

Abstract
Amides have been widely studied for decades, but their synthetic utility has remained limited in reactions that proceed with rupture of the amide C–N bond. Using Ni catalysis, we have found that amides can now be strategically employed in several important transformations: esterification, transamidation, Suzuki–Miyaura couplings, and Negishi couplings. These methodologies provide exciting new tools to build C–heteroatom and C–C bonds using an unconventional reactant (i.e., the amide), which is ideally suited for use in multi-step synthesis. It is expected that the area of amide C–N bond activation using nonprecious metals will continue to flourish and, in turn, will promote the growing use of amides as synthons in organic synthesis.
Keywords: nickel, catalysis, cross-coupling, amides, nonprecious metal
Graphical abstract
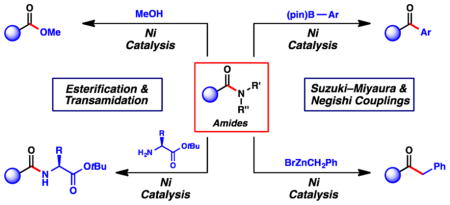
1. INTRODUCTION
Transition-metal-catalyzed cross-couplings have revolutionized the way chemists assemble small molecules.1 Despite the longstanding prevalence of palladium catalysis in this field, much interest has recently been devoted to the use of base-metal catalysis to enable coupling reactions (Figure 1).2 Our laboratory3 and others have focused on the use of nickel catalysis due to (a) nickel’s natural abundance and consequential low cost ($720/oz. for Pd vs $0.47/oz for Ni),2d–h (b) favorable toxicological profiles for orally administered drugs (permissible daily exposure: 100 μg/day for Pd vs 500 μg/day for Ni),4 (c) opportunities in green chemistry,3g–i,5 and (d) the potential to develop new chemical transformations on the basis of alternate reactivity profiles.
Figure 1.

Benefits and opportunities of nickel catalysis.
Although it is generally less appreciated in comparison to palladium catalysis, it should not go unnoticed that nickel has been employed in industrial settings, in some cases for many decades.6 Several notable examples are depicted in Figure 2. The first features DuPont’s hydrocyanation of butadiene (1). This transformation has been used to mass-produce adiponitrile (2), the precursor to Nylon-66, since 1971.6a Another notable example is the Shell Higher Olefin Process (SHOP), where ethylene (3) can be converted to α-olefins 4 en route to detergents and plasticizers. This nickel-catalyzed process is used to manufacture >1 million tonnes of olefins 4 per year.6b Finally, the use of nickel catalysis in the pharmaceutical industry is gaining traction. One notable example involves the synthesis of Lyrica (5), wherein the primary amine is introduced via Raney Ni reduction of the corresponding nitrile on a >2 tonne scale.6c With regard to cross-coupling, the Genentech process team has shown that PI3K inhibitor 6 can be accessed using a nickel-catalyzed Suzuki–Miyaura coupling performed on a >50 kg scale.6d These large-scale examples bode well for future industrial applications of nickel catalysis in chemical manufacturing processes.
Figure 2.

Industrial examples of nickel catalysis.
Given the potential benefits of nickel catalysis, it is not surprising that many academic and industrial groups have sought to develop new nickel-based methodologies (Figure 3). For example, intermolecular C–C bond forming reactions using unconventional substrates such as carbonates, carbamates, esters, and ethers have been reported for both aryl2e,3a–i,7 and benzylic8 electrophiles. Nickel has also been strategically employed in Heck reactions,9 aminations,10 and reductive couplings.11 Other notable examples involving nickel catalysis include its synergistic use in photoredox catalysis,12 in addition to efforts in C–H13 and C–F bond activation.14 Finally, the activation of amide C–N bonds3j–o,15 using nickel catalysis, which is the topic of this Perspective, has recently emerged as a powerful synthetic tool.
Figure 3.

Select examples of recent advances in nickel catalysis, including the nickel-catalyzed activation of amides.
The efficient cleavage of amide C–N bonds has remained an underdeveloped process for many decades.16 Such reactivity is challenging due to the well-known resonance stabilization of amides as postulated by Pauling in the 1950s (Figure 4).16b In fact, the repertoire of reactions involving amide C–N bond cleavage that are generally considered useful to synthetic chemists is limited. Notable transformations include (a) the conversion of Weinreb amides to ketones using basic and pyrophoric reagents (7 → 8),17 (b) the stoichiometric reduction of amides using Schwartz’s zirconium-based reagent (9 → 10),18 and (c) the hydrolysis or esterification of amides (9 → 11), which often requires harshly acidic or basic reaction conditions, a large excess of the nucleophile, high temperatures, or a combination of these reaction parameters.16a
Figure 4.

Amide stability and common synthetic methods involving amide C–N bond cleavage.
We reasoned that amides, despite being underutilized in amide C–N bond cleavage reactions, presented a unique opportunity. A new strategy to break amide C–N bonds could overcome a classical problem in organic chemistry, while also providing a new synthetic tool. For example, as suggested in Figure 5, we view amides as being ideally suited for use in multi-step synthesis. They are well-known to be stable to an array of reaction conditions (e.g., acids, bases, redox, Pd catalysis, heat, etc.) and can therefore be carried through multi-step sequences before, in principle, carrying out a late-stage transformation of the amide. As an additional feature, amides can be exploited as directing groups in C–H functionalization reactions.19 Classically, this has been achieved by ortho metalation of arene substrates, as exemplified by the lithiation of 12 to give reactive intermediate 13.20 Transition-metal-catalyzed approaches to C–H functionalization of arenes and aliphatic systems have also been developed, as exemplified by Sanford’s conversion of amide 14 to phenylated product 15.21
Figure 5.

Opportunities for amides as synthetic building blocks.
We envisioned carrying out the sequence shown in Figure 6, wherein amides 9 would undergo Ni-catalyzed amide C–N bond activation to give acyl nickel species 16.22 In situ trapping of 16 with a nucleophile would furnish acyl derivatives 17. Following this paradigm, our laboratory has recently developed several C–C and C–heteroatom bond forming reactions of amides to give ketones, esters, and amide products.3j–m,o This Perspective highlights such efforts. Although not our focus herein, many other related advances deserve high praise, such as the nickel-catalyzed decarbonylative borylation by Shi,15a Szostak’s nickel-catalyzed Negishi coupling of amides to access biaryls,15c Szostak’s decarbonylative and nondecarbonylative Pd-catalyzed reactions of twisted amides,15e,23 Zou’s Pd-catalyzed Suzuki–Miyaura coupling of amide derivatives,24 and Murakami’s insertion of alkenes into the C–N bonds of β-lactams.25
Figure 6.

