

Abstract
TMEM16A, a Ca2+-activated Cl− channel, contributes to tumor growth in breast cancer and head and neck squamous cell carcinoma (HNSCC). Here, we investigated whether TMEM16A influences the response to EGFR/HER family-targeting biological therapies. Inhibition of TMEM16A Cl− channel activity in breast cancer cells with HER2 amplification induced a loss of viability. Cells resistant to trastuzumab, a monoclonal antibody targeting HER2, showed an increase in TMEM16A expression and heightened sensitivity to Cl− channel inhibition. Treatment of HNSCC cells with cetuximab, a monoclonal antibody targeting EGFR, and simultaneous TMEM16A suppression led to a pronounced loss of viability. Biochemical analyses of cells subjected to TMEM16A inhibitors or expressing chloride-deficient forms of TMEM16A provide further evidence that TMEM16A channel function may play a role in regulating EGFR/HER2 signaling. These data demonstrate that TMEM16A regulates EGFR and HER2 in growth and survival pathways. Furthermore, in the absence of TMEM16A co-targeting, tumor cells may acquire resistance to EGFR/HER inhibitors. Finally, targeting TMEM16A improves response to biological therapies targeting EGFR/HER family members.
Introduction
Gene amplification and/or overexpression of human epithelial growth factor receptor family (Cole 2011; Yarden 2001) of tyrosine kinases occur often in solid tumors and activate pathways of growth and survival in a constitutive manner(Yarden and Sliwkowski 2001). Antibody-mediated biologic therapies targeted to suppress HER family function are known to promote patient survival by inhibiting tumor-associated signal transduction pathways. Overexpression of EGFR/HER1 and related pathways are major contributors to growth of head and neck squamous cell carcinomas (HNSCC) and the EGFR suppressing antibody, cetuximab is FDA-approved as therapy for HNSCC patients(Cassell and Grandis 2010; Leemans, et al. 2011; Vermorken, et al. 2007). Similarly, the HER2-suppressing antibody, trastuzumab is FDA-approved for treatment of breast cancer patients with ERBB2 amplification (Baselga 2000; Baselga, et al. 1999). Common challenges of these biological therapies appear to be inadequate patient responses and progression to resistance. While mechanisms are not well understood, cross-activation among EGFR/HER family members and/or alternate receptor activation appear to be involved (Mohd Sharial, et al. 2012; Nahta and Esteva 2007). Thus, it is necessary to identify regulators of HER family members and signaling mechanisms downstream from them, that when simultaneously suppressed, have the potential to improve outcome of HER-targeted therapies.
TMEM16A, also called ANO1, TAOS2, or DOG1, is the prototype of a family of Ca2+-activated Cl− channels, with eight conserved transmembrane domains (Caputo, et al. 2008; Schroeder, et al. 2008; Yang, et al. 2008). TMEM16 family members are widely expressed and contribute to fluid secretion in epithelia, contraction in smooth muscle and excitability in neurons (Gomez-Pinilla, et al. 2009; Huang, et al. 2009; Hwang, et al. 2009; Kunzelmann, et al. 2012). Early evidence identified amplification of chromosomal region 11q13 containing the TMEM16A gene in HNSCC (Akervall, et al. 1995), gastrointestinal stromal tumors (GIST) (Berglund, et al. 2014; Liu, et al. 2015), breast cancer (Wu, et al. 2015), and esophageal cancer (Huang, et al. 2006; Huang, et al. 2002) but its significance in growth pathways was not realized until recently (Duvvuri, et al. 2012; Katoh and Katoh 2003).
A handful of studies have attempted to address clinical and biochemical significance of TMEM16A in solid tumor growth. The amplification and the overexpression in HNSCC and breast cancer were shown to correlate with poor patient survival (Akervall, et al. 1995; Britschgi, et al. 2013; Duvvuri, et al. 2012; Ruiz, et al. 2012). TMEM16A was shown to activate effectors of tumor growth including, ERK1/2 and AKT1 in vitro and in vivo (Britschgi, et al. 2013; Duvvuri, et al. 2012). Subsequently, TMEM16A interaction with the ERM (ezrin/radixin/moesin) proteins (Shiwarski, et al. 2014) or EGFR (Bill, et al. 2015a; Britschgi, et al. 2013) was proposed to regulate pathways of motility or growth. TMEM16A-induced mechanisms that promote tumor growth and survival are far from being clear, but evidence points to its ability to recruit EGFR, an oncogenic receptor of the HER family.
Earlier studies suggested that the putative “pore-forming” region of TMEM16A allowing for transmembrane flux of Cl− ions was located within transmembrane domains 5–6 (amino acids 642–672), and activation of conductance function occurred by Ca2+ binding in the N-terminus and the short loops between transmembrane domains or by TMEM16A dimerization (Terashima, et al. 2013; Tien, et al. 2014). More recent findings obtained using TMEM16A mutants have suggested another view; it was proposed that amino acid sequence 728–752 following domains 5–6, forms a partial re-entrant finger within the membranous space, allowing Ca2+-sensing and Cl− flux (Bill, et al. 2015b; Tien, et al. 2014). Regardless of the reported differences in the proposed mechanism of action, the Cl− conductance function of TMEM16A appears to recruit a growth pathway.
Consistently, small molecule inhibitors of Cl− channel activity are capable of suppressing growth of HNSCC and breast cancer cells with 11q13 amplification (Britschgi, et al. 2013; Duvvuri, et al. 2012). Moreover, TMEM16A mutants (K668E, K610A and R621E) that affect Cl− conductance also affect cell viability (Britschgi, et al. 2013; Duvvuri, et al. 2012; Yang, et al. 2008). Therefore, as we begin to understand ways in which TMEM16A is regulated, biochemical evidence opens up a distinct possibility, that TMEM16A Cl− conductance function is connected to a tumor growth pathway. Moreover, with the ability to interact with EGFR, TMEM16A is a molecule of potential translational significance in solid tumor biology (Bill, et al. 2015a).
In the present study, we have explored a potential link between TMEM16A-induced Cl− conductance and pathway(s) of cell growth and survival in HER2-amplified breast cancer and HNSCC. Furthermore, we have explored the involvement of this link in the regulation of responses to EGFR/HER-family based biologically targeted therapies, namely, the anti-HER2 therapy, trastuzumab, and the anti-EGFR therapy, cetuximab.
Materials and Methods
Materials
The anti-TMEM16A antibody was from Thermo Scientific (Waltham, MA), anti-β-tubulin antibody was from Abcam (Cambridge, UK) and anti-EGFR antibody was from Santa Cruz Biotechnology (Dallas, TX). Antibodies to phospho-tyrosine Y1068 EGFR, HER2, phospho-tyrosine Y1248 HER2, plexin B1, STAT3 and phospho-tyrosine Y705 STAT3 were from Cell Signaling (Danvers, MA). Human IgG, trypan blue, crystal violet were from Sigma-Aldrich (St. Louis, MO). Trastuzumab (Genentech, San Francisco, CA) and cetuximab (Bristol-Myers Squibb, New York, NY) were purchased from the University of Pittsburgh Medical Center pharmacy. Necrostatin was from Selleck Chemicals (Houston, TX). T16A-inh (A01) was purchased from Calbiochem (EMD Millipore, Billerica, MA). The TMEM16A inhibitor, Cacc-inh, was provided by Novartis (Basel, Switzerland). K+-Cl− inhibitor R-(+)-Butylindazone; (Dihydroindenyl)oxy alkanoic acid (DIOA) and siRNA targeting control, CLC-2 and CLC-4 were purchased from Santa Cruz Biotechnology (Dallaz, TX) Matrigel was from Corning Life Sciences (Corning, NY). The CellTiter-Glo reagent was from Promega (Fitchburh, WI). RNA isolation kit was purchased from Qiagen (Hilden, Germany), primers were from Integrated DNA technologies (Coralville, IA). Complete mini tablets of protease and phosphatase inhibitors were from Roche Life Sciences (Indianapolis, IN). The Bradford protein estimation reagent, molecular weight markers, anti-rabbit or anti-mouse HRP coupled secondary antibody and iQ™SYBR® green supermix for PCR were from Bio-Rad (Hercules, CA). The EGF, Lipofectamine 2000, Opti-MEM and SuperScript III Reverse Transcriptase were from Invitrogen (Thermo-Fisher Scientific, Waltham, MA). The Ras activation kit was from Millipore (EMD Millipore, Billerica, MA). Western blots developed using the LiCor Odyssey imaging system (Lincoln, NE) were imaged using primary antibodies as above and LiCor Odyssey specific secondary antibody.
