

Abstract
Abnormal activation of the γc cytokine JAK/STAT signaling pathway assessed by STAT3 or STAT5b phosphorylation was present in a proportion of many T-cell malignancies. Activating mutations of STAT3/STAT5b and JAK1/3 were present in some but not in all cases with constitutive signaling pathway activation. Using shRNA analysis pSTAT malignant T-cell lines were addicted to JAKs/STATs whether they were mutated or not. Activating JAK/STAT mutations were not sufficient to support leukemic cell proliferation but only augmented upstream pathway signals. Functional cytokine receptors were required for pSTAT expression. Combining a JAK1/2 inhibitor with a Bcl-xL inhibitor navitoclax provided additive/synergistic activity with IL-2 dependent ATLL cell lines and in a mouse model of human IL-2 dependent ATLL. The insight that disorders of the γc/JAK/STAT system are pervasive suggests approaches including those that target gamma cytokines, their receptors or that use JAK kinase inhibitors may be of value in multicomponent therapy for T-cell malignancies.
Keywords: JAK/STAT, T-cell malignancy, JAK inhibitor
1. Introduction
The common gamma (γc) receptor-dependent cytokines and their signaling pathways play critical roles in T-cell immunity (Rochman et al., 2009, Waldmann, 2006). Activation of this pathway can be identified by the nuclear presence of pSTAT3 or pSTAT5. Recently activation of this γc/JAK/STAT system was identified in virtually all forms of T-cell leukemia/lymphoma (Waldmann and Chen, 2017). Most studies of T-cell lymphoma have focused on JAK/STAT mutations; however activation of this pathway is a unifying feature that is more pervasive (Waldmann and Chen, 2017). The demonstration of activation of the JAK/STAT pathway in a major proportion of T-cell malignancies suggests JAKs as potential therapeutic targets–an approach that may revolutionize multiagent therapy of patients with pSTAT expressing T-cell malignancies.
JAK/STAT activation in T-cell lymphoma
JAK/STAT activation was assessed by phospho-STAT3 and STAT5 expression and nuclear localization and by monitoring the negative effects of JAK inhibitors. Such JAK/STAT activation was shown to be present from rare to 86% of patients with most forms of T-cell malignancy. In particular, γc cytokine, JAK1/3, STAT3/5 pathway activation was demonstrated in NK/TCL (Bouchekioua et al., 2014, Coppo et al., 2009, Koo et al., 2012) ALK positive and ALK- ALCL, (Crescenzo et al., 2015, Chiarle et al., 2005, Khoury et al., 2003), large granular lymphocytic leukemia (LGL) (Koskela et al., 2012) prolymphocytic leukemia (PLL) (Kiel et al., 2014, Bergmann et al., 2014, Bellanger et al., 2014), Sézary Syndrome (SS) and mycosis fungoides (MF) (Eriksen et al., 2001, Zhang et al., 1996, Choi et al., 2015), as well as in smoldering and chronic ATLL (Chen et al., 2008, Chen et al., 2010, Ju et al., 2011, Kataoka et al., 2015, Elliott, et al., 2011).
STAT mutations in T-cell lymphoma
In light of the frequent activation of the JAK/STAT system in T-cell malignancies, these leukemias were examined for activating mutations of STAT proteins (Odejide et al., 2014, Koskela et al., 2012, Crescenzo et al., 2015, Coppo et al., 2009, Kücük et al., 2015, Nicolae et al., 2014, Choi et al., 2015, Zhang et al., 2012, Kataoka et al. 2015). Many T-cell malignancies manifested STAT3/STAT5b mutations that were focused predominantly in their Src homology 2-(SH2) domain – a site that is involved in docking of phosphorylated tyrosine residues, transient binding to cytokine receptors and binding to other STAT proteins that mediate dimerization and nuclear localization (Koskela et al., 2012).
