

Abstract
Pemphigus is an autoimmune blistering skin disease caused primarily by autoantibodies against desmoglein (Dsg)1 and 3. Here, we characterized the mechanisms engaged by pemphigus IgG from patients with different clinical phenotypes and autoantibody profiles. All pemphigus vulgaris (PV) and pemphigus foliaceus (PF) IgG and AK23, a monoclonal mouse antibody against Dsg3, caused loss of cell cohesion, cytokeratin retraction and p38MAPK activation. Strong alterations in Dsg3 distribution were caused by mucosal (aDsg3 antibodies), mucocutaneous (aDsg1 + aDsg3) as well as atypical (aDsg3) PV-IgG. All PV-IgG fractions and AK23 compromised Dsg3 but not Dsg1 binding and enhanced Src activity. In contrast, rapid Ca2+ influx and Erk activation were induced by mucocutaneous PV-IgG and pemphigus foliaceus (PF) IgG (aDsg1) whereas cAMP was increased by mucosal and mucocutaneous PV-IgG only. Selective inhibition of p38MAPK, Src or PKC blocked loss of keratinocyte cohesion in response to all autoantibody fractions whereas Erk inhibition was protective against mucocutaneous PV-IgG and PF-IgG only. These results demonstrate that signaling patterns parallel the clinical phenotype as some mechanisms involved in loss of cell cohesion are caused by antibodies targeting Dsg3 whereas others correlate with autoantibodies against Dsg1. The concept of key desmosome regulators may explain observations from several experimental models of pemphigus.
Introduction
Pemphigus is an autoimmune dermatosis characterized by blistering of the epidermis and/or mucous membranes1. Affected patients develop autoantibodies which bind to the cell-cell adhesion molecules desmoglein 1 (Dsg1) and/or Dsg3. These proteins belong to the cadherin superfamily of adhesion molecules and interact with corresponding molecules of the opposing cells extracellular domain. Intracellularly, they represent attachment zones for keratin filaments via the desmosomal plaque proteins desmoplakin (DP), plakoglobin (PG) and different plakophilin (PKP) isoforms2. Two main clinical variants of pemphigus exist, pemphigus vulgaris (PV) and pemphigus foliaceus (PF)3. PV has two main phenotypes: mucosal PV (m-PV), associated with autoantibodies against Dsg3 and exclusive effects on mucous membranes, and mucocutaneous PV (mc-PV), caused by autoantibodies targeting both Dsg3 and Dsg1 with additional blister formation in the suprabasal epidermis. In contrast, PF displays a phenotype restricted to blistering in the superficial epidermis with autoantibodies against Dsg1 only. Occasionally, the phenotype differs from this autoantibody pattern leading to atypical pemphigus variants (at-PV), i.e. an exclusive epidermal involvement in PV positive for anti-Dsg3 antibodies only or a combination of anti-Dsg3 and anti-Dsg14, 5. On a molecular level, autoantibody binding results in disturbed desmosome turnover and depletion of desmoglein molecules as well as severe structural alterations of desmosomes such as the reduction of desmosome size and number and uncoupling of keratin filaments6–8. The mechanisms leading to these changes are only partially understood9, 10. After identification of desmosomal adhesion molecules as the primary antigens it was proposed that the autoantibodies sterically hinder the interaction of desmogleins, leading to loss of cell cohesion and consequently to blister formation11, 12. Indeed, atomic force microscopy (AFM) studies showed that PV autoantibody fractions (PV-IgG) containing anti-Dsg3 and anti-Dsg1 block the homophilic binding of Dsg3 but, similar to PF-IgG, do not alter Dsg1 interactions13–15. Alternatively, it was shown that modulation of signaling events prevents pemphigus skin blistering in a set of different models16. These pathways include, but are not limited to, Ca2+ influx17, protein kinase C (PKC)18, sarcoma-associated kinase (Src)19, 20 and p38 protein-activated kinase (p38MAPK) signaling21. Specifically for p38MAPK it was shown that pharmacologic inhibition protected against the pathogenic effects of PV-IgG under conditions in which the interaction of Dsg3 molecules was hindered22. It is possible that loss of Dsg3 interaction by anti-Dsg3 antibody binding in turn initiates specific signaling events23 and that both mechanisms are required for pathogenicity. Until now, comprehensive insight connecting the different signaling pathways with autoantibody profiles and clinical phenotypes is lacking. In this study, we thus applied IgG fractions from patients with typical PV and PF and one atypical case of PV and evaluated (i) the potency to induce pemphigus hallmarks such as Dsg3 depletion and keratin retraction, (ii) their impact on the activity of specific signaling events and (iii) the relevance of these signaling events for loss of cell cohesion. We found signaling patterns correlating with clinical phenotype and autoantibody profile and observed key mechanisms for desmosome regulation including p38MAPK-, PKC- and Src activation which may resolve the question why in several models of pemphigus inhibition of one single signaling pathway is protective.
Results
Pathogenic effects of pemphigus autoantibodies
In this study, we compared autoantibody fractions from patients with m-PV, mc-PV, at-PV and PF (Table 1) with effects induced by the monoclonal autoantibody AK23 targeting Dsg324 in HaCaT keratinocytes expressing both Dsg1 and Dsg3 (Supplementary Fig. S1). All autoantibody fractions recognized Dsg1 and/or Dsg3 in ELISA assays depending on their antibody profile (Table 1). In addition, under the reducing conditions of the SDS-PAGE, at-PV and, to a lesser extent, m-PV also recognize Dsg3 (Supplementary Fig. 1). In keratinocytes treated with IgG from a healthy volunteer, Cytokeratin (CK)5 intermediate filaments formed a dense meshwork closely apposed to Dsg3 distributed along cell junctions in desmosomal contacts (Fig. 1a and g). All autoantibody fractions induced detachment of filaments from cell junctions, a phenomenon referred to as keratin retraction and known to be a morphological hallmark of pemphigus (Fig. 1h–y). This was paralleled by loss of intercellular cohesion detected by dispase-based dissociation assays. In contrast, alterations in Dsg3 distribution were variable. Similar to previous studies, fragmentation of Dsg3 staining with formation of linear streaks was moderate in cells incubated with AK23 or PF-IgG (Fig. 2b and f) whereas all PV-IgG fractions caused strong clustering and depletion of Dsg3 (Fig. 1c–e) closely resembling effects in human epidermis of patients13, 25, 26.
Table 1.
Antibody profiles of IgG-fractions determined by Dsg1 and Dsg3 ELISA and their clinical phenotypes.
| ELISA Score | Phenotype | ||
|---|---|---|---|
| anti-Dsg3 U/ml | anti-Dsg1 U/ml | ||
| c-IgG | — | — | healthy control |
| m-PV | 2391.0 | — | mucosal |
| m-PV2 | 162.9 | — | mucosal |
| at-PV | 153.4 | — | atypical cutaneous |
| mc-PV | 225.5 | 218.8 | mucocutaneous |
| mc-PV2 | 172.5 | 154.8 | mucocutaneous |
| PF | — | 1645.0 | cutaneous |
| PF2 | — | 266.4 | cutaneous |
Figure 1.