Cell lines and media
SKBR3 breast cancer cells were chosen for study due to HER2-positivity and because of a previously described model of trastuzumab resistance (Kulkarni, et al. 2010) and were obtained from previous work conducted by Kulkarni. Breast cancer cells were cultured in DMEM/F12 medium containing 10% fetal bovine serum (FBS); trastuzumab-resistant cells were cultured and maintained in the same medium containing trastuzumab (10 μg/ml). The head and neck cancer cell line, Cal-33, was obtained from the ATCC, while the OSC19 and UM-SCC-1 head and neck cancer cell lines were gifts from Dr. Jennifer Grandis (Department of Otolaryngology, UCSF) and Dr. Thomas Carey (Department of Otolaryngology, University of Michigan), respectively. Cal-33, OSC-19, and UM-SCC-1 were all cultured in DMEM with 10% FBS. Cal-33 cells are endogenously devoid of TMEM16A (i.e., TMEM16A−/−) and were thus used as a negative control cell line. OSC-19 cancer cells do not contain amplification of chromosomal region 11q13. UM-SCC-1 cells contain amplification of chromosomal region 11q13. Both OSC-19 and UM-SCC-1 cancer cell lines demonstrate relatively low protein expression of TMEM16A; thus, these cells were chosen for experiments involving overexpression of TMEM16A to provide maximum contrast between control and overexpressing conditions; furthermore, by comparison of OSC-19 and UM-SCC-1 allowed for evaluation of the effects of TMEM16A overexpression in the absence and presence of amplification of chromosomal region 11q13, respectively. UM-SCC-1 and OSC-19 control or TMEM16A-overerexpressing cells were generated by transduction of viruses as described previously (Duvvuri, et al. 2012). FaDu cells contain 11q13 amplification and demonstrate high endogenous protein expression of TMEM16A; thus experiments involving shRNA-knockdown of TMEM16A were conducted in FaDu cells. FaDu cells were engineered to express control, non-targeting (NT) or TMEM16A targeting shRNA in a doxycycline-inducible manner as described previously (Britschgi, et al. 2013) and were from the authors Bill and Gaither at Novartis. Induction of shRNA was performed by adding 10ng/ml doxycycline to the growth medium. These cells were cultured in DMEM containing 10% tetracycline-free FBS. For the Cl− depletion assay, Ringers solution was prepared in which Na-gluconate was substituted for NaCl at equal concentration (140 mM). HEK293T cells were purchased from Clontech (Mountain View, CA) and were cultured in DMEM containing 10% FBS.
DMEM and DMEM/F12 were purchased from Thermo-Fisher Scientific (Waltham, MA). FBS was purchased from Atlanta Biologicals (Flowery Branch, GA).
Cell viability and colony formation assays
For viability assays, (3 × 103) cells, as indicated, were plated in 96-well plates in triplicate and left untreated as controls or treated as indicated. An equal volume of the CellTiter-Glo reagent was added 2–4 days after treatments and luminescence was read to assess cell viability. Readings from the treated group were normalized to readings obtained from the control group. In other experiments, cells were cultured in 6-well plates and treated with different concentrations of Cacc-inh or T16A-inh for 4h and toxicity was assessed by trypan blue dye exclusion test. For colony formation assay, 1 × 103 cells were plated in 6-well plates and allowed to attach overnight. Plates were treated with trastuzumab alone or trastuzumab and T16A-inh together at indicated concentrations every 2 days for 18 days. Plates were fixed and stained with 0.05% crystal violet. Colonies were counted and data was normalized to the untreated control. Details of cell lines, treatment conditions/drug concentrations, drug combinations, and incubation times are included in figure captions.
Plasmids and transfection
Plasmid DNAs encoding TMEM16A mutants (E727K, L759Q) were described previously (Bill, et al. 2015b). DNA was mixed with Lipofectamine 2000 in OPTI-MEM and complexes were layered over OSC-19 cells. Cells were allowed to recover for 48h followed by solubilization and analysis in western blots.
Western blot assay
Cells were cultured and treated as indicated followed by solubilization in cold RIPA buffer containing protease and phosphatase inhibitors. Insoluble material was removed by centrifugation at 13000 rpm for 10 min at 4° C and solubilized protein was estimated by Bradford’s method. For analysis in western blots equal amounts of protein were denatured, separated in 8% acrylamide gels and transferred to nitrocellulose membrane. Membranes were incubated with the indicated primary antibodies followed by detection with HRP-coupled secondary antibody.
The blots in Figure 3 were developed using the LiCor imaging system. Briefly, equal amounts of protein from cells cultured in the indicated conditions were loaded into 8% SDS-PAGE gels and subsequently transferred to nitrocellulose membrane. These membranes were then exposed to the indicated primary antibody and then exposed to a secondary antibody for use with the LiCor system. Densitometry values were obtained using the LiCor imaging software.
Figure 3. TMEM16A-induced HNSCC tumor growth shows EGFR activation.

a, b: Tumor xenograft pairs of OSC-19 cells (control and overexpressing TMEM16A) was processed to obtain solubilized protein. Equal amounts of protein were analyzed for pY1068 EGFR, EGFR, TMEM16A and β-tubulin in western blots as shown. In b, mean tumor weight +/− SEM; significance was determined from a Student t-test (n = 4, *p < 0.05).
c: FaDu cells engineered to contain either doxycycline-inducible non-targeting (NT) control shRNA or shRNA against TMEM16A were treated with doxycycline and EGF, as indicated. Cells were harvested and processed to obtain solubilized protein. Equal amounts of protein were analyzed for pY1068 EGFR, EGFR, TMEM16A, and β-tubulin as shown. Densitometric quantification of the pY1068 EGFR signal is shown; significance was determined by a Student t-test (*p < 0.05).
Any imaging changes (brightness, contrast) performed on blots were applied uniformly to maintain image integrity. Blots are cropped for conciseness; all full length, uncropped blots are available in the supplementary data.
Tumor xenograft analysis
Equal numbers (1.5 × 106) of OSC-19 control or TMEM16A-overexpressing cells were implanted in 100 ml of matrigel subcutaneously in nude mice. All mice were euthanized, tumors were removed, and weights were recorded at an arbitrary time point in accordance with IACUC University of Pittsburgh protocols. Lysates of tumors were prepared and protein was estimated. Equal amounts of protein were separated in 8% gels and detected in western blot analyses with the indicated antibodies using the LiCor imaging system (LiCor Odyssey Classic). All experimental protocols were approved by the IACUC at the University of Pittsburgh. All animal handling was performed in accordance with guidelines approved by the IACUC at the University of Pittsburgh.
Fluorescence microscopy
Fluorescence microscopy was used to measure changes in intracellular Ca2+, using the ratiometric, fluorophore Fura-2AM or Cl− fluxes, using the fluorophore, MQAE. For both assays control of TMEM16A overexpressing UM-SCC-1 cells were plated onto poly-L-lysine coated optical petri dishes (Matek) 24 h prior to the experiment. Cells were loaded with the desired fluorophore, petri dishes were mounted on the stage of an Olympus IX-81 epifluorescence microscope and perfused with normal Ringer’s solution (in 140mM NaCl, 25mM NaHCO3, 5mM KCl, 10mM HEPES, 1mM MgCl2, 1.5mM CaCl2, and 5mM glucose) (2 ml/min) maintained at 37 °C. Fluorescence emission in response to excitation was acquired every 15 s over the course of the experiment, with brief increases in acquisition rate (every 5 s for 2 min periods) following application of agonists shown to evoke a transient response. After acquisition, the magnitude of fluorescence in each cell in the field was quantified from a circular region of interest (ROI) drawn within the cell, and the time course of emitted fluorescence was plotted as the average ± SEM of all ROIs in the field (≥ 20 cells).
