

Genomic mosaicism and novel divergence have facilitated the creation and maintenance of a hybrid species, the Italian sparrow.
Keywords: hybrid speciation, genome scan, Passer sparrow, Genome evolution, selection, immune system, reproductive isolation
Abstract
Hybridization is widespread in nature and, in some instances, can result in the formation of a new hybrid species. We investigate the genetic foundation of this poorly understood process through whole-genome analysis of the hybrid Italian sparrow and its progenitors. We find overall balanced yet heterogeneous levels of contribution from each parent species throughout the hybrid genome and identify areas of novel divergence in the hybrid species exhibiting signals consistent with balancing selection. High-divergence areas are disproportionately located on the Z chromosome and overrepresented in gene networks relating to key traits separating the focal species, which are likely involved in reproductive barriers and/or species-specific adaptations. Of special interest are genes and functional groups known to affect body patterning, beak morphology, and the immune system, which are important features of diversification and fitness. We show that a combination of mosaic parental inheritance and novel divergence within the hybrid lineage has facilitated the origin and maintenance of an avian hybrid species.
INTRODUCTION
Hybridization is increasingly recognized as a potentially creative force contributing to adaptation and species diversification (1, 2). New species may arise as a direct consequence of interbreeding between diverged taxa, in which the hybrid lineage comprises a recombinant genome with the same ploidy level and is reproductively isolated from its parent species (3, 4). This process—known as homoploid hybrid speciation—may take many forms with respect to genomic makeup, ranging from introgression of a single or few genes into a foreign genomic background (5) to balanced genomic contributions from both parent lineages (6, 7). At both ends of the spectrum, novel allelic combinations and subsequent evolution in the hybrid lineage may facilitate its escape from inferior fitness and aid in the evolution of reproductive barriers toward both parents, which is essential for maintaining its isolation. Characterizing these combinations and their genomic distribution is therefore of paramount importance for understanding the hybrid speciation process and enriching our knowledge of the role of hybridization in shaping biodiversity. However, there are few well-documented cases of homoploid hybrid species in nature, and, consequently, the genomics of this mode of speciation are poorly understood.
The Italian sparrow (Passer italiae) is a homoploid hybrid species found in mainland Italy and a few Mediterranean islands (Fig. 1A) that has arisen from hybridization between the house sparrow (Passer domesticus) and the Spanish sparrow (Passer hispaniolensis). Although ecologically more similar to the house sparrow, male Italian sparrows have plumage patterns that comprise a mosaic of male Spanish and house sparrow traits (Fig. 1A) (8). The taxonomic status of the Italian sparrow has been subject to much debate, and several hypotheses have previously been proposed for its evolutionary relationship to the other Passer sparrows (8, 9). Its intermediate appearance, in particular, led to the proposition that it is of hybrid origin (10) [see also Anderson (8)]. Only recently have genetic studies given support for this idea, demonstrating the Italian sparrow’s genetic and phenotypic mosaicism (11–13). A proposed scenario for its origin is that the Italian sparrow arose <10,000 years ago as the human commensal house sparrow expanded throughout Europe where it encountered and hybridized with the Spanish sparrow (12).
Fig. 1. Population structuring and species information.
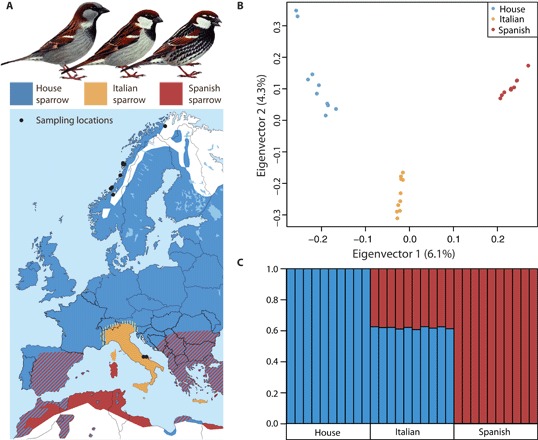
(A) Top: Illustrations of male plumage patterns in house, Italian, and Spanish sparrows modified from Svensson et al. (83). Bottom: A distribution map of house, Italian, and Spanish sparrows throughout Europe and northern Africa (9). (B) PCA of the LD-pruned high-quality SNP set. (C) Population structuring based on admixture analysis for house, Italian, and Spanish populations.
The system is unique in that the hybrid Italian sparrow remains in geographic contact with both its parent species. To the north of its range, the Italian sparrow meets the house sparrow in a narrow hybrid zone in proximity to the Alps. In addition, it lives in sympatry with the Spanish sparrow on the southeastern Italian peninsula of Gargano, with little evidence of gene flow (13). The lack of introgression in Gargano may be due to premating barriers such as habitat differentiation and timing of breeding, or to postzygotic isolating factors. Evidence for postzygotic reproductive barriers has been shown in areas of contact (13). Furthermore, hybridization is likely to have directly contributed to the development of reproductive barriers between the Italian sparrow and its parents as preexisting parental incompatibility alleles are sorted within the Italian sparrow lineage (14).
The Italian sparrow’s hybrid ancestry has thus far been characterized by a limited set of genetic markers, and the genomic architecture and regions underlying reproductive barriers in the system have yet to be identified. Here, we investigate the genetic composition of an avian hybrid species by using a whole-genome analysis of the Italian sparrow and its parent species, the house sparrow and the Spanish sparrow. Genome data provide powerful prospects for understanding hybrid speciation. Although conflicting signatures of ancestry represent a hallmark of hybrid speciation, the traces of hybridization may be obscured by processes such as backcrossing to either parent lineage, sorting of ancestral polymorphisms, genetic drift, and selection. Hence, the degree of admixture may vary widely throughout the hybrid genome (15, 16), and studying only a subset of the genome can give confounding results. Whole-genome data also offer the alluring potential for elucidating candidate regions responsible for the formation and maintenance of the hybrid species.
With the aid of a high-quality de novo reference genome created for the house sparrow, we mapped and analyzed genome data from key populations of the three focal taxa. Population genetic parameters, admixture analyses, and phylogenetic inference were used to characterize the genetic composition throughout the hybrid genome in relation to its parents. This approach also allowed us to identify areas in which the Italian sparrow segregates for alternative parental alleles and genes where the hybrid differs from both parents, which may play instrumental roles in the reproductive barriers involved in the hybrid system.
RESULTS
House sparrow reference genome
We created a high-quality de novo reference genome by sequencing and assembling the genome of a house sparrow (~130× coverage; table S1). The final assembly encompasses 1.04 Gb (gigabases), and with the aid of a medium-density linkage map, we ordered and oriented 88% of the assembly scaffolds into chromosomes, resulting in an N50 sequence size of 68.7 Mb (megabases) (table S2). We then sequenced whole genomes of 10 males of each of the focal species and one tree sparrow (Passer montanus; outgroup) at ~10× coverage per individual (table S3) and mapped them to the reference genome for downstream analysis.
Population structuring
Population genetic parameter estimates reveal genome-wide intermediacy in the Italian sparrow compared to its parents, although it is overall closer to the house sparrow (Table 1). The global average of differentiation (FST) was, as expected under a hybrid speciation model, higher between the parents, the house sparrow and the Spanish sparrow (hereinafter HS; FST = 0.33), compared to differentiation between the Italian sparrow and either the house sparrow (HI; FST = 0.18) or the Spanish sparrow (SI; FST = 0.25).
Table 1. Mean values of population genomic statistics for 100-kb sliding windows with 25-kb steps across the genome.
df, density of fixed differences.
| Parameter | Species | Autosomes | Z chromosome | Whole genome |
| FST | HS | 0.32 ± 0.10 | 0.45 ± 0.09 | 0.33 ± 0.10 |
| HI | 0.17 ± 0.07 | 0.29 ± 0.16 | 0.18 ± 0.08 | |
| SI | 0.23 ± 0.12 | 0.41 ± 0.17 | 0.25 ± 0.14 | |
| df (10−3) | HS | 0.048 ± 0.28 | 1.440 ± 1.90 | 0.146 ± 0.67 |
| HI | 0.003 ± 0.01 | 0.005 ± 0.01 | 0.003 ± 0.01 | |
| SI | 0.003 ± 0.02 | 0.211 ± 0.70 | 0.018 ± 0.19 | |
| π | House | 0.0073 ± 0.0021 | 0.0042 ± 0.0025 | 0.0071 ± 0.0023 |
| Italian | 0.0070 ± 0.0021 | 0.0048 ± 0.0025 | 0.0068 ± 0.0023 | |
| Spanish | 0.0051 ± 0.0022 | 0.0025 ± 0.0023 | 0.0049 ± 0.0023 | |
| Θ* | House | 0.0075 ± 0.002 | 0.0043 ± 0.002 | 0.0073 ± 0.002 |
| Italian | 0.0074 ± 0.002 | 0.0046 ± 0.002 | 0.0072 ± 0.002 | |
| Spanish | 0.0055 ± 0.002 | 0.0027 ± 0.002 | 0.0053 ± 0.002 |
*Watterson estimator of Θ based on the number of segregating sites.
A principal components analysis (PCA) of high-quality single-nucleotide polymorphisms (SNPs), obtained by variant calling the three focal taxa against the house sparrow reference genome and pruning for linkage disequilibrium (LD), demonstrated striking structuring between the three focal taxa. The house and Spanish sparrows separate along the first eigenvector with the Italian sparrow positioned in between with no overlap among the clusters (Fig. 1B). Furthermore, separation along eigenvector 2 indicates novel differentiation of the Italian sparrow against both parents, which may be due to extensive segregation of alternative alleles from both parents in the hybrid, differences from both parents due to selection, and/or de novo mutations in the hybrid lineage subsequent to its formation.
Parental contributions to the hybrid lineage
Whole-genome admixture analysis revealed that the Italian sparrows, on average, assign 61.9% to the house sparrow ancestry and 38.1% to the Spanish sparrow ancestry (Fig. 1C). In addition, admixture analysis across the Italian sparrow genome [100-kb (kilobase) nonoverlapping windows] revealed a large variation (σ = 19% combined for all windows) in the assignment probability to either parent (Fig. 2 and fig. S1).
Fig. 2. Phylogenetic inference and admixture analysis of the Italian sparrow.

(A) ADMIXTURE analysis of 100-kb nonoverlapping windows across the Italian sparrows’ chromosomes 1A and Z, with the house sparrow ancestry shown in blue and Spanish sparrow ancestry shown in red. (B) RAxML tree assignment results for 100-kb nonoverlapping windows across the genome for chromosomes 1A and Z. Windows depict whether the Italian sparrows grouped monophyletically with house sparrows (blue), Spanish sparrows (red), in its own clade (green), or were unresolved (white).