Nickel-catalyzed activation and reactions of amides studied in our laboratory.
2. CARBON–HETEROATOM BOND FORMING REACTIONS
Our first forays into the nickel-catalyzed manipulation of amides involved reactions with alcohols to give ester products (Figure 7).3j The esterification of amides has remained a challenging transformation for many decades, which rendered it an exciting starting point for our studies. Notable methods for the esterification of amides include classical alcoholysis under basic or acid conditions (often with heat and a large excess of nucleophile),16a the nitrosation of N-methyl amides,26 reactions of acyl aziridines,27 and Keck’s methylation/hydrolysis protocol.28 The catalytic means to convert amides (9) to esters (18) using nickel catalysis suggested in Figure 7 had not previously been demonstrated.
Figure 7.

Design of the nickel-catalyzed esterification of amides.
An extensive survey of reaction parameters led to the identification of suitable reaction conditions for the amide to ester conversion. Specifically, it was found that an array of anilides 19 underwent the desired coupling with alcohols 20 in the presence of Ni(cod)2 and the NHC ligand SIPr in toluene at 80 °C (Figure 8).3j The transformation proceeds efficiently using only 1.2 equiv of the alcohol coupling partner. With regard to the amide component, benzamides possessing electron-withdrawing, electron-donating, and ortho substituents were tolerated, as demonstrated by the formation of esters 22–24. Heterocyclic substrates, such as a furan and a quinoline, could also be used to generate esters 25 and 26, respectively. With regard to the alcohol component, primary, secondary, and tertiary alcohols could all be utilized. Select examples include the use of (−)-menthol, 1-adamantol, and N-Boc prolinol, which gave 27–29. Moreover, esters 30 and 31 were obtained when an indolyl alcohol and steroidal alcohol were used, respectively.
Figure 8.

Selected examples of the nickel-catalyzed esterification of amides methodology.
The presumed catalytic cycle for the nickel-catalyzed esterification of amides is shown in Figure 9. The reaction is thought to proceed via oxidative addition (32 + 33 → 34), ligand exchange (34 → 35), and reductive elimination (35 → 36 + 33). Our collaborators, Professor Ken Houk and coworkers, performed calculations that were consistent with this pathway, along with insightful calculations regarding the overall thermodynamics of the transformation. Oxidative addition is thought to be the rate-determining step with a calculated barrier of 26.0 kcal/mol. The three-centered oxidative addition transition state obtained from the Houk group’s calculations is shown. It should be noted that Shi’s laboratory has obtained experimental evidence for the oxidative addition step.15a
Figure 9.

Proposed mechanism of the nickel-catalyzed esterification of amides.
We also examined a series of selectivity studies, one of which is highlighted in Figure 10. In this case, we prepared bis(amide) 37 and subjected it to nickel-catalyzed esterification using 1.2 equiv of (−)-menthol (38). This led to the selective esterification of the benzamide-derived anilide, without disruption of the proline-derived anilide. Additionally, the proline-based leaving group 39 in this transformation was recovered in 83% yield, without loss of enantiopurity. Thus, this result not only demonstrates that selective cleavage of polyamides is possible but also highlights the mild nature of the reaction conditions.
Figure 10.

Selective activation of acyl amide C–N bonds over alkyl amide C–N bonds.
An additional example showcasing the mild and selective esterification of amides is provided in Figure 11. We prepared the valine derivative 40, which possesses an amide, an ester, and an epimerizable stereocenter. Upon treatment of 40 with (−)-menthol (38, 1.2 equiv) under Ni-catalyzed reaction conditions, we obtained ester 27 and liberated enantioenriched amine 41. Of note, the amide was selectively cleaved, while the ester remained intact.
Figure 11.

Ni-catalyzed esterification of valine-derived amide 40, without disturbing an ester.
Having demonstrated that amides can be employed in nickel-catalyzed couplings, we sought to examine other nucleophiles in related processes. Amines were deemed an intriguing class of nucleophiles for reaction development, as their use would allow for the unusual transamidation reaction to be achieved.3m Transamidation is a classic reaction that has seen relatively little success in the literature over the past few decades.29 Transamidation of primary amides is the most well developed process.30 With regard to the transamidation of secondary amides, breakthroughs include earlier studies by Bertrand31 and Gellman and Stahl.32
The challenges associated with the transamidation of secondary amides are summarized in Figure 12. Kinetically, the transformation is plagued by the unusually high activation barrier for amide C–N bond cleavage.16 Additionally, the overall reaction (42 + 43 → 44 + 45) is typically a thermoneutral process and may result in equilibrium mixtures, as has been seen in prior transamidation studies.32 With these considerations in mind, we proposed to achieve the net transamidation using a simple two-step sequence. Secondary amides 42 would first undergo N functionalization with an electron-withdrawing group (“Z”) to give amide derivative 46, which would presumably be activated toward oxidative addition. In the second step, treatment of 46 with an amine nucleophile under catalytic nickel conditions would lead to oxidative addition intermediate 47, which, in turn, would undergo trapping with amine 43 to yield amide 44. The reaction would be driven thermodynamically by the release of amine 48, bearing an electron-withdrawing group. From our experience with esterification chemistry, we had observed that N-Boc amides could undergo nickel-catalyzed activation, similarly to N-Ph substrates. Considering the ease by which a Boc group can be introduced onto a secondary amide, we opted to pursue N-Boc functionalization as our mode of activating the amide substrates.
Figure 12.

Challenges associated with the transamidation of secondary amides and our two-step approach.
As shown in Figure 13, we found that a range of Boc-activated secondary amides undergo nickel-catalyzed transamidation using secondary amine nucleophiles. With regard to the benzamide substrate, electron-withdrawing and electron-donating groups are tolerated, as shown by the formation of products 51–53. Additionally, heterocycles could be employed to give products such as furan 54 and thiophene 55. The scope with respect to the amine nucleophile was also found to be broad. Hindered amines could be used, as suggested by transamidation products 56 and 57. Moreover, the formation of 58–60 suggests that the methodology tolerates heterocyclic amines such as imidazolines, piperazines, pyridines, and carbazoles.
Figure 13.

