

Abstract
Selective expression of dominant negative (DN) peroxisome proliferator-activated receptor gamma (PPARγ) in vascular smooth muscle cells (SMC) results in hypertension, atherosclerosis, and increased NF-κB target gene expression. Mesenteric SMC were cultured from mice designed to conditionally express wild-type (WT) or DN-PPARγ in response to Cre-recombinase to determine how SMC PPARγ regulates expression of NF-κB-target inflammatory genes. SMC-specific overexpression of WT-PPARγ or agonist-induced activation of endogenous PPARγ blunted TNFα-induced NF-κB target gene expression and activity of a NF-κB responsive promoter. TNFα-induced gene expression responses were enhanced by DN-PPARγ in SMC. Although expression of NF-κB p65 was unchanged, nuclear export of p65 was accelerated by WT-PPARγ and prevented by DN-PPARγ in SMC. Leptomycin B, a nuclear export inhibitor, blocked p65 nuclear export and inhibited the anti-inflammatory action of PPARγ. Consistent with a role in facilitating p65 nuclear export, WT-PPARγ co-immunoprecipitated with p65, and WT-PPARγ was also exported from the nucleus after TNFα treatment. Conversely, DN-PPARγ does not bind to p65 and was retained in the nucleus after TNFα treatment. Transgenic mice expressing WT- or DN-PPARγ specifically in SMC (S-WT or S-DN) were bred with mice expressing luciferase controlled by a NF-κB-responsive promoter to assess effects on NF-κB activity in whole tissue. TNFα-induced NF-κB activity was decreased in aorta and carotid artery from S-WT, but was increased in vessels from S-DN mice. We conclude that SMC PPARγ blunts expression of pro-inflammatory genes by inhibition of NF-κB activity through a mechanism promoting nuclear export of p65, which is abolished by DN mutation in PPARγ.
Keywords: inflammation, transcription, NF-κB, vascular smooth muscle, PPARγ, nuclear export
Introduction
Peroxisome proliferator-activated receptor gamma (PPARγ) is a ubiquitously expressed ligand-activated transcription factor. PPARγ is well known to induce adipocyte differentiation and to regulate lipid metabolism, but other studies indicate roles and sites of PPARγ activity in other tissues such as macrophages and brain.1–3 PPARγ plays a protective role in the vasculature and PPARγ activators can protect against atherosclerosis and lower blood pressure.4,5 In contrast, patients carrying mutations in PPARγ exhibit severe early onset hypertension and insulin resistance and others exhibit hypertension and lipodystrophies.6,7 Taken together, experimental and clinical evidence point to a significant role for PPARγ in the regulation of cardiovascular homeostasis.
To explore the role of PPARγ in the vasculature, we generated mouse models expressing dominant negative (DN) mutations in PPARγ (either P467L or V290M) specifically in endothelium or vascular smooth muscle (SMC). Endothelial-specific interference with PPARγ led to cerebral vascular dysfunction in response to either high-fat diet (HFD) or angiotensin II,8,9 whereas overexpression of PPARγ in endothelium had a protective effect on interleukin-1β (IL-1β)-induced endothelial dysfunction.10 Transgenic mice expressing DN-PPARγ selectively in SMC (S-DN) exhibited systolic hypertension and severe vascular dysfunction through a RhoA/Rho kinase-dependent mechanism.11–14 When bred with ApoE-deficient mice and treated with a high cholesterol diet, both endothelial and SMC models exhibit exaggerated atherosclerosis associated with elevated expression of inflammatory markers in the vessel.15 Importantly, protection from atherosclerosis by a PPARγ agonist was dependent upon PPARγ activity in SMC.16 However, the precise mechanism by which DN-PPARγ function exacerbates inflammatory signals and augments atherosclerosis remains unclear.
Nuclear factor-kappa B (NF-κB) is recognized as a central regulator of inflammation, a risk factor for cardiovascular disease. Inactive NF-κB is retained in the cytoplasm through association with its inhibitory factor Iκ-B, while phosphorylation of Iκ-B promotes dissociation of NF-κB and its import into the nucleus as an active transcription factor.17 In vascular cells, NF-κB activation increases pro-inflammatory mediators such as vascular adhesion molecular 1 (VCAM1), monocyte chemotactic protein 1 (MCP1) and matrix metalloproteinase (MMP9).18,19 PPARγ has been reported to regulate NF-κB activity in macrophages by a trans-repression mechanism involving the interaction between PPARγ and NF-κB, which does not require binding of the PPARγ/RXR heterodimer to DNA.20 PPARγ was also reported to act as an E3 ubiquitin ligase regulating the ubiquitination and degradation of the p65 subunit of NF-κB.21 Here we tested the hypothesis that PPARγ controls the expression of inflammatory genes in SMC by regulating the activity of NF-κB p65. We provide evidence supporting the concept that PPARγ directly inhibits p65 in SMC, not through ubiquitination or altered expression, but by facilitating nuclear to cytoplasm transport of p65. This mechanism is impaired in SMC expressing the P467L dominant negative mutant in PPARγ which does not bind p65.
Methods
Details of the experiments using cell culture, western blotting and immunoprecipitation, real-time RT-PCR, NF-κB promoter activity, immunostaining, bioluminescence imaging and chemicals are described in the expanded Methods section of the online-only Data Supplement.
Animals
Male transgenic mice carrying wide-type (WT) or the P467L dominant negative (DN) form of human PPARγ under the control of the CAG promoter were described previously.22,23 Male transgenic mice carrying WT (S-WT) or P467L DN (S-DN) PPARγ under the control of the smooth muscle myosin heavy chain promoter were crossed with NF-κB-LUC mice expressing luciferase under the control of an NF-κB responsive promoter (the gift of Dr. Timothy Blackwell, Vanderbilt University).11,24 Experimental mice were S-WT X NF-κB-LUC and S-DN X NF-κB-LUC. Age-matched single transgenic NF-κB-LUC littermates were used as controls. In some experiments, mice were injected with TNFα (66.7 μg/kg/day) intraperitoneally for three consecutive days and were then sacrificed on the fourth day. Care of these mice met the standards set forth by the National Institutes of Health (NIH) guidelines for the care and use of experimental animals. All procedures were approved by The University of Iowa Animal Care and Use Committee.