In studies with ALK- T-cell lines using shRNA loss-of-function analysis our group demonstrated that cell lines that manifested pSTAT3 were addicted to STAT3 whether or not the STAT elements were mutated suggesting the importance of STAT3 activation in leukemic T-cell survival (Chen and Waldmann, unpublished results). Kücük and coworkers 2015 demonstrated that STAT5b mutations provided a growth advantage. Nevertheless, they demonstrated that such activating STAT mutations were not sufficient for leukemic cell proliferation but only enhanced upstream signals from the cytokine-cytokine receptor, JAK/STAT pathway. Following IL-2 withdrawal pSTAT expression disappeared in 1 hour after transient IL-2 stimulation of wild-type STAT5b transduced cells, whereas it persisted for more than 6 hours with STAT5b N642H mutant-transduced cells. In accord with this view that upstream signals were required, the growth promoting activity of the mutant was partially inhibited by the addition of the upstream JAK1/2 inhibitor AZD1480.
JAK mutations in T-cell lymphoma
Since STAT activating mutations were not sufficient but only enhanced upstream signaling, activations of JAKs were explored using shRNA loss-of-function analysis with JAK1 shRNA. JAK1 was required in phospho-STAT3 positive ALK- cell lines whereas its loss had no effect on phospho-STAT3 negative cell lines (Chen and Waldmann, unpublished results). Given the requirement for activated JAKs, especially JAK1 and JAK3 for the activation of the gamma cytokine JAK/STAT pathway, leukemic cells were examined for JAK mutations. Many mutations were found especially in the pseudokinase domain that was reported to function as a protein kinase that phosphorylates two residues that negatively regulate JAK kinases to suppress their activity (Saharinen and Silvennoinen, 2002). It is clear from our JAK1 knockdown studies that JAK1 was required in cells that manifest activation of the cytokine receptor JAK/STAT system whether mutations were present or not as with STAT mutations JAK mutations appear to be incapable of initiating but rather augment responses initiated by upstream signals.
Activation of the JAK/STAT pathway required functional cytokine receptors
In studies by Lu 2005, and Hornakova 2009 and their coworkers even with activating JAK and STAT mutations there was a requirement for expression of a functional cytokine receptor that played two roles; first as a scaffold for cross-activation of JAK kinases and second as a docking site for recruitment of STAT transcription factors. In addition to JAK/STAT mutations, mutations were present in select cytokine receptors for example the IL-7 receptor in childhood ALL (Shochat et al., 2011, Zenatti et al., 2011).
Disorders of cytokine expression in T-cell lymphoma
Although JAK3 FERM domain mutations were only rarely observed in patients with smoldering and chronic HTLV-1 associated ATLL, the gamma-c JAK/STAT pathway was usually activated (Migone et al., 1995, Chen et al., 2008). We demonstrated that the HTLV-1 encoded Tax protein transactivated two autocrine (IL-2/IL-2R alpha, IL-15/IL-15R alpha) and one paracrine (IL-9) loop in such patients (Tendler et al., 1990, Azimi et al., 1998, Chen et al., 2010). These cytokine-cytokine receptor loops led to activation of the JAK1/3 STAT signaling pathway and were associated with spontaneous proliferation of ATLL cells, a phenomenon that could be inhibited by the addition of the pan-JAK inhibitor tofacitinib (Ju et al., 2011). In further examination of cytokine disorders there was evidence supporting a role for the gamma-c cytokine IL-15 in cutaneous T-cell lymphoma (Döbbeling et al., 1998) and for IL-21 in patients with anaplastic T-cell lymphoma (Jain et al., 2015). In summary, disorders of the gamma cytokine JAK/STAT signaling pathway are pervasive in T-cell malignancy suggesting novel molecular targets and therapeutic opportunities that may be of value in the multicomponent treatment of these tumors.