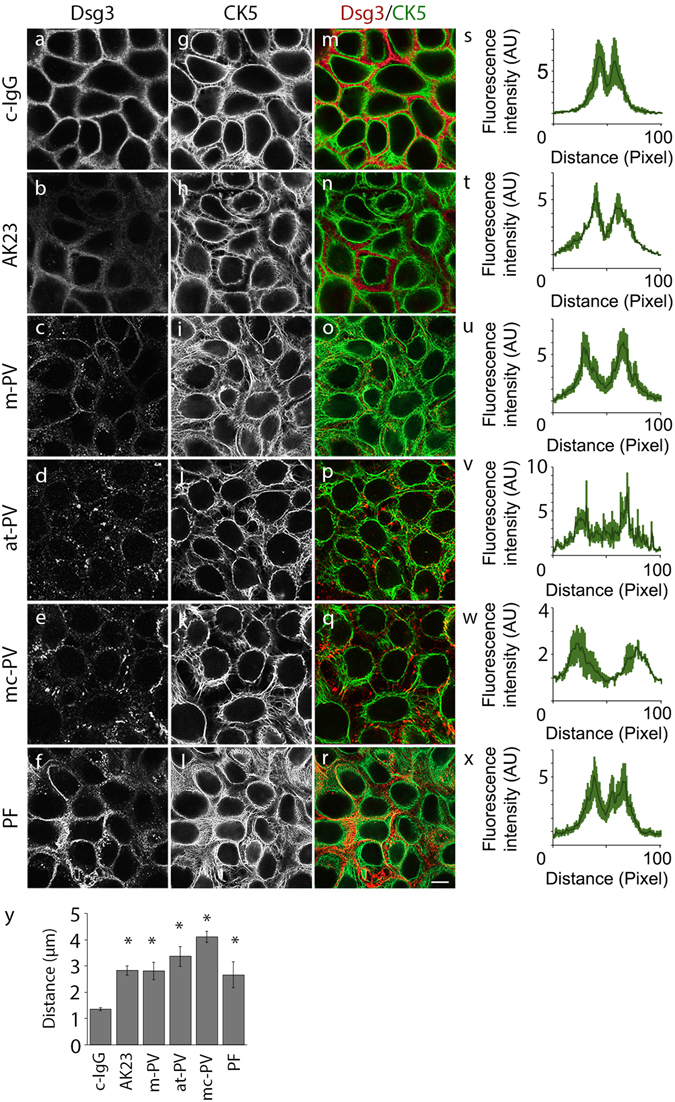
Structural changes in keratinocytes after 24 h of IgG treatment. HaCaT keratinocytes stably expressing CK5-YFP were exposed to different IgG fractions for 24 h. Images shown are representatives of four independent experiments. Dsg3 staining (a–f) and CK5 expression (g–l) after IgG treatment. (m–r) Merged images of Dsg3 staining and CK5-YFP. (s–x) CK5 fluorescence intensity of a 100 pixel long line placed centrally over the cell membrane (n = 4). (y) Analysis of the distance between fluorescence peaks shown in (s–x) in μm (n = 4, *p ≤ 0.05 vs. c-IgG). Bar represents 10 μm.
Figure 2.
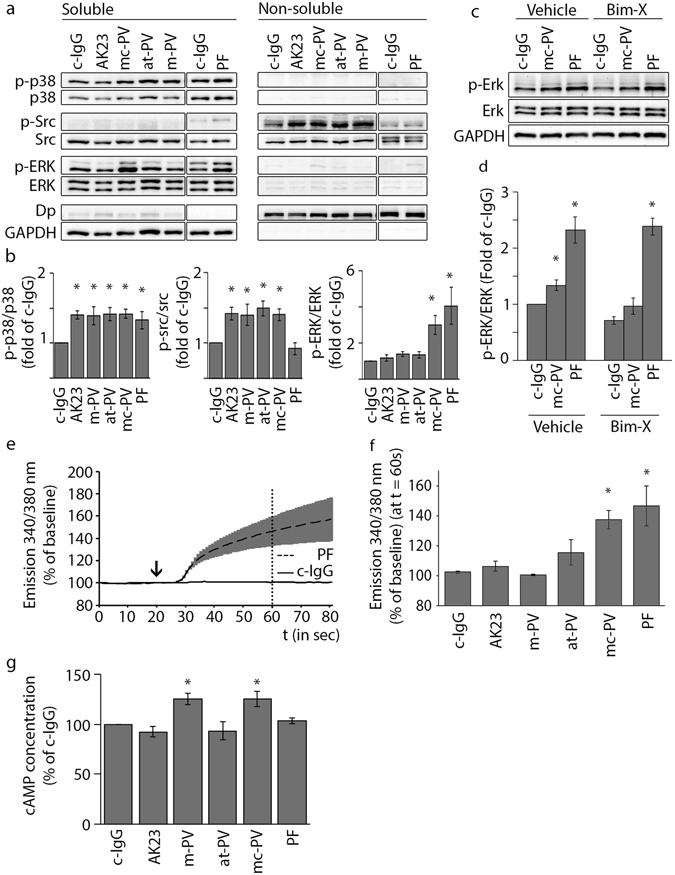
Signaling pathways activated by different IgG fractions. (a) Triton fractionation of HaCaT lysates into a TX-100 insoluble cytoskeletal and a TX-100 soluble non-cytoskeletal pool. Representative cropped Western blots of p38MAPK, src and Erk activity for each pool. (b) Densitometric analysis of signaling molecule phosphorylation normalized to the total amount in percent of control (n = 5–14, *p ≤ 0.05 vs. c-IgG). (c) Erk activation after inhibition of PKC by Bim-X and subsequent incubation of mc-PV and PF-IgG for 30 min. (d) Densitometric analysis of Erk phosphorylation corresponding to c (n = 4, *p ≤ 0.05 vs. respective c-IgG condition). Ratiometric fluorescent intensity measurement of Fura-2-AM at 340/380 nm normalized to baseline. (e) Graphs of c-IgG and PF-IgG to illustrate experimental setup. IgG incubation started at 20 s (arrow) and (f) concentrations were analyzed 60 s afterwards (dotted line) (n = 3, *p ≤ 0.05 vs. c-IgG). (g) Measurement of intracellular cAMP concentrations in keratinocytes after 6 h of IgG treatment (n = 4; *p ≤ 0.05 vs. c-IgG).
