

Abstract
Tibetan pigs that inhabit the Tibetan Plateau exhibit striking phenotypic and physiological differences from lowland pigs, and have adapted well to extreme conditions. However, the mechanisms involved in regulating gene expression at high altitude in these animals are not fully understood. In this study, we obtained transcriptomic and proteomic data from the heart tissues of Tibetan and Yorkshire pigs raised in the highlands (TH and YH) and lowlands (TL and YL) via RNA-seq and iTRAQ (isobaric tags for relative and absolute quantitation) analyses, respectively. Comparative analyses of TH vs. YH, TH vs.TL, TL vs. YL, and YH vs. YL yielded 299, 169, 242, and 368 differentially expressed genes (DEGs), and 473, 297, 394, and 297 differentially expressed proteins (DEPs), respectively. By functional annotation of these DEGs and DEPs, genes that were enriched in the HIF-1 signaling pathway (NPPA, ERK2, ENO3, and EGLN3), VEGF signaling pathway (ERK2, A2M, FGF1, CTGF, and DPP4), and hypoxia-related processes (CRYAB, EGLN3, TGFB2, DPP4, and ACE) were identified as important candidate genes for high-altitude adaptation in the Tibetan pig. This study enhances our understanding of the molecular mechanisms involved in hypoxic adaptation in pigs, and furthers our understanding of human hypoxic diseases.
Introduction
Low air pressure and low oxygen partial pressure at high altitude seriously affect the survival and development of human beings and other animals1, 2. Indigenous animals of the Tibetan Plateau exhibit heritable adaptations to this extreme environment due to natural selection; therefore, the Tibetan pig is an ideal animal model for research on the molecular ecology and pathology related to hypoxia3–5.
Adaptation to hypoxic conditions is a complex trait that is influenced by many factors6. Several genes exhibiting strongly selection have been identified in Tibetan populations by genome scanning, providing a possible genetic basis for adaptation to high-altitude conditions7–9. Likewise, a number of rapidly evolving, positively selected genes have been reported in the Tibetan pig10; however, the mechanisms involved in gene expression at high altitude are not fully understood. The development of RNA-seq and isobaric tags for relative and absolute quantitation (iTRAQ) technologies has enabled the identification of eukaryotic genes associated with complex traits via analysis of transcriptomic and proteomic profiles, with low bias, a large dynamic range, low frequency of false-positive signals, and high reproducibility. The iTRAQ method employs a set of amine-reactive isobaric tags to derivatize peptides at the N-terminus and at lysine side chains, thereby allowing for simultaneous protein identification and quantification via mass spectrometry analysis of peptide fragments (m/z range from 150 to 2,200) and signature ions (m/z values from 113 to 121), respectively. Both RNA-seq and iTRAQ have been widely used to screen for functional genes involved in muscle growth and lipid deposition in pigs and other domestic species11–14. An integrated analysis of the transcriptome and proteome of pig heart tissues at high and low altitudes would comprehensively profile hypoxic adaptations, and enable us to characterize the genes and proteins that are involved in this process.
Heart tissues were previously used to study hypoxic physiology and pathology15, 16. An increased heart rate, blood pressure, and other high-altitude responses result in changes in cardiac structure and function. In particular, animals populating the Tibetan Plateau exhibit strong cardiac function, and their cardiomyocytes show an ability to promote the expression of genes associated with adaptation to hypoxic conditions to protect cardiac cells and tissue structure, and increase blood circulation. Indeed, Tibetan pigs have adapted to high altitudes with well-developed hearts and lungs, and a blunted erythropoietic response17.
Cellular responses to hypoxia are reliant on controlled transcriptional and post-transcriptional events, in which certain genes are positively regulated and others either remain inactive or are actively repressed. In this study, we performed a comparative analysis of the transcriptomic and proteomic profiles of heart tissues obtained from Tibetan and Yorkshire pigs raised at high (TH and YH, respectively) and low (TL and YL, respectively) altitudes using RNA-seq and iTRAQ technologies. Via these analyses, we identified key genes and molecular mechanisms involved in the high- altitude adaptations of the Tibetan pig.
Results
Overview of RNA transcriptomic profiles of pig heart tissues
A total of 551 M paired-end reads were acquired from the RNA-seq experiment. Sequence alignment was performed using the pig genome (Sscrofa10.2) as a reference. Approximately 83.68% (82.07–85.18%) of the mapped reads were acquired from the RNA-seq experiment of which 76.26% (74.86–77.74%) were mapped to unique genomic locations. Additionally, of the total mapped reads, roughly 89% in each group corresponded to exons (Table S3, Fig. S1).
