

Abstract
IgE-Fc receptors and IgG-Fc receptors are expressed on hematopoietic cells, but some evidence suggests that these receptors are also found on nonhematopoietic cells, including human airway smooth muscle (hASM) cells. Our study characterizes the expression of IgE-Fc receptors (FcεRI/CD23) and IgG-Fc receptors (FcγRs-I, -II, and -III) in cultured hASM cells by flow cytometry and Western blotting, and the functional activity of receptors was determined through quantification of cell proliferation and released cytokines. Expression of Fc receptor–linked intracellular signaling proteins and phosphorylation of the mitogen-activated protein kinases (MAPKs) extracellular signal–regulated kinase 1/2 and p38MAPK in hASM cells was examined by Western blotting. Expression of FcεRI and CD23 was not detectable in hASM cells. However, FcγRI and FcγRII were shown to be expressed on these cells. Specific antibodies, validated using transfected cell lines, revealed that the inhibitory IgG receptor, FcγRIIb, was the most abundant Fc receptor subtype expressed. Although cross-linking FcγR with heat-aggregated γ globulin (HAGG) did not induce detectable cell stimulation, pretreating hASM cells with HAGG significantly inhibited IL-1α–induced increases in cytokine levels and basic fibroblast growth factor-induced cell proliferation. This inhibitory effect of HAGG was abrogated by preincubation of cells with an anti-FcγRIIb antigen-binding fragment (Fab). Expression of proteins involved in the canonical FcγRIIb inhibitory signaling pathway was established in hASM cells. Pretreatment of hASM cells with HAGG significantly inhibited IL-1α– and basic fibroblast growth factor-induced extracellular signal–regulated kinase 1/2 and p38MAPK phosphorylation. This study identifies functional expression of FcγRIIb in hASM cells, with the potential to suppress their remodeling and immunomodulatory roles.
Keywords: asthma, airway smooth muscle, Ig receptors, mitogen-activated protein kinases, airway remodeling
Ig-Fc receptor activation contributes to the initiation and exacerbation of airway inflammation (1). In allergic asthma, there is aberrant production of both antigen-specific IgE (2) and IgG (3). The role of IgE in the activation of inflammatory cells is well established, but the potential roles of IgG in asthma have not been extensively examined.
IgE can bind to both high-affinity IgE-Fc receptors (FcεRI) on mast cells and basophils and low-affinity receptors (CD23) on monocytes and B-cells (2). Allergen exposure activates these receptors to trigger release of a host of mediators, leading to immediate airway obstruction, persistent inflammation, and the manifestation of clinical asthma (2, 4).
IgG-Fc receptors (FcγR) include FcγRI, FcγRII (a and b), and FcγRIII, which are expressed primarily on macrophages, neutrophils, and dendritic cells (5). FcγR activation leads to responses such as phagocytosis, antibody-mediated cellular cytotoxicity, and the release of a range of inflammatory mediators. FcγRI, FcγRIIa, and FcγRIII, like FcεRI, signal using an immunoreceptor tyrosine–based activation motif (ITAM). FcγRIIb contains an immunoreceptor tyrosine–based inhibitory motif (ITIM), which is able to inhibit antigen-driven cell signaling in hematopoietic cells, such as B lymphocytes and mast cells (6).
Current knowledge of the functional effects of IgG in allergic asthma is somewhat limited. Several studies have shown an IgG deficiency in asthma, with low levels of IgG being associated with the severity of the asthmatic symptoms (7, 8). This may reflect diminished capacity to neutralize allergens, thus enabling more effective IgE/FcεR activation (1). The protective effects of IgG could also be mediated through immune complex binding to FcγRIIb on B lymphocytes, mast cells, and basophils (6). However, IgG immune complexes can also activate eosinophils (9) and mast cells (10), which might exacerbate airway inflammation.
The traditional view of human airway smooth muscle (hASM) is as a regulator of bronchomotor tone. In individuals with asthma, hyperplasia and hypertrophy of hASM is a feature of the airway remodeling (11). However, hASM is also now well defined as an immunomodulatory cell type that is capable of both constitutive and induced secretion of numerous cytokines (12). Thus, hASM can actively participate in inflammatory mechanisms that might also enhance the airway hyperresponsiveness and airway remodeling present in asthma (13).
The low-affinity IgE-Fc receptor, CD23, which is structurally unrelated to the other Fc receptors, has been detected in hASM (14–16). More recently, it has been reported that FcεRI exists on hASM cells, and can produce cell activation (17–19). However, the reported expression of FcεRI on hASM is not consistent with an earlier study (15). Although all three major subclasses of FcγR have been detected in hASM (15), the precise nature of the receptor expression, and their role in regulating hASM function, has not been investigated.
The present study aims to clearly determine the expression and functional activities of both FcεR and FcγR in nonasthmatic cultured hASM cells, and further establish whether remodeling and immunomodulatory roles of hASM cells can be directly regulated by antigen and/or immune complexes.
MATERIALS AND METHODS
Cell Culture
hASM was dissected from bronchi obtained during lung resection surgery (kindly provided by the Alfred and Royal Melbourne Hospitals, Melbourne, VIC, Australia), as approved by the University of Melbourne human ethics committee. hASM cells were prepared as described previously (20), and then maintained in Dulbecco's modified Eagles medium (Invitrogen, Mulgrave, Australia) supplemented with 10% vol/vol heat-inactivated FBS, 2 mM L-glutamine, 100 U/ml penicillin G, 100 μg/ml streptomycin (Sigma-Aldrich, Castle Hill, Australia), 16 mM Hepes, 0.2% vol/vol sodium bicarbonate, and 1% vol/vol nonessential amino acids (Sigma-Aldrich). Cells at passages between numbers 3 and 17 were used. A total of 17 separate specimens were obtained for this study from donors of 38–77 years of age (11 male, 5 female, 1 information not available). The indications for resection/diagnoses of the donors were: bronchial carcinoma (3); pulmonary hypertension (1); bronchiectasis (2); and pulmonary emphysema (1). Information regarding diagnoses was not available for the other donors. Telomerase-immortalized hASM cell lines (21) were cultured identically to the primary hASM cells. hASM cells were, unless stated, serum starved for between 24 and 48 hours (medium as above, but with 0.25% wt/vol BSA replacing the FBS) before use. Monomed A (SAFC Biosciences, Brooklyn, Australia), composed of insulin, transferrin, and selenium, was added to all serum-starved cells (1% vol/vol) during this period.