Gamma cytokine JAK/STAT system as a target in the treatment of T-cell malignancies
The demonstration of activation of the JAK/STAT system in T-cell malignancies provided the rationale for diverse therapeutic approaches including those that targeted the gamma-c cytokines directly, those that blocked cytokine-receptor interactions and especially JAK kinase inhibitors. The best biomarker suggesting JAKs would be a rational target is the presence of pSTAT3 or pSTAT5 and their nuclear translocation rather than the less frequent mutations affecting this system. JAK inhibitors inhibited the proliferation of cytokine dependent cell lines and ex vivo leukemic cells from patients with smoldering and chronic ATLL who manifest activation of the JAK system (Ju, et al., 2011). To translate these observations a clinical trial of ruxolitinib is underway in patients with smoldering and chronic ATLL (Conlon and Waldmann 2017). However, therapy with ruxolitinib is clearly not optimal. With rare exceptions T-cell leukemia/lymphomas were associated with activation of the JAK1/JAK3 signaling but did not involve JAK2. However, ruxolitinib and tofacitinib inhibit the off-target JAK2 kinase, therefore interfering with the cell signaling mediated by thrombopoietin, leading to thrombocytopenia. We have demonstrated that when used according to FDA guidelines, 20 micrograms twice a day orally ruxolitinib provided meaningful inhibition of the JAK/STAT pathway for only a small proportion of the 12-hour period between dosings (Dubois and Waldmann, unpublished results). In light of the limitation of the therapy possible due to toxicities with inhibitors that target JAK2, agents with greater specificity for JAK1 or JAK3 are being developed for use in T-cell malignancies. Although JAK1 inhibitors have great promise, they have activities that inhibit cytokines other than the gamma-c cytokines with consequent toxicity. JAK3 has been proposed as an alternative attractive target since its expression is restricted to cells of the hematopoietic lineage and JAK3 is associated exclusively with the common gamma chain. Inactivating mutations of JAK3 in humans or mice resulted only in severe combined immunodeficiency without non-immunological toxicities (Leonard and O’Shea, 1998). However, Haan and associates, 2011 reported that JAK1 has a dominant role over JAK3 and have concluded that JAK3 predominantly plays a role as a scaffold. These authors disagree with the proposal that selective ATP competitive JAK3 kinase inhibitors would be effective therapeutic agents. Smith, et al., 2016 have a contrasting view and have demonstrated that a selective inhibitor of JAK3 (JAK3i) blocks IL-2 stimulated T-cell proliferation at low nanomolar concentrations. Smith revealed a biphasic role for JAK3 activity in CD4+ positive T-cells. JAK3i blocked a second temporal wave of IL-2 mediated signaling. Thus Smith’s results contradict the prior conclusions of Haan who had discounted a catalytic role for JAK3. Studies are underway with JAK3i in murine xenograft models involving T-cell leukemia/lymphoma xenografts with activations of the JAK/STAT pathway.
Selective targeting of JAK/STAT signaling was augmented by combination therapy with ruxolitinib and navitoclax for Bcl-xL blockade
Although JAK-inhibitor therapy may be of benefit in T-cell leukemia/lymphoma in general and in particular in IL-2 dependent smoldering and chronic ATLL, it will have to be used in combination therapy to be of major benefit. To address this issue using a matrix-screening platform we evaluated ruxolitinib within a dose response matrix experiment with > 450 approved or investigational agents (Zhang, et al., 2015). Additive/synergistic effects were demonstrated in the IL-2 dependent ED40515 (+) cell line with ruxolitinib when combined with the inhibitors Bcl-2/Bcl-xL (navitoclax), HDACs (panobinostat, pracinostat), mTORC1/2 (AZD-8055), Aurora A/B/C/kinases (AMG 900), PI3Ks (GSK2126458) and NF-κB/IKK (Withaferin A, SPC-839, bardoxolone methyl). The following combinations also showed additive/synergistic effects in their capacity to inhibit cell proliferation in multiple IL-2 dependent ATLL cell lines AMG 900, GSK2126458, AZD-8055, and bardoxolone methyl. The Bcl-2/Bcl-xL inhibitor navitoclax was identified as a strong candidate for multicomponent therapy with ruxolitinib. The combination was noted to strongly activate BAX (Bcl-2-associated X protein), affect mitochondrial depolarization, and increase Caspase-3/7 activities in IL-2 dependent ATLL cell lines. The combination led to the cleavage of PARP and Mcl-1, the latter from a 40 kDa anti-apoptotic to a 24 kDa pro-apoptotic form (Figure 1).
Figure 1. The Combination of Ruxolitinib and Navitoclax Induced Caspase 3/7 Activation Which Resulted in Cleavage of PARP and Mcl-1 in IL-2-dependent ATL Ceils.
