

Abstract
The universally conserved enzyme CTP synthase (CTPS) forms filaments in bacteria and eukaryotes. In bacteria polymerization inhibits CTPS activity and is required for nucleotide homeostasis. Here we show that human CTPS polymerization increases catalytic activity. The cryoEM structures of bacterial and human CTPS filaments differ dramatically in overall architecture and in the conformation of the CTPS protomer, explaining the divergent consequences of polymerization on activity. The filament structure of human CTPS is the first full-length structure of the human enzyme and reveals a novel active conformation. The filament structures elucidate allosteric mechanisms of assembly and regulation that rely on a conserved conformational equilibrium. This may provide a mechanism for increasing human CTPS activity in response to metabolic state, and challenges the assumption that metabolic filaments are generally storage forms of inactivated enzymes. Allosteric regulation of CTPS polymerization by ligands likely represents a fundamental mechanism underlying assembly of other metabolic filaments.
Many metabolic enzymes undergo dynamic rearrangement into large-scale cellular structures in response to specific metabolic cues, a subset of which form defined filamentous structures in cells.1-7 While the enzymes involved are generally well characterized by extensive study of their catalytic mechanisms, monomer structures, and regulation, the filamentous forms remain largely uncharacterized. Where functional data do exist, however, it is clear that metabolic filaments are important for regulating enzyme activity and for maintaining cellular homeostasis.8-12
Filamentous polymers of CTPS appear to be universally conserved, having been observed in bacteria, yeast, flies, and human cells.1-3,13 CTPS is a focal point for regulation of pyrimidine levels, as it directly converts UTP to CTP. We recently showed in bacteria that the product CTP drives assembly of CTPS into a filament form with lower activity8, consistent with the role of CTP as an allosteric inhibitor of CTPS. The polymer is sensitive to substrate-product balance, and can rapidly depolymerize into active tetramers in response to changes in substrate concentration. These dynamics buffer the catalytic activity of CTPS to maintain a defined ribonucleotide balance. Importantly, disruption of bacterial CTPS polymerization significantly affects cell growth and metabolism, indicating that polymerization is essential for cellular homeostasis. However, the precise molecular mechanisms of CTP-induced polymerization and inhibition of CTPS have remained unclear. Moreover, while CTPS filaments appear to be universally conserved, it is unknown whether the molecular mechanisms and functional consequences of polymerization are the same across kingdoms.
CTPS directly catalyzes the conversion of UTP to CTP. The enzyme is a homotetramer, with each monomer composed of a glutamine amidotransferase (GAT) domain and a kinase-like ammonia ligase (AL) domain, joined by an α-helical linker.14 Ammonia generated in the GAT domain is transferred to the AL domain then ligated to UTP to form CTP in an ATP hydrolysis dependent reaction, with the allosteric regulator GTP playing a role in coupling the reactions. The mechanism of ammonia transfer remains unclear, although a conformational change that alters the relative orientations of the two catalytic domains has been proposed to open an ammonia channel between the active sites15. Feedback inhibition occurs through CTP binding at a site that partially overlaps with the UTP substrate-binding site.16 The catalytic mechanism, domain structures, and the tetrameric quarternary structure of the enzyme are broadly conserved between eukaryotes and bacteria.14,15,17,18
Given that the overall structures, catalytic mechanisms, and cellular polymerization of CTPS are conserved from bacteria to humans, it has been widely assumed that the mechanisms and functional consequences of polymerization are conserved for CTPS across species19. However, very little is known about the function of CTPS polymerization in eukaryotes beyond the observation that it appears to be part of a cellular stress response and to vary with developmental stage in some organisms.4,9,10,20,21 Moreover, recent cell biological studies of CTPS polymerization in eukaryotes came to opposite conclusions about the effects of assembly on enzyme activity.9,11
Here, we use cryoEM, x-ray crystallography, and kinetic assays to study the allosteric regulation of human and E. coli CTPS filaments. We demonstrate that human CTPS filaments assemble in the presence of substrates, have increased catalytic activity, and reveal a novel active conformation of CTPS. Further, we show that both human and E. coli CTPS undergo a conserved conformational cycle controlled by substrate and product binding, but have opposite determinants for filament assembly. Given the importance of human CTPS as a target for cancer and immunosuppressive drugs and of bacterial CTPS as a target for antiparasitic treatments, these molecular insights into CTPS structure and allostery may provide novel opportunities to disrupt its function and regulation.22-26
Results
Structure of the ecCTPS filament
To elucidate the mechanism of CTP-induced filament assembly in E. coli CTPS (ecCTPS) we determined the structure of CTP and ADP-bound ecCTPS filaments at 4.6 Å resolution by cryo-electron microscopy (Fig. 1, Supplementary Fig. 1). The ecCTPS filament consists of stacked tetramers that interact through their GAT and linker domains. The tetramer interface is reorganized relative to previous ecCTPS tetramer crystal structures in the apo conformation14,16, bringing the CTPS subunits into closer contact around the bound CTP (Fig. 1c,d). It was unclear, however, whether this novel ecCTPS conformation was a cause or consequence of filament assembly. To distinguish between these possibilities we solved a new ecCTPS-CTP co-crystal structure (PDB 5TKV) that reveals a tetramer with quaternary packing nearly identical to that we observe in the filament (Fig. 1e). The fact that we observe the same tetramer conformation in the filament and in a crystal without filament assembly contacts supports the notion that CTP binding allosterically controls ecCTPS assembly by directly inducing a filament-competent conformation of ecCTPS.
Figure 1. Mechanism of ecCTPS assembly.
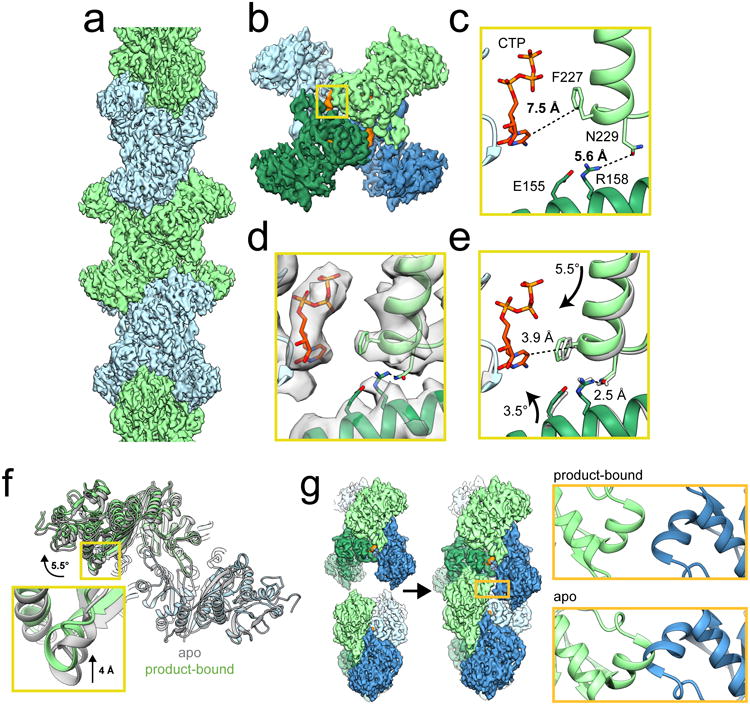
a, CryoEM reconstruction of ecCTPS filament at 4.6 Å resolution. b, A single ecCTPS tetramer from the filament, colored by protomer (blue and green) and nucleotide density highlighted in orange. c, The inhibitory CTP binding site in apo ecCTPS with CTP soaked into the crystals (PDB 2AD5). d, The inhibitory CTP binding site in the cryoEM structure shows compaction of the tetramer around CTP. e, Overlay of the filament structure (gray) with the structure of ecCTPS co-crystallized with CTP (color). Arrows indicate the rotation of subunits relative to the apo conformation in c. f, Overlay of the apo ecCTPS crystal structure (gray) with the CTP-bound filament structure (color). The protomers on the lower right are superposed, revealing a rotation of 5.5° and a 4 Å translation in the positions of the protomers on the upper left. g, Filament assembly contacts between ecCTPS tetramers can only be made in the novel CTP-bound conformation, as superposition of two apo tetramers would result in backbone clashing at the GAT contact site.