Gene expression levels were evaluated by counting the number of mapped reads per gene. In total, 18,585 expressed genes were detected in the heart tissues examined; of these, 15,701 were detected in each of the four groups (Fig. S2A). Less than 1% of the genes were expressed at greater than 1,000 FPKM (numbers of fragments per kilobase of exon per million mapped fragments); approximately 3% were expressed between 100 and 1,000 FPKM, and approximately 96% were expressed at less than 100 FPKM (Table S3, Fig. S2B). Expression value distributions were similar among the eight samples tested. Likewise, the FPKM values were evenly distributed amongst the four groups, and there were no outliers (Fig. S2C).
Analysis of differentially expressed genes (DEGs)
Pair-wise comparisons with the strict criteria of |log2 (FC)| > 1 and P < 0.01 were utilized to detect DEGs (Fig. S3A). Specifically, 299 and 242 DEGs were detected in the comparisons of TH to YH (Table S4–1, Fig. S3B) and of TL to YL (Table S4–2, Fig. S3B), respectively, including 26 DEGs that were common to each comparison (Fig. S3C). Meanwhile, we detected 169 and 368 DEGs between TH and TL (Table S4-3, Fig. S3B) and YH and YL (Table S4-4, Fig. S3B), respectively, of which 24 overlapped (Fig. S3C).
Functional analysis of DEGs
We used DAVID and Ingenuity Pathways Analysis (IPA), which have related capabilities but utilize different databases, to obtain a complete functional view of the DEGs. Using DAVID, we observed an overrepresentation of gene ontology (GO) terms related to “response to hormone stimulus” (19 genes, P = 3.15E-05) and “mitochondrion” (36 genes, P = 6.30E-05) in TH vs. YH, and “oxidation reduction” (51 genes, P = 1.77E-16) and “cofactor binding” (32 genes, P = 2.44E-15) in TL vs. YL (Table S5). We also observed an overrepresentation of the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways “ribosome” (15 genes, P = 8.52E-10) and “dilated cardiomyopathy” (7 genes, P = 9.08E-03) in TH vs. YH, and “glycolysis/gluconeogenesis” (9 genes, P = 3.02E-04) and “renin-angiotensin system” (5 genes, P = 1.22E-03) in TL vs. YL. Analysis of these two comparison groups also highlighted GOs and pathways, such as “regulation of blood pressure”, “vascular process in circulatory system”, “response to hypoxia”, and “complement and coagulation cascades pathway” (Table S5). Additionally, the mammalian target of rapamycin (mTOR) signaling pathway was detected in the TH vs. YH comparison; hypoxia-induced mTOR activation reduces mTOR targeting to enhance angiogenesis in response to hypoxia18. Moreover, we identified a variety of DEGs in pathways upstream of the mTOR pathway19, in the insulin signaling pathway (seven of the DEGs were designated to encode oxygen-independent regulators).
In the TH vs. TL analysis, the overrepresented KEGG pathways were “contractile fiber” (12 genes, P = 1.68E-08), “muscle contraction” (10 genes, P = 1.22E-03), “dilated cardiomyopathy” (6 genes, P = 5.35E-03), “complement and coagulation cascades” (5 genes, P = 1.01E-02), while in YH vs. YL, they were “oxidation reduction” (32 genes, P = 2.10E-10), “=2.10biosynthetic processl (12 genes, P = 7.72E-09), “glycolysis/gluconeogenesis” (9 genes, P = 1.37E-05), and “histidine metabolism” (6 genes, P = 1.79E-04). Notably, GOs and pathways, such as the “mitochondrion”20, “glucose metabolic process”, “dilated cardiomyopathy”16, and “cardiac muscle contraction”21 pathways, which play important roles in the hypoxia response, were also detected in TH vs. TL.
We also performed metabolic pathway analysis on the DEGs using IPA software. Based on the data obtained via this method, the main canonical pathways that were overrepresented in the TH vs. YH and TL vs. YL comparisons were related to hepatic fibrosis/hepatic stellate cell activation (FN1, CTGF, CCL5, A2M, and FGF1, and COL1A2, CTGF, AGT, and COL3A1; P = 3.85E-02 and P = 2.58E-02, respectively), and glucocorticoid receptor signaling in TH vs. YH (DUSP1, SLPI, CCL5, and A2M, and DUSP1 and AGT; P = 1.82E-01 and P = 3.66E-01, respectively). Thus, depending on the environment, Tibetan pigs modulated the expression of distinct DEGs to adapt to hypoxic conditions. In addition, interesting pathways such as the hypoxia-inducible factor 1 (HIF-1) signaling pathway (EGLN3) were detected in both the TH vs. YH and TH vs. TL comparisons (Table S6).