Maximum likelihood phylogenies created using RAxML (17) demonstrated a discordant evolutionary history throughout the hybrid genome. Individual trees were created for 100-kb nonoverlapping windows and classified according to whether the Italian sparrows grouped monophyletically with the house sparrow, Spanish sparrow, or in its own clade. The vast majority of trees (76%) remained unresolved, that is, the Italian sparrows did not form a monophyletic group alone or with either parent. This is expected because any window may harbor alleles derived from both parents as well as novel mutations in the hybrid lineage. Nonetheless, in 14% of the genomic windows, Italian sparrows grouped with house sparrows, in 9% with Spanish sparrows, and in less than 1%, they formed their own clade (Fig. 2, fig. S1, and table S4).
A 100-kb window size was chosen for sliding window analyses because LD decays within this distance (fig. S2). To test whether a smaller window size improved the proportion of resolved phylogenies by limiting the number of windows where the Italian sparrows segregate for haplotype blocks from both parents, RAxML analyses were also performed for 50- and 10-kb window sizes. Despite the reduction in window size, the proportion of resolved phylogenies remained largely similar to that of the 100-kb window analyses (table S5), and the latter was therefore kept for downstream analysis.
Incomplete lineage sorting can leave similar genomic footprints as those of hybridization. However, we find significant introgression between both parents and the Italian sparrow on the whole-genome level, as well as when the autosomes and Z chromosome are considered individually (Table 2). When ABBA BABA tests are performed in sliding windows across the genome, the fd estimator (18) showed very similar admixture proportions between the Italian sparrow and either parent, with more gene flow between the Italian and Spanish sparrows on autosomes and more introgression between the house and Italian sparrows on the Z chromosome (Table 3). In addition, the SDs of fd estimates were large, lending further support to the Italian sparrow’s mosaic composition of alternating genomic ancestry from either parent, as demonstrated in the admixture and RAxML analyses.
Table 2. Results from Patterson’s D test for introgression between lineages with block jack-knifed SE estimates and significance values.
| P1 | P2 | P3 | Patterson’s D | Jack-knifed SE | Z score | P | |
| Autosomes | House | Italian | Spanish | 0.195 | 0.001 | 27.77 | <0.00001 |
| Spanish | Italian | House | 0.035 | <0.001 | 4.98 | <0.00001 | |
| Z | House | Italian | Spanish | 0.552 | 0.004 | 8.34 | <0.00001 |
| Spanish | Italian | House | 0.575 | 0.006 | 7.62 | <0.00001 | |
| Whole genome | House | Italian | Spanish | 0.211 | <0.001 | 24.23 | <0.00001 |
| Spanish | Italian | House | 0.065 | <0.001 | 5.39 | <0.00001 |
Table 3. Mean fd values for 100-kb sliding windows with 25-kb steps across the genome.
| P1 | P2 | P3 | fd (%) Autosomes |
fd (%) Z chromosome |
fd (%) Whole genome |
| House | Spanish | Italian | 31.7 ± 15.4 | 40.3 ± 23.7 | 32.1 ± 16.0 |
| Spanish | House | Italian | 27.5 ± 21.7 | 48.7 ± 27.8 | 32.4 ± 24.9 |
To investigate the composition of mitochondrial DNA (mtDNA) in the Italian sparrow, we created a haplotype network and ran a fastSTRUCTURE (19) analysis of complete mtDNA sequences. Although the Italian sparrow has been shown to be nearly fixed for house sparrow mtDNA (12), surprisingly, two Italian sparrows demonstrate evidence of admixture from both parental mitochondrial haplotypes (Fig. 3, A and C). Further, these “admixed” individuals display intermediate estimates of sequence divergence (dXY) from the parent species along the mitochondria (Fig. 3B and fig. S3). To test for heterozygosity in mtDNA sequences, we also variant-called each individual’s mtDNA as diploid and used VCFtools (20) to calculate per individual inbreeding coefficient (F) estimates. VCFtools calculates F on the basis of the number of observed homozygotes, number of sequenced sites, and the expected number of homozygotes under the Hardy-Weinberg equilibrium. F estimates for the admixed individuals indicated an excess of heterozygosity (F = −0.25 and −0.36) compared to the other Italian individuals, which were largely homozygous (average, F = 0.67). Furthermore, the admixed individuals do not differ from the other Italian sparrows in sequence coverage (Mann-Whitney U test, P = 0.4) nor in their nuclear sequences (Mann-Whitney U test on nuclear F estimates, P = 0.09; Fig. 1A). Thus, there is no evidence for contamination of the samples. Together, all the results from the mitochondrial analyses suggest the presence of heteroplasmy, in which individuals retain mtDNA haplotypes from both parental species.
Fig. 3. Mitochondrial ancestry in the Italian sparrow.
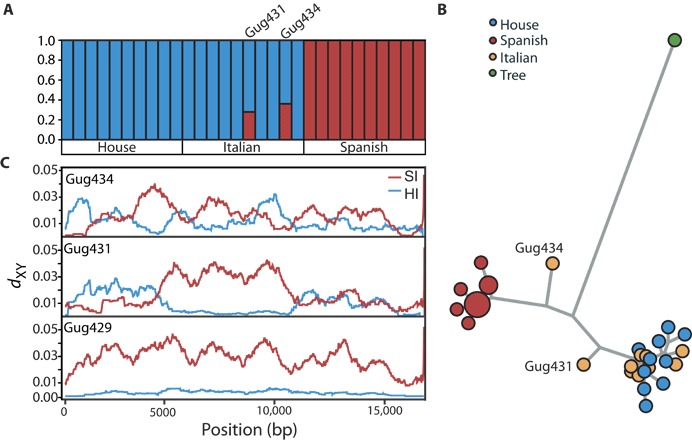
(A) fastSTRUCTURE analysis of complete mitochondrial sequences for the house, Italian, and Spanish sparrow individuals. (B) Mitochondrial haplotype network of all sparrow individuals. (C) Plots of sliding window (1-kb window and 100-bp step) sequence divergence (dXY) along the mitochondria between the house and Italian (blue) and Spanish and Italian (red) sparrows for three Italian individuals. The top and middle panels depict the mixed mitochondrial ancestry of two Italian individuals, whereas the bottom panel shows the sole house sparrow ancestry of a single Italian sparrow mitochondria that is representable for the eight remaining Italian sparrows (see fig. S3).
Signatures of selection
Genomic regions exhibiting high interspecific divergence may have been targets of species-specific selection and are often considered candidates for barriers to gene flow. For hybrid systems, this extrapolation is not straightforward because the hybrid lineage inherits evolutionary histories from two sources, thereby distorting such signals. We used disparities in FST values between lineages to identify genomic regions where the Italian sparrow displays elevated divergence from either or both of its parents. For each comparison between the Italian sparrow and either parent, we selected the top 1% 100-kb windows where the Italian sparrow showed the largest difference in FST values between one parent and the other (Eqs. 1 and 2; Fig. 4A and fig. S4)
Fig. 4. Divergence landscape and selection tests for the Passer taxa.
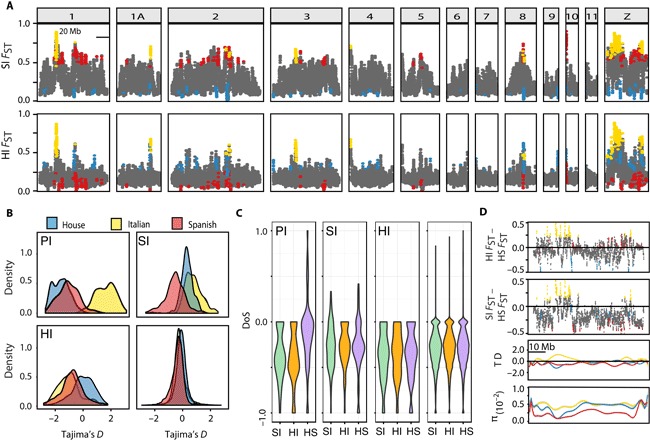
(A) FST estimates for 100-kb overlapping windows with 25-kb steps across the largest chromosomes in the genome. Windows are identified as HI divergence peaks in blue, SI in red, and PI in yellow. Microchromosomes are plotted in fig. S4. (B) Density plots of TD estimates for PI, SI, and HI outlier windows, as well as all nonoutlier windows (bottom right) for each focal species. (C) Density of DoS values within PI, SI, and HI windows and the genomic background for the three species comparisons. (D) Plots of disparities in FST values between HI/parent species (top) and SI/parent species (second from top) for all genomic windows on the Z chromosome, highlighting windows in PI peaks (yellow), SI peaks (red), and HI peaks (blue). The two bottom panels depict smoothed plots of TD and π in all 100-kb overlapping genomic windows along the Z chromosome for each focal sparrow species (house, blue; Spanish, red; Italian, yellow).
(1) House versus Italian sparrow divergence (HI) = HI FST − SI FST
(2) Spanish versus Italian sparrow divergence (SI) = SI FST − HI FST.
Concern has been raised that relative measures of divergence, such as FST, could be elevated because of reduced intraspecific variation from processes unrelated to speciation, such as background selection in low-recombining regions (21–23). Our three-taxa system enables us to account for this confounding factor. If a high FST value is driven by low nucleotide diversity due to reduced variation in the ancestral population or regions of low recombination, then the FST values would be expected to be elevated in all species comparisons. However, if one parent/hybrid comparison has a high FST, whereas the other has low FST, then this indicates a differentiated region separating the hybrid and the former parent that is not attributed to low intraspecific nucleotide diversity from background selection. Similarly, regions of higher divergence between the Italian sparrow and both parents, compared to the divergence between the parents, indicate potential regions privately isolating the Italian sparrow from both its parents. An advantage of using FST estimates in this system is that it is sensitive to recent selection events, allowing for the identification of areas of hybrid-specific evolution. Regions of novel divergence in the Italian sparrow—that is, windows in which the Italian sparrow displays high divergence against both parents (hereafter referred to as PI)—were targeted using a similar method as the HI and SI windows. For both of the hybrid/parent comparisons, parental divergence in the same window was subtracted from the Italian sparrow divergence against the respective parent. The 1% windows exhibiting the highest FST disparities, common to both hybrid/parent comparisons, were kept as PI windows (Eq. 3; Fig. 4D and fig. S4).
(3) Parents versus Italian sparrow divergence (PI) = SI FST − HS FST ∪ HI FST − HS FST.