The conformational changes between this novel tetramer conformation and the apo state are almost entirely rigid body motions, with the subunits in each state nearly the same (<0.5 Å Cα RMSD), with one exception: helix 218-228 is shifted 3.6 Å closer to the CTP bound on an adjacent monomer so that Phe227 packs against the CTP base. This shift also repositions Asn229 at the base of helix 218-228, creating a new hydrogen bond network with Arg158 and Glu155 across the tetramer interface, likely stabilizing the filament conformation (Supplementary Fig. 2). We previously reported that E155K, a mutation associated with drug resistance through relief of CTPS inhibition27, cannot assemble filaments8. Our new structure suggests this Glu to Lys mutation interferes with the hydrogen bond network that occurs across the tetramer interfaces in the inhibited conformation. These changes alter the relative orientations of the polymerization interfaces and position them to allow assembly only in the CTP-bound state. (Fig. 1f,g, Supplementary Videos 1 & 2).
Allosteric inhibition of ecCTPS in the filament
We next sought to establish the mechanism of polymerization-induced inhibition of ecCTPS. We considered three possibilities: substrates are occluded from the active site by assembly contacts, the filament sterically blocks a conformational change necessary for ammonia transfer, or the conformation of CTPS in the filament is allosterically inhibitory. To test these possibilities we first sought to decouple ecCTPS polymerization from CTP binding by engineering cysteine disulfide crosslinks at filament assembly interfaces (ecCTPSCC). Under non-reducing conditions ecCTPSCC spontaneously polymerizes without nucleotides, and its structure is identical to the wildtype structure minus nucleotides (Fig. 2a-d). Under reducing conditions unassembled ecCTPSCC is as active as the wildtype enzyme, but apo ecCTPSCC filaments preassembled under non-reducing conditions exhibit a 5-fold reduction in activity relative to free tetramers, indicating that filament assembly directly inhibits enzymatic activity (Fig. 2e).
Figure 2. Engineered disulfides drive ecCTPS assembly and inhibit activity.
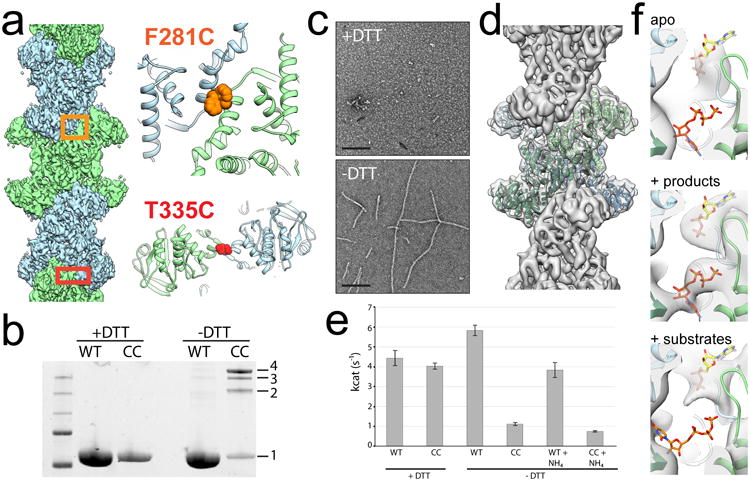
a, Design of the ecCTPSCC construct, showing the locations of cysteine mutations at F281 and T335 in the linker-linker and GAT-GAT interfaces, respectively. b, SDS-PAGE gel of ecCTPS WT and CC construct under reducing and non-reducing conditions. Under non-reducing conditions crosslinks are introduced between protomers resulting in dimers (2), trimers (3), and tetramers (4). c, Negative stain images of apo ecCTPSCC under reducing (+DTT) and non-reducing (-DTT) conditions. d, CryoEM reconstruction of ecCTPSCC at 5.7 Å resolution. e, The activity of ecCTPS WT and CC constructs were measured under reducing (+DTT) and non-reducing (-DTT) conditions, using either glutamine or ammonium phosphate (+NH4) as substrates. Values are averages of triplicate experiments +/- s.d. f, The active site cryoEM density from filament structures with no nucleotides present (apo), with products soaked in to pre-formed filaments, or substrates soaked in to pre-formed filaments. Clear density can be seen for products, while there is no density in the overlapping CTP and UTP site when substrates are added, suggesting the filament conformation is intrinsically inhibited.
When products (CTP and ADP) are soaked into pre-assembled ecCTPSCC filaments clear density is observed for the nucleotides in cryoEM reconstructions, ruling out nucleotide occlusion as a mechanism of inhibition (Fig. 2f). To test whether filaments impair ammonia transfer between active sites we tested the activity of ecCTPSCC filaments after adding ammonia directly as a substrate. Under these conditions ecCTPSCC exhibited the same reduction in activity, indicating that inhibition within the filament is not solely the result of an inability to couple the GAT and AL reactions. Finally, we tested whether ecCTPSCC filaments could bind substrates (UTP and ATP) by soaking preassembled filaments with saturating concentrations of nucleotides. Under these conditions, no density is observed for UTP in the active site (Fig. 2f), suggesting reduced affinity for substrates in the filament conformation. Together, these results indicate that polymerization allosterically regulates ecCTPS activity by stabilizing an intrinsically lower activity state upon incorporation into filaments, independent of CTP binding.
Substrates drive polymerization of hCTPS
Surprisingly, we found that purified human CTP synthase 1 (hCTPS1) polymerizes in the presence of UTP and ATP substrates, but not in the presence of CTP and ADP products, exactly the opposite behavior observed with ecCTPS (Fig. 3a). hCTPS1 filaments were stable in the presence of UTP, ATP, and GTP, but disassembled over time upon addition of glutamine, presumably due to accumulation of CTP product from the complete synthesis reaction (Supplementary Fig. 3a). Indeed, adding CTP directly to filaments assembled with substrates also resulted in disassembly, while filaments assembled with UTP, the nonhydrolyzable ATP analog AMPPNP, and GTP were stable following glutamine addition, owing to a lack of CTP synthesis (Supplementary Fig. 3b,c).
Figure 3. hCTPS1 filaments assemble with substrates and are catalytically active.