Selected examples of the nickel-catalyzed transamidation of Boc-activated amides.
The transamidation methodology is perhaps best illustrated by the results shown in Figure 14, where secondary amide 61 was converted to the series of amino acid derivatives 62 using the two-step procedure. Boc activation of 61 proceeded smoothly in 99% yield. Next, nickel-catalyzed transamidation using optically enriched amino esters delivered the desired amide products 64–69. Derivatives of alanine, phenylalanine, valine, leucine, isoleucine, and proline could be utilized in this coupling methodology. In all cases, the ester withstood the reaction conditions and the transamidation reaction proceeded without loss of stereochemical integrity. This methodology provides one of the most general solutions to the classic problem of secondary amide transamidation reported to date.
Figure 14.

Scope of the two-step, nickel-catalyzed transamidation of amides using amino acid-derived nucleophiles.
3. CARBON–CARBON BOND FORMING REACTIONS
Another exciting opportunity lay in the notion of using amides as building blocks for the construction of C–C bonds using catalysis. In contrast to popular Weinreb amide chemistry (Figure 15, 7 → 70),17 such a method could potentially sidestep the use of pyrophoric, basic reagents and offer improved functional group compatibility. Thus, we set out to develop Ni-catalyzed cross-couplings of amides 46 that would allow for the synthesis of aryl and alkyl ketones 70. We were also optimiztic that these efforts, if successful, would lend some credence to the mechanistic notion described earlier (i.e., amides could undergo oxidative addition in the presence of an appropriate nickel catalyst; see Figure 9).
Figure 15.

Comparison of Weinreb amide displacement chemistry and nickel-catalyzed cross-couplings of amides for C–C bond formation.
Of the possible C–C bond-forming cross-coupling reactions, we elected to initiate our studies by pursuing the Suzuki–Miyaura33 coupling of amides. The Suzuki–Miyaura cross-coupling, typically performed on aryl halides or pseudohalides, 34,35 has transformed the landscape of how chemists construct small molecules, especially molecules of medicinal importance.
After performing optimization studies, we were delighted to find that pinacolatoboronate esters (71) could serve as amide (49) cross-coupling partners to furnish ketone products 72 (Figure 16).3k The methodology was found to be tolerant of substitution at the ortho, meta, and para positions, as shown by the formation of products 73a–c. Additionally, substrates bearing either electron-donating or electron-withdrawing groups could be utilized, to give products 74–76. From the last two of these examples, it should be emphasized that esters and ketones withstand the reaction conditions, which is typically not the case using Weinreb amide chemistry.17 Heterocycles could also be used, as shown by the formation of furan and thiophene products 77a,b, respectively. With regard to the scope of the boronic ester component, aryl nucleophiles could be utilized, including those with ortho, meta, or para functionalities (see products 78a–c). Moreover, a series of heterocyclic boronic esters, including indoles, pyrroles, pyrazoles, and furans, underwent the cross-coupling reaction to give ketones 79–82, respectively. Finally, a robustness screen revealed that epoxides, tertiary alcohols, nitriles, secondary amides, and the free NHs of indoles could all be tolerated in this methodology.3k
Figure 16.

Selected examples of the nickel-catalyzed Suzuki–Miyaura coupling of amides.
We also pursued a series of experiments involving the Ni-catalyzed Suzuki–Miyaura cross-coupling of amides, with the hope of using this chemistry in tandem with other cross-coupling processes. The first, shown in Figure 17, shows the use of sequential nickel-catalyzed reactions of amides: the esterification reaction3j and the Suzuki–Miyaura coupling.3k Bis(amide) 83, where one of the amides bears a Boc-activating group, was treated with (−)-menthol (38) under our Ni-catalyzed esterification conditions. This led to the formation of ester 84 in 90% yield. Of note, the secondary amide was not disturbed in this process. From intermediate 84, we performed a straightforward Boc activation of the secondary amide, which set the stage for the Ni-catalyzed Suzuki–Miyaura coupling. Using heterocyclic boronic ester 85, we obtained ketone product 86 in 82% yield over two steps. The ester was not disturbed in this process. A key point to take home from this sequence is that the selectivity in the initial coupling of 83 is strictly dependent on which amide possesses the Boc-activating group. As such, we have also shown that the order of cross-couplings can be reversed (i.e., Suzuki–Miyaura coupling can take place prior to the esterification in the overall conversion of 83 to 86).3k
Figure 17.

Sequential nickel-catalyzed esterification and Suzuki–Miyaura coupling of amides.
We were also eager to execute sequential Pd- and Ni-catalyzed Suzuki–Miyaura couplings (Figure 18). Boc-activated amide 87 was readily synthesized from the corresponding carboxylic acid. With this bifunctional substrate in hand, we first performed a Pd-catalyzed Suzuki–Miyaura coupling36 with furanyl boronic acid 88 to deliver biaryl product 89. The amide was not disturbed under these Pd-catalyzed, basic reaction conditions. From amide 89, a Ni-catalyzed Suzuki–Miyaura coupling using pyrazole boronic ester 90 furnished 91. This succession of cross-couplings demonstrates how our methodology can be used in strategic combination with traditional couplings to efficiently unite heterocyclic fragments.
Figure 18.

Sequential palladium- and nickel-catalyzed Suzuki–Miyaura couplings.
Having developed a means to access aryl–aryl ketones from amides, complementary to elegant methods discovered by Szostak,15e,37,38 we sought to develop a corresponding protocol to assemble aryl–alkyl ketones. Unfortunately, the Suzuki–Miyaura couplings of aliphatic amide substrates or aliphatic boronates using the conditions described above were not successful. Thus, we turned to the use of an alternate reaction, namely the Negishi coupling.39 Ni-catalyzed Negishi couplings have previously been achieved using acyl halides,40 anhydrides, 39 and thioesters39,41 but never using amides.15c
As shown in Figure 19, we found that the identity of the amide nitrogen substituents had a pronounced effect on the success of the Ni-catalyzed Negishi coupling of amides. For example, treatment of anilide 92a (an excellent substrate for the Ni-catalyzed esterification) and benzylzinc bromide (93) with Ni(cod)2 and SIPr in THF led to the recovery of anilide 92a (entry 1). Alternatively, attempts to utilize Boc-activated secondary amide 92b, a viable Suzuki–Miyaura coupling substrate, gave the desired ketone 94 in 60% yield (entry 2). Finally, we surveyed N-tosyl substrate 92c, which underwent conversion to ketone 94 in 81% yield (entry 3). We elected to further pursue the cross-coupling of N-tosyl amide substrates when evaluating the scope of this transformation.
Figure 19.

