 5 patients (one each for A, B, C, and D subtypes and one for AD recombinants) for which their Gag-protease NL4-3 recombinant viruses represented the average replication capacity for each subtype were selected for generation of patient-derived Gag pZM246-F10 recombinant viruses. In addition, the NL43 Gag-protease sequence was also inserted into pZM246-F10. Briefly, recombinant viruses were generated by electroporation of GXR cells with patient-derived gag PCR products (amplified with primers complementary to pZM246-F10) and pZM246-F10 that was linearized by digestion with BssHII and XhoI enzymes.
Generation of mutant viruses.
A QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) and custom-designed mutagenic primers were used to introduce subtype C-specific residues 67A, 473A, and 483−/484− into the subtype B NL4-3 plasmid. Similarly, subtype B-specific residues 67S, 473P, and 483L/484Y were introduced into a patient-derived subtype C Gag-protease sequence (SK-254; GenBank accession number HM593258) of high similarity (96.4% amino acid similarity) to the consensus subtype C Gag-protease sequence. Mutant NL4-3 viruses were generated as described previously (47) via electroporation of GXR cells with 10 μg of mutated NL4-3 plasmids. Mutant SK-254 Gag-protease NL4-3 viruses were generated by amplification of mutated SK-254 Gag-protease followed by transfection of GXR cells with PCR product and Gag-protease-deleted NL4-3 plasmid (39).
Replication capacity measurement.
The replication capacities of whole virus isolates and recombinant and mutant viruses were assayed in the GXR cell line by flow cytometry as described previously (39). Briefly, virus titers were first determined by infecting GXR cells with the virus stocks and measuring GFP expression after 2 days. The titer data were used to calculate the virus volume required to obtain a multiplicity of infection (MOI) of 0.003 on day 2 of the replication capacity assay. Following infection at an MOI of 0.003, GFP expression was measured daily for a week. Replication capacity was defined as the slope of increase in the percentage of infected cells from days 3 to 6 postinfection, and results were normalized to the growth of the relevant control (NL4-3 or SK-254 Gag-protease NL4-3 or NL4-3 Gag pZM246-F10) assayed in parallel. Replication capacities were assayed at least in duplicate independently, and results were averaged.
The replication capacities of consensus C Gag pZM246-F10 and NL4-3 Gag pZM246-F10 as well as of pZM246-F10 recombinant viruses encoding patient-derived Gags of different subtypes were assayed in prestimulated PBMCs pooled from at least 2 healthy HIV-negative donors by infection of 0.5 × 106 to 1 × 106 PBMCs with 20 ng p24 as previously described (44); here, p24 ELISA was used to monitor viral replication every 2 to 3 days postinfection, up to a maximum of 21 days.
Data analysis.
Pearson's (for normally distributed variables) or Spearman's (for nonnormally distributed variables) correlation was used to assess the relationship between continuous variables. A comparison of replication capacities between subtypes B and C was performed using the Mann-Whitney U test, while analysis of variance (ANOVA) with Tukey's posttests was used to compare replication capacities between multiple subtypes in the East African cohorts. Clinical characteristics were compared between patient groups infected with different subtypes using the Kruskal-Wallis test. An exploratory codon-by-codon analysis was performed to identify specific amino acid variants that were significantly associated with altered replication capacity in subtype A. Frequencies of amino acid variants of interest were compared between subtypes using a chi-square test computer software (K. J. Preacher, Calculation for the chi-square test: an interactive calculation tool for chi-square tests of goodness of fit and independence [http://quantpsy.org]). The significance cutoff for all statistical analyses was a P value of <0.05. For the codon-by-codon analysis, multiple comparisons were addressed using q values, the P value analogue of the false-discovery rate (79). Here, associations with P values of <0.05 and q values of <0.2 were considered significant.
Accession number(s).
Sequences from the Sinikithemba cohort and HOMER cohort were previously deposited in GenBank (39, 41, 48, 80). The Gag-protease sequences from the Majengo and UARTO cohorts are available in the GenBank database under accession numbers KX233975 to KX234077 (Majengo cohort) and KX377087 to KX377116 (UARTO cohort).
ACKNOWLEDGMENTS
We thank Richard Harrigan from the BC Centre for Excellence in HIV/AIDS and David Bangsberg from the Ragon Institute of MGH, MIT and Harvard University for access to samples and their support of the study. We also acknowledge Erasha Rajkoomar and Doty Achieng' Ojwach for technical assistance.