Of the primary molecular and cellular functions that were significantly overrepresented in the four comparison groups, according to IPA, the most relevant were involved in “cardiovascular system development and function” in TH vs. YH (23 genes, P = 1.33E-09–2.40E-03), “cardiac hypertrophy” in TL vs. YL (5 genes, P = 1.393E-05–1.90E-01), “cardiovascular system development and function” in TH vs. TL (16 genes, P = 4.22E-07–4.69E-03), and “skeletal and muscular system development and function” in YH vs. YL (3 genes, P = 7.05E-06–9.10E-03).
Among the related specific functions of heart tissue, the main functions identified by IPA in TH vs. YH were “angiogenesis” (ANXA2, CCL5, CD151, CD44, CTGF, EGLN3, FGF1, FN1, HDAC9, ITGA5, JUNB, NR4A1, PFKM, SLPI, and TGM2; P = 1.33E-09), “binding of endothelial cells” (ANXA2, CCL5, CD151, CD44, FGF1, FN1, and NR4A1; P = 7.07E-09), and “development of blood vessel” (ANXA2, ATF3, CCL5, CD151, CD44, CTGF, EGLN3, FGF1, FN1, HDAC9, ITGA5, JUNB, NR4A1, and TGM2; P = 9.53E-08); in TL vs. YL were “hypertrophy of cardiomyocytes” (AGT, CRYAB, CTGF, and DUSP1; P = 1.39E-05) and “hypertrophy of heart” (AGT, CRYAB, CTGF, and DUSP1; P = 2.44E-04); in TH vs. TL were “formation of endothelial tube” (ATF3, CCL2, CD44, FN1, and PPP1R14B; P = 4.22E-07) and “angiogenesis” (CCL2, CD151, CD44, CRYAB, FGF1, FN1, HSPB8, ITGAV, JUNB, PFKM, and TGM2; P = 5.22E-06); and in YH vs. YL were “quantity of muscle cells” (AGT, CCL2, and FLNC; P = 7.05E-06) and “quantity of smooth muscle cells” (AGT and CCL2; P = 1.03E-04) (Table S6). Notably, we obtained the same pathways via functional analysis of TH vs. TL and YH vs. YL by IPA. Thus, even when inhabiting the same environment, Tibetan and Yorkshire pigs utilize distinct genes to adapt to hypoxia. All of the DEGs that were related to hypoxic adaptation in the four groups are shown in Table 1.
Table 1.
Potential key differentially expressed genes (DEGs) and their functions related to hypoxic adaption in the Tibetan pig.
| Gene | TH/YH | TL/YL | TH/TL | YH/YL | Functional analysis |
|---|---|---|---|---|---|
| A2M | 0.44 | 0.25 | 1.43 | 0.81 | Complement and coagulation cascades, blood microparticle, VEGF signaling pathway |
| AGT | 0.63 | 2.94 | 0.57 | 2.68 | Renin-angiotensin system, vasculature development, muscle contraction, blood vessel development, regulation of blood pressure |
| ANXA2 | 3.44 | 1.18 | 4.19 | 1.44 | angiogenesis, vasculature development, |
| ATF3 | 6.18 | 17.30 | 0.27 | 0.77 | gluconeogenesis, DNA binding |
| CD44 | 2.82 | 0.82 | 5.80 | 1.68 | ECM-receptor interaction, Hematopoietic cell lineage |
| CCL5 | 0.48 | 1.11 | 0.75 | 1.71 | immune response, response to insulin stimulus |
| COL3A1 | 1.58 | 4.95 | 0.50 | 1.55 | response to radiation, blood vessel development |
| CRYAB | 2.20 | 0.68 | 4.68 | 1.45 | mitochondrion, response to hypoxia, response to reactive oxygen species |
| CTGF | 5.72 | 3.31 | 1.58 | 0.91 | lung development, blood vessel development, vasculature development, angiogenesis, blood vessel morphogenesis, reactive oxygen species metabolic process, cardiovascular development, VEGF signaling pathway |
| DUSP1 | 3.92 | 2.96 | 1.50 | 1.13 | MAPK signaling pathway |
| EGLN3 | 0.47 | 0.72 | 0.78 | 1.21 | Pathways in cancer, renal cell carcinoma, oxidation-reduction process, response to hypoxia, HIF-1 signaling pathway |
| FGF1 | 4.32 | 1.55 | 0.33 | 1.59 | MAPK signaling pathway, Pathways in cancer, blood vessel morphogenesis, blood vessel development, respiratory system development, cardiovascular development, VEGF signaling pathway |
| DPP4 | 0.22 | 0.31 | 0.55 | 0.75 | response to hypoxia, response to oxygen levels, VEGF signaling pathway |
| HBB | 2.27 | 1.94 | 2.00 | 1.71 | cardiac myofibril assembly, hemoglobin complex, oxygen transport |