Statistical analysis
Experiments were performed in similar numbers in both male and female mice. There was no difference between male and female mice; therefore, all data were merged. Results are expressed as mean ± SEM. Statistical evaluation of the data was performed using GraphPad Prism. Where appropriate, a paired or unpaired Student’s t-test was used to compare between two groups. In other studies, ANOVA followed by Tukey’s test for comparisons was performed. Differences were considered significant when P value was less than 0.05.
Results
Analysis of SMC from transgenic mice inducibly expressing WT- or DN-PPARγ
We previously reported the generation of transgenic mice carrying conditionally activatable transgenes designed to express either WT- or DN-PPARγ, each also expressing tdTomato (Figure 1A).22,23 First, we cultured mesenteric SMC from these mice and activated the transgene by infecting the cells with an adenovirus encoding Cre-recombinase (AdCre). Transgene expression remained silent in SMC infected with control adenovirus (AdGFP) but was induced in AdCre-infected SMC (Figure 1B–D). As evidence of PPARγ activation, expression of FABP4, a known PPARγ target, was induced in SMC derived from WT-PPARγ (Figure 1E). Consistent with transcriptional impairment of DN-PPARγ, there was no induction of FABP4 expression in mesenteric SMC derived from DN-PPARγ mice (Figure 1F). Expression of tdTomato, which is co-activated in response to AdCre, was also induced in both groups (Figure 1B).
Figure 1. Experimental Model.
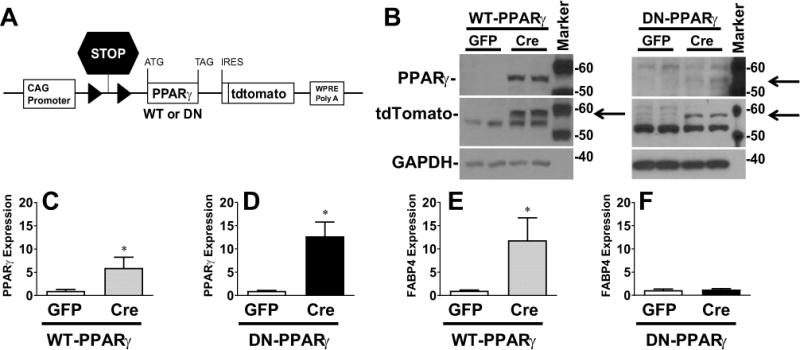
A) Schematic of the transgene inducibly expressing either WT-PPARγ or DN-PPARγ and tdTomato in response to Cre-recombinase. B) Western blot detecting the indicated protein in mesenteric artery SMC derived from the transgenic mice. Cells were infected with either control (AdGFP) or AdCre. This is representative of 4–6 experiments. Size markers transferred from the blots are shown. C–E) Relative mRNA expression of human PPARγ (C, n=10–12; D, n=6) and mouse FABP4 (E, n=10–12; F, n=6) were determined by quantitative real-time RT-PCR in these cells. Data were normalized to the control value, set to 1.0. All data are mean ± SEM. *P<0.05, control vs. AdCre.
WT-PPARγ antagonizes NF-κB-mediated inflammatory pathways in SMC
To examine if PPARγ can inhibit expression of NF-κB target genes in SMC, expression of the pro-inflammatory markers VCAM1, MCP1 and MMP9 was evaluated in AdCre- or AdGFP-treated SMC cultured from S-WT and S-DN mice after stimulation by tumor necrosis factor α (TNFα), IL-1β or lipopolysaccharide (LPS). Expression of all three genes were robustly induced by TNFα in AdGFP-treated mesenteric SMC from both WT-PPARγ and DN-PPARγ mice (Figure 2A–C). Similarly, expression of all three genes were robustly induced by IL-1β or LPS in AdGFP-treated mesenteric SMC from WT-PPARγ mice (Figure S1). AdCre-mediated activation of WT-PPARγ blunted the induction of VCAM1, MCP1 and MMP9 by all three cytokines. Similarly, WT-PPARγ prevented the TNFα-mediated increase in activity of an NF-κB-responsive promoter in SMC (Figure 2D). By contrast, expression of DN-PPARγ augmented the TNFα-induced expression of NF-κB target genes (Figure 2A–C) and TNFα-induced activity of a NF-κB-responsive promoter (Figure 2D). There was no significant change in the level of mRNA expression of TNFα receptor 1a or 1b in cultured SMC from transgenic mice inducibly expressing WT-PPARγ or DN-PPARγ compared with control cells (data not shown).
Figure 2. NF-κB Target Gene and Promoter Activity in SMC.