Signaling mechanisms triggered by pemphigus autoantibodies
Triton-X-100-mediated protein extraction was used to detect phosphorylation of p38MAPK, Src and Erk by Western blotting. Compared to control IgG, all autoantibodies increased p38MAPK activity, which was almost exclusively observed in the Triton-soluble fraction containing molecules not linked to the cytoskeleton (Fig. 2a and b). In contrast, Src activation only occurred in the desmosome-containing, non-soluble fraction, but was restricted to cells incubated with autoantibody fractions that contain aDsg3 and was absent in cells treated with PF-IgG lacking autoantibodies against Dsg3. In contrast, increased Erk phosphorylation was detected in the soluble fraction under conditions in which autoantibody fractions contained aDsg1 such as in mc-PV-IgG and PF-IgG compared to control. Because PKC activation was shown to be dependent on Ca2+ influx and we previously used Erk phosphorylation as a read-out for PKC activation17, 18, 27, we studied whether Erk activation was dependent on PKC activity (Fig. 2c and d). However, in the Triton-soluble fraction the PKC inhibitor Bim-X alone reduced both baseline Erk activity and Erk activation in response to mc-PV-IgG but not PF-IgG indicating that Erk activation was not primarily affected downstream of PKC. Next, we measured intracellular Ca2+ levels via the fluorescence ratio of FURA-2-AM emission (Fig. 2e and f). In line with the data aforementioned, significant Ca2+ increase was detectable after incubation with mc-PV-IgG and PF-IgG only. These data indicate that p38MAPK and Src can be activated by antibodies targeting Dsg3 whereas the Ca2+/PKC pathway as well as Erk is activated by IgG fractions containing aDsg1 only. These results were confirmed in normal human epithelial keratinocytes (NHEK) (Supplementary Fig. 2). Furthermore, we applied one additional IgG fraction for each phenotype yielding a similar outcome in regard to p38 and Erk activity as well as Ca2+ influx (Supplementary Fig. 3). Finally, since previously reports showed that intracellular cAMP levels can be augmented by PV-IgG28 we measured cAMP by ELISA under the conditions used for the present study (Fig. 2g). Increased cAMP was detected in response to m-PV-IgG and mc-PV-IgG selectively.
Effects of autoantibodies on Dsg3 and Dsg1 interaction measured by AFM
PV-IgG, but not PF-IgG, have been shown to interfere with interaction of Dsg3 but not Dsg1 on a single molecule level13–15. In line with this, in this study AK23, at-PV-IgG and mc-PV-IgG reduced Dsg3 binding as revealed by AFM (Fig. 3). In contrast, reduced number of Dsg1 binding events was observed by incubation with the commercially available inhibitory antibody p124 targeting Dsg1, but not with at-PV-IgG, mc-PV-IgG or PF-IgG although the latter two contained aDsg1. These data show that direct inhibition of Dsg3 binding correlated with the occurrence of aDsg3 and thus Src activation, whereas IgG fractions containing aDsg1 had no effects on Dsg1 interactions but nevertheless induced Ca2+/PKC and Erk-activation.
Figure 3.
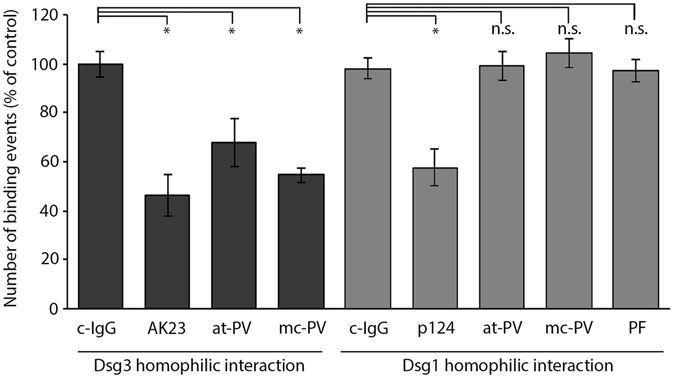
Inhibition of Dsg3 but not Dsg1 homophilic interactions by pemphigus autoantibodies. Cell free atomic force microscopy measurements on a coated mica sheet with an AFM tip functionalized with either recombinant Dsg1-Fc or Dsg3-Fc respectively. A grid with 10 × 10 points (2.5 × 2.5 μm) was measured 5 times followed by incubation with IgG-fractions, AK23 or the mouse monoclonal Dsg1 antibody p124. The identical area was scanned again and normalized to the respective measurement before incubation (n = 3–5, *p ≤ 0.05 vs. c-IgG).
Relevance of signaling pathways stimulated by autoantibodies for keratinocyte cohesion
Finally, we addressed the relevance of signaling events triggered by autoantibodies for intercellular cohesion of keratinocytes. The in vivo relevance of p38MAPK, PKC and tyrosine kinases including Src for pemphigus pathogenesis has been shown before21, 29–31. Similar to most studies in the past, we incubated inhibitors for p38MAPK (SB202190) and PKC (Bim-X) with autoantibody fractions for 24 h (Fig. 4a). Loss of keratinocyte cohesion induced by all antibody fractions was abolished by SB202190 and Bim-X. Because inhibition of Erk for 24 h using U0126 was toxic, we incubated autoantibodies with U0126 or with the Src inhibitor PP2 for 2 h only, similar to a recent report on p38MAPK22. In contrast to PP2, which was protective under all conditions, U0126 reduced effects of mc-PV-IgG and PF-IgG only, i.e. the autoantibody fractions containing aDsg1 and leading to Erk activation (Fig. 4b).
Figure 4.
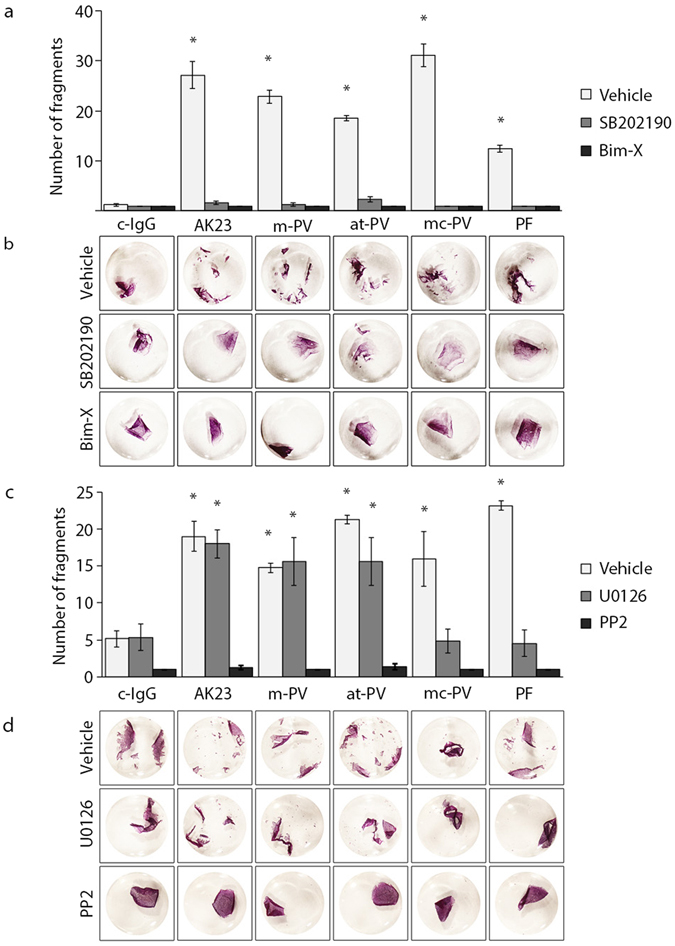
Adhesion measurements with modulation of signaling molecules. Dispase-based dissociation assays in HaCaT keratinocytes after applying the specific inhibitors SB202190 (p38MAPK), Bim-X (PKC), U0126 (Erk) or PP2 (Src) for 1 h, followed by either (a) 24 h incubation of IgG fractions or (c) 2 h incubation of IgG fractions (n = 4; *p ≤ 0.05 vs. c-IgG with the respective mediator). (b,d) Representative images of monolayers incubated with 10 μm MTT to visualize cell fragments.