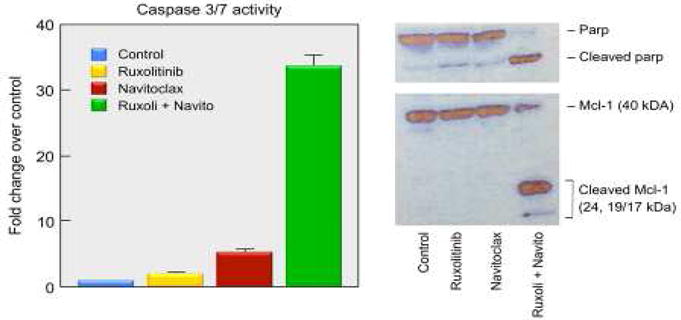
Western blot analysis of the cleavage of PARP and Mcl-1 proteins at 24 or 48 hours after different treatments as indicated. The concentrations of ruxolitinib and navitoclax using the studies shown in this Figure were 200 nM and 100 nM respectively. Reproduced with modification from Figure 3D from the Proceedings of the National Academy of Sciences. Zhang M, Mathews Griner LA, Ju W, Duveau DY, Guha R, et al., 2015. Selective targeting of JAK/STAT signaling is potentiated by Bcl-xL blockade in IL-2-dependent adult T-cell leukemia. Proc Natl Acad Sci USA, volume 112 (40), pp. 12480–12485.
The combination of ruxolitinib and navitoclax also was shown to significantly inhibit tumor growth and its administration resulted in prolonged survival of mice bearing the ED(+)/IL-2 transgenic tumor (Figure 2).
Figure 2. The Combination of Ruxolitinib and Navitoclax Significantly Inhibited Tumor Growth and Resulted in Prolonged Survival of Tumor-bearing Mice.
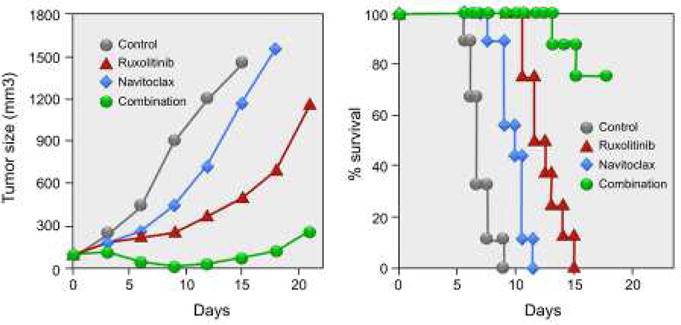
Combination therapy of ruxolitinib with navitoclax significantly inhibited ED(+)/IL-2 tumor growth in vivo. Female NSG mice were injected s.c. with 1 × 107 ED(+)/IL-2 cells. When the average tumor volume reached approximately 100 mm3, the tumor-bearing mice were divided into 4 groups (N = 8–9) with comparable average tumor volumes when the therapy was started. Ruxolitinib was continuously administered by a s.c. infusion pump at a dose of 50 mcg/kg/day for 14 days, and navitoclax was given orally at a dose of 40 mg/kg/day for 6 days. Average tumor volumes during the therapeutic course for reach group. Kaplan-Meier survival plot of the mice in the therapeutic study. Reproduced with modification from Figure 4B from the Proceedings of the National Academy of Sciences. Zhang M, Mathews Griner LA, Ju W, Duveau DY, Guha R, et al., 2015. Selective targeting of JAK/STAT signaling is potentiated by Bcl-xL blockade in IL-2-dependent adult T-cell leukemia. Proc Natl Acad Sci USA, volume 112 (40), pp. 12483.
Analysis of drug responses using in vitro culture in murine models were of value but may not reflect a facsimile of human disease states. Spontaneous proliferations of ex vivo PBMCs from patients with smoldering/chronic ATLL represented a model to assess the therapeutic potential of drug combinations in a human system. Proliferation of ATLLs in 6-day PBMC cultures assessed by 3H-thymidine uptake were determined from 5 patients with smoldering/chronic ATLL who were in the autocrine IL-2 dependent phase of their leukemia. Spontaneous proliferations of these PBMCs were partially inhibited by the addition of individual antibodies to IL-2 and to a lesser extent to IL-9 or IL-15. Profound inhibition of ex vivo proliferation was achieved by the simultaneous addition of antibodies against all 3 cytokines. Furthermore, addition of either ruxolitinib or navitoclax inhibited 6-day ex vivo proliferations of PBMCs from these patients. The combination of ruxolitinib with navitoclax provided enhanced inhibition (to 90%) of ex vivo proliferation of PBMCs from the 5 patients that was significantly greater than either drug alone (p < 0.01). Thus, the combination of ruxolitinib with navitoclax provided additive/synergistic activity with IL-2 dependent ATLL cell lines and in a mouse model of human IL-2 dependent ATLL as well as on ex vivo 6-day cultures of PBMCs from ATLL patients. These findings provide support for a therapeutic trial in patients with smoldering and chronic ATLL using a combination regimen that inhibits JAK1 and Bcl-xL. With further study it should be possible to provide effective multicomponent therapies for many T-cell malignancies that utilize as one of their elements an inhibitor of JAK1 or JAK3 kinase.