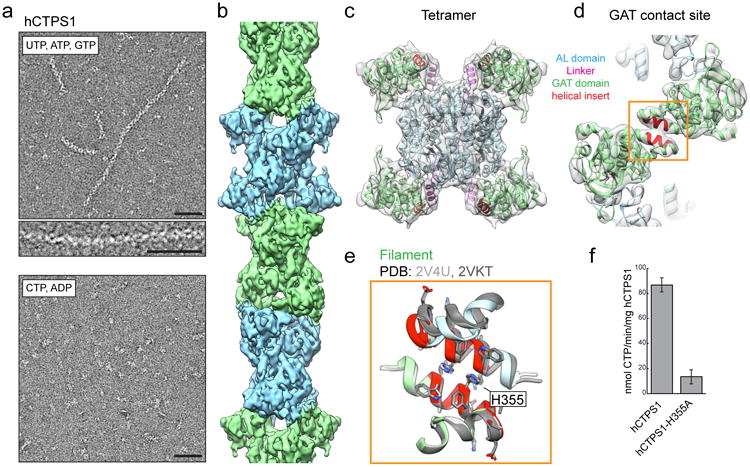
a, hCTPS1 polymerizes in the presence of substrates, but not in the presence of products. b, 6.1Å cryoEM map of the hCTPS1 filament, colored by tetramer subunit. c, Model of the hCTPS1 tetramer fit into the cryoEM map, colored by domain. d, GAT domain contact site in the hCTPS1 filament, with the eukaryotic helical insert shown in red. e, The hCTPS1 filament GAT contact site (color) is also observed in crystal structures of the hCTPS2 GAT domain (grey; PDB 2V4U and 2VKT). f, Wild-type hCTPS1 is more active than the hCTPS1-H355A non-polymerizing mutant. Values are averages of triplicate experiments +/- s.d. Scale bars are 50 nm.
hCTPS filament structure and activity
To understand how the universally conserved CTPS enzyme could have opposite determinants for filament assembly in bacteria and humans, we solved the cryoEM structure of hCTPS1 filaments assembled in the presence of substrates UTP and ATP and the allosteric effector GTP at 6.1 Å resolution (Fig. 3b,c, Supplementary Fig. 4). The UTP, ATP, and GTP combination was selected for structure determination as it gave the most robust polymerization of hCTPS1. Like ecCTPS, hCTPS1 is composed of stacked tetramers, but beyond this the structures differ in their assembly contacts, tetramer interfaces, and protomer conformations. hCTPS1 filament assembly is mediated primarily by an insert in the GAT domain that appeared early in eukaryotic evolution (Fig. 3d, Supplementary Fig. 5a). This GAT-GAT assembly interaction, which is also observed in the crystal packing of existing structures of isolated hCTPS1 GAT domains (Fig. 3e), uses a completely different interface from the E. coli enzyme and accounts for the drastic differences in filament architecture between species. There may be additional assembly contacts between C-terminal densities that are predicted to be disordered and are poorly resolved in the cryoEM structure (Supplementary Fig. 4g). Like ecCTPS, hCTPS1 assembly is allosterically controlled, but in this case a novel substrate-bound conformation drives polymerization.
To test the biochemical consequences of polymerization, we mutated a single conserved histidine in the hCTPS1 GAT domain helical insert (H355A). Unlike the wild-type enzyme, hCTPS1-H355A completely failed to polymerize in the presence of UTP, ATP, and GTP (Fig. 3a, Supplementary Fig. 5b). A negative stain reconstruction of hCTPS1-H355A under these conditions confirmed that the overall tetramer structure, which is necessary for CTPS activity, was not affected by the H355A mutation (Supplementary Fig. 5c). However, the H355A mutation led to a 6-fold reduction in hCTPS1 activity (Fig. 3f), indicating that hCTPS1 activity is enhanced upon assembly into filaments, likely because it is locked in a more active conformation. Assembly of active polymers is in contrast with the suggestion from previous work, including our own description of the ecCTPS filament, that these structures generally function as inactive reservoirs of metabolic capacity1,8,10,20. Rather, in some instances, it seems that there may be more complex regulatory schemes in which large-scale polymerization functions to boost enzyme activity.
The hCTPS1 filament structure provides the first view of any CTPS in the substrate-bound, active state, which differs from all existing CTPS structures in both the tetramerization interface and the conformation of the protomer (Fig. 4). Density for ATP and UTP nucleotide substrates is clearly visible in the cryoEM map, indicating the hCTPS1 filaments are primed for catalysis upon addition of glutamine (Fig. 4b). This is consistent with the observation that metabolic filaments assemble in response to glutamine deprivation in cells21. Compared with CTPS structures in the inactive state, including the crystal structure of the hCTPS1 AL domain tetramer, which exhibits analogous packing to 5TKV helices 221-228 and 149-164, the active hCTPS1 tetramer is extended along the filament axis and compressed perpendicular to the filament axis, by approximately 6Å and 5Å, respectively (Supplementary Videos 3 &4), owing to rearrangements of the tetramer interface (Fig. 4c).
Figure 4. hCTPS1 filaments reveal the active conformation of CTP synthase.
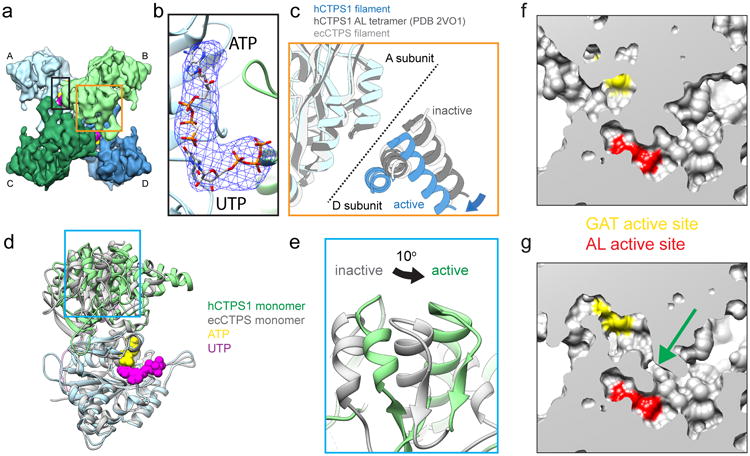
a , CryoEM structure of the hCTPS1 tetramer in the active, filament conformation. Protomers A-D are shown in different colors. b, Zoomed-in view of the black box in panel a, showing the active site with atomic models fit into the cryoEM map. A difference map (blue mesh) calculated between an apo model of the hCTPS1 filament and the hCTPS1 filament cryoEM map reveals strong density in the active site, which was modeled to UTP and ATP. c, Zoomed-in view of the orange box from panel a, showing the tetramer interface with three structures aligned on the AL domain of subunit A. The active hCTPS1 structure is extended across the tetramer interface when compared with structures of the inhibited ecCTPS filament as well as the hCTPS1 AL domain tetramer (PDB 2VO1). d, Atomic models of monomers fit to the active hCTPS1 filament (color) and the inhibited ecCTPS filament (grey), aligned on the AL domain. In the active hCTPS1 conformation, the GAT domain is rotated relative to the AL domain, bringing the two active sites closer together. e, Zoomed-in view of the blue box in panel d, showing the 10° rotation of the GAT domain in the active hCTPS1 structure. f,g Comparison of surface representations of the atomic models of hCTPS1 in the inhibited (f) and active (g) conformations reveals an opening (green arrow) between the GAT (yellow) and AL (red) active sites.