Amide N-substituent survey data for the nickel-catalyzed alkylation of amides.
A brief selection of examples that highlight the scope of this methodology, particularly with respect to the organozinc coupling partner, is shown in Figure 20. Nonbranched nucleophiles could be used, as shown by the formation of ketone 98 in 80% yield. The syntheses of ketones 99–102 demonstrate that α-branched nucleophiles could also be employed to give products of sp2–sp3 cross-coupling. Moreover, β-branching is tolerated, as shown by the coupling of a neopentylic nucleophile to give ketone 103. It should also be noted that the methodology is tolerant of substitution on the arene (e.g., −NMe2, −CF3, −F, and −OMe).3l
Figure 20.

Scope of the organozinc halide coupling partner for the nickel-catalyzed alkylation of amides.
The utility of the Ni-catalyzed Negishi coupling of amides can be seen in the example provided in Figure 21. Substrate 104, bearing an amide and a methyl ester, was coupled with cyclohexyl zinc iodide (105) under our typical reaction conditions. This reaction led to the formation of ketone 106 in 71% yield using 1 g of substrate. Of note, the ester was not disturbed under the reaction conditions. 106 is a precursor to Pfizer’s glucagon receptor modulator 107.42 Our synthesis of 106 compares favorably to the literature protocol, which proceeded in 34% yield using Weinreb amide displacement chemistry.42
Figure 21.

Application of the Ni-catalyzed alkylation of amides to the gram-scale synthesis of 106.
4. FROM THE GLOVEBOX TO THE BENCHTOP
The methods described thus far provide exciting new tools for the manipulation of amides that allow for the assembly of C–O, C–N, and C–C bonds. However, one limitation of this methodology is that Ni(cod)2, the precursor used throughout our studies, typically requires glovebox handling. Similarly, the free NHC ligand, SIPr, is unstable to benchtop conditions. Given this limitation of our amide coupling methodology and the general utility of Ni(cod)2 in unrelated catalytic reactions,43 we sought to develop a means to handle Ni(cod)2 on the benchtop.
Our approach3n is inspired by and mimics a recent methodology reported by Buchwald, wherein catalysts and ligands could be encapsulated in paraffin wax capsules to enable benchtop delivery.44,45 After developing a robust means to prepare paraffin capsules, we found that Ni(cod)2 or Ni(cod)2/SIPr mixtures could be readily encapsulated and used to promote reactions on the bench. Figure 22 highlights the Ni-catalyzed esterification (108 + 38 → 27) and transamidation (109 + 110 → 111) reactions using the paraffin capsules. Both reactions proceeded smoothly on the benchtop and with yields comparable to those reported using the typical glovebox conditions. Similar success was seen in our couplings of amides to form C–C bonds. For example, the Suzuki–Miyaura coupling of 112 and 113 delivered ketone 114 in 82% yield, whereas the Negishi coupling of 104 and 105 furnished keto ester 106 in 55% yield.
Figure 22.