Highlights.
Abnormal activation of JAK/STAT signaling is present in many T-cell malignancies.
Mutations of STAT3/STAT5b and JAK1/3 are common.
JAK/STAT mutations are insufficient but augment upstream pathway signals.
JAK kinase inhibitors may be of value in therapy of T-cell malignancies.
Acknowledgments
This research was supported by the Intramural Research Program of the Lymphoid Malignancies Branch, National Cancer Institute (NCI), Center for Cancer Research, National Institutes of Health.
Abbreviations
- ATLL
adult T-cell leukemia/lymphoma
- γc
common gamma receptor dependent cytokine
- HTLV-1
human T-cell lymphotropic virus-1
- IL-2/IL-2R
interleukin 2/interleukin 2 receptor
- JAK
Janus kinase
- JAK3i
Janus kinase 3 inhibitor
- PBMCs
peripheral blood mononuclear cells
- pSTAT3
phospho-STAT3
- STAT signal transducers
activators of transcription
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests:
None. The author does not have any actual or potential conflicts of interest.
References
- Azimi N, Brown K, Bamford RN, Tagaya Y, Siebenlist U, Waldmann TA. Human T cell lymphotropic virus type I Tax protein trans-activates interleukin 15 gene transcription through an NF-kappa B site. Pro Natl Acad Sci USA. 1998;95(5):2452–2457. doi: 10.1073/pnas.95.5.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellanger D, Jacquemin V, Chopin M, Pierron G, Bernard OA, et al. Recurrent JAK1 and JAK3 somatic mutations in T-cell prolymphocytic leukemia. Leukemia. 2014;28(2):417–419. doi: 10.1038/leu.2013.271. [DOI] [PubMed] [Google Scholar]
- Bergmann AK, Schneppenheim S, Seifert M, Betts MJ, Haake A, et al. Recurrent Mutation of JAK3 in T-Cell Prolymphocytic Leukemia. Genes Chromosomes & Cancer. 2014;53(4):309–316. doi: 10.1002/gcc.22141. [DOI] [PubMed] [Google Scholar]
- Bouchekioua A, Scourzic L, de Wever O, Zyang Y, Cervera P, et al. JAK3 deregulation by activating mutations confers invasive growth advantage in extranodal nasal-type natural killer cell lymphoma. Leukemia. 2014;28(2):338–348. doi: 10.1038/leu.2013.157. [DOI] [PubMed] [Google Scholar]
- Chen J, Petrus M, Bryant BR, Nguyen VP, Stamer M, et al. Induction of the IL-9 gene by HTLV-1 Tax stimulates the spontaneous proliferation of primary adult T-cell leukemia cells by a paracrine mechanism. Blood. 2008;111(10):5163–5172. doi: 10.1182/blood-2007-09-113654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Petrus M, Bryant BR, Nguyen VP, Goldman CK, et al. Autocrine/paracrine cytokine stimulation of leukemic cell proliferation in smoldering and chronic adult T-cell leukemia. Blood. 2010;116(26):5948–5956. doi: 10.1182/blood-2010-04-277418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Simmons WJ, Cai HY, Dhall G, Zamo A, et al. Stat3 is required for ALKmediated lymphomagenesis and provides a possible therapeutic target. Nat Med. 2005;11(6):623–629. doi: 10.1038/nm1249. [DOI] [PubMed] [Google Scholar]
- Choi J, Goh G, Walradt T, Hong BS, Bunick CG, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015;47(9):1011–19. doi: 10.1038/ng.3356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon KC, Waldmann TA. Ruxolitinib for adult T-cell leukemia. Clin. Study Rec. NCT01712659, updated Sept. 28. Natl Inst Health, NIH. 2017 https://clinicaltrials.gov/ct2/show/NCT01712659.