M.W.K., J.K.M., and D.C. received pilot grants from the Canada-Sub Saharan Africa (CANSSA) HIV/AIDS Network through funding provided by the Global Health Research Initiative (GHRI), itself a collaborative research funding partnership of the Canadian Institutes for Health Research (CIHR), the Canadian International Development Agency (CIDA), and the International Development Research Centre (IDRC). M.W.K. was partially funded by a doctoral award program of the CIHR to strengthen sub-Saharan Africa leadership in HIV prevention research. This research was further funded by grants from the South African Department of Science and Technology through the National Research Foundation, the Victor Daitz Foundation, and the Howard Hughes Medical Institute to T.N. J.N.M. was supported by the National Institutes of Health (R01 MH054907, UM1 CA181255, and P30 AI027763). Generation of the subtype B recombinant viruses was funded by a grant from the Canadian Institutes for Health Research (MOP-93536 to M.A.B. and Z.L.B.). Z.L.B. is supported by a scholar award from the Michael Smith Foundation for Health Research (MSFHR). M.A.B. holds a Canada Research Chair in Viral Pathogenesis and Immunity. This work was also partially supported through the DELTAS Africa Initiative (grant DEL-15-006). The DELTAS Africa Initiative is an independent funding scheme of the African Academy of Sciences (AAS)’s Alliance for Accelerating Excellence in Science in Africa (AESA) and is supported by the New Partnership for Africa’s Development Planning and Coordinating Agency (NEPAD Agency) with funding from the Wellcome Trust (grant 107752/Z/15/Z) and the UK government. The views expressed in this publication are those of the author(s) and not necessarily those of the AAS, NEPAD Agency, Wellcome Trust, or UK government. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Sharp PM, Hahn BH. 2008. Prehistory of HIV-1. Nature 455:605–606. doi: 10.1038/455605a. [DOI] [PubMed] [Google Scholar]
- 2.Tebit DM, Arts EJ. 2011. Tracking a century of global expansion and evolution of HIV to drive understanding and to combat disease. Lancet Infect Dis 11:45–56. doi: 10.1016/S1473-3099(10)70186-9. [DOI] [PubMed] [Google Scholar]
- 3.Hemelaar J. 2012. The origin and diversity of the HIV-1 pandemic. Trends Mol Med 18:182–192. doi: 10.1016/j.molmed.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Hemelaar J. 2013. Implications of HIV diversity for the HIV-1 pandemic. J Infect 66:391–400. doi: 10.1016/j.jinf.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 5.Los Alamos National Laboratory. HIV sequence database. https://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html Accessed 7 April 2014.
- 6.Hemelaar J, Gouws E, Ghys PD, Osmanov S, WHO-UNAIDS Network for HIV Isolation and Characterisation. 2011. Global trends in molecular epidemiology of HIV-1 during 2000-2007. AIDS 25:679–689. doi: 10.1097/QAD.0b013e328342ff93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.UNAIDS. Fact sheet 2015. http://aidsinfo.unaids.org/ Accessed 19 January 2016.