Discussion
For the first time we investigated under strictly the same settings whether signaling pathways activated by pemphigus IgG correlate with autoantibody profile and thus with clinical phenotype. Previously, it was shown that p38MAPK and RhoA signaling were modulated by both PV-IgG and PF-IgG, suggesting that autoantibodies may have similar effects in signaling21, 32. In contrast, we observed signaling patterns different for mucosal, atypical and mucocutaneous PV-IgG as well as for PF-IgG, which only partially paralleled the effects on Dsg3 interaction, in AFM experiments. The second important outcome of this study is that the potency of signaling pathway modulation to protect against loss of keratinocyte cohesion varies for different signaling mechanisms. From the latter observation, we conclude that the relevance of signaling mechanisms in the regulation of desmosomal binding is different and propose that some pathways including Src and PKC may have the role of key desmosome regulators under basal conditions whereas others are autoantibody-specific.
The signaling patterns identified here appear to be complex. Activation of p38MAPK was found under all pathological conditions which is in line with all publications where a role of this pathway in pemphigus pathogenesis has been reported downstream of PV-IgG, PF-IgG and AK2316, 19, 23, 25, 33–41. This indicates that antibodies against Dsg3 as well as autoantibody fractions containing aDsg1 but not aDsg3 can activate p38MAPK signaling (Fig. 5). In contrast, Src appears to be activated by aDsg3 specifically because both the monoclonal antibody AK23 and PV-IgG but not PF-IgG stimulated this pathway as shown here. On the other hand, Ca2+ and Erk signaling pathways were restricted to IgG fractions containing aDsg1 only. Importantly, we cannot definitely conclude whether autoantibodies directed against Dsg1 were primarily responsible for signaling because additional antibodies present in the polyclonal IgG fractions may also be involved42. The identification of signaling pathways activated by specific desmosomal and non-desmosomal autoantibodies should be a primary focus for future studies. The data presented here suggest that autoantibodies targeting Dsg3 cause direct inhibition of Dsg3 binding as well as activation of some pathways such as p38MAPK and Src which contributes to keratin retraction and loss of keratinocyte cohesion at least under mechanical stress in the dissociation assay. Moreover, it is possible that p38MAPK and Src are relevant for Dsg3 clustering as proposed before25, 33, 43 although the role of this process leading to loss of cell cohesion is not clear since it was detected in unaffected skin of m-PV and mc-PV patients as well26. Thus, it can be speculated that p38MAPK- and Src-mediated loss of keratinocyte adhesion may be sufficient to cause mucosal PV but not blistering in human skin where usually acantholysis does not occur when antibodies targeting Dsg3 but not Dsg1 are present44, 45. Rather, Ca2+ and Erk signaling may be additionally required for epidermal blistering. However, since pemphigus pathogenesis is complex and autoantibodies against other antigens can be present as well10, 42, 46, 47, we do not expect that the signaling patterns are similar in each patient. For cAMP signaling, which was proposed to be a salvage pathway in PV by limiting p38MAPK activation and Dsg3 depletion28, we observed that levels of cAMP were elevated in response to m-PV-IgG and mc-PV-IgG only but not to AK23, at-PV-IgG or PF-IgG. This may indicate that some but not all antibodies targeting Dsg3 may activate cAMP signaling or that other autoantibodies are responsible. However, it is possible that the capability of autoantibodies to stimulate cAMP signaling may modulate the clinical phenotype as well because it could contribute to the fact that in m-PV skin blistering is absent but occurred in the patient with at-PV-IgG in our study in the absence of aDsg1. Accordingly, failure to induce this protective pathway may enhance the pathogenicity of AK23, which can cause epidermal blistering in mice23, 24, 40.
Figure 5.
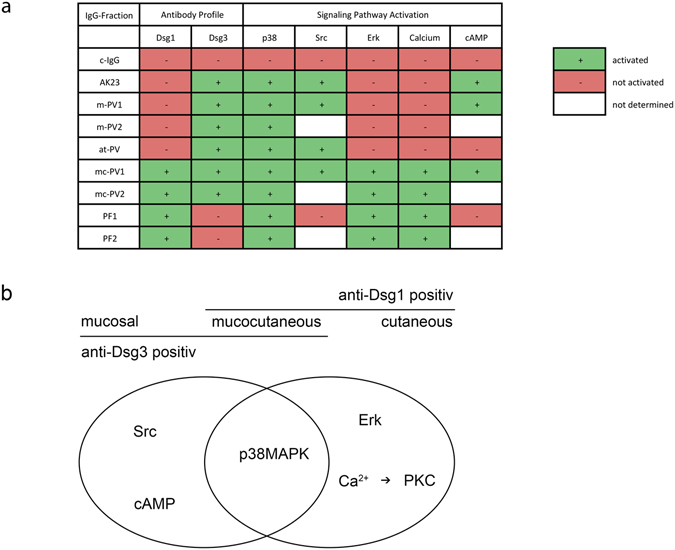
Signaling patterns associated with autoantibody profiles. Summary (a) and schematic (b) representation of signaling patterns associated with different autoantibody fractions and their respective clinical phenotypes.