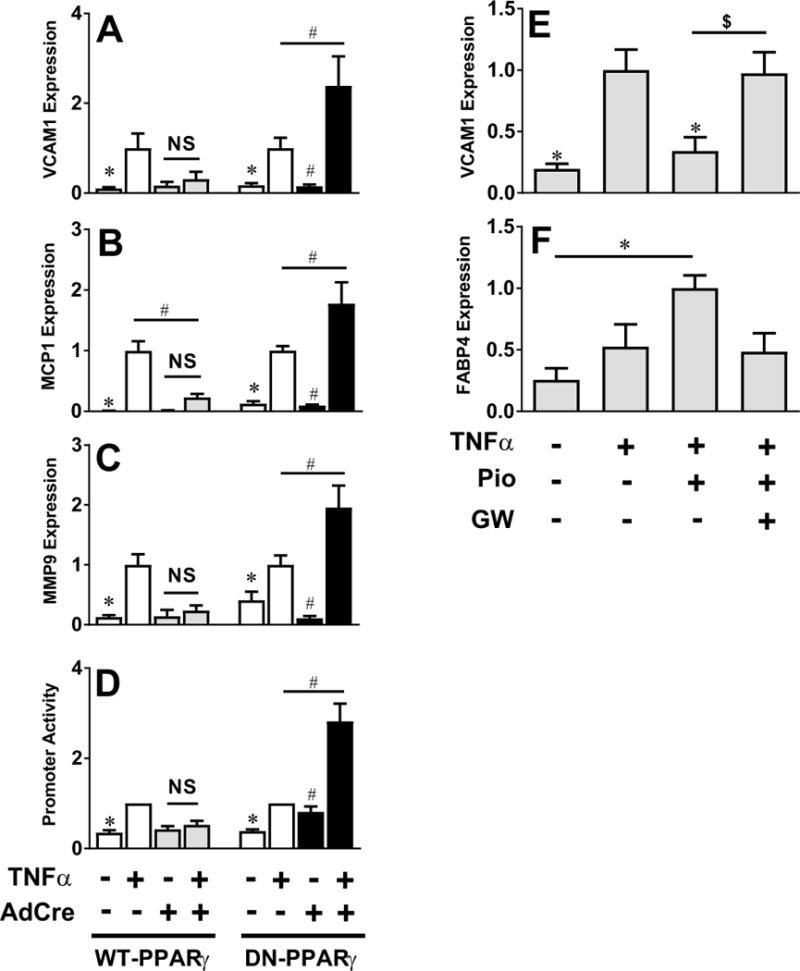
A–C) Relative mRNA expression of mouse VCAM-1 (A, n=5), MCP1 (B, n=5) and MMP9 (C, n=5) were determined by quantitative real-time RT-PCR in TNFα-treated (5 ng/ml, 6 hr) primary mesenteric SMC from mice with inducible expression of WT-PPARγ or DN-PPARγ infected with either AdGFP or AdCre. D) Activity of an NF-κB-responsive promoter was determined by luciferase assay in TNFα-treated WT-PPARγ or DN-PPARγ expressing mesenteric SMC infected with NF-κB-LUC adenovirus (72 hr, n=6–8). E–F) mRNA expression of VCAM1 (E, n=6) and FABP4 (F, n=6) in control mesenteric SMC either left untreated or treated with pioglitazone (1 μM) or GW9662 (10 μM) for 1 hr prior to TNFα. Data were normalized to TNFα (E) or pioglitazone (F) treated cells. All data are mean ± SEM. *P<0.05 vs. AdGFP + TNFα; #P<0.05 vs. AdCre + TNFα; $P<0.05 Pio vs. GW
We next used pioglitazone, a PPARγ agonist, and GW9662, a PPARγ antagonist, to assess the activity of endogenous PPARγ in SMC. Consistent with effects of overexpression, pioglitazone decreased TNFα-induced gene expression in SMC, an effect that was blocked by GW9662 (Figure 2E). Pioglitazone also induced the expression of the canonical PPARγ target gene FABP4, an effect blocked by GW9662 (Figure 2F). These data suggest that PPARγ activity in SMC attenuates NF-κB-dependent gene expression.
PPARγ does not decrease expression of p65 in SMC and HEK cells
Previous reports have found that PPARγ can act as an E3 ubiquitin ligase which targets p65 for ubiquitination.21 To determine if this mechanism was operant in our model, we assessed the effect of WT-PPARγ overexpression on p65 protein levels during TNFα-induced NF-κB signaling. TNFα-induced activation of NF-κB in SMC was evidenced by degradation of Iκ-Bα (Figure 3A). There was no alteration in the level of p65 protein or mRNA by WT-PPARγ (Figure 3A–C). To assess if PPARγ affects p65 turnover, we employed HEK293 cells transfected with p65. Overexpression of WT-PPARγ in HEK293 cells failed to elicit a change in p65 protein expression levels under baseline conditions or alter p65 stability in cells treated with cycloheximide (CHX) (Figure 3D). Similarly, there was no change in phospho-p65 or total p65 in the presence or absence of TNFα in response to overexpression of either WT-PPARγ or DN-PPARγ (Figure 3E). Addition of rosiglitazone, another PPARγ agonist, had no effect on phospho-p65 or total p65 levels. Combined treatment with rosiglitazone and TNFα had no effect on phospho-65 but modestly blunted total p65 irrespective of the presence of PPARγ. Contrary to a previous report, we were not able to detect any increase in p65 ubiquitination under any of these conditions (Figure S2).21 Our results suggest that degradation of p65 was not responsible for the PPARγ-mediated inhibition of expression of NF-κB-target genes.
Figure 3. NF-κB p65 Expression in PPARγ-expressing SMC.
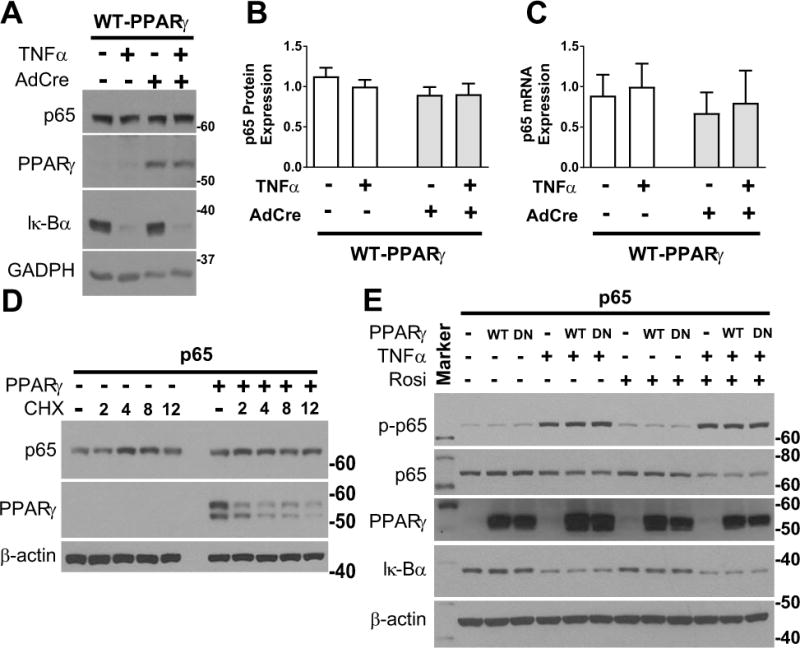
A) Western blot detecting the indicated proteins (representative of 6 experiments). B) Quantification of Western blots such as the representative shown in A (n=6). C) Quantitative RT-PCR detecting mouse p65 mRNA (n=6) in mesenteric artery SMC infected with either AdGFP or AdCre from transgenic mice with inducible expression of WT-PPARγ. Data were normalized to the TNFα-treated control. All data are mean ± SEM. D-E) Western blots detecting the indicated proteins in HEK293T cells transfected with p65 and/or WT-PPARγ or DN-PPARγ before treatment with cycloheximide (30 mg/ml, 0–12 hr), TNFα (50 ng/ml, 30 min) or rosiglitazone (1 μM, 1 hr) as indicated. p-p65 refers to the phosphorylated form of p65. Size markers transferred from the blots are shown.