| HBA | 2.26 | 2.73 | 1.60 | 1.93 | cardiac myofibril assembly, hemoglobin complex, oxygen transport |
| NPPA | 37.60 | 2.31 | 27.27 | 1.68 | regulation of blood vessel size, cardiac muscle hypertrophy in response to stress, HIF-1 signaling pathway, regulation of blood vessel size, circulatory system process, regulation of blood pressure, hypertrophy in response to stress |
| NPPB | 41.96 | 0.17 | 138.23 | 0.57 | regulation of blood vessel size, regulation of blood vessel size, circulatory system process |
| DECR1 | 0.42 | 1.02 | 0.40 | 0.97 | Mitochondrion, reductase (NADPH) activity, NADPH binding |
| TGFB2 | 9.24 | 1.74 | 2.03 | 0.38 | heart development, vasculature development, cardiac muscle tissue development, response to oxygen levels, response to hypoxia, angiogenesis, regulation of heart contraction, immune system development, pathways in cancer, TGF-beta signaling pathway |
| PDLIM3 | 2.03 | 2.58 | 4.33 | 5.49 | heart development, cardiovascular development |
| FOS | 14.93 | 24.97 | 0.54 | 0.91 | DNA binding, cellular response to reactive oxygen species, pertussis, pathways in cancer |
TH, Tibetan highland pig; TL, Tibetan lowland pig; YH, Yorkshire highland pig; YL, Yorkshire lowland pig.
Based on functional annotation, 21 DEGs were associated with hypoxia (Table 1), which involved the GO terms of “response to hypoxia”, “blood vessel development”, “response to reactive oxygen species”, “circulatory system process”, “regulation of heart contraction”, and “immune system development”, as well as the HIF-1 signaling pathway, the mitogen-activated protein kinase (MAPK) signaling pathway, pathways in cancer, glycolysis/gluconeogenesis, and the TGF-beta signaling pathway. Three genes, NPPA (natriuretic peptide type A, also known as ANP), NPPB (natriuretic peptide type B, also known as BNP), and FOS (FBJ murine osteosarcoma viral oncogene homolog), exhibited substantial differential expression, with fold changes greater than 14 in TH vs. YH (Table 1). In particular, the expression level of NPPB in TH was 41.96, which was 138.23-fold higher than that in YH and TL, and may therefore exert a positive effect on hypoxic adaptation in these animals. Similarly, NPPA was also highly expressed in this group, with an expression level of 37.60, which was 27.27-fold higher than that in YH and TL. In humans, ANP and BNP constitute the dual natriuretic peptide system of the heart. Numerous studies have clearly shown that hypoxia stimulates atrial natriuretic peptide (ANP) secretion, resulting in cellular adaptation to hypoxia and protection of the ischemic heart22–24.
Overview of protein identification and quantification by iTRAQ
A total of 128,390 spectra were obtained from the 8PLEX liquid chromatography tandem-mass spectrometry (LC-MS/MS) analysis (Fig. S4A). Pooling of the samples from both groups resulted in identification of 12,880 peptides at the peptide level, including 2578 proteins that had been originally identified with false discovery rates (FDR) ≤0.01, and with high confidence of the correct peptide sequence assignment. The numbers of proteins identified at distinct molecular weight ranges were as follows: 10–20 kDa (433), 20–30 kDa (443), 30–40 kDa (370), 40–50 kDa (318), 50–60 kDa (246), and 60–70 kDa (138). In total, these proteins accounted for 75.56% of those identified (Fig. S4B). In addition, the majority of the proteins were identified with high peptide coverage; 95.30% of the proteins had less than 50% sequence coverage, while 4.70% had greater than 50% sequence coverage (Fig. S4C). Among the proteins identified, 64.08% were represented by fewer than five peptides (Fig. S4D), indicating good sequence coverage of the proteins identified.