We found that selective inhibition not only of p38MAPK but also of Src and PKC was effective to completely abrogate loss of cell cohesion in response to PV-IgG and PF-IgG. This was surprising because PKC was activated by mc-PV-IgG and PF-IgG only whereas Src activation was confined to PV-IgG and AK23 treatment. This demonstrates that inhibition of some signaling molecules is protective although the activity state of the molecule is not altered by autoantibody incubation. One may conclude that Src and PKC are in general critical for desmosome function and thus may represent key regulators of desmosomal adhesion, e.g. by induction of hyperadhesion as outlined below. The efficacy of p38MAPK inhibition to protect against loss of keratinocyte cohesion as observed here is well established in response to pemphigus autoantibodies or depletion of Dsg3 and plakoglobin9, 21, 34, 37, 43, 48, 49. The underlying mechanisms may involve keratin retraction, depletion of Dsg3 from junctions as well as interference with formation of new desmosomes16, 27, 33, 34. For Src activation in response to PV-IgG, phosphorylation of PKP3 and detachment from Dsg3 may destabilize cell cohesion19, 20, 50. On the other hand, PKC was demonstrated to regulate keratin filament anchorage to DP and thereby sequestration of PKC to keratin filaments51–53. Moreover, inhibition of PKC-mediated DP phosphorylation as well as PKP1 overexpression rendered desmosomes hyperadhesive, i.e. Ca2+-independent51, 52, 54. Since PKC in this scenario was shown to be regulated by PKP355, a crosstalk with Src signaling is conceivable. For Erk, which has also previously been reported to be activated in response to PV-IgG56, the situation appears to be different because Erk was activated when aDsg1 were present and contributed to loss of cell cohesion under this condition only. In contrast, inhibition of Erk did not prevent loss of cell cohesion induced by autoantibodies containing aDsg3 but not aDsg1. Future studies are required to unravel mechanisms by which Erk regulates desmosomal binding and to elucidate why Erk is not of equal importance for keratinocyte cohesion as p38MAPK, Src and PKC. The concept of central desmosomal regulators may explain why in many experimental models for pemphigus modulation of a single signaling mechanism was found to be protective. Taken together, our study enhances the understanding on pemphigus pathogenesis because it proposes that a pattern of signaling events, which are of different importance for regulation of desmosomal adhesion, may define the clinical phenotype of pemphigus.
Methods
Cell culture and test reagents
The human immortalized keratinocyte cell line HaCaT57 was cultured in Dulbecco’s Modified Eagle Medium containing 10% FCS (Biochrom, Berlin, Germany), 50 U/ml penicillin and 50 g/ml streptomycin (both AppliChem, Darmstadt, Germany) in a humidified atmosphere of 5% CO2 at 37 °C HaCaT cells stably transfected with keratin5-YFP were used for immunostaining experiments (kind gift of Reinhard Windoffer and Nicole Schwarz, Institute of Molecular and Cellular Anatomy, RWTH Aachen University58). The cells were used 1d after confluency and medium was routinely changed the evening before experimental procedure. Adult Normal Human Epithelial Keratinocytes (NHEK) were purchased and cultured in Keratinocyte Growth Medium 2 (both Promocell, Heidelberg, Germany) in low calcium conditions (0.06 mM) until confluency and eventually switched to 1.2 mM calcium for 24 h. Experiments were performed in between passages 3 and 6.
Pemphigus sera and IgG purification
The pathogenic monoclonal antibody AK23 against the EC1 domain of Dsg3 was purchased (Biozol, Eching, Germany) and used at a concentration of 75 g/ml. To inhibit homophilic interactions of Dsg1, the monoclonal antibody Dsg1 p124 (Progen, Heidelberg, Germany) directed against the extracellular domain of Dsg1 was used in a 1:20 dilution. For purification of sera, patients gave written consent for research use and institutional approval of the ethics committee was given. The phenotype was characterized in the process of disease diagnosis via clinical and histological evaluation and the antibody profile was determined by Dsg1 and Dsg3 ELISA. IgG fractions were purified using protein A affinity chromatography as described previously15. Briefly, antibodies from patient sera were immobilized to protein A agarose (ThermoFisher, Waltham, USA) in purification columns for 2 h at room temperature. After washing with phosphate-buffered saline (PBS), the antibody fractions were eluted by sodium citrate buffer (20 mM, pH 2.4), neutralized with Na2CO3 and concentrated in a filter unit (Amicon Ultra–4, 100k; Merck Millipore, Darmstadt, Germany) at 19000 g for 20 min. Afterwards, the IgG-fractions were retrieved with PBS and tested for functionality in a dispase-based dissociation assay.
Immunostaining
Cells were washed with PBS and fixed with 2% formalin (freshly prepared from paraformaldehyde) in PBS for 10 min. Afterwards they were permeabilized with 1% Triton X-100 in PBS for 5 min. Subsequently, cells were blocked by 3% bovine serum albumin and 1% normal goat serum in PBS for 60 min and incubated with anti-Dsg3 mAb (clone 5G11, Invitrogen, Carlsbad, CA) at 4 °C overnight. The Cy3-labeled secondary goat anti-rabbit antibody (Dianova, Hamburg, Germany) was incubated for 1 h at room temperature and samples were mounted with n-propyl gallate as an antifade compound on glass slides. Images were acquired with a 63x NA 1.4 PL APO objective on a SP5.II confocal microscope (both Leica, Mannheim, Germany).
Triton X-100 protein fractionation, electrophoresis and Western blotting
After washing with ice-cold PBS, cell lysates were retrieved by incubation in extraction buffer (0.5% Triton X-100, 50 mmol/L MES, 25 mmol/L EGTA, 5 mmol/L MgCl2) for 10 min on ice containing a mixture of 0,1% leupeptin, aprotinin and pepstatin as well as 1% phenylmethylsulfonylfluorid under gentle shaking. Afterwards cells were scraped and centrifuged at 19000 g for 10 min at 4 °C to separate the triton-soluble, non cytoskeletal-bound fraction from the cytoskeletal bound insoluble fraction (below named pellet). The pellet was suspended in SDS-lysis buffer (25 mmol/l HEPES, 2 mmol/l EDTA, 25 mmol/l NaF and 1% SDS, pH 7,4) and sonicated. Eventually, the protein amount was measured using the BCA method (Thermo Fisher Scientific, Schwerte, Germany). Both fractions were mixed with laemmli buffer containing 50 mM dithiothreitol. Electrophoresis and wet blotting were performed according to standard procedures. Membranes were incubated at 4 °C overnight with the following primary antibodies in 5% bovine serum albumin (BSA) in tris-buffered-saline with 0.05% tween (TBS-T): phospho-p38 MAPK pAB (Thr180/Tyr182) (Cell Signaling Technologies, Cambridge, United Kingdom), p38 MAPK pAB (Cell Signaling Technologies), p-Erk mAB (E-4) (Santa Cruz, Heidelberg, Germany), p44/42 MAPK pAB (Erk1/2) (Cell Signaling Technologies), p-src pAB (Tyr416) (Cell Signaling Technologies), Src pAB (Cell Signaling Technologies), Dsg3 mAB (5H10) (Novus Biological, Littleton, USA), Dsg1 mAB (p124) (Progen), GAPDH mAB (Santa Cruz), and Desmoplakin I/II pAB (H-300) (Santa Cruz). Secondary antibodies were either a HRP-coupled goat anti-mouse, a HRP-coupled goat anti-rabbit or a HRP-coupled goat anti-human mAB (Dianova, Hamburg, Germany). Chemiluminescence was detected with the ECL system (GE Healthcare, Munich, Germany). Western Blots were cropped at the appropriate kDa size. Full-length versions can be found in Supplementary Fig. 4.