WT-PPARγ accelerates nuclear export of p65 in SMC
To explore the molecular mechanism mediating the effects of PPARγ on NF-κB signaling, we determined the subcellular localization of p65 protein. Treatment with TNFα induced rapid nuclear import of p65 in control (untransfected) primary SMC and in primary SMC induced to express WT-PPARγ (Figure 4A). In control cells, nuclear export of p65 was evident by 2 hours, and by 3 hours there was little evidence of nuclear p65. Interestingly, nuclear export of p65 was accelerated in WT-PPARγ compared to control cells. In marked contrast, nuclear p65 was preserved 3 hours after TNFα-treatment in cultured SMC induced to express DN-PPARγ, a time point at which most nuclear p65 had been exported in control cells. The difference in nuclear export of p65 is evident when the data was quantified (Figure 4B). Pretreatment with leptomycin B, a nuclear export inhibitor, decreased the export of p65 and preserved significant levels of nuclear p65 3 hours after TNFα-treatment in control and WT-PPARγ SMC (Figure 4C). Notably, leptomycin B also increased expression of the NF-κB target gene MCP1 in WT-PPARγ and control SMC treated with TNFα (Figure 4D), indicating that nuclear export restricts NF-κB-mediated gene expression in SMC. It is interesting that the effect of leptomycin phenocopies the effect of DN-PPARγ, suggesting that accelerated nuclear export of p65 may contribute to the inhibitory effect of PPARγ on expression of pro-inflammatory genes, and thus, this may contribute to the anti-inflammatory effects of PPARγ.
Figure 4. p65 Subcellular Localization.
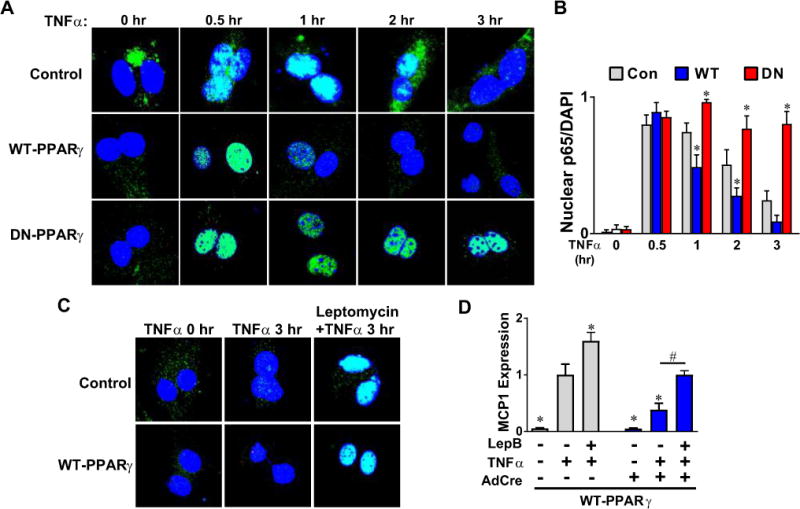
A) p65 immunostaining (green) in TNFα (0–3 hr)-treated cultured mesenteric artery SMC infected with AdCre from transgenic mice with inducible expression of either WT-PPARγ or DN-PPARγ. B) The fraction of cells with nuclear p65 was determined in a blinded fashion (50–200 cells counted per condition, n=7). C) p65 immunostaining (green) in control and WT-PPARγ expressing mesenteric SMC treated with leptomycin B (10 nM, 1 hr) before TNFα treatment (5 ng/ml, 3 hr). D) Relative mRNA expression of mouse MCP1 was determined by quantitative real-time RT-PCR in WT-PPARγ expressing cells treated with an inhibitor of nuclear export, leptomycin B (10 nM, 1 hr) prior to TNFα (5 ng/ml, 6 hr, n=6). Data were normalized to the TNFα-treated control. All data are mean ± SEM. *P<0.05 vs. TNFα-treated control cells. #P<0.05 vs. TNFα-treated AdCre-infected cells.
WT-PPARγ binds p65 and promotes cytoplasmic export in TNFα-treated SMC
That PPARγ facilitated nuclear export of p65 suggested there might be a direct interaction between the proteins. Thus, we next determined if PPARγ binds to p65 using HEK293 cells transfected with p65 and WT- or DN-PPARγ. Co-immunoprecipitation showed that WT-PPARγ but not DN-PPARγ was associated with p65 (Figure 5A). Consistent with this association, treatment of SMC with TNFα induced nuclear export of WT-PPARγ but not DN-PPARγ (Figure 5B). This export was blocked by either leptomycin B or by a p65-specific (pSer529, pSer536) inhibitor peptide. These data support the hypothesis that WT-PPARγ accelerates nuclear export of p65 by directly binding to p65.
Figure 5. PPARγ Association with p65.
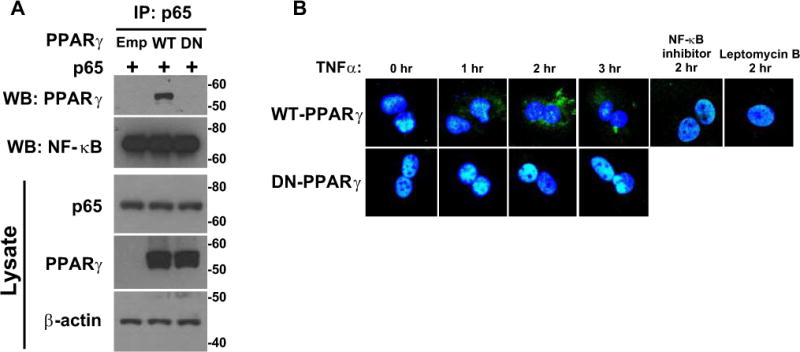
A) HEK293T cells transfected with p65, WT-PPARγ or DN-PPARγ were treated with a proteasome inhibitor, MG132 (5 μM, 12 hr). Proteins were immunoprecipitated with p65 antibody and immunoprecipitated proteins were Western blotted for the indicated protein. The top 2 blots represent immunoprecipitation with p65 and Western blot with the indicated antibody. The bottom 3 blots represent Western blots for the indicated protein from cell lysates. Size markers transferred from the blots are shown. B) PPARγ immunostaining (green) of WT-PPARγ or DN-PPARγ expressing SMC treated with TNFα for the indicated times. Where indicated, cells were treated with an NF-κB inhibitor (50 μMr) or leptomycin B (5 nM) for 1 hour prior to TNFα.