For Ca2+ measurements, emission at 510 nm was acquired in response to excitation at 340 and 380 nm, with intracellular Ca2+ changes taken as proportional to the 340/380 ratio. For loading, cells were incubated for 30 min at room temperature (RT) in normal Ringer’s solution containing 5 μM Fura-2AM, then rinsed and incubated an additional 15 min at RT in the absence of Fura-2. Loaded cells were mounted on the microscope and treated with 50 ng/ml EGF or vehicle for 10 min, re-equilibrated with normal Ringer’s for 10 min, then stimulated with 200 μM ATP for 2 min as a control for Ca2+ response. EGF-induced Ca2+ changes were measured as the percentage change in the ratio over the 10 min treatment, while ATP-induced changes were measured at the peak of the transient response. For measurement of Cl− fluxes, emission at 445 nm was acquired in response to excitation at 340 nm. Cells were loaded with 10 mM MQAE using a hypo-osmotic shock (50% normal Ringer’s solution) for 20 min at RT, then rinsed and incubated for an additional 10 min at RT with normal Ringer’s solution ± 50 ng/ml EGF. Loaded cells were mounted on the microscope, and Cl− fluxes were induced by switching to low Cl− Ringers ± 50 ng/ml EGF for 5 min followed by a return to normal Ringers. Cl− flux was calculated as the rate of change in fluorescence during the transition to low (and normal) chloride, using a linear fit of the slope.
Whole cell patch clamp
Whole-cell patch clamp was conducted as described (Zhou, et al. 2015). Briefly, NMDG-Cl solutions were used to isolate Cl− currents from UM-SCC-1 cells overexpressing TMEM16A (bath solution was 140mM NMDG-Cl, 10mM HEPES, 1mM MgCl2, 1.5mM CaCl2, 5mM glucose; pipette solution was 140mM NMDG-Cl, 10mM HEPES, 1mM MgCl2, 5mM glucose, 1mM EGTA, 1mM Mg-ATP, and 0.1mM Na-GTP). Membrane current (Im) was measured using a periodic current-voltage (I/V) protocol, which stepped the holding potential from 100 to +100 mV in 20 mV steps. Bath solution ± agonists heated to 37° C was perfused over the cell at 2 ml/min. The percent change in Im was calculated at the 60 mV holding potential. All cells tested exhibited outward rectified I/V curves with a reversal potential equal to the Cl− equilibrium potential, consistent with TMEM 16A activity.
Statistical analysis
All data are expressed as mean +/− SEM. Statistical analyses were performed with GraphPad Prism software using the Student’s t-test or ANOVA, as appropriate (*p<0.05; **p<0.01; ***p<0.001; ns. not significant).
Results
Inhibition of TMEM16A Cl− channel function induces a loss of expression and activity of EGFR and HER2 and decreases survival of HER2-amplified breast cancer cells
Previously, TMEM16A knockdown in breast cancer cells with 11q13 amplification was shown to suppress EGFR activity and colony formation (Britschgi, et al. 2013). A distinct aggressive subtype of breast cancer that affects ~25% patients harbors HER2 amplification. HER2’s ability to connect to pathways of tumor growth depends on a close partnership with EGFR. The EGFR-HER2 partnership stimulates activity of the transcription factor, STAT3, an effector of cell survival and growth. To understand the significance of TMEM16A in the regulation of EGFR and HER2, HER2-amplified SKBR3 cells were treated with T16A-inhAO1, a small molecule inhibitor of TMEM16A Cl− channel activity or untreated as controls. EGFR phosphorylation and expression was found to decrease in T16A-inhAO1-treated cells compared to controls (Fig. 1a). Since TMEM16A inhibition suppressed EGFR, the effect on HER2 was also assessed - both HER2 expression and activity were reduced.
Figure 1. TMEM16A influences expression and activation of EGFR/HER family members and a pathway of survival in ErbB2/HER2 amplified breast cancer cells.

a: SKBR3 cells were plated on 60 mm plates and allowed to attach for 2 h. Cells were left untreated as a control or treated with T16A-inh (40 μM) for 1.5 h at 37° C. Cells were lysed to obtain solubilized protein and equal amounts of protein were analyzed for pY1068 EGFR, EGFR, β-tubulin, pY1248 HER2, HER2, pY705 STAT3, STAT3, TMEM16A and Plexin B1 in western blots.
b: SKBR3 cells (3 × 103/well) were plated in 96-well plates in quadruplicate and left untreated as controls or treated with human IgG or trastuzumab (20 μg/ml each) or T16A-inh (5 μM). All treatments were carried out for 3 days at 37° C followed by measurements of viability with the CellTiter-Glo reagent. Readings from the treated group were normalized to the readings from control. Representative data from triplicate results (n = 3) are presented as mean +/− SEM. The significance (***p < 0.001) was determined from one-way ANOVA analysis.
c: SKBR3 cells (3 × 103/well) were plated in 96-well plates in quadruplicate and left untreated as controls or treated with trastuzumab (20 μg/ml) or trastuzumab plus varying concentrations of Cacc-inh (0.5–30 μM). All treatments were carried out for 3 days at 370 C followed by measurements of viability with the CellTiter-Glo reagent. Readings from the treated group were normalized to the readings from controls. Representative data from triplicate results (n = 3) are presented as mean +/− SEM. The significance (***p < 0.001) was determined from one-way ANOVA analysis.
To investigate the effect of TMEM16A inhibition downstream from EGFR-HER2, activity of pY705 STAT3 was assessed. Activity was found to decrease without affecting STAT3 levels. Recently inhibitors of TMEM16A Cl− channel activity were proposed to function by influencing protein stability and/or internalization (Bill, et al. 2014). Consistently, some loss of TMEM16A too was detectable in SKBR3 cells exposed to T16A-inh without causing significant changes in Plexin B1, another membrane-bound protein.
As a negative control, Cal-33 HNSCC cells, which are endogenously devoid of TMEM16A, were treated with T16A-inhAO1 (Supplementary Fig. 1). While T16A-inhAO1 did affect the phosphorylation of EGFR, it did not alter the total EGFR expression, as demonstrated in the SKBR3 cells. Furthermore, phosphorylation of STAT3 was unaffected by T16A-inhAO1 suggesting that minor changes in phosphorylation of membrane receptors did not affect intracellular signaling.
Taken together these data suggest that TMEM16A Cl− channel activity is linked to the regulation of expression and activity of EGFR/HER family members and STAT3 and therefore any alterations of its function are likely to influence not only tumor growth but also responses to EGFR/HER family based therapy.
Whether HER2 and TMEM16A could be co-targeted was evaluated by measuring survival of SKBR3 cells treated with trastuzumab and T16A-inhAO1, individually or in combination (Fig. 1b). Individual treatments with trastuzumab or T16A-inhAO1 reduced cell survival, whereas IgG, used as a control, had no effect. Combining trastuzumab and T16A-inhAO1 further inhibited cell survival in a cooperative manner. Cacc-inhAO1, a Ca2+-activated Cl− channel inhibitor, also reduced cell viability when combined with trastuzumab (Fig. 1c).
Previous studies have demonstrated that RNAi inhibition of TMEM16A (Britschgi, et al. 2013) leads to apoptotic activation and cell death as evidenced by increased PARP and caspase cleavage. To extend these studies, we engineered FaDu cells, with high endogenous TMEM16A expression, to contain doxycycline inducible control, non-targeting (NT) shRNA or shRNA against TMEM16A(Britschgi, et al. 2013) and assessed for viability following TMEM16A knockdown (Supplementary Fig. 2). Genetic knockdown of TMEM16A resulted in ~40% reduction in viability; the addition of the RIP-kinase inhibitor, necrostatin, which has been shown to inhibit caspase-independent necrosis (Ofengeim and Yuan 2013), did not rescue cell viability following TMEM16A knockdown.
Together, these results imply that combinatorial targeting of TMEM16A and HER2 can improve the efficacy of HER2-targeted biological therapy, likely, by activating cellular apoptosis.