Paraffin encapsulation of Ni(cod)2 and SIPr allowing amide couplings to take place on the benchtop.
Beyond the examples shown in Figure 22, several features of the benchtop reactions should be noted. (A) The capsules can be stored open to air, without appreciable loss of catalytic activity. For example, the esterification to access 27 proceeded in 95% yield using a Ni(cod)2/SIPr-paraffin capsule that had been stored on the benchtop for 2 months.3n (B) The same transformation was performed on a gram scale (using a larger capsule), which delivered the ester product 27 in 97% yield.3n (C) The Ni(cod)2 capsules could be used to enable a host of other Ni-catalyzed reactions, such as Dong’s oxidative esterification and amidation methodologies,46 our laboratory’s aryl C–N bond forming reactions,3c and Fu’s sp2–sp3 cross-coupling of alkyl halides.47 (D) Paraffin capsules containing only Ni(cod)2 or Ni(cod)2/SIPr combinations are being commercialized, which we expect will enable their more widespread use.48
5. SUMMARY
Amides have been widely studied for decades, but their synthetic utility has remained limited in reactions that proceed with rupture of the amide C–N bond. Using Ni catalysis, we have found that amides can now be strategically employed in several new reactions: esterification,3j transamidation,3m Suzuki–Miyaura,3k and Negishi couplings.3l These methodologies, and those related breakthroughs described by other key players in the field,15,23–25 provide efficient new tools to build C–heteroatom and C–C bonds using an unconventional reactant (i.e., the amide), which is ideally suited for use in multi-step synthesis.
The chemistry described herein lays the foundation for many opportunities when it comes to future development. Can the scope of the amide N-substituents be improved to encompass dialkyl amides, nonactivated secondary amides, or primary amides? Can conditions be developed to enable the coupling of amides derived from aliphatic precursors, rather than aryl and hetaryl substrates? Can other nucleophiles or trapping reagents be used? Can computations, kinetic studies, and other experiments be used to uncover mechanistic details? Can the methods discussed herein be utilized to construct molecules of importance, such as drug candidates or natural products?49 Finally, now that amides can be activated using catalysis, are there new transformations involving amides that remain to be discovered? Although only time will tell, recent studies by our laboratory3o and others15 suggest the answer to many of these questions is “yes”.
It is our hope and expectation that the area of amide C–N bond activation using nonprecious metals will continue to flourish and, in turn, will promote the growing use of amides as synthons in organic synthesis.
Acknowledgments
The authors thank the University of California, Los Angeles, the NIH (T32-GM067555 and R01-GM117016), and the Foote Family (J.E.D.) for financial support. The Houk laboratory is gratefully acknowledged for their insight throughout our collaboration.
Footnotes
Notes
The authors declare no competing financial interest.
Funding
The authors declare no competing financial interests.
References
- 1.(a) Johansson Seechurn C, Kitching MO, Colacot TJ, Snieckus V. Angew Chem, Int Ed. 2012;51:5062–5085. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]; (b) Hassan J, Sevignon M, Gozzi C, Schulz E, Lemaire M. Chem Rev. 2002;102:1359–1470. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]; (c) Muci AR, Buchwald SL. In: Topics in Current Chemistry. Miyaura N, editor. Vol. 219. Springer Verlag; New York: 2002. pp. 131–209. [Google Scholar]; (d) Jiang L, Buchwald SL. In: Metal-Catalyzed Cross-Coupling Reactions. 2. Meijere A, Diederich F, editors. Wiley-VCH; Weinheim, Germany: 2004. pp. 699–760. [Google Scholar]; (e) Corbet JP, Mignani G. Chem Rev. 2006;106:2651–2710. doi: 10.1021/cr0505268. [DOI] [PubMed] [Google Scholar]; (f) Negishi E. Bull Chem Soc Jpn. 2007;80:233–257. [Google Scholar]; (g) Shen HC. In: In Application of Transition Metal Catalysis in Drug Discovery and Development: An Industrial Perspective. Crawley ML, Trost BM, editors. Wiley; Hoboken, NJ: 2012. pp. 25–96. [Google Scholar]
- 2.(a) Bolm C, Legros J, Le Paih J, Zani L. Chem Rev. 2004;104:6217–6254. doi: 10.1021/cr040664h. [DOI] [PubMed] [Google Scholar]; (b) Enthaler S, Junge K, Beller M. Angew Chem, Int Ed. 2008;47:3317–3321. doi: 10.1002/anie.200800012. [DOI] [PubMed] [Google Scholar]; (c) Bauer EB. Curr Org Chem. 2008;12:1341–1369. [Google Scholar]; (d) Yu DG, Li BJ, Shi ZJ. Acc Chem Res. 2010;43:1486–1495. doi: 10.1021/ar100082d. [DOI] [PubMed] [Google Scholar]; (e) Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita AM, Garg NK, Percec V. Chem Rev. 2011;111:1346–1416. doi: 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Mesganaw T, Garg NK. Org Process Res Dev. 2013;17:29–39. [Google Scholar]; (g) Li BJ, Yu DG, Sun CL, Shi ZJ. Chem -Eur J. 2011;17:1728–1759. doi: 10.1002/chem.201002273. [DOI] [PubMed] [Google Scholar]; (h) Tasker SZ, Standley EA, Jamison TF. Nature. 2014;509:299–309. doi: 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Ananikov VP. ACS Catal. 2015;5:1964–1971. [Google Scholar]