WT-PPARγ inhibits NF-κB promoter activity in vessels
In order to examine the anti-inflammatory role of SMC PPARγ in whole vessels, we injected TNFα intraperitoneally in our previously reported mouse models expressing DN-PPARγ (S-DN) selectively in SMC.11 Injection with TNFα (66.7 μg/kg/day) for three consecutive days equivalently increased total leukocytes (CD45+) and monocytes/macrophages (CD45+/F4/80+) in aorta from non-transgenic and S-DN (Figure S3). Total T lymphocytes (CD3+), T helper (CD3+CD4+) and cytotoxic T cells (CD3+CD8+) were not altered in either group. These data indicate that the degree of leukocyte infiltration in aorta induced by systemic inflammatory activity is not altered by interference with SMC PPARγ.
To assess direct effects on inflammation-stimulated NF-κB activity in SMC of intact vessels, we bred transgenic mice expressing WT-PPARγ (S-WT) or DN-PPARγ (S-DN) specifically in SMC with mice expressing luciferase under control of a NF-κB-responsive promoter.24 TNFα-induced NF-κB activity was decreased in aorta and particularly in carotid artery from S-WT X NF-κB-Luc mice compared to mice expressing only NF-κB-Luc (Figure 6A). In contrast, S-DN exhibited enhanced TNFα-induced NF-κB activity (Figure 6B).
Figure 6. NF-κB Activity in Aorta and Carotid Artery.
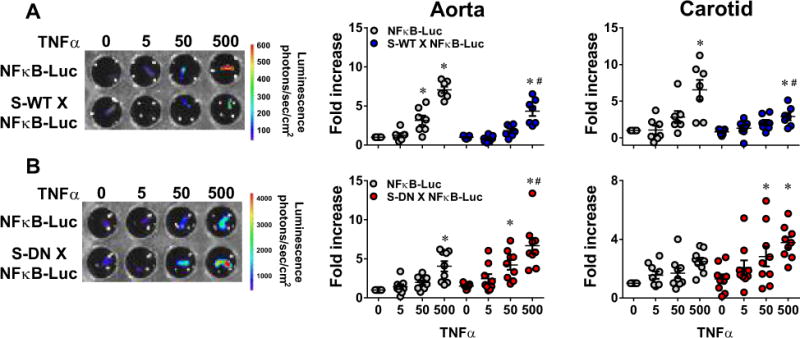
NF-κB activity was measured by luciferase assay in TNFα (0–500 pg/ml, 16–24 h)-treated aorta and carotid arteries. Experimental mice are double transgenic mice carrying NF-κB-LUC reporter mice and either S-WT (A) or S-DN (B). Age- and sex-matched single NF-κB-LUC littermates from each breeding (n=8–9) were used as controls. Data were normalized to the samples from untreated single NF-κB-LUC mice. All data are mean ± SEM. *P<0.05 vs. untreated; #P<0.05, TNFα (500 μg/ml) NF-κB-LUC vs. S-WT X NF-κB-LUC or NF-κB-LUC vs. S-DN X NF-κB-LUC.
Discussion
Thiazolidinediones such as rosiglitazone and pioglitazone are potent activators of PPARγ and were previously used to improve glycemic control in type 2 diabetes. The PROactive clinical trial reported that pioglitazone decreased macrovascular events and lowered blood pressure and cardiovascular risk.4 In contrast, patients with PPARγ mutations exhibit hypertension.6 We previously showed that loss of PPARγ function in SMC exaggerated aortic atherosclerosis with increased NF-κB target gene expression in ApoE-deficient mice fed with western diet.15 Because NF-κB-induced proinflammatory signals drive initiation, progression and development of atherosclerotic lesions,25 the present study determined the effect of SMC-PPARγ on NF-κB activity using transgenic mice inducibly over-expressing WT- or DN-PPARγ in SMC. The main findings from our study are: 1) cytokine-induced NF-κB activity in cultured SMC, as measured by expression of NF-κB target genes and the activity of a NF-κB-responsive promoter, was blunted by overexpression of WT-PPARγ, but was increased by DN-PPARγ, 2) SMC-PPARγ protects against cytokine-induced NF-κB activity by facilitating nuclear export of p65 subunit, 3) PPARγ facilitates nuclear export of p65 by direct interaction, which is blocked by the DN P467L mutation in PPARγ, and 4) WT-PPARγ blunts whereas DN-PPARγ augments TNFα-induced activity of an NF-κB responsive promoter in aorta and carotid artery.
NF-κB is well-known as a central regulator of inflammation, and several lines of evidence suggest that PPARγ protects against inflammation by interfering with NF-κB activity. Hou et al reported that PPARγ has a RING domain similar to E3 ubiquitin ligases, can directly bind to the p65 subunit of NF-κB, and induce ubiquitination and degradation of p65.21 Although we can confirm that PPARγ and p65 can directly interact, we did not find evidence supporting ubiquitination of p65 by PPARγ. First, steady state levels of p65 protein were not altered by overexpression of WT-PPARγ or by rosiglitazone. Second, PPARγ did not alter the rate of p65 degradation in cells treated with the protein synthesis inhibitor cycloheximide. Third, PPARγ did not induce ubiquitination of p65. This suggests that an alternative mechanism accounts for PPARγ-mediated interference with NF-κB activity, at least in vascular SMC.