Overall, we find higher FST values, lower nucleotide diversity, more extreme Tajima’s D (TD) values, and elevated levels of LD within the outlier windows relative to the nonoutlier windows (Fig. 4 and Table 4). These patterns bear signatures of selection, suggesting that the outlier windows may harbor or are located adjacent to genes with a role in species divergence. Particularly striking are the strongly positive TD values in windows of novel divergence in the Italian sparrow, whereas both parents exhibit negative TD values in the corresponding windows (Fig. 4B). High TD values result from an excess of medium frequency alleles, consistent with balancing selection or a population bottleneck, whereas low values indicate an excess of low-frequency polymorphisms following a selective sweep or population expansion (24). These contrasting values suggest that the Italian sparrow segregates for alleles that have undergone selection in the parents and subsequently have been subject to balancing selection within the hybrid lineage. Because the nonoutlier windows exhibit an overall negative tendency in TD, indicating a genome-wide demographic signal of expansion and/or background selection in the three species, the strongly positive TD density distribution in PI divergence windows is particularly compelling.
Table 4. Average population genomic statistics for the high-divergence windows and the genomic background.
| Parameter | Species | PI | HI | SI | Background |
| FST | Parents | 0.380 | 0.468 | 0.504 | 0.329 |
| HI | 0.651 | 0.389 | 0.109 | 0.178 | |
| SI | 0.703 | 0.125 | 0.574 | 0.241 | |
| TD | House | −1.516 | 0.010 | 0.292 | −0.132 |
| Italian | 1.625 | −1.007 | 0.750 | −0.206 | |
| Spanish | −0.979 | −0.785 | −0.576 | −0.374 | |
| π | House | 0.0014 | 0.0046 | 0.0057 | 0.0072 |
| Italian | 0.0038 | 0.0030 | 0.0057 | 0.0069 | |
| Spanish | 0.0008 | 0.0023 | 0.0021 | 0.0050 | |
| r2* | House | 0.4415 | 0.4901 | 0.4427 | 0.3155 |
| Italian | 0.5527 | 0.4309 | 0.3957 | 0.2658 | |
| Spanish | 0.3071 | 0.3896 | 0.3395 | 0.2736 | |
| Θ† | House | 0.0021 | 0.0046 | 0.0054 | 0.0073 |
| Italian | 0.0029 | 0.0037 | 0.0050 | 0.0073 | |
| Spanish | 0.0010 | 0.0027 | 0.0024 | 0.0054 |
*Mean estimates of LD in the form of pairwise r2 estimates for all SNPs within 1 kb of each other in the specified genomic windows.
†Watterson’s estimator of Θ based on the number of segregating sites.
The direction of selection (DoS) statistic was also estimated for all 100-kb genomic windows to test for selection in protein-coding sequence between the three species comparisons. The DoS statistic is conceptually similar to the McDonald-Kreitman test and uses protein-coding SNPs to measure the direction and extent of selection on the basis of nonsynonymous and synonymous fixed differences and polymorphisms between lineages (25). DoS estimates revealed more extreme signals of selection within the outlier windows compared to the genomic background. Furthermore, the extent and DoS differ between the species comparisons within the outlier windows, whereas genomic background values are very similar between the species comparisons. Stark differences were revealed in the DoS statistic between the parents and the HI and SI comparisons in the PI regions (Fig. 4C). Although the parents exhibit the full range of possible DoS values with most of the genes near neutrality, both HI and SI comparisons are largely negative, indicating an excess of nonsynonymous polymorphisms. This is expected under balancing or weak purifying selection (26). Together, the excess of nonsynonymous polymorphism and high TD values suggest the presence of balancing selection in the PI windows. Parental differentiation is, on average, only slightly higher than the genomic background in PI windows (Table 4); however, there is a higher density of genes under positive divergent selection in the parents within these windows compared to all other genomic regions (Fig. 4C). These DoS patterns may be driven by low numbers of nonsynonymous fixed sites that do not necessarily elevate the FST values across the entire genomic window between the parent taxa. Hence, it appears that some genes within PI windows are under divergent selection in the parents.
Within the SI windows, the TD value distributions are shifted to the left in the Spanish sparrow and to the right in the house sparrow and Italian sparrow (Fig. 4B). DoS estimates are variable in the SI windows but reveal a higher density of genes with an excess of nonsynonymous fixed differences between the Spanish and Italian sparrows compared to all other genomic regions (Fig. 4C), indicating that some genes are under positive divergent selection. Similarly, there is an excess of nonsynonymous fixed differences between the parent taxa within the SI windows. This suggests that these regions, in which the Italian sparrow may be assumed to have inherited from the house sparrow, harbor genes under divergent selection in the parent taxa. Moreover, the TD values indicate that this selection has mainly occurred in the Spanish sparrow lineage.
The HI windows exhibit strongly negative TD values in the Spanish and Italian sparrows, with more neutral values in the house sparrow (Fig. 4B). Although the strongly negative values could be driven by selective sweeps or purifying selection, DoS estimates indicate that there are no genes with an excess of nonsynonymous fixed differences in any of the species comparisons (Fig. 4C). This suggests that the high divergence within HI windows is attributed to the Italian sparrow’s inheritance of regions, which are under background selection or have undergone a selective sweep in the Spanish sparrow but are near neutrality in the house sparrow.
The Z chromosome versus autosomes
For all comparisons, we find overall higher divergence, lower nucleotide diversity (Table 1), and a larger proportion of resolved phylogenies on the Z chromosome relative to autosomes (table S4, Fig. 2, and fig. S1). Contrasting patterns of divergence and polymorphisms are consistent with a faster rate of evolution and/or reduced gene flow on the Z chromosome. Furthermore, although we observe a mosaic pattern of parental inheritance across the Italian sparrow genome, this pattern is strikingly more pronounced on the Z chromosome with large genomic regions alternating in inheritance from one parent or the other (Fig. 2 and fig. S1) in a block-like fashion.
Among the outlier windows, there is a significant overrepresentation on the Z chromosome (SI, χ2 = 87.064, P < 0.0001; HI, χ2 = 79.794, P < 0.0001; PI, χ2 = 787.461, P < 0.0001). Because sex chromosomes have a lower effective population size than autosomes, it can be difficult to parse out signals of selection from increased rates of genetic drift. However, DoS estimates on all genes throughout the genome for the three species comparisons revealed more extreme estimates of selection on Z-linked genes compared to autosomes, as well as a lower density of genes evolving neutrally, although the density distributions were only significantly different in the HI and SI comparisons (Fig. 5, two-sided permutation test: HS, P = 0.0649; HI, P < 0.00001; SI, P = 0.0013).
Fig. 5. Signatures of selection on the Z chromosome versus autosomes.
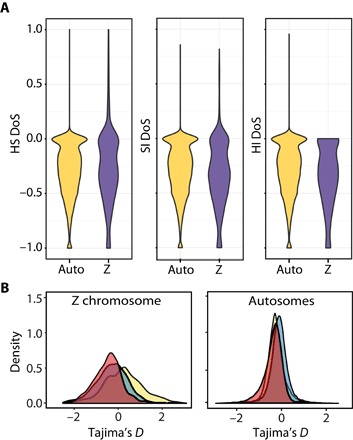
(A) Density of DoS values for all autosomal and Z-linked genes for the HS, SI, and SI species comparisons. (B) Density of TD values for all autosomal and Z-linked genes in the Spanish (red), house (blue), and Italian (yellow) sparrows.
Gene ontology analysis of high-divergence regions
Annotated genes residing within the outlier windows were extracted for ontology analyses, resulting in a total of 83 PI, 159 HI, and 76 SI outlier genes (table S6). Gene enrichment analysis revealed ontology classes significantly overrepresented for each of the three comparisons (Table 5 and tables S7 and S9), several of which have bearing on key traits separating the species in the system. For the PI genes, one of the significantly enriched classes was “dorsoventral pattern formation,” which encompasses a wide range of anatomical features, from body plan to color patterning. Plumage coloration constitutes a strong mating barrier in many bird species and is the most conspicuous phenotypic trait separating males of the focal species (27). Furthermore, the PI windows included four genes known to be associated with melanogenesis in vertebrates (table S10) (28), and a total of 13 such genes were identified in the other species comparisons, significantly more than expected by chance (one-sided permutation test; P < 0.044). Also enriched in PI windows were genes involved in the “negative regulation of immune response.”
Table 5. Significantly enriched functional groups within outlier windows.
| Window type | Functional group | Bonferroni step-down corrected P |
| HI | Amino acid transport | 9.0995 × 10−4 |
| HI | Negative regulation of G protein–coupled receptor protein signaling pathway | 1.6878 × 10−4 |
| HI | Positive regulation of translation | 0.0492 |
| HI | Regulation of guanosine triphosphatase activity | 1.5561 × 10−5 |
| HI | Regulation of autophagy | 0.0012 |
| HI | Regulation of muscle system process | 1.4758 × 10−5 |
| SI | Behavioral response to nicotine | 1.1522 × 10−4 |
| SI | Cell differentiation in the spinal cord | 0.0026 |
| SI | Cell differentiation involved in kidney development | 0.0016 |
| SI | Erythrocyte homeostasis | 0.0205 |
| SI | Palate development | 0.0201 |
| SI | Regulation of BMP signaling pathway | 0.0257 |
| SI | Regulation of mRNA processing | 0.0069 |
| SI | Regulation of organ growth | 0.0116 |
| PI | Cellular response to retinoic acid | 0.0042 |
| PI | Dorsal/ventral pattern formation | 0.0027 |
| PI | Negative regulation of innate immune response | 0.0081 |
| PI | Negative regulation of stress-activated mitogen-activated protein kinase cascade | 0.0027 |
| PI | Regulation of anion transmembrane transport | 0.0017 |
Two of the eight significantly enriched functional groups in SI divergence regions include “palate development” and “regulation of bone morphogenetic protein (BMP) signaling pathway” (Table 5). BMP proteins have been shown to play a key role in beak morphology and diversification among Darwin’s finches (29). Another SI gene, PTCH1, is a craniofacial signaling gene involved in adaptive variation in lower jaw shape in cichlids (30). These findings suggest that evolution of craniofacial structures has been an important component driving the Italian-Spanish divergence. The functional group with the lowest corrected P values and the highest number of assigned HI genes was the “regulation of G protein–coupled receptor signaling,” which has been shown to directly modify behavioral and morphological variation in birds (Table 5 and table S7) (31).
To test whether the significant gene ontologies found within the outlier regions are likely to be observed by chance when sampling windows from the genome, gene ontology (GO) permutations were also run. For each outlier window category (SI, PI, and HI), 50 permutations were performed by randomly sampling the same number of windows as the category being tested and extracting genes from the resulting windows. No overlap was found between the permutations and the outlier windows in significant GOs. Thus, we find that the outlier windows differ from the genomic background and that the significant GO groups are unlikely to be identified randomly.