Within each hCTPS1 protomer there is a large conformational change arising from an approximately 10° rotation between the GAT and AL domains relative to all previously reported full-length CTPS structures (Fig. 4d,e). The rotation is necessary to position the GAT domains for polymerization. In addition, the rotation appears to align two cavities within the GAT and AL domains to form a tunnel. The resolution of our cryo-EM structure is insufficient to clearly describe the nature of this tunnel, but it may function to facilitate ammonia transfer between the two active sites, similar to ammonia tunnels described in other enzymes28 (Fig. 4f,g). This would be consistent with earlier predictions that a conformational change is required for ammonia transfer and for coupling the GAT and AL reactions14,15.
CTPS conformational states are conserved across kingdoms
We hypothesized that the conformation observed in the hCTPS1 filament is a generally conserved active conformation of the enzyme. However, as there were no previous full-length structures of hCTPS1, we could not rule out the possibility that the unique conformation was specific to the human enzyme. To determine whether ecCTPS adopts the same conformation on substrate binding we determined the structure of the ecCTPS tetramer bound to UTP and AMPPNP by cryoEM at 8 Å (Fig. 5a,b). The structure has the same overall conformation as the active hCTPS1 filament, both at the tetramer interface (Fig. 5c, Supplementary Videos 3 &4) and in the rotation between the AL and GAT domains (Fig. 5d,e), confirming that the conformational states of CTPS are conserved from bacteria to humans.
Figure 5. ecCTPS and hCTPS1 undergo a conserved conformational cycle controlled by substrate and product binding.
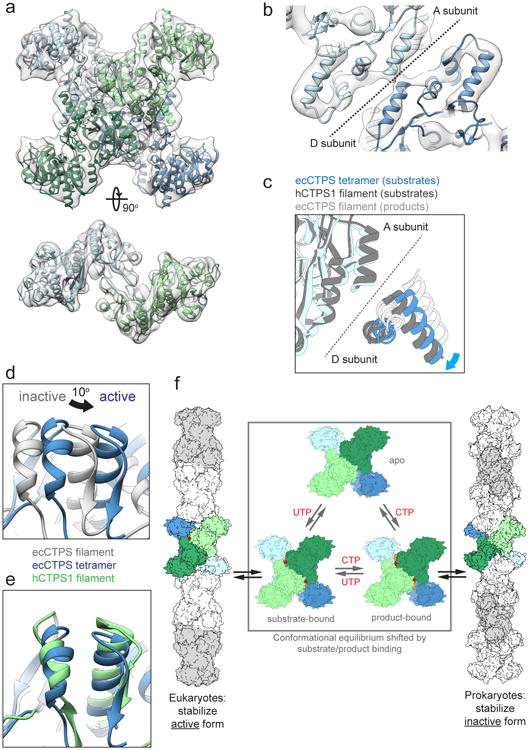
a , 8Å cryoEM map of substrate-bound ecCTPS (grey), fit with an atomic model (colored by protomer). b, The tetramer interface in the ecCTPS cryoEM map (grey) fit with an atomic model (color). Individual helices are clearly visible. c, Comparison of the tetramer interface in the active ecCTPS tetramer conformation (blue), active hCTPS1 filament conformation (dark grey), and inactive ecCTPS1 filament conformation (light grey). Corresponds to the same view shown in Figure 4c. d, 10° rotation of the ecCTPS GAT domain in the active tetramer conformation (blue) relative to the inactive filament conformation (grey). Corresponds to the same view shown in Figure 4e. e, The same view as panel d, comparing the active ecCTPS (blue) and hCTPS1 (green) GAT domain rotations. f, Model for the allosteric regulation of CTPS filament assembly. ecCTPS and hCTPS1 undergo a conserved conformation cycle, but have opposite determinants for filament assembly.
Discussion
The structures of human and bacterial CTPS filaments suggest a model for the allosteric regulation of CTPS polymerization. The conformational equilibrium between apo, substrate-bound, and product-bound states of the CTPS tetramer is universally conserved, while evolutionary divergence at polymerization interfaces has reversed the determinants for filament assembly. The inactive, product-bound conformation is stabilized in bacterial filaments, while the active, substrate-bound conformation is stabilized in eukaryotic filaments (Fig. 5f). For both filament types polymerization may mediate a more cooperative transition between high and low activity states, providing more switch-like behavior in response to small changes in substrate and product concentrations.8 What remains unclear is the relative advantage of stabilizing hCTPS in an active conformation in the polymer. One possibility is that stabilizing the polymer under cellular stress conditions keeps the bulk of hCTPS in a state primed for maximal activity immediately on return to normal growth conditions.
Both hCTPS and ecCTPS polymerize through association of their GAT domains. A short helical insertion at this interface both provides the primary assembly interface in hCTPS and prevents hCTPS from assembling with ecCTPS-like contacts (Fig. 4d,e, Supplementary Figure 4f). The approximately 10-residue insertion appeared during early eukaryotic evolution, as only a few early-diverging single-celled lineages lack the insert. This raises the interesting question of how the two different filament forms evolved. One possibility is that CTPS of early eukaryotes formed inhibitory filaments like those of bacteria, and the appearance of the GAT insert switched the filaments to an active state. An alternative is that CTPS polymerization arose independently in bacterial and eukaryotic lineages, so that the appearance of polymerization to be a deeply conserved feature of CTPS actually reflects the independent evolution of CTPS filaments. This may be the more likely scenario, as the appearance of filamentous forms of so many different metabolic enzymes in many different lineages suggests this is a relatively common evolutionary strategy. Investigating the role of polymerization in earlier diverging eukaryotes, many of them human pathogens, will be necessary to clarify the evolutionary history of CTPS filaments.
For the many different metabolic enzymes that form filaments it seems likely that in many cases, like CTPS, binding of substrates or other ligands allosterically regulates assembly. Moreover, the ability to interconvert between dispersed and polymeric enzyme forms with different intrinsic activities may provide a general mechanism for regulating or localizing metabolic activity under complex cellular conditions. For example, CTPS activity is also regulated by phosphorylation29-32, raising the possibility that post-translational modifications could modulate CTPS activity by influencing filament assembly, shifting the equilibrium between polymers and free tetramers to tune the total level of enzyme activity independent of substrate and product concentrations. Similarly, binding partners may influence assembly or disassembly of CTPS filaments. This would be analogous to cytoskeletal systems like actin filaments and microtubules, where substrate binding and hydrolysis drive intrinsic polymer dynamics but multiple factors interact to influence the timing and location of assembly. Indeed, CTPS filaments co-localize with other metabolic and signaling enzymes4,9, suggesting that cellular factors may specifically recognize and interact with metabolic polymers.