Even though the abundance of total cellular p65 was not altered by PPARγ, important changes in the temporal localization of p65 in SMC were observed. There was no difference between groups in the import of p65 into the nucleus at 30 min after TNFα treatment. However, 1–2 hrs after treatment, the amount of p65 remaining in the nucleus was significantly lower in WT-PPARγ overexpressing SMC compared to control cells. This finding is consistent with previous reports in Caco-2 cells and HUVECs.26,27 IL-1β-induced nuclear p65 protein was also decreased by pioglitazone in vascular SMC from hypertensive rats.28 The functional importance of PPARγ to accelerate nuclear export of p65 is further illustrated by our finding that WT-PPARγ-mediated reduction in NF-κB target gene expression can be impaired by pharmacological inhibition of nuclear export. Moreover, that nuclear export of PPARγ was occurring with the same time course and could be blocked with an NF-κB inhibitor lends support for a PPARγ-dependent mechanism.
Interestingly, nuclear export of p65 was severely impaired in SMC expressing DN-PPARγ. The DN mutation in PPARγ also affects its association with p65, which could co-immunoprecipitate with WT-PPARγ but not the P467L mutant form of PPARγ. The P467L mutation resides in the ligand binding domain of PPARγ. The P467L-PPARγ protein is transcriptionally defective and acts dominant negatively. Moreover, the mutation causes hypertension in subjects carrying the mutation, and hypertension in mice expressing the mutant protein selectively in SMC.6,11 It was reported that WT-PPARγ can bind p65 in the absence of ligand, but that the ligand binding domain of PPARγ is nonetheless required.21,29 Thus, the loss of PPARγ-induced p65 nuclear export in SMC expressing the P467L mutation in PPARγ is consistent with this.
One potential limitation of our study is that the molecular details of how DN-PPARγ induces accumulation of nuclear p65 after TNFα treatment remain unclear. Expression of DN-PPARγ prevents export of both p65 and DN-PPARγ. Yet, DN-PPARγ and p65 do not physically interact, at least in a stable way detected by co-immunoprecipitation. SMC endogenously express PPARγ and pioglitazone blunted induction of an NF-κB target gene (VCAM1) in response to TNFα suggesting that DN-PPARγ interferes with effects of PPARγ to block expression of inflammatory genes. Perhaps DN-PPARγ interferes with an association between endogenous PPARγ and p65. It is also possible the effect of PPARγ on p65 involves a PPARγ target gene whose expression is altered by DN-PPARγ. Indeed, we reported that expression of the P465L (the mouse equivalent to human P467L) mutation in PPARγ results in the alteration of many genes in the aorta.30,31 Future molecular studies closely examining the interaction between WT-PPARγ and p65 in the presence or absence of DN-PPARγ are warranted to fully define the mechanism of this anti-inflammatory activity.
It is well accepted that the proliferation and migration of VSMC, which is regulated in part by NF-κB, is critically involved in progression of atherosclerosis.32 It is also known that VSMC migration is increased in association with vascular remodeling when SMC PPARγ activity is impaired via dominant negative P467L mutation.33 Activation of PPARγ is protective in atherosclerosis,5,34 and this protective action requires SMC PPARγ.16 Indeed, interference with PPARγ function by expression of DN-PPARγ in SMC enhances atherosclerosis and augments NF-κB target gene expression in aorta from ApoE-deficient mice fed a high-fat diet. Thus, it is tempting to speculate that macrophage recruitment and atherosclerotic development is inhibited by PPARγ-dependent antagonism of NF-κB activity in SMC. We did not find that expression of DN-PPARγ selectively in SMC enhanced TNFα-induced macrophage infiltration in aorta, but this infiltration was likely a result of direct action of TNFα on macrophages expressing only endogenous PPARγ. In this regard, it will be instructive to determine if development of inflammatory lesions is affected by PPARγ activity in SMC when inflammation is initiated directly within the vasculature.
Perspectives
Activation of PPARγ is clinically important because it increases insulin sensitivity and improves glycemic control in type II diabetes. PPARγ activation also lowers blood pressure and protects against vascular diseases such as atherosclerosis.5,34 However, adverse effects have also been reported. PPARγ activation, at least by TZDs, causes weight gain, water retention, bone fracture, and has also been reported to increase the risk of heart failure.35 Newer, non-agonist activators of PPARγ have been reported to have similar potent antidiabetic action but without the adverse weight gain and water retention.36 PPARγ also has anti-oxidant and anti-inflammatory properties. A number of mechanisms involving NF-κB have been proposed to explain the anti-inflammatory actions of PPARγ including transrepression and p65 turnover.20,21 Herein, we examined a mechanism for the anti-inflammatory actions of PPARγ specifically in vascular SMC. Our results suggest that PPARγ protects against cytokine-induced activation of NF-κB-dependent inflammatory gene expression through a mechanism involving direct interaction and nuclear export of the p65 subunit. Thus, PPARγ does not appear to prevent the activation of NF-κB-dependent inflammatory gene expression, but acts to terminate transcription by removal of the critical p65 subunit from the nucleus. The hypertension-causing P467L mutation in PPARγ resulted in accumulation of nuclear p65, presumably due to loss of p65 binding preventing export of a PPARγ:p65 complex, thus prolonging NF-κB-mediated inflammatory gene expression. These findings expand our understanding of SMC PPARγ activities and suggest potential beneficial actions of PPARγ independent of its well characterized role as a regulator of transcription. Thus, while the global actions of PPARγ are complex, involving multiple cell types and molecular mechanisms, results from the present study provide strong evidence that the direct actions of PPARγ in the vasculature are protective against cardiovascular disease.
Supplementary Material
Novelty and Significance.
What Is New?
We studied the mechanism by which PPARγ antagonizes cytokine-induced NF-κB target gene expression and NF-κB activity in cultured smooth muscle cells from mice conditionally expressing either wildtype or dominant negative PPARγ and in aorta and carotid artery from mice selectively expressing dominant negative PPARγ selectively in vascular smooth muscle.
What Is Relevant?
PPARγ exerts anti-inflammatory actions by modulating the activity of NF-κB and expression of NF-κB target genes in response to cytokines such as TNFα in vascular smooth muscle cells.
PPARγ terminates NF-κB target gene expression and limits NF-κB activity by promoting nuclear export of the p65 subunit of NF-κB.
Hypertension-causing dominant negative mutations in PPARγ prevent nuclear export of p65.
Summary
These data support a new paradigm for the control of NF-κB activity and expression of inflammatory genes controlled by NF-κB by PPARγ in vascular smooth muscle.