Human mast cell line (HMC)–1 cells (22) (kindly provided by Dr. Joseph Butterfield, Mayo Clinic) transfected with the human FcεRI α chain (HMC-1α) (Y.C.X. and G.A.M., unpublished data) were cultured as a positive control for expression of FcεR and intracellular signaling proteins. HMC-1α cells were cultured in Iscove's modified Dulbecco's medium (SAFC Biosciences), supplemented with 10% vol/vol FBS, 4.0 mM glutamine, 500 U/ml penicillin, 500 μg/ml streptomycin, and 0.01% vol/vol α-thioglycerol (Sigma-Aldrich), and 125 μg/ml G418 (Invitrogen).
The mouse B cell line IIA1.6 transfected with human FcγRIIa or FcγRIIb (23) were cultured as positive control cells for FcγRIIa or FcγRIIb expression, and were also used to verify the specificity/selectivity of the antibodies used. IIA1.6 cells were cultured in RPMI medium (Invitrogen), supplemented with 5% vol/vol FBS, 40 mM glutamine, 500 U/ml penicillin, 500 μg/ml streptomycin, and 10 μg/ml puromycin (Sigma-Aldrich).
FACS
hASM cells were grown to approximately 80% confluence, and then incubated with or without human 4-hydroxy-3-nitro-5-iodo-phenylacetyl (NIP)–specific IgE (hIgE) (clone JW8; 2 μg/ml) and/or IL-4 (10 ng/ml) for 48 hours. Staining methods were performed on ice with two washes in FACS buffer (PBS/0.2% wt/vol BSA) between each step. hASM cells, HMC-1α cells, and IIA1.6 cells were labeled with various mouse monoclonal antibodies (mAbs) (10 μg/ml) or hIgE (2 μg/ml) for 1 hour. The mAb clones used were 3B4 (FcεRIα), MHM6 (CD23; Dako, Campbellfield, Australia), 32.2 (FcγRI), 8.7 (FcγRII), IV.3 (FcγRIIa selective), 4F5 (FcγRIIb, a gift from Dr. Robert Kimberly, University of Alabama, Birmingham), 3G8 (FcγRIII), AK7 (CD49b; Dako), mopc21 (IgG1 isotype control; Sigma-Aldrich), and PY20 (IgG2b isotype control; Sigma-Aldrich). When not attributed, mAbs were generated in house. Cells were then incubated with either biotinylated anti-mouse IgG (Jackson Immunoresearch Laboratories, West Grove, PA) or biotinylated anti-human IgE (Vector Laboratories, Burlingame, CA) for 1 hour, and then incubated with streptavidin-phycoerythrin (BD Biosciences, North Ryde, Australia) for an additional hour. Samples were analyzed by flow cytometry (FACScalibur; BD Biosciences) using Cellquest software (BD Biosciences). The fluorescence associated with 5,000–10,000 cells was measured.
Cell Stimulation and Cytokine ELISA
hASM cells were grown to approximately 80% confluence in multiwell plates and sensitized with or without 1 μg/ml hIgE for 24 hours or 2 μg/ml hIgE for 4 days. IL-4 (10 ng/ml) was also added in some cases. Cells were then washed with PBS, fresh, serum-free medium added, and challenged with or without antigen (NIP-BSA; 10 or 100 ng/ml) for a further 24 hours. Cell stimulation with IL-1α (1 ng/ml) was used as a positive control.
Heat-aggregated γ globulin (HAGG) was generated as previously described, and preparations used were shown to trigger cell activation in FcγRIIa transfected IIA1.6 cells (23). After 24-hour starvation, hASM cells were stimulated with various concentrations of HAGG for a further 24 hours. To test possible inhibitory actions, cells were pretreated with or without HAGG (3 μg/ml) for 30 minutes, and then incubated with or without IL-1α (0.01 ng/ml) for 24 hours. In studies designed to examine the receptor responsible for HAGG activity, cells were treated with or without antigen-binding fragments (Fab) of either the anti-FcγRIIb–specific mAb X63 or anti-FcγRIIa–specific mAb IV.3 before HAGG treatment. Both antibodies have been shown to block IgG interactions with their respective receptors.
Supernatants were harvested and assayed for IL-8, eotaxin, and IL-6 levels using commercially available ELISA kits (OptEIA; BD Bioscience) according to the manufacturer's instructions, and absorbance measured using a plate reader (Multiskan Ascent; Thermo, Hudson, NH).
Measurement of Cell Proliferation
hASM cells were grown to approximately 60% confluence in 12-well plates, pretreated with or without HAGG for 30 minutes, and then incubated with or without basic fibroblast growth factor (bFGF) (300 pM) for 30 hours. Cells were detached from the culture plate using trypsin-EDTA (Invitrogen), and neutralized with 10% FBS. Cells were kept on ice, incubated with trypan blue (0.2%), and counted in a hemocytometer chamber.
Western Blot Analysis
Receptor and signaling protein expression.
hASM cells were grown to approximately 80% confluence and washed twice with ice-cold PBS, followed by cell lysis in reducing SDS sample buffer (0.05 M Tris-HCL, 0.1% wt/vol SDS, 10% vol/vol glycerol, 50 mM dithiotheritol [DTT]). Lysates were separated by 10% SDS-PAGE, and resolved proteins transferred onto nitrocellulose or polyvinylidene fluoride (PVDF) membranes (GE Healthcare, Rydalmere, Australia). The membranes were blocked in 5% (wt/vol) skimmed milk protein in TBS/0.05% Tween20 for 1 hour, and then probed with anti-FcγRIIa (tail specific), anti-FcγRIIb–specific mAb 4F5, anti-downstream of tyrosine kinase (Dok)-1 (gift from Dr. Peter Lock, University of Melbourne), mouse mAb anti-spleen tyrosine kinase (Syk) (Abcam, Cambridge, UK), anti-Lyn (a Src family tyro sine kinase), anti–Src homology (SH) 2–containing inositol phosphatase (SHIP)–1, or anti–protein tyrosine phosphatase SHP–1 (all rabbit polyclonals from Dr. Margaret Hibbs, Ludwig Institute, Melbourne) overnight at 4°C. Subsequently, membranes were washed in TBS with 0.05% Tween20 and incubated with the appropriate horseradish peroxidase–conjugated secondary antibodies (Millipore, North Ryde, Australia) at room temperature for 2 hours. Immunoreactive proteins were visualized by enhanced chemiluminescence (ECL; GE Healthcare) using an ImageQuant 350 imager (GE Healthcare).