- 8.Gräf T, Pinto AR. 2013. The increasing prevalence of HIV-1 subtype C in Southern Brazil and its dispersion through the continent. Virology 435:170–178. doi: 10.1016/j.virol.2012.08.048. [DOI] [PubMed] [Google Scholar]
- 9.Conroy SA, Laeyendecker O, Redd AD, Collinson-Streng A, Kong X, Makumbi F, Lutalo T, Sewankambo N, Kiwanuka N, Gray RH, Wawer MJ, Serwadda D, Quinn TC, Rakai Health Sciences Program. 2010. Changes in the distribution of HIV type 1 subtypes D and A in Rakai District, Uganda between 1994 and 2002. AIDS Res Hum Retroviruses 26:1087–1091. doi: 10.1089/aid.2010.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kiwanuka N, Laeyendecker O, Robb M, Kigozi G, Arroyo M, McCutchan F, Eller LA, Eller M, Makumbi F, Birx D, Wabwire-Mangen F, Serwadda D, Sewankambo NK, Quinn TC, Wawer M, Gray R. 2008. Effect of human immunodeficiency virus type 1 (HIV-1) subtype on disease progression in persons from Rakai, Uganda, with incident HIV-1 infection. J Infect Dis 197:707–713. doi: 10.1086/527416. [DOI] [PubMed] [Google Scholar]
- 11.Kaleebu P, French N, Mahe C, Yirrell D, Watera C, Lyagoba F, Nakiyingi J, Rutebemberwa A, Morgan D, Weber J, Gilks C, Whitworth J. 2002. Effect of human immunodeficiency virus (HIV) type 1 envelope subtypes A and D on disease progression in a large cohort of HIV-1-positive persons in Uganda. J Infect Dis 185:1244–1250. doi: 10.1086/340130. [DOI] [PubMed] [Google Scholar]
- 12.Vasan A, Renjifo B, Hertzmark E, Chaplin B, Msamanga G, Essex M, Fawzi W, Hunter D. 2006. Different rates of disease progression of HIV type 1 infection in Tanzania based on infecting subtype. Clin Infect Dis 42:843–852. doi: 10.1086/499952. [DOI] [PubMed] [Google Scholar]
- 13.Baeten JM, Chohan B, Lavreys L, Chohan V, McClelland RS, Certain L, Mandaliya K, Jaoko W, Overbaugh J. 2007. HIV-1 subtype D infection is associated with faster disease progression than subtype A in spite of similar plasma HIV-1 loads. J Infect Dis 195:1177–1180. doi: 10.1086/512682. [DOI] [PubMed] [Google Scholar]
- 14.Palm AA, Esbjörnsson J, Månsson F, Kvist A, Isberg PE, Biague A, da Silva ZJ, Jansson M, Norrgren H, Medstrand P. 2014. Faster progression to AIDS and AIDS-related death among seroincident individuals infected with recombinant HIV-1 A3/CRF02_AG compared with sub-subtype A3. J Infect Dis 209:721–728. doi: 10.1093/infdis/jit416. [DOI] [PubMed] [Google Scholar]
- 15.Easterbrook PJ, Smith M, Mullen J, O'Shea S, Chrystie I, de Ruiter A, Tatt ID, Geretti AM, Zuckerman M. 2010. Impact of HIV-1 viral subtype on disease progression and response to antiretroviral therapy. J Int AIDS Soc 13:4. doi: 10.1186/1758-2652-13-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neilson JR, John GC, Carr JK, Lewis P, Kreiss JK, Jackson S, Nduati RW, Mbori-Ngacha D, Panteleeff DD, Bodrug S, Giachetti C, Bott MA, Richardson BA, Bwayo J, Ndinya-Achola J, Overbaugh J. 1999. Subtypes of human immunodeficiency virus type 1 and disease stage among women in Nairobi, Kenya. J Virol 73:4393–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ariën KK, Vanham G, Arts EJ. 2007. Is HIV-1 evolving to a less virulent form in humans? Nat Rev Microbiol 5:141–151. doi: 10.1038/nrmicro1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silveira J, Santos AF, Martínez AM, Góes LR, Mendoza-Sassi R, Muniz CP, Tupinambás U, Soares MA, Greco DB. 2012. Heterosexual transmission of human immunodeficiency virus type 1 subtype C in southern Brazil. J Clin Virol 54:36–41. doi: 10.1016/j.jcv.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 19.Ball SC, Abraha A, Collins KR, Marozsan AJ, Baird H, Quiñones-Mateu ME, Penn-Nicholson A, Murray M, Richard N, Lobritz M, Zimmerman PA, Kawamura T, Blauvelt A, Arts EJ. 2003. Comparing the ex vivo fitness of CCR5-tropic human immunodeficiency virus type 1 isolates of subtypes B and C. J Virol 77:1021–1038. doi: 10.1128/JVI.77.2.1021-1038.