Among the genes within outlier windows, 13 have been previously identified as candidate reproductive barrier genes between the focal species (table S10), a significant overrepresentation (one-sided permutation test; P < 0.0001). Two of these genes (RPS4 and HSDL2) exhibited steep genomic clines over the Italian sparrow’s range boundaries, indicating involvement in reproductive barriers against the parents (13). In addition, five PI genes (REEP5, A2M, A2ML1, APC, and MIA3) have been shown to exhibit steep clines within the Italian sparrow range (13), making them strong candidates for genes under selection in the hybrid species.
DISCUSSION
Genomic mosaicism in a hybrid species
We demonstrate extensive genomic admixture in an avian homoploid hybrid species, with significant contributions from both parent species. The genetic intermediacy of the Italian sparrow is evident through population structure analyses, estimates of population genetic parameters, and phylogenetic inference. Together with tests confirming introgression, our data suggest that hybridization has been the main process behind the evolution of the Italian sparrow.
The Italian sparrow exhibits an overall closer affinity to the house sparrow. Although hybrid speciation at the outset involves an equal mixing of the two genomes, the contributions from the progenitors will rarely be balanced in the hybrid species if subsequent backcrossing is involved (4), causing a genetic bias toward one of the parents. Furthermore, local levels of admixture throughout the genome will vary considerably and largely depend on the factors that rendered allele combinations compatible yet also allowed for barriers to gene flow against the parents. Hence, the house sparrow bias in the hybrid genome may result from a range of processes. House sparrows may simply have outnumbered the Spanish sparrow during the initial hybridization events, resulting in an overrepresentation of the former species’ genomic background. In addition, a bias may be explained by the hybrid overall experiencing selection that favored alleles from one parent more than the other, as a result of either adaptive evolution or purging of incompatible allele combinations. Intriguingly, the Italian sparrow shares an almost identical ecology to the human commensal house sparrow, whereas the Spanish sparrow occupies more mesic habitats. One could therefore speculate that the Italian sparrow has been exposed to a selective regime more similar to that experienced by the house sparrow, thereby causing a bias toward house sparrow alleles at many genes. Moreover, the extent of intermediacy throughout the Italian sparrow genome may indicate that it originated through bursts of parental interbreeding, which would retain admixture despite the hybrid being in contact with either parent, as has been argued for the tiger swallowtail hybrid system (7).
The mitochondrial analyses of the Italian sparrow revealed surprising evidence of heteroplasmy in two individuals. Although heteroplasmy is uncommon in animals, it has been detected in a range of species (32), particularly in interspecific hybrids (33, 34), including birds (35). It has been proposed that the mechanisms destroying paternal mtDNA in eggs may break down in hybrids, leading to paternal leakage during heterospecific crosses (33, 36). Heteroplasmy can be difficult to detect because nuclear mitochondrial pseudogenes (numts) can map to mitochondrial sequences (32), distorting the analysis of true mtDNA. However, we would have expected sequence divergence patterns to be more similar between the two admixed individuals if this was the case, and that sequence coverage then would be higher compared to the other Italian sparrows. In addition, such large mitochondrial regions would not be expected to alternate in higher sequence divergence from either parent throughout the entire mitochondrial genome, as we have observed. Hence, we find that heteroplasmy best explains the mitochondrial patterns seen in the two Italian sparrow individuals. Further analysis is required to determine its prevalence and potential fitness effects in the hybrid lineage.
The role of the Z chromosome in hybrid speciation
Sex chromosomes are known to play a prominent role in the evolution of reproductive isolation (RI) and speciation and have been suggested to be where genomic incompatibilities, such as hybrid inviability or sterility, first develop (3, 37). This has been attributed to faster rate of adaptive evolution (faster X/Z), reduced recombination, and overrepresentation of genes related to sex and reproduction (38–40). Its role in hybrid speciation has been discussed in previous work [see also Kunte et al. (7) and Elgvin et al. (11)] but has not yet been extensively investigated. The current observation of contrasting patterns of divergence and polymorphism on Z chromosomes versus autosomes mirrors results for many other bird taxa, supporting the Z chromosome as a hotspot in avian speciation. We also found more conspicuous patterns of mosaicism and stronger selection signals on the Z chromosome relative to autosomes. An important consideration of Z chromosome evolution is its reduced effective population size relative to that of autosomes (Ne at Z is three-fourth that of autosomes) due to female heterogamety. This may significantly affect the sorting of parental alleles through increased rates of fixation via drift or selection, especially in the initial stages of speciation when the population size is expected to have been small. However, our DoS estimates on protein-coding substitutions revealed fewer genes under neutrality and more genes subject to both positive and purifying selection on the Z chromosome. Overall, and in line with earlier work on the system (11, 13), our data further support the hypothesis that the Z chromosome has an important role also in hybrid speciation.
Selection within high-divergence windows
Homoploid hybrid speciation is thought to require rapid development of isolating barriers because the process is sympatric and the hybrid lineage thereby risks getting swamped by the homogenizing effect of gene flow from either parent species (4). Potential mechanisms of escaping such swamping include the emergence of trait combinations that instantly yield incompatibilities toward the parents via deleterious epistatic effects (41), assortative mating (42, 43), or transgressive effects that allow for adaptation to novel ecological conditions in the hybrid (6, 44).
The SI and HI genomic windows are areas of the genome where the Italian sparrow has strongly sorted for one parent’s genetic variation and are consequently candidate regions for barriers against the other parent. The strongest patterns of selection were observed in the SI windows where there is evidence for positive divergent selection between the parent taxa, as well as between the Italian and Spanish sparrows in coding regions. This is particularly interesting because two of the significant gene ontologies within this region relate to craniofacial development and include genes known to affect beak morphology and diversification among Darwin’s finches (29). Because beaks are the main food processing tool in birds, their morphology is often related to individual fitness (45) and subject to divergent selection to suit different foraging ecologies (46), as has been shown in Italian sparrows (47). Because the Italian sparrow ecologically resembles the house sparrow, whereas the Spanish sparrow occupies more mesic habitats, the SI comparison may be expected to show increased divergence in genes controlling beak size and shape, assuming that habitat preferences affect diet and that the hybrid followed a similar genetic trajectory to its human commensal parent. Among the HI windows, the lack of nonsynonymous fixed differences in both HI and HS comparisons is perhaps unsurprising because these regions appear to be largely neutral in house sparrows and subject to selection in the Italian and Spanish lineages. The GO analyses within HI windows revealed significant enrichment of genes involved in the regulation of G protein–coupled receptor signaling that are known to directly modify behavioral and morphologic variation in birds (31). However, this group of genes encompasses a wide range of potential functions, and further investigation is needed to determine the phenotypic effects of these genes in sparrows.
Regions where the hybrid differentiated from both parents are candidate areas of novel divergence in the hybrid lineage. Within these areas, we find evidence for balancing selection in the Italian sparrow, which suggests heterozygote advantage (overdominance) in the hybrid. However, there are multiple processes that have confounding effects on sequence variation in a hybrid species and thereby the interpretation of selection signatures, including demography and recombination. It may be the case that background selection, potentially in areas of low recombination, is occurring within these regions, resulting in negative TD values in the parents. Reduced recombination is characterized by decreased nucleotide diversity in surrounding areas, because positive selection and purifying selection are expected to affect larger genomic regions due to stronger linkage among sites (48, 49). In a hybrid, the recombinational landscape may be altered, breaking up haplotype blocks that are largely conserved in the parental lineages. Under this scenario, TD values are expected to increase and contrast the values seen in the source lineages, consistent with the positive TD values in the Italian sparrow. Furthermore, reduced recombination in the parents may also have led to an accumulation of slightly deleterious alleles, which cannot easily be purged from the population. With the release of recombinational blocks in the hybrid, heterosis may occur from the masking of these deleterious alleles.
Although there are a variety processes that may drive the observed TD patterns within the outlier windows, the combination of DoS estimates and TD values suggests a role for balancing selection in at least a portion of the genes within the PI regions. Hybridization can boost genetic variance (6), and through complementary gene action of additive alleles in parental lineages, transgressive phenotypes outside both parents’ ranges can arise in hybrids (50). This is one proposed mechanism for how speciation can occur via hybridization as transgressive traits may allow hybrids to occupy novel ecological niches, in turn, impeding gene flow from its parents (4). Hybrids may even displace their parents ecologically if they experience higher fitness in a given habitat (51). Furthermore, traits with a history of balancing selection are expected to be more likely to result in hybrid transgression because traits with an intermediate optimum maintain alleles with effects in opposing directions in the parental lineages, allowing for the additive effects of complementary genes in the hybrid (50). We found a significant enrichment of genes involved in the regulation of the immune system within PI genomic regions. The immune system may have a large impact on individual fitness, and several of its genetic components are expected to be subject of balancing selection (52). It is possible that the combination of such alleles in an admixed genome, such as the Italian sparrow, could have an advantageous effect on the general fitness of the hybrids, thereby facilitating their spread.
We acknowledge that caution should be taken in drawing conclusions about the functional effects of candidate genes from genome comparisons alone (53). However, the study highlights candidate regions that harbor genes with known associations to traits that are likely to have a bearing on reproduction in the Passer system. In addition, the concordance of genes recognized in the current study and the genes exhibiting steep clines in the study by Trier et al. make them—and/or their adjacent locations—strong candidates for the involvement in the hybrid speciation process.
CONCLUSION
To our knowledge, our study represents the first detailed investigation of the genomic admixture in a hybrid species in relation to its parents. We demonstrate substantial parental contributions throughout an avian hybrid species that maintains its integrity despite contact with its parent species. Our study also highlights candidate regions that potentially affect key traits in the system and thereby may have had instrumental roles in the formation of the hybrid species. Overall, we argue that the Italian sparrow serves as a well-documented case of the striking potential of hybridization as a creative force contributing to species diversity.
MATERIALS AND METHODS
Experimental design
The main objective of this study comprised a comparative genomic analysis of the homoploid hybrid species the Italian sparrow and its parents, the house sparrow and Spanish sparrow. The analytical framework included whole-genome sequencing of key populations of the three focal taxa and mapping these to the closely related house sparrow reference genome assembly. The individual chosen for the reference genome assembly was an inbred (pedigree F = 0.3125) female house sparrow (individual ID 8887266) sampled in 2002 on the small and inbred island of Aldra (54) in northern Norway (66°24′N, 13°6′E; Lurøy kommune, Nordland). A previous study of genome-wide SNP-chip genotyping of this population showed that this individual has a low level of heterozygosity (0.161) compared to the population mean, which is advantageous in the reference assembly process.