Mitogen-Activated Protein Kinase Phosphorylation
hASM cells were pre-treated with or without HAGG for 30 minutes and then stimulated with either bFGF (300 pM) or IL-1α (0.01 ng/ml) for 2 hours. Cells were washed twice with ice-cold PBS, and then lysed with lysis buffer (1% vol/vol Brij-96, 150 mM NaCl, 10 mM Tris-HCl [pH 7.5], pervanadate [1 mM], and protease inhibitor cocktail [Sigma]). The lysate was collected and clarified by centrifugation before addition of reducing SDS sample buffer, and processing as above. The membranes were probed with either anti–phospho–extracellular signal–regulated kinase (ERK) 1/2 (Thr202/Tyr204) or anti–phospho-p38 mitogen-activated protein kinase (MAPK) (Thr180/Tyr182) (Cell Signaling Technology, Danvers, MA), then visualized as described previously here. Blots were then stripped (1.5% wt/vol glycine, 0.1% wt/vol SDS, 1% vol/vol Tween20 [pH 2.2]), and reprobed with either anti-p38MAPK or anti-ERK1/2 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA), and visualized again. Band intensities were quantified by densitometry using the Image J program (National Institutes of Health, Bethesda, MD).
mRNA Extraction and Quantitative PCR
mRNA was extracted from hASM cells and peripheral blood leukocytes (PBLs) using an RNeasy Mini Kit (Qiagen, Doncaster, Australia), according to the manufacturer's protocol. Reverse transcription and quantitative PCR were performed as described previously (24). 18S ribosomal RNA was used as a housekeeping gene. The primer sequences (Table E1) were designed using Primer Express software (Applied Biosystems, Mulgrave, Australia). The mRNA expression level was expressed as 1/(threshold cycle determined for each gene − threshold cycle of 18S ribosomal RNA) (1/ΔCt).
Immunohistochemistry
Parraffin-embedded blocks of human lung tissue or bronchi were sectioned at a thickness of 5 μm on a microtome (LKB Historange; LKB Instruments, Bromma, Sweden), stretched on a water bath, and then collected on 3-aminoprophyl silane (8%)–precoated microscope slides. The slides were dried overnight, deparaffinized in histolene, and rehydrated through a descending alcohol series. After two 5-minute washes in PBS, the slides were incubated with 1.5% normal horse serum for 20 minutes at room temperature, and then incubated with mAb 8.7, mAb anti–smooth muscle α-actin, or mouse IgG1 isotype control (Dako overnight at 4°C. After two 5-minute washes in PBS, the slides were exposed to a biotinylated horse anti-mouse IgG secondary antibody for 30 minutes at room temperature. The endogenous peroxidase activity was blocked by immersion in methanol containing 0.4% hydrogen peroxide for 10 minutes. After another two washes with PBS, the slides were incubated for 30 minutes with avidin-biotinylated horseradish peroxidase complex (Vectastain Elite ABC kit; Vector Laboratories). Immunoreactivity was visualized by addition of the metal enhanced diaminobenzidine (immunoPure metalenhanced DAB substrate kit; Thermo Fisher Scientific, Scoresby, Australia). Slides were then counterstained with hematoxylin, and visualized by light microscopy.
Statistical Analysis
Data from ELISA and FACS analysis are expressed as the mean (±SEM), and reported n values represent the number of independent primary hASM cell cultures used or number of experiments repeated using the immortalized hASM cell cultures. For ELISA data and Western blotting data, differences between treatment groups were tested using one-way repeated measures ANOVA and Bonferroni's post hoc test for multiple comparisons. A two-tailed, paired Student's t test and linear regression analysis was also used to compare certain groups of ELISA data. A P value less than 0.05 was considered significant. Statistical analysis was performed using GraphPad Prism for Windows (version 5.00; GraphPad Software, La Jolla, CA).
RESULTS
Absence of Detectable IgE-Fc Receptor Expression in hASM Cells
Cell surface expression of FcεR was examined by FACS analysis. hASM cells were studied under both serum-depleted and serum-complete culture conditions. In addition, in an attempt to partly mimic conditions occurring in the asthmatic airway, and to potentially up-regulate any expression of FcεR, some cells were also incubated in the presence of both hIgE (2 μg/ml) and IL-4 (10 ng/ml). As expected, hASM cells strongly expressed the α2β1-integrin CD49b (Figure 1C). However, although FcεRI and CD23 were detected on HMC-1α cells (Figures 1A and 1B), no expression of these receptors was shown on hASM cells using specific mAbs (Figures 1D and 1E), or hIgE (Figure 1F). Identical results were obtained when hASM cells were cultured in serum-free (Figure 1) or serum-complete media, or when hASM cells were treated with hIgE and IL-4 for 2 days (data not shown). Two independent cultures of telomerase-immortalized hASM cells were also analyzed, and, likewise, no surface expression of FcεRI (Figure E1A in the online supplement) or CD23 (Figure E1B) was detected. Moreover, we were unable to detect the FcεRIα subunit using Western blotting (n = 6; data not shown).
Figure 1.
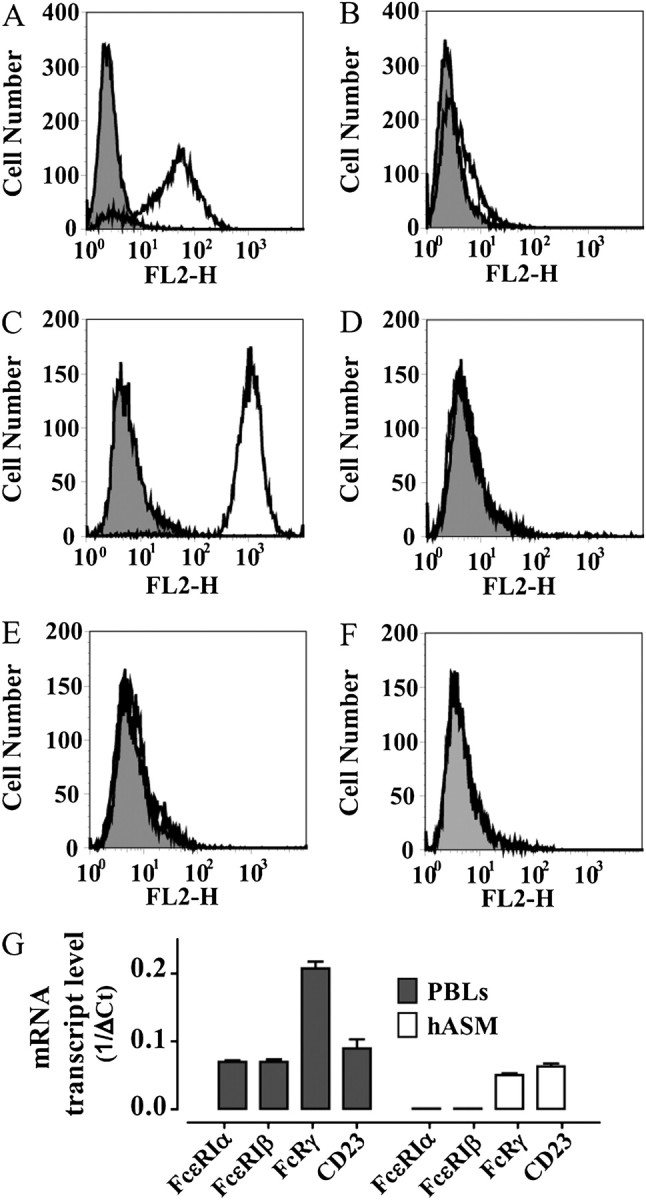
No detectable IgE-Fc receptor (FcεR) I and CD23 expression on primary human airway smooth muscle (hASM) cells. Positive control human mast cell line (HMC)–1α cells showed expression of FcεRI (A) and low expression of CD23 (B). Whereas CD49b was strongly expressed in hASM cells (C), no expression of FcεRI (D) or CD23 (E) was observed, which was confirmed by hIgE labeling (F). Results shown are representative of experiments conducted using eight different primary hASM cell cultures. (G) The mRNA of the three subunits of FcεRI (αβγ) and CD23 were detected in peripheral blood leukocytes (PBLs), whereas a low-level expression of mRNA for FcRγ and CD23, but not FcεRI α or β subunits, was detected in hASM cells (n = 6).