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abraha A, Nankya IL, Gibson R, Demers K, Tebit DM, Johnston E, Katzenstein D, Siddiqui A, Herrera C, Fischetti L, Shattock RJ, Arts EJ. 2009. CCR5- and CXCR4-tropic subtype C HIV-1 isolates have a lower level of pathogenic fitness than the other dominant group M subtypes: implications for the epidemic. J Virol 83:5592–5605. doi: 10.1128/JVI.02051-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Renjifo B, Gilbert P, Chaplin B, Msamanga G, Mwakagile D, Fawzi W, Essex M, Tanzanian Vitamin and HIV Study Group. 2004. Preferential in-utero transmission of HIV-1 subtype C as compared to HIV-1 subtype A or D. AIDS 18:1629–1636. doi: 10.1097/01.aids.0000131392.68597.34. [DOI] [PubMed] [Google Scholar]
- 22.John-Stewart GC, Nduati RW, Rousseau CM, Mbori-Ngacha DA, Richardson BA, Rainwater S, Panteleeff DD, Overbaugh J. 2005. Subtype C is associated with increased vaginal shedding of HIV-1. J Infect Dis 192:492–496. doi: 10.1086/431514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodriguez MA, Ding M, Ratner D, Chen Y, Tripathy SP, Kulkarni SS, Chatterjee R, Tarwater PM, Gupta P. 2009. High replication fitness and transmission efficiency of HIV-1 subtype C from India: implications for subtype C predominance. Virology 385:416–424. doi: 10.1016/j.virol.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 24.Kiwanuka N, Laeyendecker O, Quinn TC, Wawer MJ, Shepherd J, Robb M, Kigozi G, Kagaayi J, Serwadda D, Makumbi FE, Reynolds SJ, Gray RH. 2009. HIV-1 subtypes and differences in heterosexual HIV transmission among HIV-discordant couples in Rakai, Uganda. AIDS 23:2479–2484. doi: 10.1097/QAD.0b013e328330cc08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Konings FA, Burda ST, Urbanski MM, Zhong P, Nadas A, Nyambi PN. 2006. Human immunodeficiency virus type 1 (HIV-1) circulating recombinant form 02_AG (CRF02_AG) has a higher in vitro replicative capacity than its parental subtypes A and G. J Med Virol 78:523–534. doi: 10.1002/jmv.20572. [DOI] [PubMed] [Google Scholar]
- 26.Njai HF, Gali Y, Vanham G, Clybergh C, Jennes W, Vidal N, Butel C, Mpoudi-Ngolle E, Peeters M, Ariën KK. 2006. The predominance of human immunodeficiency virus type 1 (HIV-1) circulating recombinant form 02 (CRF02_AG) in West Central Africa may be related to its replicative fitness. Retrovirology 3:40. doi: 10.1186/1742-4690-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ariën KK, Abraha A, Quiñones-Mateu ME, Kestens L, Vanham G, Arts EJ. 2005. The replicative fitness of primary human immunodeficiency virus type 1 (HIV-1) group M, HIV-1 group O, and HIV-2 isolates. J Virol 79:8979–8990. doi: 10.1128/JVI.79.14.8979-8990.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quiñones-Mateu ME, Ball SC, Marozsan AJ, Torre VS, Albright JL, Vanham G, van der Groen G, Colebunders RL, Arts EJ. 2000. A dual infection/competition assay shows a correlation between ex vivo human immunodeficiency virus type 1 fitness and disease progression. J Virol 74:9222–9233. doi: 10.1128/JVI.74.19.9222-9233.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marozsan AJ, Moore DM, Lobritz MA, Fraundorf E, Abraha A, Reeves JD, Arts EJ. 2005. Differences in the fitness of two diverse wild-type human immunodeficiency virus type 1 isolates are related to the efficiency of cell binding and entry. J Virol 79:7121–7134. doi: 10.1128/JVI.79.11.7121-7134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iordanskiy S, Waltke M, Feng Y, Wood C. 2010. Subtype-associated differences in HIV-1 reverse transcription affect viral replication. Retrovirology 7:85. doi: 10.1186/1742-4690-7-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Velazquez-Campoy A, Todd MJ, Vega S, Freire E. 2001. Catalytic efficiency and vitality of HIV-1 proteases from African viral subtypes. Proc Natl Acad Sci U S A 98:6062–6067. doi: 10.1073/pnas.111152698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwabu Y, Kinomoto M, Tatsumi M, Fujita H, Shimura M, Tanaka Y, Ishizaka Y, Nolan D, Mallal S, Sata T, Tokunaga K. 2010. Differential anti-APOBEC3G activity of HIV-1 Vif proteins derived from different subtypes. J Biol Chem 285:35350–35358. doi: 10.1074/jbc.M110.173286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeeninga RE, Hoogenkamp M, Armand-Ugon M, de Baar M, Verhoef K, Berkhout B. 2000. Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J Virol 74:3740–3751. doi: 10.1128/JVI.74.8.3740-3751.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naghavi MH, Schwartz S, Sönnerborg A, Vahlne A. 1999. Long terminal repeat promoter/enhancer activity of different subtypes of HIV type 1. AIDS Res Hum Retroviruses 15:1293–1303. doi: 10.1089/088922299310197. [DOI] [PubMed] [Google Scholar]
- 35.Ng OT, Laeyendecker O, Redd AD, Munshaw S, Grabowski MK, Paquet AC, Evans MC, Haddad M, Huang W, Robb ML, Reynolds SJ, Gray RH, Wawer MJ, Serwadda D, Eshleman SH, Quinn TC. 2014. HIV type 1 polymerase gene polymorphisms are associated with phenotypic differences in replication capacity and disease progression. J Infect Dis 209:66–73. doi: 10.1093/infdis/jit425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mann JK, Byakwaga H, Kuang XT, Le AQ, Brumme CJ, Mwimanzi P, Omarjee S, Martin E, Lee GQ, Baraki B, Danroth R, McCloskey R, Muzoora C, Bangsberg DR, Hunt PW, Goulder PJ, Walker BD, Harrigan PR, Martin JN, Ndung'u T, Brockman MA, Brumme ZL. 2013. Ability of HIV-1 Nef to downregulate CD4 and HLA class I differs among viral subtypes. Retrovirology 10:100. doi: 10.1186/1742-4690-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mann JK, Chopera D, Omarjee S, Kuang XT, Le AQ, Anmole G, Danroth R, Mwimanzi P, Reddy T, Carlson J, Radebe M, Goulder PJ, Walker BD, Abdool Karim S, Novitsky V, Williamson C, Brockman MA, Brumme ZL, Ndung'u T. 2014. Nef-mediated down-regulation of CD4 and HLA class I in HIV-1 subtype C infection: association with disease progression and influence of immune pressure. Virology 468-470C:214–225. doi: 10.1016/j.virol.2014.1008.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wright JK, Novitsky V, Brockman MA, Brumme ZL, Brumme CJ, Carlson JM, Heckerman D, Wang B, Losina E, Leshwedi M, van der Stok M, Maphumulo L, Mkhwanazi N, Chonco F, Goulder PJ, Essex M, Walker BD, Ndung'u T. 2011. Influence of Gag-protease-mediated replication capacity on disease progression in individuals recently infected with HIV-1 subtype C. J Virol 85:3996–4006. doi: 10.1128/JVI.02520-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wright JK, Brumme ZL, Carlson JM, Heckerman D, Kadie CM, Brumme CJ, Wang B, Losina E, Miura T, Chonco F, van der Stok M, Mncube Z, Bishop K, Goulder PJR, Walker BD, Brockman MA, Ndung'u T. 2010. Gag-protease-mediated replication capacity in HIV-1 subtype C chronic infection: associations with HLA type and clinical parameters. J Virol 84:10820–10831. doi: 10.1128/JVI.01084-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prince JL, Claiborne DT, Carlson JM, Schaefer M, Yu T, Lahki S, Prentice HA, Yue L, Vishwanathan SA, Kilembe W, Goepfert P, Price MA, Gilmour J, Mulenga J, Farmer P, Derdeyn CA, Tang J, Heckerman D, Kaslow RA, Allen SA, Hunter E. 2012. Role of transmitted Gag CTL polymorphisms in defining replicative capacity and early HIV-1 pathogenesis. PLoS Pathog 8(11):e1003041. doi: 10.1371/journal.ppat.1003041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brockman M, Brumme Z, Brumme C, Miura T, Sela J, Rosato P, Kadie C, Carlson J, Markle T, Streeck H, Kelleher A, Markowitz M, Jessen H, Rosenberg E, Altfeld M, Harrigan P, Heckerman D, Walker B, Allen T. 2010. Early selection in Gag by protective HLA alleles contributes to reduced HIV-1 replication capacity that may be largely compensated for in chronic infection. J Virol 84:11937–11949. doi: 10.1128/JVI.01086-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Freed EO, Martin MA. 2007. HIVs and their replication, p 2107–2185. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 43.Troyer RM, Collins KR, Abraha A, Fraundorf E, Moore DM, Krizan RW, Toossi Z, Colebunders RL, Jensen MA, Mullins JI, Vanham G, Arts EJ. 2005. Changes in human immunodeficiency virus type 1 fitness and genetic diversity during disease progression. J Virol 79:9006–9018. doi: 10.1128/JVI.79.14.9006-9018.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chopera DR, Cotton LA, Zawaira A, Mann JK, Ngandu NK, Ntale R, Carlson JM, Mlisana K, Woodman Z, de Assis Rosa D, Martin E, Miura T, Pereyra F, Walker BD, Gray CM, Martin DP, Ndung'u T, Brockman MA, Karim SA, Brumme ZL, Williamson C, the CAPRISA 002 Study Team. 2012. Intersubtype differences in the effect of a rare p24 Gag mutation on HIV-1 replicative fitness. J Virol 86:13423–13433. doi: 10.1128/JVI.02171-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prado JG, Prendergast A, Thobakgale C, Molina C, Tudor-Williams G, Ndung'u T, Walker BD, Goulder P. 2010. Replicative capacity of human immunodeficiency virus type 1 transmitted from mother to child is associated with pediatric HIV-1 disease progression rate. J Virol 84:492–502. doi: 10.1128/JVI.01743-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crawford H, Lumm W, Leslie A, Schaefer M, Boeras D, Prado JG, Tang J, Farmer P, Ndung'u T, Lakhi S, Gilmour J, Goepfert P, Walker BD, Kaslow R, Mulenga J, Allen S, Goulder PJR, Hunter E. 2009. Evolution of HLA-B*5703 HIV-1 escape mutations in HLA-B*5703-positive individuals and their transmission recipients. J Exp Med 206:909–919. doi: 10.1084/jem.20081984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wright JK, Naidoo VL, Brumme ZL, Prince JL, Claiborne DT, Goulder PJR, Brockman MA, Hunter E, Ndung'u T. 2012. Impact of HLA-B*81-associated mutations in HIV-1 Gag on viral replication capacity. J Virol 86:3193–3199. doi: 10.1128/JVI.06682-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miura T, Brockman MA, Brumme ZL, Brumme CJ, Pereyra F, Trocha A, Block BL, Schneidewind A, Allen TM, Heckerman D, Walker BD. 2009. HLA-associated alterations in replication capacity of chimeric NL4-3 viruses carrying gag-protease from elite controllers of human immunodeficiency virus type 1. J Virol 83:140–149. doi: 10.1128/JVI.01471-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Serwadda D, Wawer MJ, Musgrave SD, Sewankambo NK, Kaplan JE, Gray RH. 1992. HIV risk factors in three geographic strata of rural Rakai District, Uganda. AIDS 6:983–989. doi: 10.1097/00002030-199209000-00012. [DOI] [PubMed] [Google Scholar]
- 50.Fellay J, Shianna KV, Ge D, Colombo S, Ledergerber B, Weale M, Zhang K, Gumbs C, Castagna A, Cossarizza A, Cozzi-Lepri A, De Luca A, Easterbrook P, Francioli P, Mallal S, Martinez-Picado J, Miro JM, Obel N, Smith JP, Wyniger J, Descombes P, Antonarakis SE, Letvin NL, McMichael AJ, Haynes BF, Telenti A, Goldstein DB. 2007. A whole-genome association study of major determinants for host control of HIV-1. Science 317:944–947. doi: 10.1126/science.1143767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weiss HA, Quigley MA, Hayes RJ. 2000. Male circumcision and risk of HIV infection in sub-Saharan Africa: a systematic review and meta-analysis. AIDS 14:2361–2370. doi: 10.1097/00002030-200010200-00018. [DOI] [PubMed] [Google Scholar]
- 52.Zhu T, Korber BT, Nahmias AJ, Hooper E, Sharp PM, Ho DD. 1998. An African HIV-1 sequence from 1959 and implications for the origin of the epidemic. Nature 391:594–597. doi: 10.1038/35400. [DOI] [PubMed] [Google Scholar]
- 53.Robertson D, Anderson J, Bradac J, Carr J, Foley B, Funkhouser R, Gao F, Hahn B, Kalish M, Kuiken C. 2000. HIV-1 nomenclature proposal. Science 288:55–55. doi: 10.1126/science.288.5463.55d. [DOI] [PubMed] [Google Scholar]