Several cell biological studies have shown that eukaryotic CTPS filaments assemble in response to nutrient stress and at particular developmental stages.4,9-11,20,21 Although some of these studies have suggested that eukaryotic CTPS filaments may be composed of catalytically inactive dimers11,20, our study demonstrates that isolated human CTPS filaments are polymers of active, substrate-bound tetramers. Consistent with our results, CTPS in Drosophila germ cells forms filaments at developmental stages with a high demand for CTP, and a constitutively active mutant of human CTPS forms filaments under conditions where the wild-type protein is diffuse throughout the cytoplasm.9 Experiments in human cells have also shown that metabolic filaments assemble in response to glutamine deprivation, and disassemble upon glutamine addition21, an effect which we observe here with purified hCTPS1.
Phosphorylation at various sites is known to modulate eukaryotic CTPS activity, leading to changes in cellular concentrations of CTP and phospholipids.29,31,32 Multiple phosphorylation sites are located in the C-terminal tail of hCTPS130, which may form assembly contacts in the hCTPS1 filament. It will therefore be interesting to investigate whether or not phosphorylation has a direct effect on hCTPS1 polymerization, and how enzyme regulation by polymerization is integrated into other regulatory pathways. The CTPS filament structures presented here will provide a mechanistic basis for future investigations of the cellular consequences of polymerization.
Methods
Purification of ecCTPS
Wild-type ecCTPS was purified as described previously.3,14 The ecCTPSCC mutant was purified in the same manner, except 10 mM DTT was included throughout the purification.
Purification of hCTPS1
hCTPS1 was expressed in S. cerevisiae strain GHY55, as described by Han et al. 2005.32 This strain lacks the endogenous S. cerevisiae CTPS genes URA7 and URA8, and contains plasmid pDO105-hCTPS1, which directs expression of 6His-tagged (C-terminal) hCTPS1 from the ADH1 promoter. GHY55 cells were grown in 4× YPD media and harvested by freezing cell pellets in liquid nitrogen. Cell pellets were ground to a powder while frozen, and 15g of cell powder was resuspended in 100 mL of lysis buffer (50 mM Tris-HCl, 200 mM NaCl, 0.3M sucrose, 20 mM imidazole, 0.5 mM PMSF, pH 8.0). Lysates were clarified by centrifugation at 14,000 RPM for 40 minutes at 4°C in a Thermo Scientific Fiberlite F14-14×50cy rotor. Clarified lysates were applied to a 5mL HisTrap FF Crude column (GE) on an ÄKTA Start chromatography system (GE), and the column was washed with 30 column volumes (CV) wash buffer (20 mM Tris-HCl, 0.5M NaCl, 45 mM imidazole, 10% glycerol, pH 7.9). Protein was eluted as 1mL fractions with 5CV elution buffer (20 mM Tris-HCl, 0.5M NaCl, 250 mM imidazole, 10% glycerol, pH 7.9). Fractions containing hCTPS1 were pooled and dialyzed into storage buffer (20 mM Tris-HCl, 0.5M NaCl, 10% glycerol, 7 mM β-mercaptoethanol, pH 7.9) using Snakeskin 3500 MWCO dialysis tubing (Thermo Scientific). Dialyzed protein was concentrated ∼6-fold by centrifugation in a 3 kDa cut-off centrifugal filter unit (Millipore). hCTPS1 prepared for cryoEM was purified in the same manner, except protein was concentrated prior to dialysis, and then dialyzed into 20 mM Tris-HCl, 50 mM NaCl, 7 mM β-mercaptoethanol, pH 7.9. The hCTPS1-H355A mutant was generated by site-directed mutagenesis of plasmid pDO105-hCTPS1, and purified in the same manner as wild-type hCTPS1.
Site-directed mutagenesis
Site-directed mutagenesis of the E. coli and human CTPS expression constructs was performed using the QuickChange (Agilent Technologies) and Q5 (NEB) systems, respectively.
CTPS activity assays
CTPS activity was determined at 37°C by measuring Δotimes;Abs291nm over time using a NanoDrop 2000c spectrophotometer (Thermo Scientific). Absolute CTP production was calculated using the change in extinction coefficient between UTP and CTP at 291 nm (1338 M-1cm-1).33 Kcat values were determined from the initial linear region of the Abs291nm versus time measurements. For ecCTPS, assays were performed with 0.5-2.5 μM ecCTPS or ecCTPSCC in a standard reaction buffer containing 50 mM Na-HEPES (pH 8.0) and 10 mM MgCl2, with or without 10 mM DTT as appropriate. Final substrate concentrations were 0.6 mM UTP, 1.5 mM ATP, 0.2 mM GTP and 10 mM glutamine or ammonia. Assays were carried out using the “annealing” method described by Habrian, et al34: ecCTPS was incubated at 21°C for 3 minutes, and then combined with pre-warmed (37°C) nucleotides. The enzyme-nucleotide mix was then incubated for a further 4 minutes at 37°C, after which pre-warmed glutamine or ammonia was added. The complete reaction mixture was then immediately transferred to a pre-warmed UV cuvette and Δotimes;Abs291nm over time measured at 37°C. For hCTPS1, assays were performed with 8 μM hCTPS1 in a standard reaction buffer containing 20 mM Tris-HCl (pH 7.9) and 10 mM MgCl2, with final substrate concentrations of 2 mM UTP, 2 mM ATP, 0.2 mM GTP, and 10 mM glutamine. The annealing method described above was also used for hCTPS1 activity assays, except the enzyme-nucleotide mix was incubated for 1 hour at 37°C to allow for hCTPS1 polymerization.
Negative stain EM
Samples for negative stain EM were prepared by applying CTPS to carbon-coated grids and staining with 0.7% uranyl formate. For ecCTPSCC, protein was dialyzed overnight into 50 mM Na-HEPES (pH 8.0) with or without 10 mM DTT before being coated onto grids at a final concentration of 2.5 μM. For hCTPS1 and hCTPS1-H355A, samples were prepared with 2.5 μM protein in 20 mM Tris-HCl (pH 7.9) and 10 mM MgCl2, supplemented with appropriate nucleotides at the following concentrations: UTP (2 mM), ATP (2 mM), AMP-PNP (2mM), GTP (0.2 mM), ADP (2 mM), and CTP (2 mM). hCTPS1 was incubated with nucleotides for 1 hour at 37°C before being coated onto grids. For depolymerization experiments, glutamine (10 mM) was added following incubation with nucleotides, and negative stain samples were prepared 30 and 120 minutes following glutamine addition. Electron microscopy was performed on a Tecnai G2 Spirit (FEI co.) operating at 120 kV, and images were acquired at 52,000× magnification on a US4000 4k × 4k CCD camera (Gatan, Inc.).