The IgE Fc receptor expression was further investigated at the mRNA level by quantitative PCR. mRNA from PBLs was used as a positive control. Although mRNA for FcεRI α, β, and γ subunits and CD23 were readily detected in PBLs, no expression of mRNA for FcεRI α or β subunits was detected in hASM cells. However, a low-level expression of mRNA for the Fc receptor common γ subunit (FcRγ) and CD23 was detected in hASM cells (n = 6; Figure 1G).
We also tested whether IgE/antigen treatment of hASM cells could elicit cytokine production from these cells. Whereas IL-1α produced robust release of IL-8 and eotaxin from hASM cells, IgE/antigen produced no significant change in either IL-8 or eotaxin release (n = 5), even when cells were sensitized with hIgE and IL-4 for 4 days (n = 3; Figure E2).
Expression of FcγRI and FcγRIIb in hASM Cells
We next examined cell surface expression of the IgG receptors, FcγRI, FcγRII, and FcγRIII, in hASM cells by FACS analysis. Whereas FcγRIII could not be detected (Figure 2C), there was moderate expression of FcγRII (Figure 2B), and a low expression level of FcγRI (Figure 2A) in these cells.
Figure 2.

Expression of FcγRI and FcγRIIb in hASM cells. There was low expression of FcγRI (A), moderate expression of FcγRII (B), and no detectable FcγRIII (C) on hASM cells. Using the anti-FcγRIIa–selective monoclonal antibodies (mAb) IV.3 (D) and anti-FcγRIIb–specific mAb 4F5 (E), FcγRIIb was shown to be the FcγRII isoform expressed. This expression pattern was confirmed by Western blotting (F). Results shown are representative of experiments conducted using five to eight different hASM cell cultures.
Identifying which isoform of FcγRII was expressed on hASM cells is important, as activation of these receptors can lead to different functional consequences (5). The extracellular regions of FcγRIIa and FcγRIIb are highly homologous. To distinguish between them, we used an anti-FcγRIIa–selective mAb IV.3 (25) and the recently developed anti-FcγRIIb–specific mAb 4F5 (26) and X63 (P.S.T. and P.M.H., unpublished data). IIA1.6 cells that had been transfected with either human FcγRIIa or FcγRIIb were used to validate the specificity and selectivity of the antibodies used (Figure E3). Using these antibodies, we could show that it is the inhibitory receptor, FcγRIIb, that is expressed on hASM cells (Figure 2E), with cells having no detectable FcγRIIa expression (Figure 2D).
The selective expression of FcγRIIb, but not FcγRIIa, in hASM cells was confirmed by Western blotting using an anti-FcγRIIa intracellular tail-specific polyclonal antibody and mAb 4F5. Whereas FcγRIIa (Figure 2F, lane 2) and FcγRIIb (Figure 2F, lane 1) were selectively expressed in FcγRIIa- and FcγRIIb-transfected cells, respectively, FcγRIIb, but not FcγRIIa, was observed in protein extracts from five independent primary hASM cultures (Figure 2F, lanes 3–7). The expression of FcγRI (Figure E4A), FcγRII (Figure E4B), and the lack of detectable FcγRIII expression (Figure E4C) was also shown in the immortalized hASM cell lines by FACS analysis. Using the anti-FcγRIIa–selective mAb IV.3 and anti-FcγRIIb–specific mA b4F5, FcγRIIb (Figure E4E), but not FcγRIIa (Figure E4D), was again shown to be the FcγRII isoform expressed. FcγRII expression in ASM was examined in situ using tissue from normal human lung or bronchi. Consistent with our findings in cultured hASM cells, FcγRII expression was shown in ASM tissue by immunostaining using mAb 8.7 (Figure 3C). ASM bundles were clearly identified by anti–smooth muscle α-actin immunostaining (Figure 3B), and no observable staining was found with the isotype control antibody (Figure 3A).
Figure 3.

Expression of FcγRII in ASM in situ by immunohistochemistry. FcγRII immunoreactivity was observed in the smooth muscle cells of human airway sections (C). An anti–smooth muscle α-actin mAb was used to clearly identify the ASM (B), whereas the isotype control mAb showed no immunostaining (A). The findings are representative of results generated from three donors without asthma.
Activation of FcγRIIb Inhibits IL-1α–Induced Cytokine Production and bFGF-Induced hASM Cell Proliferation
In light of the expression of the stimulatory FcγRI and the classically inhibitory FcγRIIb receptors on hASM cells, we assessed whether engagement of these receptors by HAGG resulted in stimulation or inhibition of hASM function.
First, hASM cells were treated with HAGG under conditions known to stimulate hematopoietic cells (23). Although IL-1α produced robust cytokine release from hASM cells and bFGF induced a significant mitogenic response, treatment of hASM cells with HAGG did not evoke any cytokine secretion (n = 5), or induce cell proliferation (n = 4) (Figure E5).
We next sought to determine whether inhibitory actions of FcγR were evident in hASM cells. Interestingly, HAGG treatment significantly inhibited IL-1α–induced IL-8 and IL-6, but not eotaxin production from hASM cells (Figure 4A). HAGG treatment also significantly inhibited bFGF-induced hASM cell proliferation (Figure 4B). As HAGG is a polyclonal antibody preparation, our results might be confounded by other effects of the antibody preparation, such as neutralization of released cytokines (IL-8, IL-6, or eotaxin) or the cytokine stimuli (IL-1α and bFGF). However, using a series of ELISA assays, there was no evidence of significant HAGG-mediated neutralization of any of these cytokines (data not shown).
Figure 4.