An increase in TMEM16A expression accompanies resistance to HER2-targeted biologic therapy and TMEM16A inhibition reverses resistance
Although biologic therapies targeted against the EGFR/HER family members are known to suppress tumor growth and improve survival in breast cancer and HNSCC patients, clinical responses are transient and resistance is common. To investigate whether TMEM16A plays a role in resistance, we chose to explore whether cancer cells with acquired-resistance to biologic therapy respond to TMEM16A inhibition. A SKBR3 breast cancer cell model (Kulkarni, et al. 2010) of acquired-resistance to the anti-HER2 antibody trastuzumab was utilized in the study.
To confirm resistance, viability of parent and resistant SKBR3 cells was assessed after treatment with trastuzumab (Supplementary Fig. 3a). A growth-inhibitory response to trastuzumab was detected in parent cells, but not in resistant cells. Cells were lysed to obtain solubilized protein for western blot analysis of TMEM16A. Compared to parent cells (Supplementary Fig. 3b), resistant cells exhibited an increase in the TMEM16A expression. Quantitation indicated an average of about two-fold increase in TMEM16A expression (Supplementary Fig. 3c). The upregulation in the resistant SKBR3 cells was specific to TMEM16A, as no increase in EGFR or HER2 (Nahta, et al. 2007) was detectable by western blot (Supplementary Fig. 3d).
To investigate whether the change in TMEM16A expression influences sensitivity to TMEM16A inhibition, viability was assessed in parent and trastuzumab-resistant cells treated with T16A-inhAO1. TMEM16A inhibition led to a loss of viability in both cell types, however, the resistant cells showed a two-fold greater inhibition compared to parent cells (Fig. 2a). Furthermore, combined treatment with trastuzumab and T16A-inhAO1 suppressed colony formation in both cell types, whereas treatment with trastuzumab alone had a little or no effect (Fig. 2b, c).
Figure 2. Trastuzumab-resistant breast cancer cells are increasingly sensitive to TMEM16A inhibition.
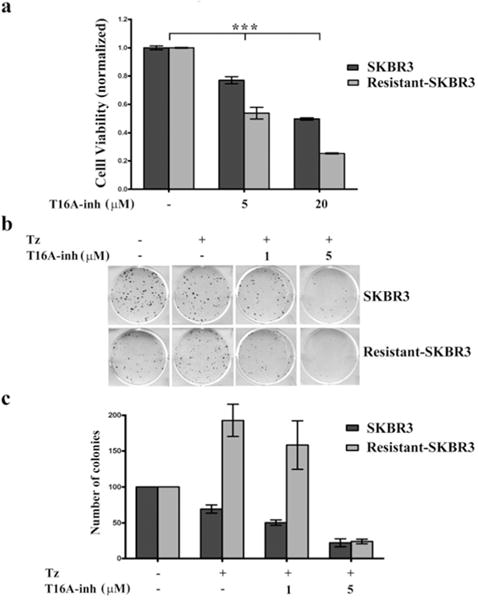
a: SKBR3 cells, parent or trastuzumab-resistant, (3 × 103/well) were plated in quadruplicate in 96-well plates and left untreated as controls or treated with T16A-inh (5 or 20 μM) for 3 days at 37° C. Viability was measured with the CellTiter-Glo reagent and readings from the treated group were normalized to readings from the controls. Representative data from triplicate results (n = 3) are presented as mean +/− SEM. The significance (***p < 0.001) was determined from one-way ANOVA analysis.
b: SKBR3 cells, parent or trastuzumab-resistant, (1 × 103/well) were plated in 6-well plates. Cells were allowed to attach overnight after which cells were left untreated as controls or treated with trastuzumab alone (10 μg/ml) or trastuzumab (10 μg/ml) combined with T16A-inh (1 or 5 μM). Treatments were repeated every 2–3 days for a period of 2–3 weeks. Plates were fixed and stained with crystal violet. A representative plate from triplicate results (n = 3) is shown. c: The number of colonies for each condition from independent experiments (n = 3) was counted. The numbers obtained for the treated groups were normalized to the control group. Data are presented as mean +/− SEM of triplicate results (n = 3).
Together, these data support the conclusion that HER2-resistant cells increasingly rely on TMEM16A-induced survival mechanisms and therefore, are more sensitive to its inhibition. These data also raise the possibilities that in certain tumor cells, TMEM16A cooperates with members of the EGFR/HER family, imparting sensitivity to EGFR/HER family-based therapy. Furthermore, combined suppression of TMEM16A and EGFR/HER receptors may inhibit growth and reverse/delay resistance more effectively, thus, providing a clinical advantage.
TMEM16A interacts with and activates EGFR in non-11q13 amplified HNSCC cells
Interactions between TMEM16A and EGFR play a critical role in tumor growth in 11q13 amplified HNSCC (Britschgi, et al. 2013). To evaluate the influence of TMEM16A on non-11q13-amplified tumors, OSC-19 cells were transfected with either a control or TMEM16A-overexpressing plasmid. OSC-19 cells were chosen due to low endogenous expression of TMEM16A, which allowed for enhanced contrast between the control and TMEM16A-overexpressing conditions. OSC-19 control or TMEM16A-overexpressing cells were inoculated subcutaneously in nude mice. Tumors arising were weighed and processed to obtain protein that was analyzed in western blots for pY1068 EGFR, EGFR and TMEM16A. OSC-19 TMEM16A-overexpressing tumor xenografts demonstrated an increase in both pY1068 EGFR and total EGFR levels compared to controls (Fig. 3a). In addition, average end weights of TMEM16A-overexpressing tumors were ~2–3-fold higher than control tumors (Fig. 3b). These in vivo data provide further support for the hypothesis that TMEM16A, independent of the other 11q13 amplified genes and their proteins, is involved in the promotion of pathways of tumor growth and facilitates EGFR signaling in HNSCC cells.
Having demonstrated that overexpression of TMEM16A correlates with in vivo upregulation of EGFR in non-11q13 amplified cells, the effect of shRNA knockdown of TMEM16A on EGFR was investigated in FaDu HNSCC cells. TMEM16A knockdown significantly decreased the overall levels of EGFR along with the phosphorylated fraction relative to cells with NT shRNA (Fig. 3c). Furthermore, following TMEM16A knockdown, EGF stimulation did not significantly increase the phosphorylated fraction of EGFR (Fig. 3c), suggesting that TMEM16A may play a role in sensitizing EGFR to EGF stimulation. Taken together, TMEM16A appears to interact with and promote EGFR-induced early signaling events in HNSCC growth pathways.
EGF does not have a direct influence on TMEM16A activity or function in HNSCC
Having shown that TMEM16A regulates EGFR activity and sensitivity to EGFR-targeted therapy in HNSCC, we explored whether TMEM16A is affected by EGF. Initially, whether EGF treatment causes a change in intracellular Ca2+ and activates TMEM16A was assessed by fluorescent dyes and a patch clamp assay. Stimulation of EGFR-signaling with EGF in UM-SCC1 cells did not produce a change in intracellular Ca2+ flux or activation of TMEM16A, whereas ATP, a known inducer of intracellular Ca2+ flux, produced a robust increase in Ca2+ and stimulated TMEM16A-induced Cl− flux (Supplementary Fig. 4). Cl− flux was greater in cells overexpressing TMEM16A, consistent with the presence of a higher number of chloride channels. Taken together, these data indicate that EGF-induced EGFR signaling is not directly involved in regulation of TMEM16A channel activity or Cl− conductance function.
Inhibition of TMEM16A improves response to EGFR targeting antibody in HNSCC
EGFR is highly expressed and involved in promotion of oncogenic growth in ~80–90% HNSCC, but cetuximab is the only FDA-approved biologic therapy targeting EGFR (Vermorken, et al. 2007). It is now known that ~10–13% HNSCC patients benefit from cetuximab therapy and the remaining are nonresponsive (Vermorken, et al. 2007). Since our data support involvement of TMEM16A in the regulation of EGFR activation and expression in HNSCC, we sought to investigate whether simultaneous inhibition of TMEM16A and EGFR can improve response to cetuximab. Therefore, OSC-19 cells were treated with cetuximab, T16A-inhAO1 or a combination of both inhibitors and cell viability was analyzed (Fig. 4a, b); the combination decreased cell viability more effectively. Another TMEM16A inhibitor, Cacc-inhAO1 also decreased viability in cells treated with cetuximab as compared to cetuximab alone (Fig. 4c). Together these data suggest that the co-inhibition of TMEM16A and EGFR enhances response to EGFR-targeted therapy in HNSCC.