We used male house sparrows from island populations in northern Norway (n = 10), Italian sparrows from Guglionesi (n = 10), and Spanish sparrows from Lesina (n = 10), both of the latter locations in central eastern Italy (Fig. 1 and table S3). In addition, one tree sparrow from Giardini Naxos in Sicily was added to the sampling scheme to serve as an outgroup. The sparrows were caught with mist nets, and ~25 μl of blood was extracted through venipuncture of the left brachial vein and stored in either 1 ml of standard lysis buffer (P. italiae, P. hispaniolensis, and P. montanus samples) or 100% ethanol (P. domesticus samples). The appropriate catching and sampling permits were obtained from the appropriate authorities for the respective locations.
Reference genome assembly
DNA from the house sparrow chosen for the reference genome assembly was extracted using the protocol described by Hagen et al. (55). Detailed information on the reference individual has been deposited in the National Center for Biotechnology Information (NCBI) BioSample database under accession number SAMN02929199.
Sequencing for the de novo assembly of the house sparrow reference genome was performed on an Illumina platform using HiSeq 2000 instruments at the Norwegian Sequencing Centre at the University of Oslo (www.sequencing.uio.no) and at Génome Québec at McGill University (www.genomequebec.com/en/home.html). The sequencing strategy, including platform choice, fragment size, and coverage, was chosen following recommendations of the ALLPATHS-LG assembly software (Broad Institute, Cambridge, MA). ALLPATHS-LG has proven to be a robust assembler for larger eukaryotic genomes (56), and it uses a combination of short reads from paired-end and various mate pair (MP) libraries. For a complete list of the library construction and sequence yield, see table S1.
All MP library reads were trimmed for adaptor sequences using cutadapt (v. 1.5) (57). Read files were trimmed for the specific adaptors used for the various library construction protocols, including external adapters and junction adapters. The adapter trimming resulted in between 17.71 and 20.93% of the total bases being discarded before assembly. All adapter trimmed reads were used as input for ALLPATHS-LG (v. 46923) (Broad Institute, Cambridge, MA). File preparation was conducted according to the manufacturer’s recommendations. The main run was performed using the TARGETS=submission option to make a submission prepared assembly version. The resulting assembly comprised a total of 1.04 Gb divided into 2766 scaffolds ≥1 kb (N50 = 66.3 Mb). Each scaffold was blasted against the NCBI nucleotide database using BLAST+ (v 2.2.29) (58). All scaffolds with a top hit that was not avian or reptilian were removed from the assembly. Only top hits with an e value greater than e−5 and an alignment length greater than 100 base pairs (bp) were considered reliable. This resulted in the removal of 195 scaffolds from the assembly.
For gene annotation of the reference assembly, we used the MAKER (v. 2.31.8) pipeline (59). This pipeline used RNA sequencing (RNA-seq) data to collect physical evidence for genes and incorporated additional gene predictions from other programs [see Yandell et al. (60)]. To obtain physical evidence for gene annotations, we used RNA-seq data from a previous project (13), and all of these were downloaded and assembled with Newbler (v. 3.0; -cdna option) to aid in the genome annotation. GeneMark-ES (61), with the min_contig option set to 10,000, and CEGMA (62) were first applied on the genome assembly file. The resulting CEGMA.gff file was then used to train a SNAP .hmm file (63). Both SNAP and GeneMark .hmm files, in addition to a transcriptome assembly and UniProt database (www.UniProt.org), were then fed into a first-pass run of MAKER with the following modifications to the maker_opts.ctl file: est2genome=1, protein2genome=1, keep_preds=1, single_exon=1, max_dna_len=300000, and min_contig=10000. AUGUSTUS (v.3.0.2) (64) was trained on the transcriptome assembly and a snap model of the MAKER .gff first run predictions. MAKER was finally run a second round, including the trained predictions from previous steps, with the CEGMA SNAP .hmm file replaced by the predictions SNAP model from the MAKER first run. Only the MAKER second-pass predictions were used for further analysis. After quality filtering (annotation edit distance score, ≥0.5), the resulting annotation included 13,685 protein-coding genes.
Linkage mapping and construction of chromosome sequences
Populations of P. domesticus on four different islands in the northern part of Norway (Aldra, Hestmannøy, Leka, and Vega) have been extensively monitored on an individual basis since 1993 [Hestmannøy; for example, Jensen et al. (45)], 1998 [Aldra; Billing et al. (54)], or 2001 [Leka and Vega; Hagen et al. (55)]. A total of 2290 house sparrows on these four islands were genotyped on a 10K SNP array developed for P. domesticus (55), and data were used to construct a complex pedigree using the program Cervus (65). A subset of the pedigree, which included genotype data on 6491 SNPs for 862 individuals included in 105 families, was used in the map construction.
A modified version of the CRIMAP 2.4 software (66), including added utilities provided by X. Liu and M. Grosz (Monsanto, St. Louis, MO), was used for the map construction. Initially, SNPs were assigned to linkage groups (LGs) on the basis of pairwise linkages and the grouping algorithm implemented in the AUTOGROUP option of the program. The analysis assigned 6491 SNPs to 29 larger autosomal LGs (macrochromosomes), 456 SNPs to an LG corresponding to the Z chromosome, and 37 SNPs to nine smaller LGs. Four of the smaller LGs contained one or more SNPs showing weak linkage to markers on other LGs, suggesting that they could be merged into other linkage groups, whereas the remaining five LGs built groups on their own, suggesting that they represent microchromosomes. Hence, because the house sparrow karyotype was expected to consist of 38 pairs of chromosomes [2n = 76; Bulatova et al. (67)], we identified LGs that probably correspond to 35 of 38 chromosomes.
After the initial grouping of SNPs, markers on the 29 larger autosomal LGs were ordered using the BUILD and FLIPSN options in CRIMAP. Following this, 120 bp flanking each SNPs were positioned in the house sparrow assembly and used to assign scaffolds to LGs, order and orientate scaffolds within LGs, and build sequences for 29 autosomal chromosomes in P. domesticus. If a scaffold contained only one marker in the linkage map, then the scaffold was assigned the same orientation as in the zebra finch genome.
Following the construction of chromosome sequences, the SNP order within each LG was fine-tuned using physical positions of the SNPs in the chromosome. The CHROMPIC option in CRIMAP was then used to phase genotypes within LGs, and a script was written to correct or remove erroneous genotypes on the basis of unlikely tight double recombination events. Finally, multipoint linkage maps for the 29 autosomal LGs were constructed using the FIXED option of CRIMAP and the Kosambi correction function (68).
Chromosome nomenclature in the house sparrow was determined by alignments against the zebra finch, flycatcher, and chicken genomes. To avoid potential confusion by adding new chromosome names for the smaller linkage groups (microchromosomes), these LGs were not included as separate chromosomes in the current assembly. Because no linkage map was constructed for the Z chromosome, scaffolds on this chromosome were ordered and orientated according to the zebra finch genome. Moreover, other avian mitochondrial genomes were used to identify the scaffold containing the house sparrow mitochondrial genome.
Sequencing
DNA was isolated using either Qiagen DNeasy 96 Blood and Tissue Kits (Qiagen N.V.), according to the manufacturer’s instructions, with the exception of eluting the isolate in EB buffer instead of AE buffer (P. italiae, P. hispaniolensis, and P. montanus samples), or the ReliaPrep Large Volume HT gDNA Isolation System (Promega) automated on a Biomek NXp robot (Beckman Coulter), as described by Hagen et al. (P. domesticus samples).
For each sample, an Illumina TruSeq gDNA 180-bp library was created and quality-controlled for high-throughput massive parallel sequencing. The libraries were then sequenced on the Illumina HiSeq 2000 platform with 100 bp read length and three individuals per lane. All sequencing was performed by the Génome Québec at McGill University (Montreal, Canada) (www.genomequebec.com/en/home.html).
Mapping to the reference genome and variant calling
Before population genomic analyses, interspersed repeat elements were masked from the assembly with RepeatMasker (v 4.0.5) (A. F. A. Smit, R. Hubley, and P. Green; RepeatMasker at http://repeatmasker.org). The default settings were used with the exception of adding the “Do not mask simple” option, which conservatively masked only interspersed repeats and left regions of low diversity unmasked. The whole-genome sequences were mapped to the house sparrow assembly with BWA-MEM (v 0.7.5a-r405) (69) using default settings with the exception of adding read group identifiers and a -M parameter to enable Picard (http://picard.sourceforge.net) compatibility for downstream analyses. With the mapped reads, we then removed polymerase chain reaction duplicates with MarkDuplicates in Picard tools (v 1.72) (http://picard.sourceforge.net) with the default settings for every parameter except validation stringency, which was set to lenient. We then realigned the reads mapped to the house sparrow reference genome using GATK’s IndelRealigner tool with the default settings.
An SNP set was required for some analyses. Thus, to create an SNP set from the wgs reads, we used GATK’s Genome Analysis Toolkit (v 3.2.2) (70, 71). The realigned .bam files were run in GATK’s HaplotypeCaller to create a genomic variant call format (gVCF) file for each individual using the default settings. Next, the gVCF files were genotyped using GATK’s GenotypeGVCFs function. This resulted in a large VCF file of 37,526,610 SNPs and 5,784,223 indels for all individuals across the genome.
Variants from the VCF file were further quality-filtered using VCFtools (v 0.1.12b) (32). SNPs with a genotype quality >20, a quality value >20, and a mean depth >5 across all individuals were kept as a set of high-quality variants. In addition, because the downstream analyses focused on the chromosomes and mitochondria, unplaced scaffolds were removed, leaving 35,867,119 SNPs and 5,389,326 indels. For some analyses, further filtering was applied and detailed in the appropriate methods section.
Ancestry estimates and population structuring
We used NgsAdmix to estimate whole-genome admixture of each sparrow individual from the focal populations because it estimated admixture on the basis of genotype likelihoods from the realigned reads (.bam files) rather than genotype calls. We first calculated genotype likelihoods from the .bam files mapped to the reference genome in ANGSD (v 0.911-47-g4705d60) (72) with the parameters -doGlf 2, -doMajorMinor 1, -SNP_pval 1e-6, -doMaf 1. We also filtered the reads for quality scores >20. The resulting file was then input into NgsAdmix and run with K = 2 and K = 3 ancestral populations to calculate genome-wide admixture for each individual (K = 2 shown in Fig. 1; K = 3 shown in fig. S6).
The PCA was run on the filtered SNP set excluding the tree sparrow using the R package SNPRelate (v 1.6.2) (73). Before the PCA, the SNP set was LD-pruned for r2 values >0.5 in SNPRelate (l.d.threshold = 0.5). The PCA was then run for the full nuclear genome for all biallelic sites (268,962 SNPs) using default settings.