CryoEM
Samples for cryoEM were prepared by applying protein to glow-discharged C-Flat holey-carbon grids (Protochips Inc.), blotting with a Vitrobot (FEI co.), and rapidly plunging into liquid ethane. For ecCTPSCC, protein was dialyzed into non-reducing buffer (50 mM Na-HEPES pH 8.0) to promote filament assembly. Samples for cryoEM were then prepared with 2.5 μM ecCTPSCC in the absence of nucleotides or following 1 hour incubation of filaments with 10 mM MgCl2 in addition to UTP (0.6 mM) and ATP (1.5 mM) or CTP (0.6 mM) and ADP (1.5 mM). hCTPS1 (5 μM) was incubated for 1 hour with 10 mM MgCl2, 2 mM UTP, 2 mM ATP, and 0.2 mM GTP before preparing cryoEM samples. For ecCTPS tetramers, 5 μM ecCTPS was incubated with 10 mM MgCl2, 0.6 mM UTP, 1.5 mM AMP-PNP, 0.2 mM GTP and 10 mM glutamine before preparing cryoEM samples. CryoEM data for the wildtype ecCTPS filament were acquired in a Tecnai Polara operating at 300 kV and recorded on a K2 Summit Direct Detect camera with a total dose of ∼34 e-/Å2 with 36 frames per exposure. For all other samples, cryoEM data was collected on a Tecnai G2 F20 (FEI co.) operating at 200 kV with a K2 Summit Direct Detect camera (Gatan Inc.) with a pixel size of 1.26 Å/pixel. Movies were acquired in counting mode with 36 frames and a total dose of ∼45 e-/Å2 (filaments) or ∼68 e-/Å2 (ecCTPS tetramers), and with a defocus range between 0.8 and 2.5 μm (filaments) or 1.0 and 4.5 μm (ecCTPS tetramers). Leginon software35 was used to automate data collection.
Image processing and 3D reconstruction
Movie frames were aligned using DOSEFGPU DRIFTCORR36 and CTF parameters were estimated using CTFFIND337. For helical samples, lengths of helices were manually defined using Appion38 manual picker, and overlapping segments were extracted along the length of each helix. Particle stacks were CTF corrected by phase flipping in SPIDER39, using parameters determined by CTFFIND3. 3D reconstruction of helices was performed by iterative helical real space reconstruction (IHRSR)40,41 in SPIDER, using hsearch_lorentz42 to refine helical symmetry parameters. Cylinders were used as starting models, and the D2 point group symmetry of the CTPS tetramer was enforced. Iterative gold-standard refinement was performed with increasingly smaller angular increments (minimum 1.5°). For single particle (tetramer) samples, particles were picked automatically using DoG Picker43 and then extracted using Appion. Relion44 was used for CTF correction (using CTFFIND3 parameters) and subsequent classification and refinement. We performed reference-free 2D classification, and selected 7700 and 39000 particles for 3D classification of the hCTPS1-H355A and ecCTPS tetramers, respectively. For 3D classification, initial reference volumes were generated from the homology model of the hCTPS1 tetramer or crystal structure of the product-bound ecCTPS tetramer. Both reference volumes were low-pass filtered to 60Å, and D2 symmetry was enforced during 3D classification. Following 3D classification, 4400 and 6400 particles were selected for hCTPS1-H355A and ecCTPS, respectively, for gold-standard 3D refinement. Again, reference volumes were low-pass filtered to 60Å and D2 symmetry was enforced during 3D refinement. The gold-standard FSC=0.143 criterion was used for estimating resolution. Raw volumes were amplitude corrected and low and high-pass filtered using SPIDER. Details of each 3D reconstruction are summarized in Table 1.
Table 1. EM data collection and refinement statistics.
| ecCTPS filament (EMD-8504, PDB 5U3C) | hCTPS1 filament (EMD- 8474 PDB 5U03) | ecCTPS tetramer (EMD- 8475 PDB 5U05) | ecCTPS-CC (products)(EMD-8513, PDB 5U6R | ecCTPS-CC (substrates)(EMD-8491) | ecCTPS-CC (apo)(EMD-8490) | hCTPS1-H355A tetramer (EMD- 8476) | |
|---|---|---|---|---|---|---|---|
| Data collection | |||||||
| Microscope | Polara | TF20 | TF20 | TF20 | TF20 | TF20 | Spirit |
| Voltage (kV) | 300 | 200 | 200 | 200 | 200 | 200 | 120 |
| Detector | K2 summit | K2 summit | K2 summit | K2 summit | K2 summit | K2 summit | Ultrascan CCD |
| Electron exposure (e–/Å2) | 34 | 45 | 68 | 45 | 45 | 45 | 30 |
| Pixel size (Å) | 1.22 | 1.26 | 1.26 | 1.26 | 1.26 | 1.26 | 2.07 |
| Reconstruction | |||||||
| Point group symmetry | D2 | D2 | D2 | D2 | D2 | D2 | D2 |
| Refined helical symmetry | 48.5°, 81.6 Å | 60.6°, 104.1 | 48.2°, 82.1Å | 50.6°, 83.5Å | 48.2°, 82.3 Å | ||
| Particles | 8622 | 24880 | 6407 | 31170 | 7135 | 13715 | 4413 |
| Resolution (0.143 fsc) (Å) | 4.6 | 6.1 | 7.9 | 5.7 | 8.6 | 7.6 | 17 |
| Refinement | |||||||
| Initial model used (PDB code) | 2AD5 | 2VO1, 2VKT | 2AD5 | 2AD5 | |||
| Model composition | |||||||
| Protein residues | 534 | 559 | 534 | 534 | |||
| Ligands | CTP, ADP, Mg | UTP, ATP, Mg | CTP, ADP, Mg | ||||
| Validation | |||||||
| Clashscore | 2 | 4 | 14 | 2 | |||
| Poor rotamers (%) | 0.1 | 0.2 | 0.1 | 0.1 | |||
| Ramachandran plot | |||||||
| Favored (%) | 94 | 96 | 94 | 94 | |||
| Allowed (%) | 4 | 4 | 4 | 4 | |||
| Outliers (%) | 1 | 1 | 1 | 1 |
Building and fitting of atomic models
An ecCTPS monomer (PDB 2AD5) was initially fit as a single rigid body into the cryo-EM structure of the filament. For the human filament an initial homology model of the full-length hCTPS monomer was generated from the crystal structures of the individual domains (PDB 2VO1 and 2VKT) aligned to ecCTPS (PDB 2AD5) using MODELLER45, then fit to cryoEM density as three rigid bodies: GAT domain, linker domain, and ALase domain. For both structures atomic models were refined with the Rosetta Relax application46, using the cryoEM density as a constraint. Adjacent protomers within the tetramer and at filament contacts sites were included during the Relax procedure, to allow for refinement at subunit interfaces.