- 54.Björndal A, Sönnerborg A, Tscherning C, Albert J, Fenyö EM. 1999. Phenotypic characteristics of human immunodeficiency virus type 1 subtype C isolates of Ethiopian AIDS patients. AIDS Res Hum Retroviruses 15:647–653. doi: 10.1089/088922299310944. [DOI] [PubMed] [Google Scholar]
- 55.Campbell TB, Schneider K, Wrin T, Petropoulos CJ, Connick E. 2003. Relationship between in vitro human immunodeficiency virus type 1 replication rate and virus load in plasma. J Virol 77:12105–12112. doi: 10.1128/JVI.77.22.12105-12112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Claiborne DT, Prince JL, Scully E, Macharia G, Micci L, Lawson B, Kopycinski J, Deymier MJ, Vanderford TH, Nganou-Makamdop K, Ende Z, Brooks K, Tang J, Yu T, Lakhi S, Kilembe W, Silvestri G, Douek D, Goepfert PA, Price MA, Allen SA, Paiardini M, Altfeld M, Gilmour J, Hunter E. 2015. Replicative fitness of transmitted HIV-1 drives acute immune activation, proviral load in memory CD4+ T cells, and disease progression. Proc Natl Acad Sci U S A 112:E1480–E1489. doi: 10.1073/pnas.1421607112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bachu M, Yalla S, Asokan M, Verma A, Neogi U, Sharma S, Murali RV, Mukthey AB, Bhatt R, Chatterjee S, Rajan RE, Cheedarla N, Yadavalli VS, Mahadevan A, Shankar SK, Rajagopalan N, Shet A, Saravanan S, Balakrishnan P, Solomon S, Vajpayee M, Satish KS, Kundu TK, Jeang KT, Ranga U. 2012. Multiple NF-κB sites in HIV-1 subtype C long terminal repeat confer superior magnitude of transcription and thereby the enhanced viral predominance. J Biol Chem 287:44714–44735. doi: 10.1074/jbc.M112.397158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blaak H, Brouwer M, Ran LJ, de Wolf F, Schuitemaker H. 1998. In vitro replication kinetics of human immunodeficiency virus type 1 (HIV-1) variants in relation to virus load in long-term survivors of HIV-1 infection. J Infect Dis 177:600–610. doi: 10.1086/514219. [DOI] [PubMed] [Google Scholar]
- 59.Amornkul PN, Karita E, Kamali A, Rida WN, Sanders EJ, Lakhi S, Price MA, Kilembe W, Cormier E, Anzala O, Latka MH, Bekker LG, Allen SA, Gilmour J, Fast PE, IAVI Africa HIV Prevention Partnership. 2013. Disease progression by infecting HIV-1 subtype in a seroconverter cohort in sub-Saharan Africa. AIDS 27:2775–2786. doi: 10.1097/QAD.0000000000000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bergstrom CT, McElhany P, Real LA. 1999. Transmission bottlenecks as determinants of virulence in rapidly evolving pathogens. Proc Natl Acad Sci U S A 96:5095–5100. doi: 10.1073/pnas.96.9.5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lazaro E, Escarmis C, Perez-Mercader J, Manrubia SC, Domingo E. 2003. Resistance of virus to extinction on bottleneck passages: study of a decaying and fluctuating pattern of fitness loss. Proc Natl Acad Sci U S A 100:10830–10835. doi: 10.1073/pnas.1332668100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elena SF, Gonzalez-Candelas F, Novella IS, Duarte EA, Clarke DK, Domingo E, Holland JJ, Moya A. 1996. Evolution of fitness in experimental populations of vesicular stomatitis virus. Genetics 142:673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deymier MJ, Ende Z, Fenton-May AE, Dilernia DA, Kilembe W, Allen SA, Borrow P, Hunter E. 2015. Heterosexual transmission of subtype C HIV-1 selects consensus-like variants without increased replicative capacity or interferon-α resistance. PLoS Pathog 11(9):e1005154. doi: 10.1371/journal.ppat.1005154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Derdeyn CA, Hunter E. 2008. Viral characteristics of transmitted HIV. Curr Opin HIV AIDS 3:16–21. doi: 10.1097/COH.0b013e3282f2982c. [DOI] [PubMed] [Google Scholar]