Figure 4. TMEM16A inhibition improves response to biologic therapy of EGFR in HNSCC.

a: OSC-19 cells (3 × 103/well) were plated in 96-well plates in quadruplicate and left untreated as controls or treated with human IgG or cetuximab (20 μg/ml each) or T16A-inh (20 μM). Cells were treated also with cetuximab (20 μg/ml) and T16A-inh (20 μM) together. All treatments were carried out for 4 days at 370 C. Viability was measured with the CellTiter-Glo reagent. Readings from the treated groups were normalized to the readings from the control. Representative data from triplicate results (n = 3) are presented as mean +/− SEM. The significance (***p < 0.001) was determined from one-way ANOVA analysis.
b: OSC-19 cells (3 × 103/well) were plated in 96-well plates in quadruplicate and left untreated as controls or treated with cetuximab (20 μg/ml) or cetuximab along with varying concentrations of T16A-inh (1–20 μM). All treatments were carried out for 3 days at 37° C followed by measurements of viability with the CellTiter-Glo reagent. Readings from the treated groups were normalized to the readings from the controls. Representative data from triplicate results (n = 3) are presented as mean +/− SEM. The significance (***p < 0.001) was determined from one-way ANOVA analysis.
c: OSC-19 cells (3 × 103/well) were plated in 96-well plate in quadruplicate and left untreated as controls or treated with cetuximab (20 μg/ml) or cetuximab along with varying concentrations of Cacc-inh (0.5–30 μM). All treatments were carried out for 3 days at 37° C followed by measurements of viability with the CellTiter-Glo reagent. Readings from the treated group were normalized to readings from the controsl. Representative data from triplicate results (n = 3) are presented as mean +/− SEM. The significance (***p < 0.001) was determined from one-way ANOVA analysis.
To investigate if TMEM16A inhibitors cause cytotoxicity during early signaling events, OSC-19 and SKBR3 cells were treated with different concentrations (0, 5, 20 or 40 μM) of Cacc-inhAO1 or T16A-inhAO1 for 4h and toxicity was assessed with the trypan blue dye exclusion test. Under these conditions, 97–99% cells were viable indicating that the efficacy of TMEM16A on signaling and response to HER family inhibitors did not lead to toxicity. Previously, in cells with 11q13 amplification, Cacc-inhAO1 was implicated in the regulation of EGFR expression and internalization (Terashima, et al. 2013). To assess if Cacc-inhAO1 functions in a similar manner in other cell types, OSC-19 cells treated with Cacc-inhAO1 for 0–2h were analyzed on western blots (Supplementary Fig. 5a); loss of EGFR expression was also accompanied by loss of EGFR phosphorylation. Similarly, SKBR3 cells treated with the TMEM16A inhibitor, T16A-inhAO1 for 0–4h showed loss of expression and phosphorylation of both EGFR and HER2 (Supplementary Fig. 5b). These findings strongly support the possibility that TMEM16A interacts with and plays a role in regulating EGFR and HER2.
To investigate the effects of TMEM16A, we used FaDu cells that have 11q13 amplification. The effects of shRNA-mediated TMEM16A suppression on cetuximab response were assessed in FaDu cells expressing NT or TMEM16A shRNA in the absence or the presence of IgG, cetuximab and doxycycline. As shown Supplementary Fig. 6, TMEM16A shRNA induction and cetuximab each led to some loss of cell viability (~ 20–25%); IgG did not produce an additional effect. However, a pronounced cooperative inhibition of viability (~ 55%) was observed when TMEM16A shRNA and cetuximab were combined. These data suggest that the combined suppression of TMEM16A and EGFR inhibited the cell survival pathway more effectively than either individually. Thus, inhibition of TMEM16A Cl− channel activity by small molecule inhibitors or by shRNA suppress the EGFR pathway and improve response to targeted therapy. These data open up the possibility that combined inhibition of members of the EGFR/HER family and TMEM16A may improve therapeutic efficacy.
TMEM16A Cl− channel activity is involved in the molecular regulation of the EGFR/HER pathway
Although we found that inhibition of TMEM16A Cl− channel activity with T16A-inhAO1 or Cacc-inhAO1 decreased EGFR signaling, it was not clear whether the inhibition of Cl− conductance or the decrease in TMEM16A expression was responsible for the observed effects. Therefore, to understand whether Cl− flux by itself influences EGFR signaling in cancer cells and whether its suppression causes molecular changes similar to those induced by T16A-inh or Cacc-inh exposure, SKBR3 cells were subjected to a physiologically low Cl− medium for a brief period of time and activation of EGFR pathway was analyzed. Western blot analysis revealed that a loss of EGFR activity and expression occurred specifically under Cl− depleted conditions (Fig. 5a). Similar results were obtained in OSC-19 cells in which incubation in a low Cl− medium or treatment with T16A-inh resulted in an inhibition of EGFR signaling (Fig. 5b). These data suggest that one molecular target of Cl− flux is EGFR itself, which implies that perturbation of the TMEM16A Cl− channel function has potential to influence response to EGFR/HER-targeted therapy.
Figure 5. Cl− channel function of TMEM16A regulates activity of EGFR pathway.

a: SKBR3 cells were plated in 60 mm plates and allowed to attach for 2 h. Cells were subjected to medium containing a physiologically normal level of Cl− as a control (N) or medium in which Cl− was depleted (Low) for 3 h at 37° C. Cells were lysed to obtain solubilized protein and equal amounts of protein were analyzed for pY1068 EGFR, EGFR, pY705 STAT3 and β-tubulin in western blots.
b: OSC-19 cells were plated in 60 mm plates and allowed to attach for 2 h. Cells were subjected to medium containing physiologically normal level of Cl− as a control (N) or T16A-inh (40 μM) and also to medium in which Cl− was depleted (Low). All treatments were carried out for 2.5 h at 37° C at which time cells were lysed to obtain solubilized protein. Equal amounts of protein were analyzed for pY1068 EGFR, EGFR, pY705 STAT3 and β-tubulin in western blots.
c: OSC-19 cells were plated in 100 mm plates in triplicate. One plate was mock-transfected as a control and the other two were transfected with plasmids encoding TMEM16A mutants (E727K or L759Q) known to be deficient in Cl− channel activity. Cells were lysed to obtain solubilized protein that was analyzed for TMEM16A, EGFR, pY705 STAT3 and β-tubulin in western blots.
Manipulation of extracellular chloride concentration can affect the entire plasma membrane and potentially influence other channels in addition to TMEM16A. Therefore, to gain further evidence for involvement of the TMEM16A Cl− conductance function in the regulation of EGFR signaling, OSC-19 cells were transiently transfected with two different plasmids expressing TMEM16A mutants (E727K, L759Q) that are known to be deficient in Cl− channel activity29.
Biochemical analysis of control and transfected cells using western blots (Fig. 5c) indicated that expression of the TMEM16A mutants induced a loss of EGFR expression. These data support the involvement of the Cl− conductance function of TMEM16A in the regulation of EGFR.
Although the physical location of or exact primary sequence that conforms to the TMEM16A Cl− “pore forming region” is not known (Bill, et al. 2015b; Britschgi, et al. 2013; Terashima, et al. 2013; Tien, et al. 2014; Yang, et al. 2008), a link between TMEM16A Cl− conductance function and EGFR expression begins to emerge based on the evidence presented here. In this connection, TMEM16A activation introduces a Cl− flux across the plasma membrane. The flux appears to regulate expression of EGFR. Thus, dysregulation of the TMEM16A channel function, due to TMEM16A overexpression or gene amplification often observed in human tumors, is expected to retain molecular dependency of the EGFR/HER2-induced pathways of tumor cell viability and growth. The inhibition of the TMEM16A Cl− channel function relieves the dependency by suppressing EGFR/HER family expression and consequently enhances response or reverses resistance to targeted therapy.