Measurements of cellular cAMP levels
Cellular cAMP levels were determined by cAMP enzyme linked immunosorbent assay (CA-200) (Sigma-Aldrich, St. Louis, MO). After 6 h of IgG incubation cells were washed and lysed by 0.1 M HCl. The assay was further performed according to manufacturer’s instructions.
Ca2+ concentration measurements
Cells were washed with measurement buffer (140 mM NaCl, 3.6 mM KCl, 2.6 mM CaCl2(H2O)2, 0.5 mM NaH2PO4(H2O)2, 2 mM NaHCO3, HEPES and 5 mM D + Glucose) prior to experiment performance. A mixture of 0.4% Fura-2-AM (Molecular Probes, Thermo Fisher) and 0.4% Pluronic F-127 (Life Technologies, Thermo Fisher) diluted in measurement buffer were afterwards incubated for 20 min at 37 °C. Cells were subsequently incubated in fresh measurement buffer for another 15 min before analysis was started. The experiments were performed with an inverted microscope (Carl Zeiss Microscopy) equipped with a polychrome V light source (TILL Photonics, Martinsried, Germany) a CoolSNAP-Hq2 digital camera (Photometrics) and a Fura-2 filter set (AHF Analysetechnik, Tuebingen, Germany).
Atomic force microscopy (AFM)
Measurements were performed as previously described by our group41 under cell-free conditions using a NanoWizard® 3 AFM (JPK-Instruments, Berlin, Germany). Cleaved mica sheets (SPI-Supplies, West Chester, USA) and pyramidal-shaped D-Tips of Si3N4 MLCT cantilevers with a nominal spring constant of 0.03 N/m and a 20 nm tip radius (Bruker, Mannheim, Germany) were coated with a flexible heterobifunctional acetal-polyethylenglycol linker (PEG, Hermann Gruber Lab, Institute of Biophysics, Linz, Austria) and in turn functionalized with purified Dsg1-Fc and Dsg3-Fc (both 0.15 mg/ml). In force spectroscopy mode (FS-mode), the cantilever was repeatedly lowered onto and retracted from the mica sheet with a pulling speed of 1 μm/s, a z-length of 0.3 μm and a resting contact time of 0.1 s. An area of 25 × 25 μm was selected and 500 measurement positions were probed in a grid of 10 × 10 readings for five times in HBSS buffer. The same area on the mica sheet was scanned for binding events with the same specifications before and after addition of IgG-fractions for 1 h. By calibration using the thermal-noise method59, deflections of the cantilever were converted into forces and plotted as force-distance curves. These force curves were evaluated for unbinding events, indicating interactions between tip and mica-attached Dsg molecules. The number of unbinding events per total number of approach/retract cycles was determined as binding frequency. Binding frequency after addition of pemphigus autoantibodies was normalized to the value before measurement at the same area.
Dispase-based dissociation assay
The cell monolayer was detached from the well bottom by a 20 min incubation step with Dispase-II (Sigma-Aldrich) after thorough washing with HBSS (Sigma Aldrich). Afterwards, Dispase-II solution was substituted by HBSS and mechanical stress was applied by shearing the monolayer 10 times with an electrical 1 ml pipette. The cell fragments, counted under a binocular microscope (Leica), are a measure for loss of cell cohesion. Cells were incubated for 10 min in 10 μm thiazolyl blue tetrazolium bromide (MTT) (Sigma Aldrich) for better visibility.
Data analysis and statistics
Images were processed using Photoshop CC (Adobe Systems, San Jose, CA). Densitometric measurements were performed using the gel analysis function in ImageJ (Wayne Rasband, www.nih.gov). Data were analyzed in Excel (Microsoft, Redmond, WA). Statistical analysis was performed using one way ANOVA followed by Dunn-Šidák correction using Graphpad Prism (Graphpad Software, LaJolla, CA). Significance was presumed with p ≤ 0.05. Data are shown as mean ± SEM. Each n represents one independent experiment.
Electronic supplementary material
Acknowledgements
We thank Martina Hitzenbichler, Thomas Korbica, Sabine Mühlsimer and Linda Jacobi for excellent technical assistance and Maryia Radeva for helpful scientific discussions as well as Marie Wanuske and Jan Hoffmann for proof reading the manuscript. Experiments were funded by the Deutsche Forschungsgesellschaft (FOR 2497).
Author Contributions
E.W. performed the experiments. F.V. supervised the AFM studies and L.R. helped with establishment of FURA measurements. M.S. and O.H. determined the antibody profiles. M.G., E.S, R.E. and M.H. provided the pemphigus sera. E.W., V.S. and J.W. wrote the paper and interpreted the data. V.S. and J.W. supervised the work.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-03697-7
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Volker Spindler, Email: Volker.Spindler@med.uni-muenchen.de.
Jens Waschke, Email: Jens.Waschke@med.uni-muenchen.de.