Our data suggests that impairment of PPARγ activity in vascular smooth muscle promotes inflammation by augmenting NF-κB-dependent expression of inflammatory genes.
This PPARγ-NF-κB mechanism may influence the degree of inflammatory activity in the blood vessel in hypertension and atherosclerosis.
Acknowledgments
The authors thank Bill Paradee, Norma Sinclair, JoAnne Schwarting, and Patricia Yarolem for genotyping mice. Transgenic mice were generated at the University of Iowa Genome Editing Facility supported in part by grants from the National Institutes of Health (NIH) and from the Roy J. and Lucille A. Carver College of Medicine. We would like to thank Drs. Steven Lentz and John Engelhardt for the NF-κB-Luciferase adenovirus, Dr. Jianqiang Shao for assistance with p65 staining, and Dr. Justin L Grobe for discussions. We thank Dr. Mark Anderson (currently at Johns Hopkins University) for facilitating the transfer and gift of NF-κB transgenic mice from Dr. Timothy Blackwell (Vanderbilt University). We also acknowledge use of equipment and assistance from the Central Microscopy Facility at University of Iowa.
Funding Source
This work was supported through research grants from the National Institutes of Health (NIH) to C.D.S. (HL084207, HL125603, HL131689), K.R. (HL084207), and grants from the American Heart Association to C.D.S. (15SFRN23480000) and K.R. (14EIA18860041), and the University of Iowa Fraternal Order of Eagles Diabetes Research Center to K.R. The authors gratefully acknowledge the generous research support of the Roy J. Carver Trust.
Footnotes
Disclosures:
None
References
- 1.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature. 1998;391:79–82. doi: 10.1038/34178. [DOI] [PubMed] [Google Scholar]
- 2.Lu M, Sarruf DA, Talukdar S, Sharma S, Li P, Bandyopadhyay G, Nalbandian S, Fan W, Gayen JR, Mahata SK, Webster NJ, Schwartz MW, Olefsky JM. Brain PPAR-gamma promotes obesity and is required for the insulin-sensitizing effect of thiazolidinediones. Nature Medicine. 2011;17:618–622. doi: 10.1038/nm.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ryan KK, Li B, Grayson BE, Matter EK, Woods SC, Seeley RJ. A role for central nervous system PPAR-gamma in the regulation of energy balance. Nature Medicine. 2011;17:623–626. doi: 10.1038/nm.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dormandy JA, Charbonnel B, Eckland DJ, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–1289. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- 5.Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106:523–531. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, Chatterjee VK, O’Rahilly S. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402:880–883. doi: 10.1038/47254. [DOI] [PubMed] [Google Scholar]
- 7.Caron-Debarle M, Auclair M, Vigouroux C, Boccara F, Capel E, Vigarel C, Guerci B, Lascols O, Capeau J. PPARG mutations responsible for lipodystrophy with severe hypertension activate the cellular renin-angiotensin system. Arterioscler Thromb Vasc Biol. 2013;33:829–838. doi: 10.1161/ATVBAHA.112.300962. [DOI] [PubMed] [Google Scholar]
- 8.Beyer AM, de Lange WJ, Halabi CM, Modrick ML, Keen HL, Faraci FM, Sigmund CD. Endothelium-specific interference with peroxisome proliferator activated receptor gamma causes cerebral vascular dysfunction in response to a high-fat diet. Circ Res. 2008;103:654–661. doi: 10.1161/CIRCRESAHA.108.176339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu C, Lu KT, Mukohda M, Davis DR, Faraci FM, Sigmund CD. Interference with PPARgamma in endothelium accelerates angiotensin II-induced endothelial dysfunction. Physiol Genomics. 2016;48:124–134. doi: 10.1152/physiolgenomics.00087.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukohda M, Stump M, Ketsawatsomkron P, Hu C, Quelle FW, Sigmund CD. Endothelial PPAR-gamma provides vascular protection from IL-1beta-induced oxidative stress. Am J Physiol Heart Circ Physiol. 2016;310:H39–48. doi: 10.1152/ajpheart.00490.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halabi CM, Beyer AM, de Lange WJ, Keen HL, Baumbach GL, Faraci FM, Sigmund CD. Interference with PPARg Function in Smooth Muscle Causes Vascular Dysfunction and Hypertension. Cell Metabolism. 2008;7:215–226. doi: 10.1016/j.cmet.2007.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ketsawatsomkron P, Lorca RA, Keen HL, Weatherford ET, Liu X, Pelham CJ, Grobe JL, Faraci FM, England SK, Sigmund CD. PPARg Regulates Resistance Vessel Tone Through a Mechanism Involving RGS5-Mediated Control of PKC and BKCa Channel Activity. Circ Res. 2012;111:1446–1458. doi: 10.1161/CIRCRESAHA.112.271577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pelham CJ, Ketsawatsomkron P, Groh S, Grobe JL, de Lange WJ, Ibeawuchi SR, Keen HL, Weatherford ET, Faraci FM, Sigmund CD. Cullin-3 Regulates Vascular Smooth Muscle Function and Arterial Blood Pressure via PPARg and RhoA/Rho-Kinase. Cell Metabolism. 2012;16:462–472. doi: 10.1016/j.cmet.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agbor LN, Ibeawuchi SC, Hu C, Wu J, Davis DR, Keen HL, Quelle FW, Sigmund CD. Cullin-3 mutation causes arterial stiffness and hypertension through a vascular smooth muscle mechanism. JCI Insight. 2016;1:e91015. doi: 10.1172/jci.insight.91015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pelham CJ, Keen HL, Lentz SR, Sigmund CD. Dominant negative PPARg promotes atherosclerosis, vascular dysfunction, and hypertension through distinct effects in endothelium and vascular muscle. Am J Physiol Regul Integr Comp Physiol. 2013;304:R690–R701. doi: 10.1152/ajpregu.00607.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Subramanian V, Golledge J, Ijaz T, Bruemmer D, Daugherty A. Pioglitazone-induced reductions in atherosclerosis occur via smooth muscle cell-specific interaction with PPARg. Circ Res. 2010;107:953–958. doi: 10.1161/CIRCRESAHA.110.219089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nolan GP, Ghosh S, Liou HC, Tempst P, Baltimore D. DNA binding and I kappa B inhibition of the cloned p65 subunit of NF-kappa B, a rel-related polypeptide. Cell. 1991;64:961–969. doi: 10.1016/0092-8674(91)90320-x. [DOI] [PubMed] [Google Scholar]