ADMIXTURE (v 1.23) (74) was run in 100-kb stepping windows across the genome on the SNP set that was further filtered to include only SNPs with genotypes in at least one individual of each species (34,776,981 SNPs). A window size of 100 kb was chosen as LD tends to decay within this distance in the genome (fig. S2). The SNP set was converted to a BED file format using PLINK (v 1.07) (75). The ADMIXTURE analysis was run with K = 2 set as the number of ancestral populations for the analysis. To visualize the admixture of the parent taxa’s ancestry in the Italian sparrow, a prior was set fixing the parent species as K1 and K2, so that ancestry estimates to either the house or the Spanish cluster were inferred for each window across the Italian sparrow genome. Aside from this, the default settings were used.
In addition, RAxML (v 8.0.26) (17) was run for 100-kb stepping windows across the genome for SNPs genotyped in at least one individual in each species. The model was set to “GTRGAMMA,” and the tree sparrow was designated as the outgroup. The resulting trees for each window were then categorized on the basis of whether all Italian sparrow individuals grouped monophyletically with one of its parents, in its own clade, or was “unresolved,” meaning the Italian sparrow individuals did not form a monophyletic group. The same method was used for 50- and 10-kb stepping windows to test whether window size affected the proportion of resolved phylogenies.
Mitochondrial analyses
A gVCF file of mitochondrial variants was created separately by running GATK (v 3.3.0) HaplotypeCaller with its haploid option and, aside from that, default settings. The gVCF file was then genotyped with GATK’s GenotypeGVCFs tool. The resulting VCF file was filtered in the same manner as the main VCF file in VCFtools (v 0.1.12b) by removing SNPs with a minimum genotype quality and/or minimum quality threshold <20 and a minimum depth of 5. The mitochondrial haplotype network was created in Fitchi (76) using the filtered mitochrondrial SNP set. A mitochondrial fastSTRUCTURE (19) analysis was also performed on the filtered mitochondrial SNP set and was further filtered for SNPs with a maximum depth of 50 and genotypes in at least 30% of individuals using the default settings and with K = 2 to show the admixture of the parent taxa’s mitochondria in the Italian sparrow.
A separate VCF file, including calls for every site, was made using GATK’s GenotypeGVFs (v 3.3.0) under its default setting with the exception of its –includeNonVariantSites option and ploidy=1 to calculate dXY values for the mitochondria. dXY was calculated in sliding 1-kb sliding windows with 100-bp steps for each sparrow individual along the mitochondria using Martin et al.’s script (v. August 2014) with –minimumExploitableData set to 0.3 and the minimum SNPs per window set to 3. The Mann-Whitney U test to determine whether the apparent heteroplasmic individuals differed in coverage was tested in R on the mean depth per Italian sparrow individual for the mitochondrial sequences. Similarly, a Mann-Whitney U test was implemented in R to determine whether nuclear F estimates for the Italian sparrows with heteroplasmic mtDNA differed from values of the Italian sparrows with house mtDNA. Nuclear F values and mtDNA coverage were calculated on a per-individual basis using VCFtools (v 0.1.12b).
A mitochrondrial SNP set run as diploid was also created using GATK’s (v 3.3.0) GenotypeGVCFs tool under default setting so that the heterozygosity of each individual’s mtDNA sequence could be estimated. Overall, F values were calculated for each individual using VCFtools (v 0.1.12b) with the diploid SNP set filtered for a minimum depth >3 and a maximum depth of 15.
Population genomic analyses in sliding windows
Estimates for FST, nucleotide diversity, and TD were calculated in overlapping sliding windows 100 kb in size with 25-kb steps with ANGSD with a minimum quality filter set to >20, minimum mapping quality >20, and genotype likelihood model of 2. FST values are the weighted mean FST for each window across the genome. ANGSD calculated nucleotide diversity as θD, which was divided by the number of sites in the window to obtain a nucleotide diversity value per site for the window. TD was calculated using a folded site frequency spectrum that treats the ancestral state as unknown.
To calculate the density of fixed differences (df) and identify fixed differences in the genome, the SNP set created with GATK’s HaplotypeCaller was further filtered so that each population had to have at least three individuals genotyped for each SNP to ensure that SNPs genotyped for very few individuals in a population were not considered fixed differences between species. Fixed differences were found using the R package PopGenome’s (v 2.1.6) (77) biallelic matrix function to identify sites where a population is monomorphic and another population is monomorphic with a different value.
Incomplete lineage sorting complicates inference of hybridization as the two processes leave similar signatures in the genome and has therefore been emphasized as a critical test when evaluating hybrid species. To distinguish between these processes and quantify the extent of gene flow between the parent and hybrid lineages, we used a four-taxon ABBA BABA test for introgression (D-statistics) and fd estimator (18, 78, 79). The analysis uses patterns of ancestral and derived alleles in the ingroups and outgroups to distinguish between incomplete lineage sorting and hybridization and has been shown to be a robust, although conservative, method for identifying introgressed loci (18). Whole-genome ABBA BABA (D-statistics) and the jack-knifed SE values were calculated for two topologies {(house, Italian), Spanish [tree]} and {(Spanish, Italian), house [tree]} with the tree sparrow individual used as the outgroup in ANGSD’s ABBA BABA multipopulation tool. The tree sparrow fasta file was created directly from the .bam file in ANGSD with the -doFasta 3 option. The fd estimator used to quantify the gene flow based on ABBA BABA statistics was calculated in 100-kb sliding windows with 25-kb steps with the script from Martin et al. (v. August 2014) using the high-quality SNP set with –minimumExploitableData set to 0.3 and the minimum SNPs per window set to 10. Again, the tree sparrow was designated as the outgroup.
The decay of LD was calculated across each chromosome for the three focal taxa by first filtering the SNP set for variants genotyped for at least 80% of the individuals. Pairwise r2 values were calculated between all SNPs within a 100-kb window in PLINK (v 1.09b) (75). Decay plots were created by binning the distance between SNPs in 1-kb increments and averaging the r2 values within each bin.
To provide a comparison of LD estimates within and outside of outlier divergence windows, pairwise r2 values were also calculated in PLINK (v 1.09b) (75) for all SNPs (genotyped in at least 80% of individuals) within 1 kb of each other and binned into 100-kb genomic windows with 25-kb steps where each bin represents the average r2 values for all pairwise comparisons within that window.
DoS estimates were calculated separately for all genes on across the genome, as well as for sliding windows across the genome between all three species comparisons (HI, SI, and HS). Nonsynonymous and synonymous fixed differences and polymorphisms were identified in PopGenome (v 2.1.6) (77) using the MKT-methods function, and their values were used to calculate DoS according to the formula by Stoletzki and Eyre-Walker (25).
Identification of high-divergence windows and genes
To select regions with FST values high against one parent and low against the other, we subtracted the HI FST value for every sliding window from the SI FST value and vice versa. We then took the top 1% windows for each hybrid/parent comparison as high-divergence windows. Regions where the Italian sparrow was divergent against both parent species were identified by subtracting the parent’s FST value for every sliding window from both hybrid/parent comparisons, and the top 1% windows common to both comparisons were identified as private Italian regions of high differentiation (see Eqs. 1 to 3). Genes were then extracted from the sets of HI, SI, and PI high-divergence windows (table S6). A gene was considered to be in the genomic window if a portion of it falls within the region.
Statistical analysis: Gene enrichment analysis and simulations
To investigate whether there were any significantly enriched GO groups or network pathways within the high-divergence windows (SI, HI, and PI), we ran gene enrichment analyses with the ClueGo (v 2.2.5) plugin (80) implemented in Cytoscape (v 3.3.0) (81). The lists of genes found in each category of our high-divergence windows were input separately and tested with a right-sided hypergeometric enrichment test using both humans (tables S7 to S9) and, next, chickens (tables S11 to S13) as the model organisms. The GO database BiologicalProcess-GOA (09 February 2016, humans; 09 March 2016, chickens) was used along with the network specificity set to medium, Bonferroni step-down method of P value correction and a minimum P < 0.05 for reporting results. All other settings were set as default. The results between human and chicken analyses were comparable with the chickens being a subset of the human results (tables S11 to S13) because the human genome was a better-annotated genome. Thus, the results based on the analysis with the human reference genome are discussed in the main text.
Gene enrichment analysis permutations were run to test whether the enriched gene ontologies from our outlier windows could also be found when choosing the same number of windows randomly across the genome. To do this, we used BEDTools (v 2.17.0) (82) shuffle to randomly select the same number of 100-kb windows as found for the top SI, HI, and PI regions 50 times for each comparison and excluded windows that were already selected as high-divergence windows. We then extracted the genes from all the window selections in the same manner as genes were extracted from our outlier windows. For each permutation, we also randomly sampled genes from each gene list so that the number of genes matched that of genes found in the outlier comparison it was simulating. We then ran ClueGo (v 1.8.0) for each permutation against the human biological process database (09 February 2016, humans) under the default settings with Bonferroni correction and counted the number of times the significant gene ontologies from the outlier windows also showed up in the randomly selected windows. None of the enriched gene ontologies from our outlier analyses reoccurred during our permutations.
Because important genes for species divergence within our outlier windows may not fall into single enriched gene groups, we also cross-checked our candidate genes with genes known to be involved in pigmentation and SNPs previously identified as being candidate RI genes in the system (table S10). Candidate genes previously identified for the system were the SNP set of species diagnostic markers between the house and Spanish sparrows by Trier et al. (13). The color gene candidate markers were taken from the list identified by Poelstra et al. (28). We used a permutation approach to test whether the number of previously identified RI candidate genes and/or candidate color genes among outlier genes was likely to occur by chance. We randomly sampled outlier sets (n = 318 genes) 10,000 times and counted RI/color genes occurring in each sample to generate a null distribution. We then used a one-sided test to examine whether the observed number was greater than the null distribution.