References
- 1.Stanley JR, Amagai M. Pemphigus, bullous impetigo, and the staphylococcal scalded-skin syndrome. N. Engl. J. Med. 2006;355:1800–10. doi: 10.1056/NEJMra061111. [DOI] [PubMed] [Google Scholar]
- 2.Johnson JL, Najor NA, Green KJ. Desmosomes: regulators of cellular signaling and adhesion in epidermal health and disease. Cold Spring Harb. Perspect. Med. 2014;4:a015297–a015297. doi: 10.1101/cshperspect.a015297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kneisel, A. & Hertl, M. Autoimmune bullous skin diseases. Part 1: Clinical manifestations. J. der Dtsch. Dermatologischen Gesellschaft = J. Ger. Soc. Dermatology JDDG9, 844–56; quiz 857 doi:10.1111/j.1610-0387.2011.07793.x (2011). [DOI] [PubMed]
- 4.Carew B, Wagner G. Cutaneous pemphigus vulgaris with absence of desmoglein 1 autoantibodies. An example of the extended desmoglein compensation theory. Australas. J. Dermatol. 2014;55:292–5. doi: 10.1111/ajd.12154. [DOI] [PubMed] [Google Scholar]
- 5.Yoshida K, et al. Cutaneous type pemphigus vulgaris: a rare clinical phenotype of pemphigus. J. Am. Acad. Dermatol. 2005;52:839–45. doi: 10.1016/j.jaad.2005.01.106. [DOI] [PubMed] [Google Scholar]
- 6.Aoyama Y, Kitajima Y. Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the Triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J. Invest. Dermatol. 1999;112:67–71. doi: 10.1046/j.1523-1747.1999.00463.x. [DOI] [PubMed] [Google Scholar]
- 7.Sokol E, et al. Large-Scale Electron Microscopy Maps of Patient Skin and Mucosa Provide Insight into Pathogenesis of Blistering Diseases. J. Invest. Dermatol. 2015;135:1763–70. doi: 10.1038/jid.2015.109. [DOI] [PubMed] [Google Scholar]
- 8.Wilgram GF, Caulfield JB, Lever WF. An electron microscopic study of acantholysis in pemphigus vulgaris. J. Invest. Dermatol. 1961;36:373–82. doi: 10.1038/jid.1961.58. [DOI] [PubMed] [Google Scholar]
- 9.Spindler V, Dehner C, Hübner S, Waschke J. Plakoglobin but Not Desmoplakin Regulates Keratinocyte Cohesion via Modulation of p38MAPK Signaling. J. Invest. Dermatol. 2014;134:1655–64. doi: 10.1038/jid.2014.21. [DOI] [PubMed] [Google Scholar]
- 10.Stahley SN, Kowalczyk AP. Desmosomes in acquired disease. Cell Tissue Res. 2015;360:439–56. doi: 10.1007/s00441-015-2155-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991;67:869–77. doi: 10.1016/0092-8674(91)90360-B. [DOI] [PubMed] [Google Scholar]
- 12.Mahoney MG, et al. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J. Clin. Invest. 1999;103:461–8. doi: 10.1172/JCI5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heupel W-M, Zillikens D, Drenckhahn D, Waschke J. Pemphigus vulgaris IgG directly inhibit desmoglein 3-mediated transinteraction. J. Immunol. (Baltimore, Md. 1950) 2008;181:1825–34. doi: 10.4049/jimmunol.181.3.1825. [DOI] [PubMed] [Google Scholar]
- 14.Spindler V, et al. Desmocollin 3-mediated binding is crucial for keratinocyte cohesion and is impaired in pemphigus. J. Biol. Chem. 2009;284:30556–64. doi: 10.1074/jbc.M109.024810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waschke J, Bruggeman P, Baumgartner W, Zillikens D, Drenckhahn D. Pemphigus foliaceus IgG causes dissociation of desmoglein 1-containing junctions without blocking desmoglein 1 transinteraction. J. Clin. Invest. 2005;115:3157–65. doi: 10.1172/JCI23475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waschke J, Spindler V. Desmosomes and extradesmosomal adhesive signaling contacts in pemphigus. Med. Res. Rev. 2014;34:1127–45. doi: 10.1002/med.21310. [DOI] [PubMed] [Google Scholar]
- 17.Seishima M, et al. Pemphigus IgG, but not bullous pemphigoid IgG, causes a transient increase in intracellular calcium and inositol 1,4,5-triphosphate in DJM-1 cells, a squamous cell carcinoma line. J. Invest. Dermatol. 1995;104:33–7. doi: 10.1111/1523-1747.ep12613469. [DOI] [PubMed] [Google Scholar]
- 18.Osada K, Seishima M, Kitajima Y. Pemphigus IgG activates and translocates protein kinase C from the cytosol to the particulate/cytoskeleton fractions in human keratinocytes. J. Invest. Dermatol. 1997;108:482–7. doi: 10.1111/1523-1747.ep12289726. [DOI] [PubMed] [Google Scholar]
- 19.Chernyavsky AI, Arredondo J, Kitajima Y, Sato-Nagai M, Grando SA. Desmoglein versus non-desmoglein signaling in pemphigus acantholysis: characterization of novel signaling pathways downstream of pemphigus vulgaris antigens. J. Biol. Chem. 2007;282:13804–12. doi: 10.1074/jbc.M611365200. [DOI] [PubMed] [Google Scholar]
- 20.Cirillo N, AlShwaimi E, McCullough M, Prime SS. Pemphigus vulgaris autoimmune globulin induces Src-dependent tyrosine-phosphorylation of plakophilin 3 and its detachment from desmoglein 3. Autoimmunity. 2014;47:134–40. doi: 10.3109/08916934.2013.866100. [DOI] [PubMed] [Google Scholar]
- 21.Berkowitz P, et al. p38MAPK inhibition prevents disease in pemphigus vulgaris mice. Proc. Natl. Acad. Sci. USA. 2006;103:12855–60. doi: 10.1073/pnas.0602973103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vielmuth F, Waschke J, Spindler V. Loss of Desmoglein Binding Is Not Sufficient for Keratinocyte Dissociation in Pemphigus. J. Invest. Dermatol. 2015;135:3068–3077. doi: 10.1038/jid.2015.324. [DOI] [PubMed] [Google Scholar]
- 23.Spindler V, et al. Peptide-mediated desmoglein 3 crosslinking prevents pemphigus vulgaris autoantibody-induced skin blistering. J. Clin. Invest. 2013;123:800–11. doi: 10.1172/JCI60139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsunoda K, et al. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J. Immunol. (Baltimore, Md. 1950) 2003;170:2170–8. doi: 10.4049/jimmunol.170.4.2170. [DOI] [PubMed] [Google Scholar]
- 25.Saito M, et al. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PLoS ONE. 2012;7:e50696. doi: 10.1371/journal.pone.0050696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Wier G, Pas HH, Kramer D, Diercks GFH, Jonkman MF. Smaller desmosomes are seen in the skin of pemphigus patients with anti-desmoglein 1 antibodies but not in patients with anti-desmoglein 3 antibodies. J. Invest. Dermatol. 2014;134:2287–90. doi: 10.1038/jid.2014.140. [DOI] [PubMed] [Google Scholar]
- 27.Rötzer V, Breit A, Waschke J, Spindler V. Adducin is required for desmosomal cohesion in keratinocytes. J. Biol. Chem. 2014;289:14925–40. doi: 10.1074/jbc.M113.527127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spindler V, Vielmuth F, Schmidt E, Rubenstein DS, Waschke J. Protective endogenous cyclic adenosine 5′-monophosphate signaling triggered by pemphigus autoantibodies. J. Immunol. (Baltimore, Md. 1950) 2010;185:6831–8. doi: 10.4049/jimmunol.1002675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pretel M, et al. An imbalance in Akt/mTOR is involved in the apoptotic and acantholytic processes in a mouse model of pemphigus vulgaris. Exp. Dermatol. 2009;18:771–80. doi: 10.1111/j.1600-0625.2009.00893.x. [DOI] [PubMed] [Google Scholar]