FcγR aggregation inhibits stimulus-induced hASM cell activation. Treatment of hASM cells with heat-aggregated γ globulin (HAGG) inhibits IL-1α–induced IL-8 and IL-6 release (A), and basic fibroblast growth factor (bFGF)-induced cell proliferation (B). Cytokine level or cell numbers are normalized to the mean percentage (±SEM) of the IL-1α–only responses (IL-8, 71.5 ± 21.2 ng/ml; IL-6, 12.0 ± 3.4 ng/ml; eotaxin, 409.8 ± 66.8 pg/ml), or bFGF-only cell number per milliliter (1.4 × 105 ± 0.1 × 105 cells). Student's t test was used to compare the HAGG pretreated groups to the IL-1α– or bFGF-only group (**P < 0.01; ***P < 0.001). Using linear regression analysis, the inhibitory effects were directly correlated to the cell expression of FcγRIIb (C) (*P < 0.05; n = 6–7).
The level of inhibition that we observed with HAGG varied depending upon the hASM culture used, as did the expression level of FcγRIIb determined by FACS. When FcγRIIb expression level was compared with the level of inhibition of cytokine production and cell proliferation, a positive correlation was evident, providing strong evidence that engagement of FcγRIIb underlies the inhibitory effect of HAGG (Figure 4C).
To further demonstrate that HAGG engagement of FcγRIIb was responsible for the inhibitory effects, we used the specific anti-FcγRIIb mAb X63 that is known to block IgG binding to the receptor (P.S.T. and P.M.H., unpublished data). mAb X63 Fab fragments, but not Fab fragments generated from the anti-FcγRIIa–specific mAb IV.3, significantly reduced HAGG-mediated inhibition of IL-1α–induced IL-8 production (Figure 5).
Figure 5.

Blockade of FcγRIIb inhibits HAGG activity on IL-1α–induced responses in hASM cells. Treating hASM cells with antigen-binding fragment (Fab) X63, Fab IV.3, and/or HAGG did not change the basal IL-8 production. Whereas HAGG significantly inhibited IL-1α–induced response, pretreating the cells with Fab X63, but not Fab IV.3, blocked this inhibitory effect. Experiments were conducted using one immortalized hASM cell line (and repeated three times), and on one primary hASM cell culture. IL-8 level is normalized to the mean percentage (±SEM) of the IL-1α–only responses (62.8 ± 19.1 ng/ml). Differences were identified using one-way repeated measurement ANOVA and Bonferroni's multiple comparison test (**P < 0.01 compared with IL-1α treatment alone; ##P < 0.01 compared with HAGG + IL-1α treatment).
Expression of FcγRIIb-Associated Signaling Proteins in hASM Cells
FcγRIIb is a receptor classically expressed in hematopoietic cells, and its intracellular signaling properties have been largely examined in this context. However, it is not clear whether the key FcγRIIb-associated signaling proteins are expressed in mesenchymal cells, such as hASM. Western blot analysis (Figure 6) showed that the Src-kinase, Lyn (53/56 kD), the inhibitory adapter protein, Dok-1 (62 kD), SHIP-1 (145 kD), and SHP-1 (65 kD) are present in hASM cells. Syk (72 kD) was also present in the hASM cells; however, large quantities of a likely 50-kD proteolytic fragment dominated in the majority of cultures. This might be due to Syk (72 kD) being highly susceptible to proteolysis, as shown in previous works (27). HMC-1α cells were used as control cells throughout, and, as expected, showed expression of all the above proteins (Figure 6, lane1).
Figure 6.

Expression of FcγRIIb-associated signaling proteins in hASM cells. Lysates of positive control HMC-1α cells (lane 1) and hASM cultures were analyzed by Western blotting after 24-hour serum starvation. To avoid possible ambiguity introduced by repetitive stripping and reprobing of blots, results from the Src tyrosine kinase Lyn, spleen tyrosine kinase (Syk), and downstream of tyrosine kinase (Dok)-1 were generated on separate blots. In addition, Lyn and Dok-1 results are a composite of two separate experiments. Results shown for Src homology (SH)2–containing inositol phosphatase (SHIP)–1 and protein tyrosine phosphatase SHP–1 are from the same immunoblot. The expression of β-actin was used as a loading control throughout, with all immunoblots showing equivalent protein loading. The blot for β-actin shown was generated after reprobing of the SHIP-1/SHP-1 blot. Representative results of five (from a total of eight) independent hASM cultures are shown.
Activation of FcγRIIb Inhibits IL-1α–Induced or bFGF-Induced MAPK Phosphorylation in hASM Cells
Given the importance of MAPK pathways in regulating cytokine production and mitogenic responses in hASM (28), we examined whether engagement of FcγRIIb by HAGG would affect the activation of ERK1/2 and p38MAPK in these cells. As expected, both bFGF and IL-1α were able to induce p38MAPK and ERK1/2 phosphorylation in hASM cells (Figure 7). Importantly, pretreatment of hASM cells with HAGG significantly inhibited bFGF-induced p38MAPK (Figure 7A) and ERK1/2 phosphorylation (Figure 7C), and also inhibited IL-1α–induced ERK1/2 (Figure 7D), but not p38MAPK phosphorylation (Figure 7B).
Figure 7.

FcγR aggregation inhibits stimulus-induced mitogen-activated protein kinase (MAPK) phosphorylation in hASM cells. Pretreating hASM cells with HAGG significantly inhibited bFGF (300 pM)-induced p38MAPK (A) and extracellular signal–regulated kinase (ERK) 1/2 phosphorylation (C), and also inhibited IL-1α (0.01 ng/ml)–induced ERK1/2 (D), but not p38MAPK phosphorylation (B). Differences were identified using one-way repeated measurement ANOVA and Bonferroni's multiple comparison test (*P < 0.05; **P < 0.01; ***P < 0.001 compared with IL-1α treatment alone or bFGF treatment alone).
DISCUSSION
Increasing evidence suggests that hASM cells are not only involved in airway obstruction, but can also participate in inflammatory mechanisms that lead to the airway hyperresponsiveness and airway remodeling present in asthma (12). Antibodies are capable of inducing potent pro- or anti-inflammatory responses via their interaction with specific cell surface Fc receptors (29). FcεR and FcγR, classically expressed on immune cells, have also been detected on hASM cells (14, 15, 17), raising the possibility that these cells could be directly modulated by antigen and/or immune complexes. The present study aimed to confirm FcεR and FcγR expression, and to determine the functional activities of these receptors.