- 3.(a) Quasdorf KW, Tian X, Garg NK. J Am Chem Soc. 2008;130:14422–14423. doi: 10.1021/ja806244b. [DOI] [PubMed] [Google Scholar]; (b) Quasdorf KW, Riener M, Petrove KV, Garg NK. J Am Chem Soc. 2009;131:17748–17749. doi: 10.1021/ja906477r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ramgren SD, Silberstein AL, Yang Y, Garg NK. Angew Chem, Int Ed. 2011;50:2171–2173. doi: 10.1002/anie.201007325. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mesganaw T, Silberstein AL, Ramgren SD, Fine Nathel NF, Hong X, Liu P, Garg NK. Chem Sci. 2011;2:1766–1771. doi: 10.1039/c1sc00230a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Mesganaw T, Fine Nathel NF, Garg NK. Org Lett. 2012;14:2918–2921. doi: 10.1021/ol301275u. [DOI] [PubMed] [Google Scholar]; (f) Hie L, Ramgren SD, Mesganaw T, Garg NK. Org Lett. 2012;14:4182–4185. doi: 10.1021/ol301847m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ramgren SD, Hie L, Ye Y, Garg NK. Org Lett. 2013;15:3950–3953. doi: 10.1021/ol401727y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Hie L, Chang JJ, Garg NK. J Chem Educ. 2015;92:571–574. doi: 10.1021/ed500158p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Fine Nathel NF, Kim J, Hie L, Jiang X, Garg NK. ACS Catal. 2014;4:3289–3293. doi: 10.1021/cs501045v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Hie L, Fine Nathel NF, Shah TK, Baker EL, Hong X, Yang Y-F, Liu P, Houk KN, Garg NK. Nature. 2015;524:79–83. doi: 10.1038/nature14615. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Weires NA, Baker EL, Garg NK. Nat Chem. 2015;8:75–79. doi: 10.1038/nchem.2388. [DOI] [PubMed] [Google Scholar]; (l) Simmons BJ, Weires NA, Dander JE, Garg NK. ACS Catal. 2016;6:3176–3179. doi: 10.1021/acscatal.6b00793. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Baker EL, Yamano MM, Zhou Y, Anthony SM, Garg NK. Nat Commun. 2016;7:11554. doi: 10.1038/ncomms11554. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Dander JE, Weires NA, Garg NK. Org Lett. 2016;18:3934–3936. doi: 10.1021/acs.orglett.6b01758. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Hie L, Baker EL, Anthony SM, Garg NK. Angew Chem, Int Ed. 2016;55:15129–15132. doi: 10.1002/anie.201607856. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Hie L, Fine Nathel NF, Hong X, Yang Y-F, Houk KN, Garg NK. Angew Chem, Int Ed. 2016;55:2810–2814. doi: 10.1002/anie.201511486. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Desrosiers J-N, Hie L, Biswas S, Zatolochnaya OV, Rodriguez S, Heewon l, Grinberg N, Haddad N, Yee NK, Garg NK, Senanayake CH. Angew Chem. 2016;128:12100–12103. doi: 10.1002/anie.201606955. [DOI] [PubMed] [Google Scholar]
- 4.United States Pharmacopeial Convention. <232> Elemental Impurities–Limits Revision. 2013. pp. 1–3. [Google Scholar]
- 5.(a) Anastas PT, Warner JC. In Green Chemistry: Theory and Practice. Oxford University Press; New York: 1998. [Google Scholar]; (b) Anastas PT, Kirchhoff MM. Acc Chem Res. 2002;35:686–694. doi: 10.1021/ar010065m. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kirk-Othmer Encyclopedia of Chemical Technology. 4. Vol. 1. Wiley; New York: 1998. p. 446. 1991–present; p SUPP. [Google Scholar]; (b) Keim W. Angew Chem, Int Ed. 2013;52:12492–12496. doi: 10.1002/anie.201305308. [DOI] [PubMed] [Google Scholar]; (c) Martinez CA, Hu S, Dumond Y, Tao J, Kelleher P, Tully L. Org Process Res Dev. 2008;12:392–398. [Google Scholar]; (d) Tian Q, Cheng Z, Yajima HM, Savage SJ, Green KL, Humphries T, Reynolds ME, Babu S, Gosselin F, Askin D, Kurimoto I, Hirata N, Iwaskai M, Shimasaki Y, Miki M. Org Process Res Dev. 2013;17:97–107. [Google Scholar]
- 7.For nickel-catalyzed aryl electrophile couplings, see: Blakey SB, MacMillan DWC. J Am Chem Soc. 2003;125:6046–6047. doi: 10.1021/ja034908b.Zhang XQ, Wang ZX. Org Biomol Chem. 2014;12:1448–1453. doi: 10.1039/c3ob41989d.Li BJ, Yu DG, Sun CL, Shi ZJ. Chem - Eur J. 2011;17:1728–1759. doi: 10.1002/chem.201002273.Yamaguchi J, Muto K, Itami K. Eur J Org Chem. 2013;2013:19–30.Cornella J, Zarate C, Martin R. Chem Soc Rev. 2014;43:8081–8097. doi: 10.1039/c4cs00206g.Tobisu M, Chatani N. Acc Chem Res. 2015;48:1717–1726. doi: 10.1021/acs.accounts.5b00051.
- 8.For select examples of nickel-catalyzed benzylic couplings, see: Guan BT, Xiang SK, Wang BQ, Sun ZP, Wang Y, Zhao KQ, Shi ZJ. J Am Chem Soc. 2008;130:3268–3269. doi: 10.1021/ja710944j.Taylor BLH, Swift EC, Waetzig JD, Jarvo ER. J Am Chem Soc. 2011;133:389–391. doi: 10.1021/ja108547u.Harris MR, Hanna LE, Green MA, Moore CE, Jarvo ER. J Am Chem Soc. 2013;135:3303–3306. doi: 10.1021/ja311783k.Zhou Q, Srinivas HD, Dasgupta S, Watson MP. J Am Chem Soc. 2013;135:3307–3310. doi: 10.1021/ja312087x.Yu DG, Wang X, Zhu RY, Luo S, Zhang XB, Wang BQ, Wang L, Shi ZJ. J Am Chem Soc. 2012;134:14638–14641. doi: 10.1021/ja307045r.León T, Correa A, Martin R. J Am Chem Soc. 2013;135:1221–1224. doi: 10.1021/ja311045f.Maity P, Shacklady-McAtee DM, Yap GPA, Sirianni ER, Watson MP. J Am Chem Soc. 2013;135:280–285. doi: 10.1021/ja3089422.
- 9.For select examples of nickel-catalyzed Heck reactions, see: Gøgsig TM, Kleimark J, Nilsson Lill SO, Korsager S, Lindhart AT, Norrby P-O, Skrydstrup T. J Am Chem Soc. 2012;134:443–452. doi: 10.1021/ja2084509.Matsubara R, Gutierrez AC, Jamison TF. J Am Chem Soc. 2011;133:19020–19023. doi: 10.1021/ja209235d.Tasker SZ, Gutierrez AC, Jamison TF. Angew Chem, Int Ed. 2014;53:1858–1861. doi: 10.1002/anie.201308391.
- 10.For select examples of nickel-catalyzed aminations, see refs 3c,d and the following: Wolfe JP, Buchwald SL. J Am Chem Soc. 1997;119:6054–6058.Park NH, Teverovskiy G, Buchwald SL. Org Lett. 2014;16:220–223. doi: 10.1021/ol403209k.Shimasaki T, Tobisu M, Chatani N. Angew Chem, Int Ed. 2010;49:2929–2932. doi: 10.1002/anie.200907287.Barker TJ, Jarvo ER. J Am Chem Soc. 2009;131:15598–15599. doi: 10.1021/ja907038b.
- 11.For select examples of nickel-catalyzed reductive couplings, see: Everson DA, Shrestha R, Weix DJ. J Am Chem Soc. 2010;132:920–921. doi: 10.1021/ja9093956.Everson DA, Jones BA, Weix DJ. J Am Chem Soc. 2012;134:6146–6159. doi: 10.1021/ja301769r.Biswas S, Weix DJ. J Am Chem Soc. 2013;135:16192–16197. doi: 10.1021/ja407589e.Cherney AH, Kadunce NT, Reisman SE. J Am Chem Soc. 2013;135:7442–7445. doi: 10.1021/ja402922w.Oblinger E, Montgomery J. J Am Chem Soc. 1997;119:9065–9066.Mahandru GM, Liu G, Montgomery J. J Am Chem Soc. 2004;126:3698–3699. doi: 10.1021/ja049644n.Miller KM, Huang WS, Jamison TF. J Am Chem Soc. 2003;125:3442–3443. doi: 10.1021/ja034366y.Chaulagain MR, Sormunen GJ, Montgomery J. J Am Chem Soc. 2007;129:9568–9569. doi: 10.1021/ja072992f.