X-ray structure of an alternate CTP-inhibited ecCTPS conformation
N-terminal His-tagged ecCTPS mutant C268A was expressed and purified using metal chelate chromatography as previously described.16 ecCTPS(C268A) had identical kcat and CTP inhibition as wildtype. Protein for crystallization was stored in 10 mM Tris-Cl, 0.5 mM TCEP, pH 8.0 at -80°C at 10-15 mg/mL. P21212 crystals isomorphous to the published apo- and CTP/ADP-liganded structures (1S1M and 2AD5) were grown using vapor diffusion from 7.5 mg/mol EcCTPS-C268A, 0.8-1.2 M ammonium sulfate, 0.1 M Tris-Cl, pH 8.0 as previously described14 with drops supplemented with 5 mM CTP, 5 mM magnesium sulfate and 10 mM glutamine. For cryoprotection, crystals were briefly rinsed in 1:1 mixture of 50% MPD:mother liquor (25% MPD final), wicked and flash-cooled in liquid nitrogen. Reflection data were acquired at Stanford Synchrotron Radiation Lightsource Beamline 1-5 at 100°K at a wavelength of 0.979610 Å and 1° oscillation/image, using a ADSC Quantum 315 detector. Eighty eight frames were processed using DENZO/Scalepack (HKL2000 package). Phases were derived from the water-free published CTP and ADP complex structure (PDB 2AD5). Maximum-likelihood structure factor refinement was carried out on non-hydrogen atoms using Refmac 5.6 with Babinet scaling with an anisotropic B-factor correction (BSOL fixed at 125.00 and final anisotropic scaling parameters B11 = -1.82 B22 = 2.83 B33 = -1.01 B12 = 0.00 B13 = 0.00 B23 = 0.00). Quaternary changes were accounted for by initial rigid body refinement for individual monomers and then for three separate domains of each monomer (1-266, 267-287, 288-544). Cycles of manual model building with Coot 0.6.2 and combined positional and B-factor refinement led to the final model. The distribution of favored, allowed and outlier Ramachandran angles were 94%, 5% and 1%, respectively (51st percentile for structures at this resolution). The data collection and final model statistics are given in Table 2.
Table 2. X-ray data collection and refinement statistics.
| ecCTPS CTP-bound (PDB 5TKV) | |
|---|---|
| Data collection | |
| Space group | P21212 |
| Cell dimensions | |
| a, b, c (Å) | 159.16, 110.68, 129.49 |
| α, β, γ (°) | 90.00, 90.00, 90.00 |
| Resolution (Å) | 30.0 - 2.70 (2.82 - 2.70)a |
| Rsym or Rmerge (HKL2000) | 0.092 (0.310) |
| I / σI | 11.2 (3.1) |
| Completeness (%) | 98.3 (99.0) |
| Redundancy | 3.4 (3.2) |
| Refinement (Refmac 5.6) | |
| Resolution (Å) | 29.8 - 2.70 |
| No. reflections | 59184/3159 |
| Rwork / Rfree | 0.158/0.208 |
| No. atoms | 8827 |
| Protein | 8305 |
| Ligand/ion (CTP/Mg) | 158 |
| Water | 364 |
| B-factors (Å2) | |
| Protein | 46 |
| Ligand/ion (CTP/Mg) | 43 |
| Water | 47 |
| R.m.s. deviations (Refmac5.6) | |
| Bond lengths (Å) | 0.0156 |
| Bond angles (°) | 1.94 |
Single crystal. Values in parentheses are for highest-resolution shell.
Sequence comparison
CTPS sequences were obtained from BLAST searches47, and multiple sequence alignments of several hundred sequences was performed with MAFFT48.
Data availability
Coordinates and structure factors for the CTP-inhibited ecCTPS crystal structure have been deposited in the Protein Data Bank under accession code PDB 5TKV. EM structures and associated atomic models have been deposited in the Electron Microscopy Data Bank and Protein Data Bank with the following accession codes: ecCTPS filament (EMD-8504; PDB 5U3C), hCTPS1 filament (EMD-8474; PDB 5U03), hCTPS1-H355A tetramer (EMD-8476), ecCTPS tetramer (EMD-8475; PDB 5U05), ecCTPS-CC (apo) (EMD-8490), ecCTPS-CC (substrates) (EMD-8491), ecCTPS-CC (products) (EMD-8513; PDB 5U6R). Other data supporting this study are available from the corresponding authors upon reasonable request.
Supplementary Material
Acknowledgments
The authors are grateful to G. Carman (Rutgers University, United States) for the hCTPS1-expressing S. cerevisiae strain. This work was supported by the US National Institutes of Health (R01 GM118396 to J.M.K.) and the Human Frontier Science Program (RGY0076/2013 to Z.G. and J.M.K.). Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Footnotes
Contributions: E.M.L. and J.M.K. wrote the manuscript. E.M.L., J.M.K, and D.R.H. solved the EM structures. J.A.E. and E.P.B. solved the crystal structure of ecCTPS. M.S. and J.M.H. generated disulfide cross-linked ecCTPS constructs and optimized purification and assembly conditions. E.M.L. and A.M carried out CTPS activity assays. R.M.B. and Z.G. performed initial characterization of ecCTPS filaments. E.M.L., Z.G., E.P.B. and J.M.K. contributed to experimental design and data analysis and interpretation.
Competing Financial Interests: The authors declare no competing financial interests.
References
- 1.Noree C, Sato BK, Broyer RM, Wilhelm JE. Identification of novel filament-forming proteins in Saccharomyces cerevisiae and Drosophila melanogaster. J Cell Biol. 2010;190:541–51. doi: 10.1083/jcb.201003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu JL. Intracellular compartmentation of CTP synthase in Drosophila. J Genet Genomics. 2010;37:281–96. doi: 10.1016/S1673-8527(09)60046-1. [DOI] [PubMed] [Google Scholar]
- 3.Ingerson-Mahar M, Briegel A, Werner JN, Jensen GJ, Gitai Z. The metabolic enzyme CTP synthase forms cytoskeletal filaments. Nat Cell Biol. 2010;12:739–46. doi: 10.1038/ncb2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carcamo WC, et al. Induction of cytoplasmic rods and rings structures by inhibition of the CTP and GTP synthetic pathway in mammalian cells. PLoS One. 2011;6:e29690. doi: 10.1371/journal.pone.0029690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen QJ, et al. Filamentation of Metabolic Enzymes in Saccharomyces cerevisiae. J Genet Genomics. 2016;43:393–404. doi: 10.1016/j.jgg.2016.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Narayanaswamy R, et al. Widespread reorganization of metabolic enzymes into reversible assemblies upon nutrient starvation. Proc Natl Acad Sci U S A. 2009;106:10147–52. doi: 10.1073/pnas.0812771106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.An S, Kumar R, Sheets ED, Benkovic SJ. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science. 2008;320:103–6. doi: 10.1126/science.1152241. [DOI] [PubMed] [Google Scholar]
- 8.Barry RM, et al. Large-scale filament formation inhibits the activity of CTP synthetase. Elife. 2014;3:e03638. doi: 10.7554/eLife.03638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strochlic TI, et al. Ack kinase regulates CTP synthase filaments during Drosophila oogenesis. EMBO Rep. 2014;15:1184–91. doi: 10.15252/embr.201438688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petrovska I, et al. Filament formation by metabolic enzymes is a specific adaptation to an advanced state of cellular starvation. Elife. 2014 doi: 10.7554/eLife.02409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noree C, Monfort E, Shiau AK, Wilhelm JE. Common regulatory control of CTP synthase enzyme activity and filament formation. Mol Biol Cell. 2014;25:2282–90. doi: 10.1091/mbc.E14-04-0912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang CC, et al. Cytoophidium assembly reflects upregulation of IMPDH activity. J Cell Sci. 2015;128:3550–5. doi: 10.1242/jcs.175265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen K, et al. Glutamine analogs promote cytoophidium assembly in human and Drosophila cells. J Genet Genomics. 2011;38:391–402. doi: 10.1016/j.jgg.