Supplementary Material
Acknowledgments
We thank F. Eroukhmanoff and A. Runemark for helpful comments on the early versions of the manuscript and A. Mazzarella, S. Jentoft, A. Tooming-Klunderud, R. Røsbak, and K. Yttersian Sletta for technical assistance. All computational work was performed on the Abel Supercomputing Cluster [Norwegian Metacenter for High Performance Computing (NOTUR) and the University of Oslo] operated by the Research Computing Services group at The University Center for Information Technology (www.hpc.uio.no/). Sequencing library creation and high-throughput sequencing were carried out at the NSC, University of Oslo, Norway, and McGill University and Génome Québec Innovation Centre, Canada. This project received support from RCN project 208481, and we especially acknowledge the work carried out by J. K. A. Samy in establishing the genome browser, available at http://cees-genomes.hpc.uio.no/gb2/gbrowse/house_sparrow or just cees-genomes.hpc.uio.no/gb2/gbrowse/house_sparrow. Funding: This work was supported by The Research Council of Norway grants 240557, 221956, and 223257. Author contributions: T.O.E., C.N.T., and G.-P.S. designed the research. T.O.E., C.N.T., O.K.T., and A.J.N. created the house sparrow reference genome. S.L., I.J.H., and H.J. created the house sparrow linkage map. T.O.E., C.N.T., O.K.T., and M.R. analyzed the data. T.O.E. and C.N.T. wrote the first manuscript draft. All authors contributed to the writing of the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The raw data produced for this project have been deposited at the NCBI Sequence Read Archive under BioProject PRJNA255814 accession numbers SRR5407744–SRR5407749 (house sparrow reference assembly) and SRR5369936–SRR5369966 (population whole-genome sequencing). The house sparrow reference assembly has been deposited at DDBJ/ENA/GenBank under accession MBAE00000000. The version described in this paper is version MBAE01000000.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1602996/DC1
fig. S1. Stepping window ADMIXTURE and RAxML analyses across the Italian sparrow genome.
fig. S2. LD decay across all chromosomes.
fig. S3. Mitochondrial sequence divergence between each Italian sparrow and the parent populations.
fig. S4. Divergence peaks between the Italian sparrow and either parent taxa on microchromosomes.
fig. S5. Divergence peaks for PI regions on large chromosomes.
fig. S6. Population structuring analysis of sparrow individuals.
table S1. Sequencing scheme for the house sparrow reference genome assembly.
table S2. Final assembly statistics.
table S3. Sample information.
table S4. Results from genome-wide RAxML analysis.
table S5. Comparison of resolved RAxML phylogenies at variable window sizes.
table S6. ENSEMBLE gene IDs for genes within top divergence windows.
table S7. Significantly enriched GO pathways from ClueGo analysis in HI outlier windows.
table S8. Significantly enriched GO pathways from ClueGo analysis in SI outlier windows.
table S9. Significantly enriched GO pathways from ClueGo analysis in PI outlier windows.
table S10. Genes among outlier windows identified as candidates for the involvement in melanogenesis and RI in sparrows.
table S11. Significantly enriched GO pathways from ClueGo analysis HI outlier windows with chickens as the reference genome.
table S12. Significantly enriched GO pathways from ClueGo analysis in SI outlier windows with chickens as the reference genome.
table S13. Significantly enriched GO pathways from ClueGo analysis in PI outlier windows with chickens as the reference genome.
REFERENCES AND NOTES
- 1.Seehausen O., Butlin R. K., Keller I., Wagner C. E., Boughman J. W., Hohenlohe P. A., Peichel C. L., Sætre G.-P., Bank C., Brännström Å., Brelsford A., Clarkson C. S., Eroukhmanoff F., Feder J. L., Fischer M. C., Foote A. D., Franchini P., Jiggins C. D., Jones F. C., Lindholm A. K., Lucek K., Maan M. E., Marques D. A., Martin S. H., Matthews B., Meier J. I., Möst M., Nachman M. W., Nonaka E., Rennison D. J., Schwarzer J., Watson E. T., Westram A. M., Widmer A., Genomics and the origin of species. Nat. Rev. Genet. 15, 176–192 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Abbott R., Albach D., Ansell S., Arntzen J. W., Baird S. J. E., Bierne N., Boughman J., Brelsford A., Buerkle C. A., Buggs R., Butlin R. K., Dieckmann U., Eroukhmanoff F., Grill A., Cahan S. H., Hermansen J. S., Hewitt G., Hudson A. G., Jiggins C., Jones J., Keller B., Marczewski T., Mallet J., Martinez-Rodriguez P., Möst M., Mullen S., Nichols R., Nolte A. W., Parisod C., Pfennig K., Rice A. M., Ritchie M. G., Seifert B., Smadja C. M., Stelkens R., Szymura J. M., Väinölä R., Wolf J. B. W., Zinner D., Hybridization and speciation. J. Evol. Biol. 26, 229–246 (2013). [DOI] [PubMed] [Google Scholar]
- 3.J. A. Coyne, H. A. Orr, Speciation (Sinauer Associates Inc., 2004). [Google Scholar]
- 4.Mallet J., Hybrid speciation. Nature 446, 279–283 (2007). [DOI] [PubMed] [Google Scholar]
- 5.Heliconius Genome Consortium, Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487, 94–98 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rieseberg L. H., Raymond O., Rosenthal D. M., Lai Z., Livingstone K., Nakazato T., Durphy J. L., Schwarzbach A. E., Donovan L. A., Lexer C., Major ecological transitions in wild sunflowers facilitated by hybridization. Science 301, 1211–1216 (2003). [DOI] [PubMed] [Google Scholar]
- 7.Kunte K., Shea C., Aardema M. L., Scriber J. M., Juenger T. E., Gilbert L. E., Kronforst M. R., Sex chromosome mosaicism and hybrid speciation among tiger swallowtail butterflies. PLOS Genet. 7, e1002274 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.T. R. Anderson, Biology of the Ubiquitous House Sparrow: From Genes to Populations (Oxford Univ. Press Inc., 2006). [Google Scholar]
- 9.J. D. Summers-Smith, The Sparrows: A Study of the Genus Passer (T & AD Poyser, 1988). [Google Scholar]
- 10.Meise W., Zur Systematik und Verbreitungsgeschichte der Haus- und Weidensperlinge, Passer domesticus (L.) und hispaniolensis (T.). J. Ornithol. 84, 631–672 (1936). [Google Scholar]
- 11.Elgvin T. O., Hermansen J. S., Fijarczyk A., Bonnet T., Borge T., Sæther S. A., Voje K. L., Sætre G.-P., Hybrid speciation in sparrows II: A role for sex chromosomes? Mol. Ecol. 20, 3823–3837 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Hermansen J. S., Sæther S. A., Elgvin T. O., Borge T., Hjelle E., Sætre G.-P., Hybrid speciation in sparrows I: Phenotypic intermediacy, genetic admixture and barriers to gene flow. Mol. Ecol. 20, 3812–3822 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Trier C. N., Hermansen J. S., Sætre G.-P., Bailey R. I., Evidence for mito-nuclear and sex-linked reproductive barriers between the hybrid Italian sparrow and its parent species. PLOS Genet. 10, e1004075 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hermansen J. S., Haas F., Trier C. N., Bailey R. I., Nederbragt A. J., Marzal A., Sætre G.-P., Hybrid speciation through sorting of parental incompatibilities in Italian sparrows. Mol. Ecol. 23, 5831–5842 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Schumer M., Rosenthal G. G., Andolfatto P., How common is homoploid hybrid speciation? Evolution 68, 1553–1560 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Mallet J., Hybridization as an invasion of the genome. Trends Ecol. Evol. 20, 229–237 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Stamatakis A., RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin S. H., Davey J. W., Jiggins C. D., Evaluating the use of ABBA–BABA statistics to locate introgressed loci. Mol. Biol. Evol. 32, 244–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raj A., Stephens M., Pritchard J. K., fastSTRUCTURE: Variational inference of population structure in large SNP data sets. Genetics 197, 573–589 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DePristo M. A., Handsaker R. E., Lunter G., Marth G. T., Sherry S. T., McVean G., Durbin R. 1000 Genomes Project Analysis Group , The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cruickshank T. E., Hahn M. W., Reanalysis suggests that genomic islands of speciation are due to reduced diversity, not reduced gene flow. Mol. Ecol. 23, 3133–3157 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Noor M. A. F., Bennett S. M., Islands of speciation or mirages in the desert? Examining the role of restricted recombination in maintaining species. Heredity 103, 439–444 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Charlesworth B., Measures of divergence between populations and the effect of forces that reduce variability. Mol. Biol. Evol. 15, 538–543 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Tajima F., Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123, 585–595 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stoletzki N., Eyre-Walker A., Estimation of the neutrality index. Mol. Biol. Evol. 28, 63–70 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Charlesworth J., Eyre-Walker A., The McDonald–Kreitman test and slightly deleterious mutations. Mol. Biol. Evol. 25, 1007–1015 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Bailey R. I., Tesaker M. R., Trier C. N., Sætre G.-P., Strong selection on male plumage in a hybrid zone between a hybrid bird species and one of its parents. J. Evol. Biol. 28, 1257–1269 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Poelstra J. W., Vijay N., Hoeppner M. P., Wolf J. B. W., Transcriptomics of colour patterning and coloration shifts in crows. Mol. Ecol. 24, 4617–4628 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Abzhanov A., Protas M., Grant B. R., Grant P. R., Tabin C. J., Bmp4 and morphological variation of beaks in Darwin’s finches. Science 305, 1462–1465 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Roberts R. B., Hu Y., Albertson R. C., Kocher T. D., Craniofacial divergence and ongoing adaptation via the hedgehog pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 13194–13199 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abe H., Inoue-Murayama M., Structural variation of G protein-coupled receptor in birds. Recept. Clin. Invest. 1, e162 (2014). [Google Scholar]
- 32.Kmiec B., Woloszynska M., Janska H., Heteroplasmy as a common state of mitochondrial genetic information in plants and animals. Curr. Genet. 50, 149–159 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Radojičić J. M., Krizmanić I., Kasapidis P., Zouros E., Extensive mitochondrial heteroplasmy in hybrid water frog (Pelophylax spp.) populations from Southeast Europe. Ecol. Evol. 5, 4529–4541 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shitara H., Hayashi J.-I., Takahama S., Kaneda H., Yonekawa H., Maternal inheritance of mouse mtDNA in interspecific hybrids: Segregation of the leaked paternal mtDNA followed by the prevention of subsequent paternal leakage. Genetics 148, 851–857 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kvist L., Martens J., Nazarenko A. A., Orell M., Paternal leakage of mitochondrial DNA in the great tit (Parus major). Mol. Biol. Evol. 20, 243–247 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Bromham L., Eyre-Walker A., Smith N. H., Smith J. M., Mitochondrial Steve: Paternal inheritance of mitochondria in humans. Trends Ecol. Evol. 18, 2–4 (2003). [Google Scholar]
- 37.Qvarnström A., Bailey R. I., Speciation through evolution of sex-linked genes. Heredity 102, 4–15 (2009). [DOI] [PubMed] [Google Scholar]