- 65.Votteler J, Neumann L, Hahn S, Hahn F, Rauch P, Schmidt K, Studtrucker N, Solbak SM, Fossen T, Henklein P, Ott DE, Holland G, Bannert N, Schubert U. 2011. Highly conserved serine residue 40 in HIV-1 p6 regulates capsid processing and virus core assembly. Retrovirology 8:11. doi: 10.1186/1742-4690-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fujii K, Munshi UM, Ablan SD, Demirov DG, Soheilian F, Nagashima K, Stephen AG, Fisher RJ, Freed EO. 2009. Functional role of Alix in HIV-1 replication. Virology 391:284–292. doi: 10.1016/j.virol.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Patil A, Bhattacharya J. 2012. Natural deletion of L35Y36 in p6 gag eliminate LYPXnL/ALIX auxiliary virus release pathway in HIV-1 subtype C. Virus Res 170:154–158. doi: 10.1016/j.virusres.2012.08.020. [DOI] [PubMed] [Google Scholar]
- 68.Strack B, Calistri A, Craig S, Popova E, Göttlinger HG. 2003. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114:689–699. doi: 10.1016/S0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- 69.Alexander L, Weiskopf E, Greenough TC, Gaddis NC, Auerbach MR, Malim MH, O'Brien SJ, Walker BD, Sullivan JL, Desrosiers RC. 2000. Unusual polymorphisms in human immunodeficiency virus type 1 associated with nonprogressive infection. J Virol 74:4361–4376. doi: 10.1128/JVI.74.9.4361-4376.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aralaguppe SG, Winner D, Singh K, Sarafianos SG, Quiñones-Mateu ME, Sönnerborg A, Neogi U. 2017. Increased replication capacity following evolution of PYxE insertion in Gag-p6 is associated with enhanced virulence in HIV-1 subtype C from East Africa. J Med Virol 89:106–111. doi: 10.1002/jmv.24610. [DOI] [PubMed] [Google Scholar]
- 71.Fowke KR, Nagelkerke NJ, Kimani J, Simonsen JN, Anzala AO, Bwayo JJ, MacDonald KS, Ngugi EN, Plummer FA. 1996. Resistance to HIV-1 infection among persistently seronegative prostitutes in Nairobi, Kenya. Lancet 348:1347–1351. doi: 10.1016/S0140-6736(95)12269-2. [DOI] [PubMed] [Google Scholar]
- 72.Hunt PW, Cao HL, Muzoora C, Ssewanyana I, Bennett J, Emenyonu N, Kembabazi A, Neilands TB, Bangsberg DR, Deeks SG, Martin JN. 2011. Impact of CD8+ T-cell activation on CD4+ T-cell recovery and mortality in HIV-infected Ugandans initiating antiretroviral therapy. AIDS 25:2123–2131. doi: 10.1097/QAD.0b013e32834c4ac1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ndung'u T, Sepako E, McLane MF, Chand F, Bedi K, Gaseitsiwe S, Doualla-Bell F, Peter T, Thior I, Moyo SM, Gilbert PB, Novitsky VA, Essex M. 2006. HIV-1 subtype C in vitro growth and co-receptor utilization. Virology 347:247–260. doi: 10.1016/j.virol.2005.11.047. [DOI] [PubMed] [Google Scholar]
- 74.Brockman MA, Tanzi GO, Walker BD, Allen TM. 2006. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J Virol Methods 131:134–142. doi: 10.1016/j.jviromet.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 75.de Oliveira T, Deforche K, Cassol S, Rambaut A, Vandamme AM. Stanford University HIV Drug Resistance Database: Rega HIV-1 subtyping tool—version 2.0. http://www.bioafrica.net/rega-genotype/html/subtyping.html Accessed 7 April 2014.
- 76.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 77.Huang KH, Goedhals D, Carlson JM, Brockman MA, Mishra S, Brumme ZL, Hickling S, Tang CS, Miura T, Seebregts C, Heckerman D, Ndung'u T, Walker B, Klenerman P, Steyn D, Goulder P, Phillips R, Bloemfontein-Oxford Collaborative Group, van Vuuren C, Frater J. 2011. Progression to AIDS in South Africa is associated with both reverting and compensatory viral mutations. PLoS One 6(4):e19018. doi: 10.1371/journal.pone.0019018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Graham FL, Smiley J, Russell WC, Nairn R. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- 79.Storey JD, Tibshirani R. 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brumme ZL, John M, Carlson JM, Brumme CJ, Chan D, Brockman MA, Swenson LC, Tao I, Szeto S, Rosato P, Sela J, Kadie CM, Frahm N, Brander C, Haas DW, Riddler SA, Haubrich R, Walker BD, Harrigan PR, Heckerman D, Mallal S. 2009. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS One 4(8):e6687. doi: 10.1371/journal.pone.0006687. [DOI] [PMC free article] [PubMed] [Google Scholar]