Our study does not support previous suggestions that nonasthmatic hASM cells express either CD23 or FcεRI. We were unable to detect the IgE-binding FcεRIα subunit in nonasthmatic hASM cells at either the mRNA or the protein level. These results are consistent with the study of Hakonarson and colleagues (15), who also failed to detect FcεRI expression in either asthmatic or nonasthmatic hASM tissue by RT-PCR, and could also not detect CD23 in nonasthmatic hASM tissue by Western blotting. However, the authors did show CD23 expression by RT-PCR and FACS analysis in asthmatic hASM and nonasthmatic hASM cells after treatment with human serum (15). We also detected mRNA for CD23 in hASM cells. However, we were not able to show CD23 expression by flow cytometry. Although the different antibody clones used in the studies may possibly account for some of the disparity in results, we could not measure binding of the endogenous ligand IgE either. Other studies have also found that treatment of hASM cells with hIgE or atopic asthmatic serum was able to induce cytokine production (15, 17, 19), and that the cell activation could be inhibited using a monoclonal anti-IgE antibody (19) or monoclonal anti-CD23 blocking antibody (15). However, we could not measure functional activation of hASM through IgE receptors. This lack of response did not relate to poor-quality IgE or ineffective antigen, as both the hIgE and antigen used was capable of inducing marked cytokine release from HMC-1α cells (data not shown).
A potential reason for the inconsistencies between studies examining IgE receptor expression in hASM might relate to phenotypic differences between the cells, the culture serum used, or the passage number of hASM cells. The study of Hakonarson and colleagues (15) used asthmatic hASM or nonasthmatic hASM treated with control or allergic human serum. hASM cells taken from patients with asthma exhibit increased synthetic function and mitogenesis, and subsequently produce more proinflammatory factors and express different surface proteins when compared with cells taken from donors without asthma (30). Moreover, expression of CD23 on nonasthmatic hASM cells is up-regulated in response to allergic serum (15) and cytokines that are present therein, such as IL-4 and granulocyte/macrophage colony–stimulating factor (14). Thus, an asthmatic airway wall environment might be necessary for effective IgE receptor expression in hASM. In partial support of this, in addition to mRNA for CD23, we also detected the mRNA of the common FcRγ subunit in hASM cells, which is essential for FcεRIα surface expression. However, our attempts to partially recreate “asthmatic” conditions, through sensitizing hASM cells with hIgE to stabilize FcεR expression, and by treatment with IL-4, did not have any measurable effect on FcεR expression. Therefore, if hASM does express FcεRI, likely multiple factors are required, perhaps including IL-13, which has also been shown to induce FcεRIα subunit gene expression in hASM cells (31).
In some Fc receptor expression studies, we have used hASM cells from later passages compared with those used in other published works. To ensure that receptor expression was not dependent upon passage number, surface levels of FcεR and FcγR were measured repeatedly by flow cytometry in hASM cultures over a range of passages. In these studies, there was no detectable relationship between passage number and expression of either IgE or IgG Fc receptors (Figure E6)
Previous studies have shown that all three major isoforms of FcγR can be expressed on hASM cells (15), and, likewise, expression has also been shown in other mesenchymal cells, including vascular smooth muscle (32) and endothelium (33). We found that both FcγRI and FcγRII, but not FcγRIII, was expressed on hASM cells. Importantly, by immunohistochemistry, we also observed FcγRII expression in situ in the ASM in nonasthmatic lung specimens. Combined, these findings raise the possibility that hASM cells could respond directly to IgG immune complexes. These immune complexes might be formed by classical antibody/antigen interactions, or perhaps through nonspecific aggregation of IgG (34) brought about by the heightened oxidative environment in asthmatic airways (35). Moreover, FcγR may act as receptors for the pentraxin acute-phase proteins, such as C-reactive protein (36), which is at an elevated concentration in patients with severe asthma (37).
FcγRI associates with homodimers of the FcRγ subunit, which contains an ITAM motif that triggers cell activation. FcγRII has two major isoforms, which, in immune cells, are generally considered to produce opposing effects. FcγRIIa has an integral ITAM in its cytoplasmic portion, whereas FcγRIIb contains an ITIM motif that causes inhibitory actions (best described in B lymphocytes, mast cells, and basophils [6]) when coengaged with ITAM-containing receptors. Our study demonstrates expression of the latter receptor in hASM. Consistent with the dominant expression of FcγRIIb in hASM cells, and the known negative regulatory roles of FcγRIIb, HAGG was shown to inhibit the proinflammatory or mitogenic effects of IL-1α and bFGF, with the extent of inhibition noted in individual hASM cultures being strongly correlated with the levels of FcγRIIb expressed on the cells. The inhibitory effect of HAGG was also attenuated by a specific anti-FcγRIIb antibody Fab fragment.
The mechanism through which HAGG can produce inhibition of IL-1α and bFGF has been examined in this study. In hematopoietic cells, the inhibitory actions of FcγRIIb are best characterized when coaggregated with an ITAM-containing receptor. In this situation, coaggregation leads to Src-kinase–mediated phosphorylation of the FcγRIIb ITIM and subsequent recruitment of inhibitory signaling proteins, such as Dok-1, SHIP-1, and SHP-1, thereby inhibiting the ITAM-mediated signaling (5, 6). We have shown expression of the Src-kinase, Lyn, Dok-1, SHIP, and SHP-1 in hASM cells. Others have also recently reported expression of some of these signaling proteins in hASM cells (31, 38). Thus, the canonical FcγRIIb signaling pathways demonstrated in hematopoietic cells could also operate in hASM cells. These negative signaling molecules have broad inhibitory actions on the MAPK pathways that are known to underlie hASM cell proliferation and cytokine production in response to bFGF and IL-1α respectively (39–41). The best-studied MAPKs in hASM cells are ERK1/2 and p38MAPK (28). The proliferation responses of bFGF in hASM cells have been extensively characterized by our laboratory (42), and we have previously shown bFGF-induced cell proliferation via ERK1/2 and p38MAPK pathways (41). Interestingly, our data demonstrate that activation of FcγRIIb with HAGG strongly inhibited bFGF-induced p38MAPK and ERK1/2 phosphorylation. However, HAGG only partially inhibited IL-1α–induced ERK1/2 phosphorylation, a finding that may explain the modest inhibitory effects of HAGG on IL-1α–induced cytokine production by comparison with the marked reduction in hASM cell proliferation.
But what is the initial ITAM–ITIM interaction that evokes this inhibitory effect? It is possible that coengagement of FcγRIIb with FcγRI provides enough of a trigger to activate FcγRIIb pathways, leading to inhibition of IL-1α and bFGF signaling through “bystander” activity. An alternative FcγRIIb inhibitory signaling pathway has, however, also been shown in B cells (43). This pathway is activated solely through FcγRIIb cross-linking, and it may also contribute to the FcγRIIb-mediated inhibitory effects that we observed. A clearer understanding of the FcγRIIb inhibitory mechanism in hASM cells could lead to novel therapeutic strategies that mimic or amplify the FcγRIIb pathway that could be useful for the treatment of asthma and other airway disorders.