- 12.For select examples of the use of nickel in photoredox transformations, see: Zuo Z, Ahneman DT, Chu L, Terrett JA, Doyle AG, MacMillan DWC. Science. 2014;345:437–440. doi: 10.1126/science.1255525.Tellis JC, Primer DN, Molander GA. Science. 2014;345:433–436. doi: 10.1126/science.1253647.Thoi VS, Kornienko N, Margarit CG, Yang P, Chang CJ. J Am Chem Soc. 2013;135:14413–14424. doi: 10.1021/ja4074003.Sastre F, Corma A, García H. Angew Chem, Int Ed. 2013;52:12983–12987. doi: 10.1002/anie.201307851.Noble A, McCarver SJ, MacMillan DWC. J Am Chem Soc. 2015;137:624–627. doi: 10.1021/ja511913h.Shields BJ, Doyle AG. J Am Chem Soc. 2016;138:12719–12722. doi: 10.1021/jacs.6b08397.Heitz DR, Tellis JC, Molander GA. J Am Chem Soc. 2016;138:12715–12718. doi: 10.1021/jacs.6b04789.
- 13.For select examples of nickel-catalyzed C–H activations, see: Muto K, Yamaguchi J, Itami K. J Am Chem Soc. 2012;134:169–172. doi: 10.1021/ja210249h.Amaike K, Muto K, Yamaguchi J, Itami K. J Am Chem Soc. 2012;134:13573–13576. doi: 10.1021/ja306062c.Shiota H, Ano Y, Aihara Y, Fukumoto Y, Chatani N. J Am Chem Soc. 2011;133:14952–14955. doi: 10.1021/ja206850s.Aihara Y, Chatani N. J Am Chem Soc. 2013;135:5308–5311. doi: 10.1021/ja401344e.Aihara Y, Chatani N. J Am Chem Soc. 2014;136:898–901. doi: 10.1021/ja411715v.Nakao Y, Kashihara N, Kanyiva KS, Hiyama T. Angew Chem, Int Ed. 2010;49:4451–4454. doi: 10.1002/anie.201001470.Tobisu M, Hyodo I, Chatani N. J Am Chem Soc. 2009;131:12070–12071. doi: 10.1021/ja9053509.Doster ME, Hatnean JA, Jeftic T, Modi S, Johnson SA. J Am Chem Soc. 2010;132:11923–11925. doi: 10.1021/ja105588v.
- 14.For select examples of nickel-catalyzed C–F bond activations, see: Tobisu M, Xu T, Shimasaki T, Chatani N. J Am Chem Soc. 2011;133:19505–19511. doi: 10.1021/ja207759e.Sun AD, Love JA. Org Lett. 2011;13:2750–2753. doi: 10.1021/ol200860t.Takachi M, Kita Y, Tobisu M, Fukumoto Y, Chatani N. Angew Chem, Int Ed. 2010;49:8717–8720. doi: 10.1002/anie.201004543.
- 15.For nickel-catalyzed amide C–N bond activations from other laboratories, see: Hu J, Zhao Y, Zhang Y, Shi Z. Angew Chem, Int Ed. 2016;55:8718–8722. doi: 10.1002/anie.201603068.Shi S, Meng G, Szostak M. Angew Chem, Int Ed. 2016;55:6959–6963. doi: 10.1002/anie.201601914.Shi S, Szostak M. Chem - Eur J. 2016;22:10420–10424. doi: 10.1002/chem.201602202.Shi S, Szostak M. Org Lett. 2016;18:5872–5875. doi: 10.1021/acs.orglett.6b02952.Meng G, Shi S, Szostak M. Synlett. 2016;27:2530–2540.
- 16.(a) Greenberg A, Breneman CM, Liebman JF, editors. The Amide Linkage: Structural Significance in Chemistry, Biochemistry, and Materials Science. Wiley; Hoboken, NJ: 2003. [Google Scholar]; (b) Pauling L, Corey RB, Branson HR. Proc Natl Acad Sci U S A. 1951;37:205–211. doi: 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815–3818. [Google Scholar]; (b) Balasubramaniam S, Aidhen IS. Synthesis. 2008;2008:3707–3738. [Google Scholar]
- 18.Spletstoser JT, White JM, Tunoori AR, Georg GI. J Am Chem Soc. 2007;129:3408–3419. doi: 10.1021/ja066362+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For select examples of amide-directed C–H functionalization, see: Rouquet G, Chatani N. Angew Chem, Int Ed. 2013;52:11726–11743. doi: 10.1002/anie.201301451. and references therein.
- 20.Clayden J, Davies RP, Hendy MA, Snaith R, Wheatley AEH. Angew Chem, Int Ed. 2001;40:1238–1240. [PubMed] [Google Scholar]
- 21.Topczewski JT, Cabrera PJ, Saper NI, Sanford MS. Nature. 2016;531:220–224. doi: 10.1038/nature16957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Although the Ni-catalyzed activation of amide C–N bonds was unknowns, couplings of acyl halides, anhydrides, and thioesters have been reported. See: Labadie JW, Tueting D, Stille JK. J Org Chem. 1983;48:4634–4642.Stille JK. Angew Chem, Int Ed Engl. 1986;25:508–524.Stephan MS, Teunissen AJJM, de Vries JG. Angew Chem, Int Ed. 1998;37:662–664. doi: 10.1002/(SICI)1521-3773(19980316)37:5<662::AID-ANIE662>3.0.CO;2-0.Goossen LJ, Ghosh K. Angew Chem, Int Ed. 2001;40:3458–3460. doi: 10.1002/1521-3773(20010917)40:18<3458::aid-anie3458>3.0.co;2-0.Kakino R, Yasumi S, Shimizu I, Yamamoto A. Bull Chem Soc Jpn. 2002;75:137–148.Kakino R, Narahashi H, Shimizu I, Yamamoto A. Bull Chem Soc Jpn. 2002;75:1333–1345.Bykov VV, Korolev DN, Bumagin NA. Russ Chem Bull. 1997;46:1631–1632.Zhang L, Wu J, Shi L, Xia C, Li F. Tetrahedron Lett. 2011;52:3897–3901.Kakino R, Shimizu I, Yamamoto A. Bull Chem Soc Jpn. 2001;74:371–376.Cherney AH, Reisman SE. Tetrahedron. 2014;70:3259–3265.Harada T, Kotani Y, Katsuhira T, Oku A. Tetrahedron Lett. 1991;32:1573–1576.Negishi E-i, Bagheri V, Chatterjee S, Luo F-T, Miller JA, Stoll AT. Tetrahedron Lett. 1983;24:5181–5184.Iwai T, Nakai T, Mihara M, Ito T, Mizuno T, Ohno T. Synlett. 2009;2009:1091–1094.Grey RA. J Org Chem. 1984;49:2288–2289.Bercot EA, Rovis T. J Am Chem Soc. 2002;124:174–175. doi: 10.1021/ja017086w.Bercot EA, Rovis T. J Am Chem Soc. 2004;126:10248–10249. doi: 10.1021/ja046528b.Johnson JB, Yu RT, Fink P, Bercot EA, Rovis T. Org Lett. 2006;8:4307–4310. doi: 10.1021/ol0616337.Tokuyama H, Yokoshima S, Yamashita T, Fukuyama T. Tetrahedron Lett. 1998;39:3189–3192.Mori Y, Seki M. Tetrahedron Lett. 2004;45:7343–7345.Miyazaki T, Han-ya Y, Tokuyama H, Fukuyama T. Synlett. 2004:477–480.
