

Sir,
Calpainopathy belongs to a group of limb-girdle muscular dystrophies (LGMD), characterized by proximal muscle weakness. Calpainopathy or LGMD2A is an autosomal recessive disease caused by mutations in the calpain-3 gene (CAPN3).[1]
We report the case of a 16-year-old male patient with a myopathy with proximal muscle weakness. He started tiptoeing up, at the age of 4. At 13-year-old, he began to have difficulty climbing stairs, incorporating from the ground and running, being referred to the neurologist. Physical examination showed muscle weakness of shoulder and pelvic girdle, positive Gower's sign, lumbar lordosis, and joint contractions.
CK was elevated (7341 UI/L). Electromyography showed myopathic pattern, muscle biopsy, a dystrophic pattern, and the immunohistochemical analysis showed a lower intensity of staining with anti-DYS-3 antibody (Dp427m-dystrophin protein) [Figure 1a].
Figure 1.
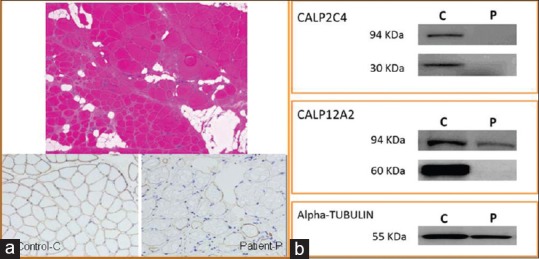
(a) Top: Hematoxylin and eosin staining: Dystrophic pattern. Bottom: DYS-3 antibody: Left, control (c) and right patient (p). The patient shows a loss of expression in some fibers. (b) Western blot: Abnormal calpain-3 protein expression. CALP2C4 antibody: absence of 94 and 30 KDa protein bands in the patient (p) relative to control. (c) CALP-12A2 antibody: No significant differences in the 94KDa protein in the P relative to C, no band observed in the 60KDa protein in P
These results focused the diagnosis on the Becker muscular dystrophy (BMD), and treatment with corticosteroids was indicated. He was operated for bilateral tendon lengthening.
A multiplex ligation-dependent probe amplification (MLPA) test was carried out to detect duplications and deletions in the dystrophin gene (DMD). MLPA results for DMD gene were normal. The study continued by next-generation sequencing (NGS) analysis, using a commercial panel of a clinical exome (TruSight One of Illumina®). No point mutations in the DMD gene were found and 23 genes associated with LGMD and Emery-Dreifuss muscular dystrophy were analyzed: TRIM32/DES/SGCB/FKTN/CAV3/FKRP/SGCG/SGCD/DNAJB6/SGCA/CAPN3/TTN/ANO5/DYSF/PLEC/DAG1/EMD/LMNA/MYOT/TCAP/POMGNT1/POMT1/POMT2. The patient had two mutations in heterozygosity in the CAPN3 gene: c. 550delA; p. Thr184Argfs*36 and c. 3261_3262delAGinsTCATCT; p. Arg788Serfs*14 (NM_000070). Sanger sequencing confirmed the mutations detected in the patient and identified the mutations in the father and the mother, respectively.
An abnormal calpain-3 protein expression was demonstrated in the skeletal muscle biopsy of the patient by Western blot [Figure 1b], and the diagnostic of calpainopathy was confirmed. Both mutations in the CAPN3 gene cause a change in the reading frame (frameshift mutation), leading to a premature stop codon and calpain-3 abnormal protein. They have been described previously as LGMD2A pathogenic in several studies.[2,3]
The wide variety of muscular dystrophies and common clinical manifestations makes necessary the immunohistochemical studies, immunoblotting, and molecular genetics to reach the definitive diagnosis of these pathologies. We report a case with a diagnostic suspicion of BMD, where the incorporation of NGS has been essential for the diagnosis of calpainopathy. NGS enables the screening of many genes at once and was chosen since the classical Sanger method is laborious and time-spending. Mutation identification is the necessary approach to an upcoming gene therapy.[4]
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We wish to thank Ruben de Sancho and Amparo García Cardenal for their technical contribution in carrying out the experiments and Rocio Mena and Maria Victoria Gomez for CAPN3 gene sequencing.
REFERENCES
- 1.Richard I, Roudaut C, Saenz A, Pogue R, Grimbergen JE, Anderson LV, et al. Calpainopathy-a survey of mutations and polymorphisms. Am J Hum Genet. 1999;64:1524–40. doi: 10.1086/302426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fanin M, Nascimbeni AC, Fulizio L, Angelini C. The frequency of limb girdle muscular dystrophy 2A in northeastern Italy. Neuromuscul Disord. 2005;15:218–24. doi: 10.1016/j.nmd.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 3.Urtasun M, Sáenz A, Roudaut C, Poza JJ, Urtizberea JA, Cobo AM, et al. Limb-girdle muscular dystrophy in Guipúzcoa (Basque Country, Spain) Brain. 1998;121(Pt 9):1735–47. doi: 10.1093/brain/121.9.1735. [DOI] [PubMed] [Google Scholar]
- 4.Bartoli M, Roudaut C, Martin S, Fougerousse F, Suel L, Poupiot J, et al. Safety and efficacy of AAV-mediated calpain 3 gene transfer in a mouse model of limb-girdle muscular dystrophy type 2A. Mol Ther. 2006;13:250–9. doi: 10.1016/j.ymthe.2005.09.017. [DOI] [PubMed] [Google Scholar]