- 38.Charlesworth B., Coyne J. A., Barton N. H., The relative rates of evolution of sex chromosomes and autosomes. Am. Natural. 130, 113–146 (1987). [Google Scholar]
- 39.Mank J. E., Axelsson E., Ellegren H., Fast-X on the Z: Rapid evolution of sex-linked genes in birds. Genome Res. 17, 618–624 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ellegren H., The different levels of genetic diversity in sex chromosomes and autosomes. Trends Genet. 25, 278–284 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Rieseberg L. H., Hybrid origins of plant species. Annu. Rev. Ecol. Syst. 28, 359–389 (1997). [Google Scholar]
- 42.Salazar C., Baxter S. W., Pardo-Diaz C., Wu G., Surridge A., Linares M., Bermingham E., Jiggins C. D., Genetic evidence for hybrid trait speciation in Heliconius butterflies. PLOS Genet. 6, e1000930 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Melo M. C., Salazar C., Jiggins C. D., Linares M., Assortative mating preferences among hybrids offers a route to hybrid speciation. Evolution 63, 1660–1665 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Gross B. L., Rieseberg L. H., The ecological genetics of homoploid hybrid speciation. J. Hered. 96, 241–252 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jensen H., Steinsland I., Ringsby T. H., Sæther B.-E., Evolutionary dynamics of a sexual ornament in the house sparrow (Passer domesticus): The role of indirect selection within and between sexes. Evolution 62, 1275–1293 (2008). [DOI] [PubMed] [Google Scholar]
- 46.P. R. Grant, B. R. Grant, How and Why Species Multiply: The Radiation of Darwin’s Finches (Princeton Univ. Press, 2008). [Google Scholar]
- 47.Eroukhmanoff F., Hermansen J. S., Bailey R. I., Sæther S.-A., Sætre G.-P., Local adaptation within a hybrid species. Heredity 111, 286–292 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Charlesworth B., Morgan M. T., Charlesworth D., The effect of deleterious mutations on neutral molecular variation. Genetics 134, 1289–1303 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burri R., Nater A., Kawakami T., Mugal C. F., Olason P. I., Smeds L., Suh A., Dutoit L., Bureš S., Garamszegi L. Z., Hogner S., Moreno J., Qvarnström A., Ružić M., Sæther S.-A., Sætre G.-P., Török J., Ellegren H., Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers. Genome Res. 25, 1656–1665 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rieseberg L. H., Archer M. A., Wayne R. K., Transgressive segregation, adaptation and speciation. Heredity 83, 363–372 (1999). [DOI] [PubMed] [Google Scholar]
- 51.Buerkle C. A., Morris R. J., Asmussen M. A., Rieseberg L. H., The likelihood of homoploid hybrid speciation. Heredity 84, 441–451 (2000). [DOI] [PubMed] [Google Scholar]
- 52.Ferrer-Admetlla A., Bosch E., Sikora M., Marqués-Bonet T., Ramírez-Soriano A., Muntasell A., Navarro A., Lazarus R., Calafell F., Bertranpetit J., Casals F., Balancing selection is the main force shaping the evolution of innate immunity genes. J. Immunol. 181, 1315–1322 (2008). [DOI] [PubMed] [Google Scholar]
- 53.Payseur B. A., Rieseberg L. H., A genomic perspective on hybridization and speciation. Mol. Ecol. 25, 2337–2360 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Billing A. M., Lee A. M., Skjelseth S., Borg Å. A., Hale M. C., Slate J., Pärn H., Ringsby T. H., Sæther B.-E., Jensen H., Evidence of inbreeding depression but not inbreeding avoidance in a natural house sparrow population. Mol. Ecol. 21, 1487–1499 (2012). [DOI] [PubMed] [Google Scholar]
- 55.Hagen I. J., Billing A. M., Rønning B., Pedersen S. A., Pärn H., Slate J., Jensen H., The easy road to genome-wide medium density SNP screening in a non-model species: Development and application of a 10 K SNP-chip for the house sparrow (Passer domesticus). Mol. Ecol. Resour. 13, 429–439 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Bradnam K. R., Fass J. N., Alexandrov A., Baranay P., Bechner M., Birol I., Boisvert S., Chapman J. A., Chapuis G., Chikhi R., Chitsaz H., Chou W.-C., Corbeil J., Del Fabbro C., Docking T. R., Durbin R., Earl D., Emrich S., Fedotov P., Fonseca N. A., Ganapathy G., Gibbs R. A., Gnerre S., Godzaridis É., Goldstein S., Haimel M., Hall G., Haussler D., Hiatt J. B., Ho I. Y., Howard J., Hunt M., Jackman S. D., Jaffe D. B., Jarvis E. D., Jiang H., Kazakov S., Kersey P. J., Kitzman J. O., Knight J. R., Koren S., Lam T.-W., Lavenier D., Laviolette F., Li Y., Li Z., Liu B., Liu Y., Luo R., MacCallum I., MacManes M. D., Maillet N., Melnikov S., Naquin D., Ning Z., Otto T. D., Paten B., Paulo O. S., Phillippy A. M., Pina-Martins F., Place M., Przybylski D., Qin X., Qu C., Ribeiro F. J., Richards S., Rokhsar D. S., Ruby J. G., Scalabrin S., Schatz M. C., Schwartz D. C., Sergushichev A., Sharpe T., Shaw T. I., Shendure J., Shi Y., Simpson J. T., Song H., Tsarev F., Vezzi F., Vicedomini R., Vieira B. M., Wang J., Worley K. C., Yin S., Yiu S.-M., Yuan J., Zhang G., Zhang H., Zhou S., Korf I. F., Assemblathon 2: Evaluating de novo methods of genome assembly in three vertebrate species. GigaScience 2, 1–31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011). [Google Scholar]
- 58.Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T. L., BLAST+: Architecture and applications. BMC Bioinformatics 10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Holt C., Yandell M., MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12, 491 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yandell M., Ence D., A beginner’s guide to eukaryotic genome annotation. Nat. Rev. Genet. 13, 329–342 (2012). [DOI] [PubMed] [Google Scholar]
- 61.Lukashin A. V., Borodovsky M., GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 26, 1107–1115 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parra G., Bradnam K., Korf I., CEGMA: A pipeline to accurately annotate core genes in eukaryotic genornes. Bioinformatics 23, 1061–1067 (2007). [DOI] [PubMed] [Google Scholar]
- 63.Korf I., Gene finding in novel genomes. BMC Bioinformatics 5, 59 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stanke M., Tzvetkova A., Morgenstern B., AUGUSTUS at EGASP: Using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 7 (suppl. 1), S11.1–S11.8 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kalinowski S. T., Taper M. L., Marshall T. C., Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 16, 1099–1106 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Bidanel J.-P., Milan D., Iannuccelli N., Amigues Y., Boscher M.-Y., Bourgeois F., Caritez J.-C., Gruand J., Le Roy P., Lagant H., Quintanilla R., Renard C., Gellin J., Ollivier L., Chevalet C., Detection of quantitative trait loci for growth and fatness in pigs. Genet. Sel. Evol. 33, 289–309 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bulatova N. S., Radjabli S. I., Panov E. N., Karyological description of three species of the genus Passer. Experientia 28, 1369–1371 (1972). [Google Scholar]
- 68.Kosambi D. D., The estimation of map distances from recombination values. Ann. Hum. Genet. 12, 172–175 (1943). [Google Scholar]
- 69.Li H., Durbin R., Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.DePristo M. A., Banks E., Poplin R., Garimella K. V., Maguire J. R., Hartl C., Philippakis A. A., del Angel G., Rivas M. A., Hanna M., McKenna A., Fennell T. J., Kernytsky A. M., Sivachenko A. Y., Cibulskis K., Gabriel S. B., Altshuler D., Daly M. J., A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A., The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Korneliussen T. S., Moltke I., Albrechtsen A., Nielsen R., Calculation of Tajima’s D and other neutrality test statistics from low depth next-generation sequencing data. BMC Bioinformatics 14, 289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zheng X., Levine D., Shen J., Gogarten S. M., Laurie C., Weir B. S., A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alexander D. H., Novembre J., Lange K., Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M. A. R., Bender D., Maller J., Sklar P., de Bakker P. I. W., Daly M. J., Sham P. C., PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Matschiner M., Fitchi: Haplotype genealogy graphs based on the Fitch algorithm. Bioinformatics 32, 1250–1252 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Pfeifer B., Wittelsbürger U., Ramos-Onsins S. E., Lercher M. J., PopGenome: An efficient Swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 31, 1929–1936 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Durand E. Y., Patterson N., Reich D., Slatkin M., Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 28, 2239–2252 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Green R. E., Krause J., Briggs A. W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M. H.-Y., Hansen N. F., Durand E. Y., Malaspinas A.-S., Jensen J. D., Marques-Bonet T., Alkan C., Prüfer K., Meyer M., Burbano H. A., Good J. M., Schultz R., Aximu-Petri A., Butthof A., Höber B., Höffner B., Siegemund M., Weihmann A., Nusbaum C., Lander E. S., Russ C., Novod N., Affourtit J., Egholm M., Verna C., Rudan P., Brajkovic D., Kucan Ž., Gušic I., Doronichev V. B., Golovanova L. V., Lalueza-Fox C., de la Rasilla M., Fortea J., Rosas A., Schmitz R. W., Johnson P. L. F., Eichler E. E., Falush D., Birney E., Mullikin J. C., Slatkin M., Nielsen R., Kelso J., Lachmann M., Reich D., Pääbo S., A draft sequence of the Neandertal genome. Science 328, 710–722 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M., Kirilovsky A., Fridman W.-H., Pagès F., Trajanoski Z., Galon J., ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T., Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.L. Svensson, P. J. Grant, K. Mullarney, D. Zetterstroem, Gyldendals store fugleguide: Europas og middelhavsområdets fugler i felt (Gyldendal, 1999). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1602996/DC1
fig. S1. Stepping window ADMIXTURE and RAxML analyses across the Italian sparrow genome.
fig. S2. LD decay across all chromosomes.
fig. S3. Mitochondrial sequence divergence between each Italian sparrow and the parent populations.
fig. S4. Divergence peaks between the Italian sparrow and either parent taxa on microchromosomes.
fig. S5. Divergence peaks for PI regions on large chromosomes.
fig. S6. Population structuring analysis of sparrow individuals.
table S1. Sequencing scheme for the house sparrow reference genome assembly.
table S2. Final assembly statistics.
table S3. Sample information.
table S4. Results from genome-wide RAxML analysis.
table S5. Comparison of resolved RAxML phylogenies at variable window sizes.
table S6. ENSEMBLE gene IDs for genes within top divergence windows.
table S7. Significantly enriched GO pathways from ClueGo analysis in HI outlier windows.
table S8. Significantly enriched GO pathways from ClueGo analysis in SI outlier windows.
table S9. Significantly enriched GO pathways from ClueGo analysis in PI outlier windows.
table S10. Genes among outlier windows identified as candidates for the involvement in melanogenesis and RI in sparrows.
table S11. Significantly enriched GO pathways from ClueGo analysis HI outlier windows with chickens as the reference genome.
table S12. Significantly enriched GO pathways from ClueGo analysis in SI outlier windows with chickens as the reference genome.
table S13. Significantly enriched GO pathways from ClueGo analysis in PI outlier windows with chickens as the reference genome.