- 23.(a) Meng G, Szostak M. Org Lett. 2015;17:4364–4367. doi: 10.1021/acs.orglett.5b02209. [DOI] [PubMed] [Google Scholar]; (b) Meng G, Szostak M. Org Biomol Chem. 2016;14:5690–5705. doi: 10.1039/c6ob00084c. [DOI] [PubMed] [Google Scholar]; (c) Liu C, Meng G, Liu Y, Liu R, Lalancette R, Szostak R, Szostak M. Org Lett. 2016;18:4194–4197. doi: 10.1021/acs.orglett.6b01836. [DOI] [PubMed] [Google Scholar]; (d) Meng G, Szostak M. Angew Chem, Int Ed. 2015;54:14518–14522. doi: 10.1002/anie.201507776. [DOI] [PubMed] [Google Scholar]; (e) Meng G, Shi S, Szostak M. ACS Catal. 2016;6:7335–7339. [Google Scholar]; (f) Liu C, Meng G, Szostak M. J Org Chem. 2016;81:12023–12030. doi: 10.1021/acs.joc.6b02294. [DOI] [PubMed] [Google Scholar]
- 24.(a) Li X, Zou G. Chem Commun. 2015;51:5089–5092. doi: 10.1039/c5cc00430f. [DOI] [PubMed] [Google Scholar]; (b) Li X, Zou G. J Organomet Chem. 2015;794:136–145. [Google Scholar]
- 25.Yada A, Okajima S, Murakami M. J Am Chem Soc. 2015;137:8708–8711. doi: 10.1021/jacs.5b05308. [DOI] [PubMed] [Google Scholar]
- 26.White EH. J Am Chem Soc. 1955;77:6011–6014. [Google Scholar]
- 27.Nishimoto SI, Izukawa T, Kagiya T. Bull Chem Soc Jpn. 1982;55:1484–1488. [Google Scholar]
- 28.Keck GE, McLaws MD, Wager TT. Tetrahedron. 2000;56:9875–9883. [Google Scholar]
- 29.(a) González-Rosende ME, Castillo E, Sepúlveda-Arques J. Prog React Kinet Mech. 2004;29:311–332. [Google Scholar]; (b) Pattabiraman VR, Bode JW. Nature. 2011;480:471–479. doi: 10.1038/nature10702. [DOI] [PubMed] [Google Scholar]; (c) Sergeeva MV, Mozhaev VV, Rich JO, Khmelnitsky YL. Biotechnol Lett. 2000;22:1419–1422. [Google Scholar]
- 30.(a) Dineen TA, Zajac MA, Myers AG. J Am Chem Soc. 2006;128:16406–16409. doi: 10.1021/ja066728i. [DOI] [PubMed] [Google Scholar]; (b) Allen CL, Atkinson BN, Williams JMJ. Angew Chem, Int Ed. 2012;51:1383–1386. doi: 10.1002/anie.201107348. [DOI] [PubMed] [Google Scholar]
- 31.Bon E, Bigg DCH, Bertrand G. J Org Chem. 1994;59:4035–4036. [Google Scholar]
- 32.Eldred SE, Stone DA, Gellman SH, Stahl SS. J Am Chem Soc. 2003;125:3422–3423. doi: 10.1021/ja028242h. [DOI] [PubMed] [Google Scholar]
- 33.(a) Lennox JJA, Lloyd-Jones GC. Chem Soc Rev. 2014;43:412–443. doi: 10.1039/c3cs60197h. [DOI] [PubMed] [Google Scholar]; (b) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483. [Google Scholar]; (c) Suzuki A. Angew Chem, Int Ed. 2011;50:6722–6737. doi: 10.1002/anie.201101379. [DOI] [PubMed] [Google Scholar]
- 34.For the Pd-catalyzed Suzuki–Miyaura coupling of pyridyl esters, see: Tatamidani H, Kakiuchi F, Chatani N. Org Lett. 2004;6:3597–3599. doi: 10.1021/ol048513o.
- 35.For the decarbonylative Ni-catalyzed Suzuki–Miyaura coupling of phenyl esters, see: Muto K, Yamaguchi J, Musaev DG, Itami K. Nat Commun. 2015;6:7508. doi: 10.1038/ncomms8508.
- 36.Billingsley K, Buchwald SL. J Am Chem Soc. 2007;129:3358–3366. doi: 10.1021/ja068577p. [DOI] [PubMed] [Google Scholar]
- 37.Szostak and co-workers have made seminal contributions to the Suzuki–Miyaura coupling of amides using both Pd and Ni catalysis; see refs 15b and 23a,b,e.
- 38.Szostak and co-workers have reported an alternate Negishi coupling of amides to access aryl–aryl ketones from amides; see ref 15c.
- 39.For reviews on Negishi cross-couplings, see: Haas D, Hammann JM, Greiner R, Knochel P. ACS Catal. 2016;6:1540–1552.Negishi E. Angew Chem, Int Ed. 2011;50:6738–6764. doi: 10.1002/anie.201101380.Negishi E. Acc Chem Res. 1982;15:340–348.Phapale VB, Cárdenas DJ. Chem Soc Rev. 2009;38:1598–1607. doi: 10.1039/b805648j.Benischke AD, Ellwart M, Becker MR, Knochel P. Synthesis. 2016;48:1101–1107.
- 40.Zhang Y, Rovis T. J Am Chem Soc. 2004;126:15964–15965. doi: 10.1021/ja044113k. [DOI] [PubMed] [Google Scholar]
- 41.Shimizu T, Seki M. Tetrahedron Lett. 2002;43:1039–1042. [Google Scholar]
- 42.Apnes GE, Didluk MT, Filipski KJ, Guzman-Perez A, Lee ECY, Pfefferkorn JA, Stevens BD, Tu MM. Glucagon Receptor Modulators. 20120202834. US Patent. 2012 Aug 9;
- 43.Ni(cod)2 has been employed in >800 methodology studies and is the most common Ni(0) precatalyst used. SciFinder searching for bis(1,5-cyclooctadiene)nickel(0) being used as a reagent yielded hits corresponding to 848 original manuscripts since 1985 (accessed April 28, 2016).
- 44.Sather AC, Lee HG, Colombe JR, Zhang A, Buchwald SL. Nature. 2015;524:208–211. doi: 10.1038/nature14654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.For the use of paraffin to allow for the handling of other air-sensitive chemicals, see: Taber DF, Li H-Y. 20050288257. US Patent. 2005 Dec 29;Fang Y, Liu Y, Ke Y, Guo C, Zhu N, Mi X, Ma Z, Hu Y. Appl Catal, A. 2002;235:33–38.Taber DF, Frankowski KJ. J Org Chem. 2003;68:6047–6048. doi: 10.1021/jo030005p.Taber DF, Nelson CG. J Org Chem. 2006;71:8973–8974. doi: 10.1021/jo061420v.. For examples of reagents encapsulated in paraffin, see: Kosak KM, Kosak MK. 5413924. US Patent. 1995 May 9;Shen C, Spannenberg A, Wu X-F. Angew Chem, Int Ed. 2016;55:5067–5070. doi: 10.1002/anie.201600953.
- 46.Whittaker AM, Dong VM. Angew Chem, Int Ed. 2015;54:1312–1315. doi: 10.1002/anie.201410322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhou J, Fu GC. J Am Chem Soc. 2004;126:1340–1341. doi: 10.1021/ja039889k. [DOI] [PubMed] [Google Scholar]
- 48.Paraffin capsules containing Ni(cod)2 and Ni(cod)2/SIPr will soon be available commercially from TCI (TCIchemicals.com) and Sigma-Aldrich. The capsules and their TCI product numbers are as follows (Sigma-Aldrich product numbers are pending): bis(1,5-cyclooctadiene)nickel(0) (wax encapsulated), B5417; bis(1,5-cyclooctadiene) nickel(0)-1,3-bis(2,6-diisopropylphenyl)imidazolidin-2-yli-dene (wax encapsulated), B5418.
- 49.For the prevalence of amides in pharmaceuticals, see: Ghose AK, Viswanadhan VN, Wendoloski jJ. J Comb Chem. 1999;1:55–68. doi: 10.1021/cc9800071.Brown DG, Boström J. J Med Chem. 2016;59:4443–4458. doi: 10.1021/acs.jmedchem.5b01409.