- 18.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. 2007;7:803–815. doi: 10.1038/nri2171. [DOI] [PubMed] [Google Scholar]
- 19.Gareus R, Kotsaki E, Xanthoulea S, van dM I, Gijbels MJ, Kardakaris R, Polykratis A, Kollias G, de Winther MP, Pasparakis M. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab. 2008;8:372–383. doi: 10.1016/j.cmet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 20.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hou Y, Moreau F, Chadee K. PPARgamma is an E3 ligase that induces the degradation of NFkappaB/p65. Nature Communications. 2012;3:1300. doi: 10.1038/ncomms2270. [DOI] [PubMed] [Google Scholar]
- 22.Stump M, Guo DF, Lu KT, Mukohda M, Cassell MD, Norris AW, Rahmouni K, Sigmund CD. Nervous System Expression of PPARgamma and Mutant PPARgamma Has Profound Effects on Metabolic Regulation and Brain Development. Endocrinology. 2016;157:4266–4275. doi: 10.1210/en.2016-1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stump M, Guo DF, Lu KT, Mukohda M, Liu X, Rahmouni K, Sigmund CD. Effect of Selective Expression of Dominant Negative PPARgamma in Proopiomelanocortin Neurons on the Control of Energy Balance. Physiological Genomics. 2016;48:491–501. doi: 10.1152/physiolgenomics.00032.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng DS, Han W, Chen SM, Sherrill TP, Chont M, Park GY, Sheller JR, Polosukhin VV, Christman JW, Yull FE, Blackwell TS. Airway epithelium controls lung inflammation and injury through the NF-kappa B pathway. J Immunol. 2007;178:6504–6513. doi: 10.4049/jimmunol.178.10.6504. [DOI] [PubMed] [Google Scholar]
- 25.Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA, Neumeier D. Activated transcription factor nuclear factor-kappa B is present in the atherosclerotic lesion. JClinInvest. 1996;97:1715–1722. doi: 10.1172/JCI118598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AG, Pettersson S, Conway S. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat Immunol. 2004;5:104–112. doi: 10.1038/ni1018. [DOI] [PubMed] [Google Scholar]
- 27.Lim S, Lee KS, Lee JE, Park HS, Kim KM, Moon JH, Choi SH, Park KS, Kim YB, Jang HC. Effect of a new PPAR-gamma agonist, lobeglitazone, on neointimal formation after balloon injury in rats and the development of atherosclerosis. Atherosclerosis. 2015;243:107–119. doi: 10.1016/j.atherosclerosis.2015.08.037. [DOI] [PubMed] [Google Scholar]
- 28.Martin A, Perez-Giron JV, Hernanz R, Palacios R, Briones AM, Fortuno A, Zalba G, Salaices M, Alonso MJ. Peroxisome proliferator-activated receptor-gamma activation reduces cyclooxygenase-2 expression in vascular smooth muscle cells from hypertensive rats by interfering with oxidative stress. J Hypertens. 2012;30:315–326. doi: 10.1097/HJH.0b013e32834f043b. [DOI] [PubMed] [Google Scholar]
- 29.Chung SW, Kang BY, Kim SH, Pak YK, Cho D, Trinchieri G, Kim TS. Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-gamma and nuclear factor-kappa B. JBiolChem. 2000;275:32681–32687. doi: 10.1074/jbc.M002577200. [DOI] [PubMed] [Google Scholar]
- 30.Keen HL, Halabi CM, Beyer AM, de Lange WJ, Liu X, Maeda N, Faraci FM, Casavant TL, Sigmund CD. Bioinformatic analysis of gene sets regulated by ligand-activated and dominant-negative peroxisome proliferator-activated receptor gamma in mouse aorta. ArteriosclerThrombVascBiol. 2010;30:518–525. doi: 10.1161/ATVBAHA.109.200733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keen HL, Ryan MJ, Beyer A, Mathur S, Scheetz TE, Gackle BD, Faraci FM, Casavant TL, Sigmund CD. Gene expression profiling of potential PPARg target genes in mouse aorta. Physiological Genomics. 2004;18:33–42. doi: 10.1152/physiolgenomics.00027.2004. [DOI] [PubMed] [Google Scholar]
- 32.Navab M, Fogelman AM, Berliner JA, Territo MC, Demer LL, Frank JS, Watson AD, Edwards PA, Lusis AJ. Pathogenesis of atherosclerosis. Am J Cardiol. 1995;76:18C–23C. doi: 10.1016/s0002-9149(99)80466-4. [DOI] [PubMed] [Google Scholar]
- 33.Ketsawatsomkron P, Keen HL, Davis DR, Lu KT, Stump M, De Silva TM, Hilzendeger AM, Grobe JL, Faraci FM, Sigmund CD. Protective Role for Tissue Inhibitor of Metalloproteinase-4, a Novel Peroxisome Proliferator-Activated Receptor-gamma Target Gene, in Smooth Muscle in Deoxycorticosterone Acetate-Salt Hypertension. Hypertension. 2016;67:214–222. doi: 10.1161/HYPERTENSIONAHA.115.06391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levi Z, Shaish A, Yacov N, Levkovitz H, Trestman S, Gerber Y, Cohen H, Dvir A, Rhachmani R, Ravid M, Harats D. Rosiglitazone (PPARgamma-agonist) attenuates atherogenesis with no effect on hyperglycaemia in a combined diabetes-atherosclerosis mouse model. Diabetes ObesMetab. 2003;5:45–50. doi: 10.1046/j.1463-1326.2003.00240.x. [DOI] [PubMed] [Google Scholar]
- 35.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPARg signaling and metabolism: the good, the bad and the future. NatMed. 2013;19:557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi JH, Banks AS, Kamenecka TM, et al. Antidiabetic actions of a non-agonist PPARgamma ligand blocking Cdk5-mediated phosphorylation. Nature. 2011;477:477–481. doi: 10.1038/nature10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.