- 30.Sánchez-Carpintero I, et al. In vivo blockade of pemphigus vulgaris acantholysis by inhibition of intracellular signal transduction cascades. Br. J. Dermatol. 2004;151:565–70. doi: 10.1111/j.1365-2133.2004.06147.x. [DOI] [PubMed] [Google Scholar]
- 31.Spindler V, et al. The extent of desmoglein 3 depletion in pemphigus vulgaris is dependent on Ca(2+)-induced differentiation: a role in suprabasal epidermal skin splitting? Am. J. Pathol. 2011;179:1905–16. doi: 10.1016/j.ajpath.2011.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waschke J, et al. Inhibition of Rho A activity causes pemphigus skin blistering. J. Cell Biol. 2006;175:721–7. doi: 10.1083/jcb.200605125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bektas M, Jolly PS, Berkowitz P, Amagai M, Rubenstein DS. A pathophysiologic role for epidermal growth factor receptor in pemphigus acantholysis. J. Biol. Chem. 2013;288:9447–56. doi: 10.1074/jbc.M112.438010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berkowitz P, et al. Desmosome signaling. Inhibition of p38MAPK prevents pemphigus vulgaris IgG-induced cytoskeleton reorganization. J. Biol. Chem. 2005;280:23778–84. doi: 10.1074/jbc.M501365200. [DOI] [PubMed] [Google Scholar]
- 35.Kawasaki Y, Aoyama Y, Tsunoda K, Amagai M, Kitajima Y. Pathogenic monoclonal antibody against desmoglein 3 augments desmoglein 3 and p38 MAPK phosphorylation in human squamous carcinoma cell line. Autoimmunity. 2006;39:587–90. doi: 10.1080/08916930600971943. [DOI] [PubMed] [Google Scholar]
- 36.Luyet C, et al. Preclinical studies identify non-apoptotic low-level caspase-3 as therapeutic target in pemphigus vulgaris. PLoS ONE. 2015;10:e0119809. doi: 10.1371/journal.pone.0119809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wright PE, Dyson HJ. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao X, et al. Autoimmunity to desmocollin 3 in pemphigus vulgaris. Am. J. Pathol. 2010;177:2724–30. doi: 10.2353/ajpath.2010.100483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sayar BS, et al. EGFR inhibitors erlotinib and lapatinib ameliorate epidermal blistering in pemphigus vulgaris in a non-linear, V-shaped relationship. Exp. Dermatol. 2014;23:33–8. doi: 10.1111/exd.12290. [DOI] [PubMed] [Google Scholar]
- 40.Schulze K, et al. An adult passive transfer mouse model to study desmoglein 3 signaling in pemphigus vulgaris. J. Invest. Dermatol. 2012;132:346–55. doi: 10.1038/jid.2011.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vielmuth F, Hartlieb E, Kugelmann D, Waschke J, Spindler V. Atomic force microscopy identifies regions of distinct desmoglein 3 adhesive properties on living keratinocytes. Nanomedicine nanotechnology, Biol. Med. 2015;11:511–20. doi: 10.1016/j.nano.2014.10.006. [DOI] [PubMed] [Google Scholar]
- 42.Ahmed AR, et al. Monopathogenic vs multipathogenic explanations of pemphigus pathophysiology. Exp. Dermatol. 2016;25:839–846. doi: 10.1111/exd.13106. [DOI] [PubMed] [Google Scholar]
- 43.Mao X, Sano Y, Park JM, Payne AS. p38 MAPK activation is downstream of the loss of intercellular adhesion in pemphigus vulgaris. J. Biol. Chem. 2011;286:1283–91. doi: 10.1074/jbc.M110.172874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Amagai M, Stanley JR. Desmoglein as a target in skin disease and beyond. J. Invest. Dermatol. 2012;132:776–84. doi: 10.1038/jid.2011.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kneisel, A. & Hertl, M. Autoimmune bullous skin diseases. Part 1: Clinical manifestations. J. der Dtsch. Dermatologischen Gesellschaft = J. Ger. Soc. Dermatology JDDG9, 844–56; quiz 857 doi:10.1111/j.1610-0387.2011.07793.x (2011). [DOI] [PubMed]
- 46.Sajda T, et al. Multiplexed autoantigen microarrays identify HLA as a key driver of anti-desmoglein and -non-desmoglein reactivities in pemphigus. Proc. Natl. Acad. Sci. USA. 2016;113:1859–64. doi: 10.1073/pnas.1525448113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Waschke J. The desmosome and pemphigus. Histochem. Cell Biol. 2008;130:21–54. doi: 10.1007/s00418-008-0420-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berkowitz P, Chua M, Liu Z, Diaz La, Rubenstein DS. Autoantibodies in the autoimmune disease pemphigus foliaceus induce blistering via p38 mitogen-activated protein kinase-dependent signaling in the skin. Am. J. Pathol. 2008;173:1628–36. doi: 10.2353/ajpath.2008.080391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hartlieb E, Rötzer V, Radeva M, Spindler V, Waschke J. Desmoglein 2 compensates for desmoglein 3 but does not control cell adhesion via regulation of p38 mitogen-activated protein kinase in keratinocytes. J. Biol. Chem. 2014;289:17043–53. doi: 10.1074/jbc.M113.489336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neuber S, et al. c-Src mediated tyrosine phosphorylation of plakophilin 3 as a new mechanism to control desmosome composition in cells exposed to oxidative stress. Cell Tissue Res. 2015;359:799–816. doi: 10.1007/s00441-014-2063-x. [DOI] [PubMed] [Google Scholar]
- 51.Dehner C, Rötzer V, Waschke J, Spindler V. A desmoplakin point mutation with enhanced keratin association ameliorates pemphigus vulgaris autoantibody-mediated loss of cell cohesion. Am. J. Pathol. 2014;184:2528–36. doi: 10.1016/j.ajpath.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 52.Hobbs RP, Green KJ. Desmoplakin regulates desmosome hyperadhesion. J. Invest. Dermatol. 2012;132:482–5. doi: 10.1038/jid.2011.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kröger C, et al. Keratins control intercellular adhesion involving PKC-α-mediated desmoplakin phosphorylation. J. Cell Biol. 2013;201:681–92. doi: 10.1083/jcb.201208162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tucker DK, Stahley SN, Kowalczyk AP. Plakophilin-1 protects keratinocytes from pemphigus vulgaris IgG by forming calcium-independent desmosomes. J. Invest. Dermatol. 2014;134:1033–43. doi: 10.1038/jid.2013.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Keil R, Rietscher K, Hatzfeld M. Antagonistic Regulation of Intercellular Cohesion by Plakophilins 1 and 3. J. Invest. Dermatol. 2016;136:2022–9. doi: 10.1016/j.jid.2016.05.124. [DOI] [PubMed] [Google Scholar]
- 56.Frusić-Zlotkin M, et al. Apoptotic mechanism in pemphigus autoimmunoglobulins-induced acantholysis-possible involvement of the EGF receptor. Autoimmunity. 2006;39:563–75. doi: 10.1080/08916930600971836. [DOI] [PubMed] [Google Scholar]
- 57.Boukamp P, et al. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988;106:761–71. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moch M, Herberich G, Aach T, Leube RE, Windoffer R. Measuring the regulation of keratin filament network dynamics. Proc. Natl. Acad. Sci. USA. 2013;110:10664–9. doi: 10.1073/pnas.1306020110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Slattery AD, Blanch AJ, Quinton JS, Gibson CT. Accurate measurement of Atomic Force Microscope cantilever deflection excluding tip-surface contact with application to force calibration. Ultramicroscopy. 2013;131:46–55. doi: 10.1016/j.ultramic.2013.03.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.