- Coppo P, Gouilleux-Gruart V, Huang Y, Bouhlal H, Bouamar H, et al. STAT3 transcription factor is constitutively activated and is oncogenic in nasal-type NK/T-cell lymphoma. Leukemia. 2009;23(9):1667–1678. doi: 10.1038/leu.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crescenzo R, Abate F, Lasorsa E, Tabbo F, Gaudiano M, et al. Convergent Mutations and Kinase Fusions Lead to Oncogenic STAT3 Activation in Anaplastic Large Cell Lymphoma. Cancer Cell. 2015;27(4):516–532. doi: 10.1016/j.ccell.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döbbeling U, Dummer R, Laine E, Potoczna N, Gin JZ, Burg G. Interleukin-15 is an autocrine/paracrine viability factor for cutaneous T-cell lymphoma cells. Blood. 1998;92(1):252–258. [PubMed] [Google Scholar]
- Elliott NE, Cleveland SM, Grann V, Janik J, Waldmann TA, Dave UP. FERM domain mutations induce gain of function in JAK3 in adult T-cell leukemia/lymphoma. Blood. 2011;118(14):3911–3921. doi: 10.1182/blood-2010-12-319467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen KW, Kaltoft K, Mikkelsen G, Nielsen M, Zhang Q, et al. Constitutive STAT3-activation in Sezary syndrome: tyrphostin AG490 inhibits STAT3-activation, interleukin-2 receptor expression and growth of leukemic Sezary cells. Leukemia. 2001;15(5):787–793. doi: 10.1038/sj.leu.2402093. [DOI] [PubMed] [Google Scholar]
- Haan C, Rolvering C, Raulf F, Kapp M, Druckes P, et al. Jak1 Has a Dominant Role over Jak3 in Signal Transduction through gamma c-Containing Cytokine Receptors. Chem & Biol. 2011;18(3):314–323. doi: 10.1016/j.chembiol.2011.01.012. [DOI] [PubMed] [Google Scholar]
- Hornakova T, Staerk J, Royer Y, Flex E, Tartaglia M, et al. Acute Lymphoblastic Leukemia-associated JAK1 Mutants Activate the Janus Kinase/STAT Pathway via Interleukin-9 Receptor alpha Homodimers. J Biol Chem. 2009;284(11):6773–6781. doi: 10.1074/jbc.M807531200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Chen J, Nicolae A, Wang HS, Shin DM, et al. IL-21-driven neoplasms in SJL mice mimic some key features of human angioimmunoblastic T-cell lymphoma. Am J Pathol. 2015;185(11):3102–3114. doi: 10.1016/j.ajpath.2015.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju W, Zhang ML, Jiang JK, Thomas CJ, Oh U, et al. CP-690,550, a therapeutic agent, inhibits cytokine-mediated Jak3 activation and proliferation of T cells from patients with ATL and HAM/TSP. Blood. 2011;117(6):1938–1946. doi: 10.1182/blood-2010-09-305425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka K, Nagata Y, Kitanaka A, Shiraishi Y, Shimamura T, et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat Genet. 2015;47(11):1304–15. doi: 10.1038/ng.3415. [DOI] [PubMed] [Google Scholar]
- Khoury JD, Medeiros LJ, Rassidakis GZ, Yared MA, Tsioli P, et al. Differential expression and clinical significance of tyrosine-phosphorylated STAT3 in ALK(+) and ALK(−) anaplastic large cell lymphoma. Clin Cancer Res. 2003;9(10):3692–3699. [PubMed] [Google Scholar]
- Kiel MJ, Velusamy T, Rolland D, Sahasrabuddhe AA, Chung F, et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. 2014;124(9):1460–1472. doi: 10.1182/blood-2014-03-559542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo GC, Tan SY, Tang T, Poon SL, Allen GE, et al. Janus Kinase 3-Activating Mutations Identified in Natural/Killer/T-cell Lymphoma. Cancer Discov. 2012;2(7):591–597. doi: 10.1158/2159-8290.CD-12-0028. [DOI] [PubMed] [Google Scholar]