Voltage-gated chloride (CLC) and potassium-chloride co-transporter (KCC) have no effect on EGFR protein
Although TMEM16A interacts with EGFR and aids EGFR-signaling in cancer cells (Bill et al., 2015a), it remains unclear if other chloride channels can impact EGFR signaling in cancer cell lines. Therefore, it was of interest to study the effect of voltage-gated (CLC) and potassium- (KCC) chloride cotransporter on EGFR signaling. Recent studies have shown that voltage-gated ion channels, specifically the chloride channels (CLC) have crucial roles in regulating cell proliferation, thereby influencing the development and progression of cancer (Rao et al., 2015). Analysis of TCGA data via cBioPortal revealed that CLC-2, 4, and 7 were most frequently altered in HNSCC and invasive breast carcinoma (Supplementary Figure 7). RT-PCR profiling of cancer cell lines for CLC- 2, CLC-4 and CLC-7 showed that these genes were expressed, and that CLC-2 and CLC-4 were most abundantly expressed (Supplementary Figure 8a). We did not detect expression of CLC-1 and CLC-5 in our panel of cell line (data not shown). Therefore, we explored the effects of CLC-2 and CLC-4 knockdown on protein levels of EGFR in UM-SCC-1 and SKBR3 cells. With >50% knockdown of CLC- 2 and CLC-4 mRNA, the EGFR protein expression and phosphorylation status in these samples remained unchanged (Supplementary Figures 8b and 8c).
K–Cl cotransporter (KCC) over-expression has been associated with enhanced tumor cell malignancy and invasiveness in a variety of cancers (Kitagawa, et al. 2013). In order to evaluate whether KCC affects EGFR expression, we used DIOA, a known KCC pharmacological inhibitor in two HNSCC cell lines. However, in our results, the KCC inhibitor DIOA did not alter phosphorylated or total EGFR levels (Supplementary Figure 9).
Discussion
Chromosomal band 11q13 is frequently amplified in subtypes of GIST, breast cancer, HNSCC and esophageal cancer (Berglund, et al. 2014; Britschgi, et al. 2013; Duvvuri, et al. 2012; Liu, et al. 2015; Ruiz, et al. 2012). Situated within this chromosomal segment is the gene TMEM16A that encodes a Ca2+-activated Cl− channel that has been shown to contribute to pathways involved in solid tumor growth and progression (Ayoub, et al. 2010; Britschgi, et al. 2013; Duvvuri, et al. 2012). The molecular mechanisms that connect TMEM16A to tumor growth are not well understood, but in breast cancer and HNSCC patients, 11q13 amplification correlates with TMEM16A protein overexpression and poor clinical outcome (Britschgi, et al. 2013; Duvvuri, et al. 2012). Moreover, TMEM16A contributes to tumor growth and viability even in the absence of 11q13 amplification (Duvvuri, et al. 2012). Wu et al. (2015) have previously shown that TMEM16A expression correlates with clinical outcome in breast cancer independently of HER2 expression (Wu, et al. 2015). However, the effect of HER2 blockade on TMEM16A expression was not investigated in these studies and, hence, the present experiment was designed to explore the significance of interaction between TMEM16A and HER-family proteins. Based on our data, we suggest that TMEM16A may regulate HER-family signaling and may be altered in the context of trastuzumab treatment. Further work is needed to ascertain if this idea can be confirmed in a clinical setting.
In the present study, we have explored the possibility that the inhibition of TMEM16A Cl− channel function has the potential to improve outcome of targeted therapy in solid tumors. Our data show 1) that TMEM16A contributes to a pathway of growth and survival in breast and HNSCC tumor cells, 2) that TMEM16A plays a role in the molecular regulation of the expression and the activity of EGFR and its partner HER2 in tumor growth and survival, 3) that tumor cells with acquired resistance to HER family- targeted therapy are more sensitive to the suppression of TMEM16A Cl− channel activity and 4) that the combined suppression of TMEM16A Cl− channel activity and EGFR inhibits tumor cell survival and growth more effectively than either pathway alone and enhances response to antibody-mediated biologic therapy of the EGFR/HER family. These data support the notion that TMEM16A contributes to molecular signaling associated with solid tumor growth and that regulation of TMEM16A activity presents as a promising opportunity to improve patient responses to targeted therapy of the EGFR/HER family.
One or more members of the ErbB/HER family are usually expressed in solid tumor where they are known to contribute to tumor growth and aggressiveness. Targeted antibody-mediated biologic therapies that, unlike chemotherapy, inhibit individual ErbB/HER family members, are effective in suppressing tumor growth in a target and a pathway specific manner. These therapies however, tend to selectively target EGFR/HER homodimers while retaining the signal transduction processes through heterodimers. As a result, activity of EGFR/HER is known to persist or recur in tumors even under conditions of exposure to targeted therapy(Bill, et al. 2014; Nahta and Esteva 2007). Thus, EGFR/HER-family heterodimerization leading to constitutive cross-talk among family members is a significant challenge in overcoming therapeutic resistance in solid tumors. Our data suggest that the activation of TMEM16A may act as an important causal event in the development of resistance to EGFR/ErbB2-targeted therapy.
We have shown that the growth of human HNSCC and breast cancer cells that overexpress TMEM16A is associated with the over-activation of EGFR both in vitro and in vivo. By promoting phosphorylation/activation of EGFR and ErbB2/HER2, TMEM16A appears to decrease response to targeted, antibody-based therapy. Inhibition of TMEM16A function by small molecule inhibitors or shRNA suppresses the activation of EGFR and/or HER2 and therefore, diminishes cell growth and viability. Consistent with involvement of TMEM16A in the regulation of EGFR/HER-dependent growth pathways in these cells, tumor cell growth suppression was more pronounced upon combined inhibition of TMEM16A and EGFR/HER2. Thus, the failure to suppress the function of TMEM16A results in inadequate inhibition of EGFR/HER-induced growth pathways and may facilitate therapeutic resistance. In support of this possibility, we find that breast cancer cells that have acquired resistance to trastuzumab show elevated TMEM16A expression and an increased sensitivity to its inhibition, suggesting increased molecular dependence on TMEM16A-dependent growth pathways. Moreover, considering that cross-activation among EGFR and other members of HER family are known causes of trastuzumab-resistance (Yarden 2001), and that TMEM16A is a regulator of the EGFR pathway (Britschgi, et al. 2013), TMEM16A dysregulation may very well result in resistance to other EGFR/HER targeted therapies. Our data implicate TMEM16A as a therapeutic target in solid tumors demonstrating resistance to EGFR/HER-targeted therapies.
How TMEM16A induces resistance to EGFR/HER family-based therapies, including trastuzumab and cetuximab, is not clear, but considering that TMEM16A interacts with EGFR (Britschgi, et al. 2013), more direct regulation may be involved. The present finding that reduction of extracellular Cl− inhibits EGFR expression and activity in both HNSCC and breast cancer cells, supports involvement of regulatory dependence on a Cl− channel. Our data point to TMEM16A in regulation of this Cl− flux, since TMEM16A inhibitors or TMEM16A mutants deficient in the Cl− conductance function reduced EGFR-signaling. Moreover, changes in the Cl− flux across the membrane may affect cellular site-specific signaling by influencing internalization of total and/or phosphorylated receptor or ligand secretion (Bill, et al. 2014). However, these data should be interpreted with caution, since manipulation of ionic content can have non-specific effects on plasma membrane integrity and potential (Zhou, et al. 2015). The observed changes in the expression of total and phosphorylated forms of EGFR and HER2 in cells treated with TMEM16A inhibitors support the involvement of receptor internalization or trafficking processes (Bill, et al. 2015a; Bill, et al. 2014). Knockdown of TMEM16A in breast cancer cells reduces not only EGFR phosphorylation, but also influences the autocrine pathway of ligand secretion (Britschgi, et al. 2013). The levels of secreted cytokines, EGF and TGF, were decreased in cells in which TMEM16A expression was knocked down, whereas overexpression of TMEM16A was sufficient to promote secretion. Although a similar effect on ligand secretion was not measurable in HNSCC cell lines, its existence or activation under certain condition was not ruled out (Britschgi, et al. 2013). Notably, stimulation of EGFR with EGF did not alter the activity of TMEM16A in UM-SCC-1 cells, indicating that the TMEM16A activity may not be regulated by EGFR as shown in other cells lines (Jeulin, et al. 2008; Mroz and Keely 2012). Taken together, the activation of TMEM16A Cl− channel function influences early signaling events, including the regulation of EGFR/HER2 expression and phosphorylation and, thus, impacts the response or resistance to biologic therapy of EGFR/HER family members. Our data do not exclude the possibility that TMEM16A has functions that are independent of its channel function. It remains plausible that TMEM16A can interact with members of the EGFR family independently of its channel function, and that channel-deficient TMEM16A mutants of may have altered membrane stability.