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 14.Endrizzi JA, Kim H, Anderson PM, Baldwin EP. Crystal structure of Escherichia coli cytidine triphosphate synthetase, a nucleotide-regulated glutamine amidotransferase/ATP-dependent amidoligase fusion protein and homologue of anticancer and antiparasitic drug targets. Biochemistry. 2004;43:6447–63. doi: 10.1021/bi0496945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goto M, Omi R, Nakagawa N, Miyahara I, Hirotsu K. Crystal structures of CTP synthetase reveal ATP, UTP, and glutamine binding sites. Structure. 2004;12:1413–23. doi: 10.1016/j.str.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 16.Endrizzi JA, Kim H, Anderson PM, Baldwin EP. Mechanisms of product feedback regulation and drug resistance in cytidine triphosphate synthetases from the structure of a CTP-inhibited complex. Biochemistry. 2005;44:13491–9. doi: 10.1021/bi051282o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kursula P, et al. Structure of the synthetase domain of human CTP synthetase, a target for anticancer therapy. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2006;62:613–7. doi: 10.1107/S1744309106018136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lauritsen I, Willemoes M, Jensen KF, Johansson E, Harris P. Structure of the dimeric form of CTP synthase from Sulfolobus solfataricus. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2011;67:201–8. doi: 10.1107/S1744309110052334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aughey GN, Liu JL. Metabolic regulation via enzyme filamentation. Crit Rev Biochem Mol Biol. 2015;51:282–93. doi: 10.3109/10409238.2016.1172555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aughey GN, et al. Nucleotide synthesis is regulated by cytoophidium formation during neurodevelopment and adaptive metabolism. Biol Open. 2014;3:1045–56. doi: 10.1242/bio.201410165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Calise SJ, et al. Glutamine deprivation initiates reversible assembly of mammalian rods and rings. Cell Mol Life Sci. 2014;71:2963–73. doi: 10.1007/s00018-014-1567-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin E, et al. CTP synthase 1 deficiency in humans reveals its central role in lymphocyte proliferation. Nature. 2014;510:288–92. doi: 10.1038/nature13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hofer A, Steverding D, Chabes A, Brun R, Thelander L. Trypanosoma brucei CTP synthetase: a target for the treatment of African sleeping sickness. Proc Natl Acad Sci U S A. 2001;98:6412–6. doi: 10.1073/pnas.111139498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hindenburg AA, Taub RN, Grant S, Chang G, Baker MA. Effects of pyrimidine antagonists on sialic acid regeneration in HL-60 cells. Cancer Res. 1985;45:3048–52. [PubMed] [Google Scholar]
- 25.Kang GJ, et al. Cyclopentenylcytosine triphosphate. Formation and inhibition of CTP synthetase. J Biol Chem. 1989;264:713–8. [PubMed] [Google Scholar]
- 26.Politi PM, et al. Phase I clinical trial of continuous infusion cyclopentenyl cytosine. Cancer Chemother Pharmacol. 1995;36:513–23. doi: 10.1007/BF00685802. [DOI] [PubMed] [Google Scholar]
- 27.Trudel M, Van Genechten T, Meuth M. Biochemical characterization of the hamster thy mutator gene and its revertants. J Biol Chem. 1984;259:2355–9. [PubMed] [Google Scholar]
- 28.Raushel FM, Thoden JB, Holden HM. Enzymes with molecular tunnels. Acc Chem Res. 2003;36:539–48. doi: 10.1021/ar020047k. [DOI] [PubMed] [Google Scholar]
- 29.Chang YF, Martin SS, Baldwin EP, Carman GM. Phosphorylation of human CTP synthetase 1 by protein kinase C: identification of Ser(462) and Thr(455) as major sites of phosphorylation. J Biol Chem. 2007;282:17613–22. doi: 10.1074/jbc.M702799200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi MG, Carman GM. Phosphorylation of human CTP synthetase 1 by protein kinase A: identification of Thr455 as a major site of phosphorylation. J Biol Chem. 2007;282:5367–77. doi: 10.1074/jbc.M610993200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi MG, Park TS, Carman GM. Phosphorylation of Saccharomyces cerevisiae CTP synthetase at Ser424 by protein kinases A and C regulates phosphatidylcholine synthesis by the CDP-choline pathway. J Biol Chem. 2003;278:23610–6. doi: 10.1074/jbc.M303337200. [DOI] [PubMed] [Google Scholar]
- 32.Han GS, et al. Expression of Human CTP synthetase in Saccharomyces cerevisiae reveals phosphorylation by protein kinase A. J Biol Chem. 2005;280:38328–36. doi: 10.1074/jbc.M509622200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Long CW, Pardee AB. Cytidine triphosphate synthetase of Escherichia coli B. I. Purification and kinetics. J Biol Chem. 1967;242:4715–21. [PubMed] [Google Scholar]
- 34.Habrian C, et al. Inhibition of Escherichia coli CTP Synthetase by NADH and Other Nicotinamides and Their Mutual Interactions with CTP and GTP. Biochemistry. 2016;55:5554–5565. doi: 10.1021/acs.biochem.6b00383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suloway C, et al. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 36.Li X, et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat Methods. 2013;10:584–90. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol. 2003;142:334–47. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 38.Lander GC, et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol. 2009;166:95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frank J, et al. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–9. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 40.Egelman EH. The iterative helical real space reconstruction method: surmounting the problems posed by real polymers. J Struct Biol. 2007;157:83–94. doi: 10.1016/j.jsb.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 41.Sachse C, et al. High-resolution electron microscopy of helical specimens: a fresh look at tobacco mosaic virus. J Mol Biol. 2007;371:812–35. doi: 10.1016/j.jmb.2007.05.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Egelman EH. A robust algorithm for the reconstruction of helical filaments using single-particle methods. Ultramicroscopy. 2000;85:225–34. doi: 10.1016/s0304-3991(00)00062-0. [DOI] [PubMed] [Google Scholar]
- 43.Voss NR, Yoshioka CK, Radermacher M, Potter CS, Carragher B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J Struct Biol. 2009;166:205–13. doi: 10.1016/j.jsb.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012;180:519–30. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Current protocols in bioinformatics. 2016;54:5.6.1–5.6.37. doi: 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DiMaio F, et al. Atomic-accuracy models from 4.5-A cryo-electron microscopy data with density-guided iterative local refinement. Nat Methods. 2015;12:361–5. doi: 10.1038/nmeth.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnson M, et al. NCBI BLAST: a better web interface. Nucleic acids research. 2008;36:W5–9. doi: 10.1093/nar/gkn201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular biology and evolution. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors for the CTP-inhibited ecCTPS crystal structure have been deposited in the Protein Data Bank under accession code PDB 5TKV. EM structures and associated atomic models have been deposited in the Electron Microscopy Data Bank and Protein Data Bank with the following accession codes: ecCTPS filament (EMD-8504; PDB 5U3C), hCTPS1 filament (EMD-8474; PDB 5U03), hCTPS1-H355A tetramer (EMD-8476), ecCTPS tetramer (EMD-8475; PDB 5U05), ecCTPS-CC (apo) (EMD-8490), ecCTPS-CC (substrates) (EMD-8491), ecCTPS-CC (products) (EMD-8513; PDB 5U6R). Other data supporting this study are available from the corresponding authors upon reasonable request.