The broad inhibitory actions of FcγRIIb in hASM are consistent with a role for this receptor in the beneficial effects of FcγR ligands implied by association of asthma severity and low IgG levels (7, 8). It is unclear whether FcγR expression is disrupted in disorders such as asthma. However, changes in FcγR expression in B cells and monocytes have been observed in chronic inflammatory conditions, such as lupus and rheumatoid arthritis (26, 44). Although a previous study showed that expression of FcγR was similar between nonasthmatic and asthmatic hASM (15), relatively few samples were analyzed in that study, and the isoform of FcγRII expressed was not determined. Further study of FcγR expression in asthma is thus warranted.
In conclusion, we could not detect protein expression of either FcεRI or CD23 in cultured hASM cells derived from individuals without asthma. However, we found that FcγRIIb and FcγRI were expressed in hASM cells, and that the activation of FcγRIIb inhibited pleiotropic cytokine–induced hASM cell activities, which have strong relevance to asthma. This places FcγRIIb as a novel regulator of hASM cell function that has a potentially important role in maintaining appropriate airway function in the presence of immune complexes or other FcγR ligands.
Acknowledgments
The authors thank Drs. Robert Kimberly, Peter Lock, and Margaret Hibbs for providing antibodies used in the study. They also thank Dr. Joseph Butterfield (Mayo Clinic) for providing the human mast cell line–1 cells.
Originally Published in Press as DOI: 10.1165/rcmb.2009-0371OC on July 1, 2010
This work was supported by funding from the Asthma Foundation of Queensland, Australia (G.A.M.) and the National Health and Medical Research Council, Australia (A.G.S. [grant 509,001] and P.M.H.).
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Author Disclosure: W.T.G. received a sponsored grant from the National Institutes of Health for more than $100,001 and consultancy fees for $1,001–$5,000; P.M.H. received a patent for Trillium Pharmaceuticals (Canada) for Fc receptor DNA and for SCE drugs (no income received or likely); M. S. received lecture fees from GlaxoSmithKline for up to $1,000. None of the other authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Bruhns P, Fremont S, Daeron M. Regulation of allergy by Fc receptors. Curr Opin Immunol 2005;17:662–669. [DOI] [PubMed] [Google Scholar]
- 2.Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat Rev Immunol 2008;8:205–217. [DOI] [PubMed] [Google Scholar]
- 3.Oxelius VA. Immunoglobulin constant heavy γ subclass chain genes in asthma and allergy. Immunol Res 2008;40:179–191. [DOI] [PubMed] [Google Scholar]
- 4.Bradding P, Walls AF, Holgate ST. The role of the mast cell in the pathophysiology of asthma. J Allergy Clin Immunol 2006;117:1277–1284. [DOI] [PubMed] [Google Scholar]
- 5.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol 2001;19:275–290. [DOI] [PubMed] [Google Scholar]
- 6.Nimmerjahn F, Ravetch JV. Fcγ receptors as regulators of immune responses. Nat Rev Immunol 2008;8:34–47. [DOI] [PubMed] [Google Scholar]
- 7.de Moraes Lui C, Oliveira LC, Diogo CL, Kirschfink M, Grumach AS. Immunoglobulin γ subclass concentrations and infections in children and adolescents with severe asthma. Pediatr Allergy Immunol 2002;13:195–202. [DOI] [PubMed] [Google Scholar]
- 8.Loftus BG, Price JF, Lobo-Yeo A, Vergani D. IgG subclass deficiency in asthma. Arch Dis Child 1988;63:1434–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaneko M, Swanson MC, Gleich GJ, Kita H. Allergen-specific IgG1 and IgG3 through FcγRII induce eosinophil degranulation. J Clin Invest 1995;95:2813–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tkaczyk C, Okayama Y, Woolhiser MR, Hagaman DD, Gilfillan AM, Metcalfe DD. Activation of human mast cells through the high affinity IgG receptor. Mol Immunol 2002;38:1289–1293. [DOI] [PubMed] [Google Scholar]
- 11.Hirst SJ, Martin JG, Bonacci JV, Chan V, Fixman ED, Hamid QA, Herszberg B, Lavoie JP, McVicker CG, Moir LM, et al. Proliferative aspects of airway smooth muscle. J Allergy Clin Immunol 2004;114:S2–S17. [DOI] [PubMed] [Google Scholar]
- 12.Damera G, Tliba O, Panettieri RA Jr. Airway smooth muscle as an immunomodulatory cell. Pulm Pharmacol Ther 2009;22:353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tliba O, Amrani Y, Panettieri RA Jr. Is airway smooth muscle the “missing link” modulating airway inflammation in asthma? Chest 2008;133:236–242. [DOI] [PubMed] [Google Scholar]
- 14.Belleau JT, Gandhi RK, McPherson HM, Lew DB. Upregulation of CD23 (FcεRII) expression in human airway smooth muscle cells (huASMC) in response to IL-4, GM-CSF, and IL-4/GM-CSF. Clin Mol Allergy 2005;3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hakonarson H, Carter C, Kim C, Grunstein MM. Altered expression and action of the low-affinity IgE receptor FcεRII (CD23) in asthmatic airway smooth muscle. J Allergy Clin Immunol 1999;104:575–584. [DOI] [PubMed] [Google Scholar]
- 16.Hakonarson H, Grunstein MM. Autologously up-regulated Fc receptor expression and action in airway smooth muscle mediates its altered responsiveness in the atopic asthmatic sensitized state. Proc Natl Acad Sci USA 1998;95:5257–5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gounni AS, Wellemans V, Yang J, Bellesort F, Kassiri K, Gangloff S, Guenounou M, Halayko AJ, Hamid Q, Lamkhioued B. Human airway smooth muscle cells express the high affinity receptor for IgE (FcεRI): a critical role of FcεRI in human airway smooth muscle cell function. J Immunol 2005;175:2613–2621. [DOI] [PubMed] [Google Scholar]
- 18.Redhu NS, Saleh A, Shan L, Gerthoffer WT, Kung SK, Halayko AJ, Lamkhioued B, Gounni AS. Proinflammatory and Th2 cytokines regulate the high affinity IgE receptor (FcεRI) and IgE-dependant activation of human airway smooth muscle cells. PLoS ONE 2009;4:e6153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roth M, Tamm M. The effects of omalizumab on IgE-induced cytokine synthesis by asthmatic airway smooth muscle cells. Ann Allergy Asthma Immunol 2010;104:152–160. [DOI] [PubMed] [Google Scholar]