A majority of HNSCC patients, despite the expression of wild-type EGFR, are found nonresponsive to biologic inhibition of EGFR mediated by cetuximab (Pai and Westra 2009; Vermorken, et al. 2007). HNSCC are thus thought to harbor other unknown but essential regulators of EGFR and its downstream targets that are capable of influencing therapeutic outcome (Leemans, et al. 2011; Tian, et al. 2011). Recently, involvement of TMEM16A in the activation of RAS-RAF-ERK1/2 signal transduction pathway of tumor growth was described in HNSCC (Duvvuri, et al. 2012). Furthermore, TMEM16A function as a signal transducer was proposed to associate with its function to regulate Cl− flux (Duvvuri, et al. 2012). Our data suggest that the manipulation of Cl− flux affects activity of STAT3, the downstream effector of EGFR pathway. The ability of TMEM16A to independently regulate downstream mechanisms of growth may render tumor cells less responsive to EGFR inhibitors, including cetuximab. The increase in expression and/or activity of TMEM16A often found in HNSCC may therefore influence predictions of response or sensitivity to EGFR/HER-targeted therapy. Consistent with this, 11q13 amplification and TMEM16A overexpression in HNSCC cells was found to impart an enhanced sensitivity to small molecule inhibitors of the EGFR tyrosine kinase (Bill, et al. 2015a). Whether TMEM16A overexpression predicts sensitivity to cetuximab therapy remains to be explored in a clinical setting, but as presented here, the 11q13-amplified FaDu cells expressing TMEM16A shRNA show an increase in cetuximab-response, suggesting that combined targeting of TMEM16A and EGFR maybe efficacious in selected patients.
In summary, we have described a molecular connection between TMEM16A, a Ca2+ activated Cl− channel, and members of the EGFR/HER family that appears to be, in part, mediated by Cl− flux through the channel. By regulating the expression and the activity of EGFR/HER family members, TMEM16A impacts tumor cell growth and viability signaling and therefore, the response to antibody-mediated EGFR/HER family-targeted biologic therapies in breast cancer and HNSCC. Our data support the combined of TMEM16A along with members of the EGFR/HER family as a strategy to increase response to EGFR/HER-targeted therapy and to delay or prevent emergence of resistance.
Supplementary Material
Acknowledgments
Supported by: Funded in part by the Department of Veterans Affairs BLSR&D (IO1 BX003456), PNC Foundation, the University of Pittsburgh CMRF (UD), and the SPORE in Head and Neck Cancer (JRG).
This work does not represent the views of the US Government or the Department of Veterans Affairs.
References
- Akervall JA, Jin Y, Wennerberg JP, Zatterstrom UK, Kjellen E, Mertens F, Willen R, Mandahl N, Heim S, Mitelman F. Chromosomal abnormalities involving 11q13 are associated with poor prognosis in patients with squamous cell carcinoma of the head and neck. Cancer. 1995;76(5):853–859. doi: 10.1002/1097-0142(19950901)76:5<853::aid-cncr2820760520>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Ayoub C, Wasylyk C, Li Y, Thomas E, Marisa L, Robe A, Roux M, Abecassis J, de Reynies A, Wasylyk B. ANO1 amplification and expression in HNSCC with a high propensity for future distant metastasis and its functions in HNSCC cell lines. Br J Cancer. 2010;103(5):715–726. doi: 10.1038/sj.bjc.6605823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J. Clinical trials of single-agent trastuzumab (Herceptin) Semin Oncol. 2000;27(5 Suppl 9):20–26. [PubMed] [Google Scholar]
- Baselga J, Tripathy D, Mendelsohn J, Baughman S, Benz CC, Dantis L, Sklarin NT, Seidman AD, Hudis CA, Moore J, Rosen PP, Twaddell T, Henderson IC, Norton L. Phase II study of weekly intravenous trastuzumab (Herceptin) in patients with HER2/neu-overexpressing metastatic breast cancer. Semin Oncol. 1999;26(4 Suppl 12):78–83. [PubMed] [Google Scholar]
- Berglund E, Akcakaya P, Berglund D, Karlsson F, Vukojevic V, Lee L, Bogdanovic D, Lui WO, Larsson C, Zedenius J, Frobom R, Branstrom R. Functional role of the Ca(2)(+)-activated Cl(−) channel DOG1/TMEM16A in gastrointestinal stromal tumor cells. Exp Cell Res. 2014;326(2):315–325. doi: 10.1016/j.yexcr.2014.05.003. [DOI] [PubMed] [Google Scholar]
- Bill A, Gutierrez A, Kulkarni S, Kemp C, Bonenfant D, Voshol H, Duvvuri U, Gaither LA. ANO1/TMEM16A interacts with EGFR and correlates with sensitivity to EGFR-targeting therapy in head and neck cancer. Oncotarget. 2015a;6(11):9173–9188. doi: 10.18632/oncotarget.3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bill A, Hall ML, Borawski J, Hodgson C, Jenkins J, Piechon P, Popa O, Rothwell C, Tranter P, Tria S, Wagner T, Whitehead L, Gaither LA. Small molecule-facilitated degradation of ANO1 protein: a new targeting approach for anticancer therapeutics. J Biol Chem. 2014;289(16):11029–11041. doi: 10.1074/jbc.M114.549188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bill A, Popa MO, van Diepen MT, Gutierrez A, Lilley S, Velkova M, Acheson K, Choudhury H, Renaud NA, Auld DS, Gosling M, Groot-Kormelink PJ, Gaither LA. Variomics screen identifies the re-entrant loop of the calcium-activated chloride channel ANO1 that facilitates channel activation. J Biol Chem. 2015b;290(2):889–903. doi: 10.1074/jbc.M114.618140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britschgi A, Bill A, Brinkhaus H, Rothwell C, Clay I, Duss S, Rebhan M, Raman P, Guy CT, Wetzel K, George E, Popa MO, Lilley S, Choudhury H, Gosling M, Wang L, Fitzgerald S, Borawski J, Baffoe J, Labow M, Gaither LA, Bentires-Alj M. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci U S A. 2013;110(11):E1026–1034. doi: 10.1073/pnas.1217072110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322(5901):590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- Cassell A, Grandis JR. Investigational EGFR-targeted therapy in head and neck squamous cell carcinoma. Expert Opin Investig Drugs. 2010;19(6):709–722. doi: 10.1517/13543781003769844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole WC. ANO1-ther brick in the wall–role of Ca2+-activated Cl− channels of interstitial cells of Cajal in cholinergic motor control of gastrointestinal smooth muscle. J Physiol. 2011;589(Pt 19):4641–4642. doi: 10.1113/jphysiol.2011.218453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvvuri U, Shiwarski DJ, Xiao D, Bertrand C, Huang X, Edinger RS, Rock JR, Harfe BD, Henson BJ, Kunzelmann K, Schreiber R, Seethala RS, Egloff AM, Chen X, Lui VW, Grandis JR, Gollin SM. TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res. 2012;72(13):3270–3281. doi: 10.1158/0008-5472.CAN-12-0475-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Pinilla PJ, Gibbons SJ, Bardsley MR, Lorincz A, Pozo MJ, Pasricha PJ, Van de Rijn M, West RB, Sarr MG, Kendrick ML, Cima RR, Dozois EJ, Larson DW, Ordog T, Farrugia G. Ano1 is a selective marker of interstitial cells of Cajal in the human and mouse gastrointestinal tract. Am J Physiol Gastrointest Liver Physiol. 2009;296(6):G1370–1381. doi: 10.1152/ajpgi.00074.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Rock JR, Harfe BD, Cheng T, Huang X, Jan YN, Jan LY. Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc Natl Acad Sci U S A. 2009;106(50):21413–21418. doi: 10.1073/pnas.0911935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Godfrey TE, Gooding WE, McCarty KS, Jr, Gollin SM. Comprehensive genome and transcriptome analysis of the 11q13 amplicon in human oral cancer and synteny to the 7F5 amplicon in murine oral carcinoma. Genes Chromosomes Cancer. 2006;45(11):1058–1069. doi: 10.1002/gcc.20371. [DOI] [PubMed] [Google Scholar]