- Koskela HLM, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmaki H, et al. Somatic STAT3 Mutations in Large Granular Lymphocytic Leukemia. N Engl J Med. 2012;366(20):905–1913. doi: 10.1056/NEJMoa1114885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kücük C, Jiang B, Hu XZ, Zhang WY, Chan JKC, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from gamma delta-T or NK cells. Nat Commun. 2015;6 doi: 10.1038/ncomms7025. article number 6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard WJ, O’Shea JJ. JAKS AND STATS: Biological implications. Annu Rev Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- Lu XH, Levine R, Tong W, Wernig G, Pikman Y, et al. Expression of a homodimeric type I cytokine receptor is required for JAK2V617F-mediated transformation. Proc Natl Acad Sci USA. 2005;102(52):18962–18967. doi: 10.1073/pnas.0509714102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migone TS, Lin JX, Cereseto A, Mulloy JC, O’Shea JJ, et al. Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science. 1995;269(5220):79–81. doi: 10.1126/science.7604283. [DOI] [PubMed] [Google Scholar]
- Nicolae A, Xi L, Pittaluga S, Abdullaev Z, Pack SD, et al. Frequent STAT5B mutations in gamma delta hepatosplenic T-cell lymphomas. Leukemia. 2014;28(11):2244–2248. doi: 10.1038/leu.2014.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea JJ, Notarangelo LD, Johnston JA, Candotti F. Advances in the understanding of cytokine signal transduction: The role of Jaks and STATs in immunoregulation and the pathogenesis of immunodeficiency. J Clin Immunol. 1997;17(6):431–447. doi: 10.1023/A:1027388508570. [DOI] [PubMed] [Google Scholar]
- Odejide O, Weigert O, Lane AA. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123(9):1293–1296. doi: 10.1182/blood-2013-10-531509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by gamma(c) family cytokines. Nat Rev Immunol. 2009;9(7):480–490. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem. 2002;277(49):47954–47963. doi: 10.1074/jbc.M205156200. [DOI] [PubMed] [Google Scholar]
- Shochat C, Tal N, Bandapalli OR, Palmi C, Ganmore I, et al. Gain-of-function mutations in interleukin-7 receptor-alpha (IL7R) in childhood acute lymphoblastic leukemias. J Expl Med. 2011;208(6):1333–1333. doi: 10.1084/jem.201105802011512c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GA, Uchida K, Weiss A, Taunton J. Essential biphasic role for JAK3 catalytic activity in IL-2 receptor signaling. Nat Chem Bio. 2016;12(5):373–79. doi: 10.1038/NCHEMBIO.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tendler CL, Greenberg SJ, Blattner WA, Manns A, Murphy E, et al. Transactivation of interleukin-2 and its receptor induces immune activation in human T-cell lymphotropic virus type I-associated myelopathy: pathogenic implications and a rationale for immunotherapy. Proc Natl Acad Sci USA. 1990;87(13):5218–5222. doi: 10.1073/pnas.87.13.5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. 2006;6(8):595–601. doi: 10.1038/nri1901. [DOI] [PubMed] [Google Scholar]
- Waldmann TA, Chen J. Disorders of the JAK/STAT pathway in T cell lymphoma pathogenesis: implications for immunotherapy. Annu Rev Immunol. 2017;35:533–550. doi: 10.1146/annurev-immunol-110416-120628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenatti PP, Ribeiro D, Li WQ, Zuurbier L, Silva MC, et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat Genet. 2011;43(10):932–U31. doi: 10.1038/ng.924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Nowak I, Vonderheid EC, Rook AH, Kadin ME, et al. Activation of Jak/STAT proteins involved in signal transduction pathway mediated by receptor for interleukin 2 in malignant T lymphocytes derived from cutaneous anaplastic large T-cell lymphoma and Sezary syndrome. Pro Natl Acad Sci USA. 1996;93(17):9148–9153. doi: 10.1073/pnas.93.17.9148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Ding L, Holmfeldt L, Wu G, Heatley SL, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ML, Griner LAM, Ju W, Duveau DY, Guha R, et al. Selective targeting of JAK/STAT signaling is potentiated by Bcl-xL blockade in IL-2-dependent adult T-cell leukemia. Pro Natl Acad Sci USA. 2015;112(40):12480–12485. doi: 10.1073/pnas.1516208112. [DOI] [PMC free article] [PubMed] [Google Scholar]