- 20.Fernandes D, Guida E, Koutsoubos V, Harris T, Vadiveloo P, Wilson JW, Stewart AG. Glucocorticoids inhibit proliferation, cyclin D1 expression, and retinoblastoma protein phosphorylation, but not activity of the extracellular-regulated kinases in human cultured airway smooth muscle. Am J Respir Cell Mol Biol 1999;21:77–88. [DOI] [PubMed] [Google Scholar]
- 21.Gosens R, Stelmack GL, Dueck G, McNeill KD, Yamasaki A, Gerthoffer WT, Unruh H, Gounni AS, Zaagsma J, Halayko AJ. Role of caveolin-1 in p42/p44 MAP kinase activation and proliferation of human airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 2006;291:L523–L534. [DOI] [PubMed] [Google Scholar]
- 22.Butterfield JH, Weiler D, Dewald G, Gleich GJ. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk Res 1988;12:345–355. [DOI] [PubMed] [Google Scholar]
- 23.Powell MS, Barnes NC, Bradford TM, Musgrave IF, Wines BD, Cambier JC, Hogarth PM. Alteration of the FcγRIIa dimer interface affects receptor signaling but not ligand binding. J Immunol 2006;176:7489–7494. [DOI] [PubMed] [Google Scholar]
- 24.Schuliga MJ, See I, Ong SC, Soon L, Camoretti-Mercado B, Harris T, Stewart AG. Fibrillar collagen clamps lung mesenchymal cells in a nonproliferative and noncontractile phenotype. Am J Respir Cell Mol Biol 2009;41:731–741. [DOI] [PubMed] [Google Scholar]
- 25.Astier A, de la Salle H, Moncuit J, Freund M, Cazenave JP, Fridman WH, Hanau D, Teillaud JL. Detection and quantification of secreted soluble FcγRIIa in human sera by an enzyme-linked immunosorbent assay. J Immunol Methods 1993;166:1–10. [DOI] [PubMed] [Google Scholar]
- 26.Su K, Yang H, Li X, Li X, Gibson AW, Cafardi JM, Zhou T, Edberg JC, Kimberly RP. Expression profile of FcγRIIb on leukocytes and its dysregulation in systemic lupus erythematosus. J Immunol 2007;178:3272–3280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taniguchi T, Kobayashi T, Kondo J, Takahashi K, Nakamura H, Suzuki J, Nagai K, Yamada T, Nakamura S, Yamamura H. Molecular cloning of a porcine gene syk that encodes a 72-kda protein-tyrosine kinase showing high susceptibility to proteolysis. J Biol Chem 1991;266:15790–15796. [PubMed] [Google Scholar]
- 28.Gerthoffer WT, Singer CA. MAPK regulation of gene expression in airway smooth muscle. Respir Physiol Neurobiol 2003;137:237–250. [DOI] [PubMed] [Google Scholar]
- 29.Powell MS, Hogarth PM. Fc receptors. Adv Exp Med Biol 2008;640:22–34. [DOI] [PubMed] [Google Scholar]
- 30.Howarth PH, Knox AJ, Amrani Y, Tliba O, Panettieri RA Jr, Johnson M. Synthetic responses in airway smooth muscle. J Allergy Clin Immunol 2004;114:S32–S50. [DOI] [PubMed] [Google Scholar]
- 31.Lee JH, Kaminski N, Dolganov G, Grunig G, Koth L, Solomon C, Erle DJ, Sheppard D. Interleukin-13 induces dramatically different transcriptional programs in three human airway cell types. Am J Respir Cell Mol Biol 2001;25:474–485. [DOI] [PubMed] [Google Scholar]
- 32.Ryu J, Lee CW, Shin JA, Park CS, Kim JJ, Park SJ, Han KH. FcγRIIa mediates C-reactive protein–induced inflammatory responses of human vascular smooth muscle cells by activating NADPH oxidase 4. Cardiovasc Res 2007;75:555–565. [DOI] [PubMed] [Google Scholar]
- 33.Mineo C, Gormley AK, Yuhanna IS, Osborne-Lawrence S, Gibson LL, Hahner L, Shohet RV, Black S, Salmon JE, Samols D, et al. FcγRIIb mediates C-reactive protein inhibition of endothelial NO synthase. Circ Res 2005;97:1124–1131. [DOI] [PubMed] [Google Scholar]
- 34.Lunec J, Blake DR, McCleary SJ, Brailsford S, Bacon PA. Self-perpetuating mechanisms of immunoglobulin γ aggregation in rheumatoid inflammation. J Clin Invest 1985;76:2084–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park HS, Kim SR, Lee YC. Impact of oxidative stress on lung diseases. Respirology 2009;14:27–38. [DOI] [PubMed] [Google Scholar]
- 36.Mold C, Baca R, Du Clos TW. Serum amyloid P component and C-reactive protein opsonize apoptotic cells for phagocytosis through Fcγ receptors. J Autoimmun 2002;19:147–154. [DOI] [PubMed] [Google Scholar]
- 37.Takemura M, Matsumoto H, Niimi A, Ueda T, Matsuoka H, Yamaguchi M, Jinnai M, Muro S, Hirai T, Ito Y, et al. High sensitivity C-reactive protein in asthma. Eur Respir J 2006;27:908–912. [DOI] [PubMed] [Google Scholar]
- 38.Pertel T, Zhu D, Panettieri RA, Yamaguchi N, Emala CW, Hirshman CA. Expression and muscarinic receptor coupling of lyn kinase in cultured human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2006;290:L492–L500. [DOI] [PubMed] [Google Scholar]
- 39.Dunne A, O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal transduction during inflammation and host defense. Sci STKE 2003;2003:re3. [DOI] [PubMed]
- 40.Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 2005;16:139–149. [DOI] [PubMed] [Google Scholar]
- 41.Fernandes DJ, Ravenhall CE, Harris T, Tran T, Vlahos R, Stewart AG. Contribution of the p38MAPK signalling pathway to proliferation in human cultured airway smooth muscle cells is mitogen-specific. Br J Pharmacol 2004;142:1182–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stewart AG, Fernandes D, Tomlinson PR. The effect of glucocorticoids on proliferation of human cultured airway smooth muscle. Br J Pharmacol 1995;116:3219–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tzeng SJ, Bolland S, Inabe K, Kurosaki T, Pierce SK. The B cell inhibitory Fc receptor triggers apoptosis by a novel c-Abl family kinase–dependent pathway. J Biol Chem 2005;280:35247–35254. [DOI] [PubMed] [Google Scholar]
- 44.Wijngaarden S, van de Winkel JG, Jacobs KM, Bijlsma JW, Lafeber FP, van Roon JA. A shift in the balance of inhibitory and activating Fcγ receptors on monocytes toward the inhibitory Fcγ receptor IIb is associated with prevention of monocyte activation in rheumatoid arthritis. Arthritis Rheum 2004;50:3878–3887. [DOI] [PubMed] [Google Scholar]