- Huang X, Gollin SM, Raja S, Godfrey TE. High-resolution mapping of the 11q13 amplicon and identification of a gene, TAOS1, that is amplified and overexpressed in oral cancer cells. Proc Natl Acad Sci U S A. 2002;99(17):11369–11374. doi: 10.1073/pnas.172285799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SJ, Blair PJ, Britton FC, O’Driscoll KE, Hennig G, Bayguinov YR, Rock JR, Harfe BD, Sanders KM, Ward SM. Expression of anoctamin 1/TMEM16A by interstitial cells of Cajal is fundamental for slow wave activity in gastrointestinal muscles. J Physiol. 2009;587(Pt 20):4887–4904. doi: 10.1113/jphysiol.2009.176198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeulin C, Seltzer V, Bailbe D, Andreau K, Marano F. EGF mediates calcium-activated chloride channel activation in the human bronchial epithelial cell line 16HBE14o-: involvement of tyrosine kinase p60c-src. Am J Physiol Lung Cell Mol Physiol. 2008;295(3):L489–496. doi: 10.1152/ajplung.90282.2008. [DOI] [PubMed] [Google Scholar]
- Katoh M, Katoh M. FLJ10261 gene, located within the CCND1-EMS1 locus on human chromosome 11q13, encodes the eight-transmembrane protein homologous to C12orf3, C11orf25 and FLJ34272 gene products. Int J Oncol. 2003;22(6):1375–1381. [PubMed] [Google Scholar]
- Kitagawa M, Niisato N, Shiozaki A, Ohta-Fujimoto M, Hosogi S, Miyazaki H, Ichikawa D, Otsuji E, Marunaka Y. A regulatory role of K(+)-Cl(−) cotransporter in the cell cycle progression of breast cancer MDA-MB-231 cells. Arch Biochem Biophys. 2013;539(1):92–98. doi: 10.1016/j.abb.2013.06.014. [DOI] [PubMed] [Google Scholar]
- Kulkarni S, Reddy KB, Esteva FJ, Moore HC, Budd GT, Tubbs RR. Calpain regulates sensitivity to trastuzumab and survival in HER2-positive breast cancer. Oncogene. 2010;29(9):1339–1350. doi: 10.1038/onc.2009.422. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Schreiber R, Kmit A, Jantarajit W, Martins JR, Faria D, Kongsuphol P, Ousingsawat J, Tian Y. Expression and function of epithelial anoctamins. Exp Physiol. 2012;97(2):184–192. doi: 10.1113/expphysiol.2011.058206. [DOI] [PubMed] [Google Scholar]
- Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11(1):9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- Liu F, Cao QH, Lu DJ, Luo B, Lu XF, Luo RC, Wang XG. TMEM16A overexpression contributes to tumor invasion and poor prognosis of human gastric cancer through TGF-beta signaling. Oncotarget. 2015;6(13):11585–11599. doi: 10.18632/oncotarget.3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohd Sharial MS, Crown J, Hennessy BT. Overcoming resistance and restoring sensitivity to HER2-targeted therapies in breast cancer. Ann Oncol. 2012;23(12):3007–3016. doi: 10.1093/annonc/mds200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mroz MS, Keely SJ. Epidermal growth factor chronically upregulates Ca(2+)-dependent Cl(−) conductance and TMEM16A expression in intestinal epithelial cells. J Physiol. 2012;590(8):1907–1920. doi: 10.1113/jphysiol.2011.226126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahta R, Esteva FJ. Trastuzumab: triumphs and tribulations. Oncogene. 2007;26(25):3637–3643. doi: 10.1038/sj.onc.1210379. [DOI] [PubMed] [Google Scholar]
- Nahta R, Yuan LX, Du Y, Esteva FJ. Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling. Mol Cancer Ther. 2007;6(2):667–674. doi: 10.1158/1535-7163.MCT-06-0423. [DOI] [PubMed] [Google Scholar]
- Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14(11):727–736. doi: 10.1038/nrm3683. [DOI] [PubMed] [Google Scholar]
- Pai SI, Westra WH. Molecular pathology of head and neck cancer: implications for diagnosis, prognosis, and treatment. Annu Rev Pathol. 2009;4:49–70. doi: 10.1146/annurev.pathol.4.110807.092158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz C, Martins JR, Rudin F, Schneider S, Dietsche T, Fischer CA, Tornillo L, Terracciano LM, Schreiber R, Bubendorf L, Kunzelmann K. Enhanced expression of ANO1 in head and neck squamous cell carcinoma causes cell migration and correlates with poor prognosis. PLoS One. 2012;7(8):e43265. doi: 10.1371/journal.pone.0043265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134(6):1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiwarski DJ, Shao C, Bill A, Kim J, Xiao D, Bertrand CA, Seethala RS, Sano D, Myers JN, Ha P, Grandis J, Gaither LA, Puthenveedu MA, Duvvuri U. To “grow” or “go”: TMEM16A expression as a switch between tumor growth and metastasis in SCCHN. Clin Cancer Res. 2014;20(17):4673–4688. doi: 10.1158/1078-0432.CCR-14-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashima H, Picollo A, Accardi A. Purified TMEM16A is sufficient to form Ca2+-activated Cl− channels. Proc Natl Acad Sci U S A. 2013;110(48):19354–19359. doi: 10.1073/pnas.1312014110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Kongsuphol P, Hug M, Ousingsawat J, Witzgall R, Schreiber R, Kunzelmann K. Calmodulin-dependent activation of the epithelial calcium-dependent chloride channel TMEM16A. FASEB J. 2011;25(3):1058–1068. doi: 10.1096/fj.10-166884. [DOI] [PubMed] [Google Scholar]
- Tien J, Peters CJ, Wong XM, Cheng T, Jan YN, Jan LY, Yang H. A comprehensive search for calcium binding sites critical for TMEM16A calcium-activated chloride channel activity. Elife. 2014;3 doi: 10.7554/eLife.02772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermorken JB, Trigo J, Hitt R, Koralewski P, Diaz-Rubio E, Rolland F, Knecht R, Amellal N, Schueler A, Baselga J. Open-label, uncontrolled, multicenter phase II study to evaluate the efficacy and toxicity of cetuximab as a single agent in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck who failed to respond to platinum-based therapy. J Clin Oncol. 2007;25(16):2171–2177. doi: 10.1200/JCO.2006.06.7447. [DOI] [PubMed] [Google Scholar]
- Wu H, Guan S, Sun M, Yu Z, Zhao L, He M, Zhao H, Yao W, Wang E, Jin F, Xiao Q, Wei M. Ano1/TMEM16A Overexpression Is Associated with Good Prognosis in PR-Positive or HER2-Negative Breast Cancer Patients following Tamoxifen Treatment. PLoS One. 2015;10(5):e0126128. doi: 10.1371/journal.pone.0126128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YD, Cho H, Koo JY, Tak MH, Cho Y, Shim WS, Park SP, Lee J, Lee B, Kim BM, Raouf R, Shin YK, Oh U. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455(7217):1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001;37(Suppl 4):S3–8. doi: 10.1016/s0959-8049(01)00230-1. [DOI] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Wong CO, Cho KJ, van der Hoeven D, Liang H, Thakur DP, Luo J, Babic M, Zinsmaier KE, Zhu MX, Hu H, Venkatachalam K, Hancock JF. SIGNAL TRANSDUCTION. Membrane potential modulates plasma membrane phospholipid dynamics and K-Ras signaling. Science. 2015;349(6250):873–876. doi: 10.1126/science.aaa5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.