Analysis of differentially expressed proteins (DEPs)
Based on the selection criteria of FC > 1.2 or <0.83, we detected 473, 297, 394, and 297 DEPs in the TH vs. YH, TL vs. YL, TH vs. TL, and YH vs. YL comparisons, respectively. Specifically, 253 up-regulated and 220 down-regulated DEPs were identified in the TH vs. YH comparison, while 115 and 182, 275 and 119, and 179 and 188 up-regulated and down-regulated proteins were detected in TL vs. YL, TH vs. TL, and YH vs. YL, respectively (Table S7, Fig. S5). Notably, there were 87 and 86 overlapping proteins in the TH vs. YH and TL vs. YL, and in TH vs. TL and YH vs. YL comparisons, respectively (Table S8, Fig. 1).
Figure 1.
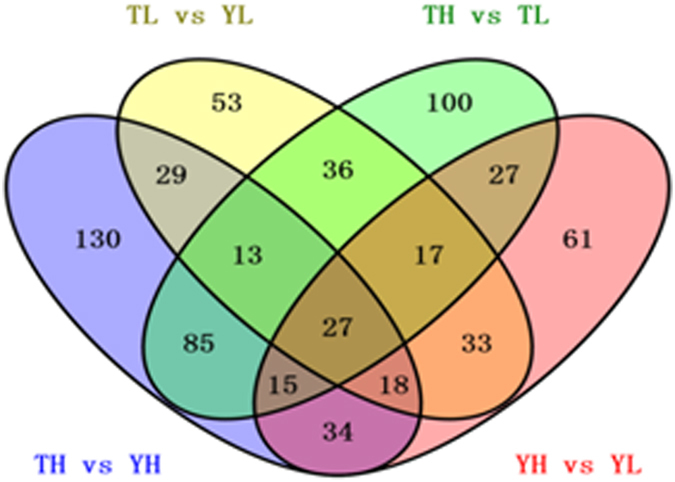
Venn diagram of differentially expressed genes (DEGs) among the four comparison groups. TH, Tibetan highland pig; TL, Tibetan lowland pig; YH, Yorkshire highland pig; YL, Yorkshire lowland pig.
Validation of DEGs and DEPs
To validate the accuracy of the DEGs detected by RNA-seq analysis, we used real-time reverse transcription-quantitative polymerase chain reaction (RT-qPCR) to evaluate the expression levels of eight DEGs: ADHFE1, CCL5, CKM, DECR1, DPP4, EGLN3, FOS, and SLA-5. The expression levels of these genes in each group are shown in Fig. S6. The eight genes selected were differentially expressed among the four comparison groups (TH vs. YH, TH vs.TL, YH vs. YL, and TL vs. YL), and the RNA-seq data were concordant with those obtained by RT-qPCR. Moreover, RT-qPCR analysis confirmed the DEG expression patterns observed in the two pig breeds at each altitude (Fig. S7).
Notably, the results of previous studies suggest that the label-free method is typically more efficient, in regard to the number of proteins identified, than the iTRAQ approach25–27. In the current study, a total of 238 of the 351 (67.81%) and 686 (34.69%) proteins identified using the iTRAQ and label-free methods, respectively, were identified by both methods (Fig. S8A). For these proteins, the fold changes observed via each method were plotted, and a correlation with an R2 value of 0.604 was calculated (Fig. S8B). Of the overlapping 238 proteins, only three were found to be differentially expressed in divergent directions (i.e., up-regulation vs. down-regulation) in the comparison of TH vs. YH via iTRAQ and label-free experiments. Indeed, the overall trends of the commonly quantified proteins were in agreement between the two methods.
Functional analysis of DEPs
To better understand the biological functions of DEPs in Tibetan and Yorkshire pigs at different altitudes, the DEPs were further classified to identify pathways in the heart tissues based on GO and KEGG functional annotations (Table S9, Fig. 2). In TH vs. YH and TL vs. YL, the GO terms were mainly associated with “mitochondrion” (involved in 39 proteins and 44 proteins, respectively), “oxidation reduction” (involved in 23 proteins and 23 proteins, respectively), “blood circulation” (contained proteins such as CAV2, ACE, RENBP, AGT, MYL1, COL1A2, EPHX2, ERAP1, STAT1, HBB, TCAP, and MYH6), “NAD or NADH binding” (involved in 5 proteins and 4 proteins, respectively), “response to oxygen levels” (involved in 8 proteins and 7 proteins, respectively), and “iron ion homeostasis” (contained proteins such as TF, TFRC, HPX, FTH1, HP, and CP). In addition, we found interesting GOs in TH vs. YH, such as “gas transport” (HBQ1, CA2, AQP1, and HBB), “regulation of blood pressure” (ACE, RENBP, AGT, COL1A2, EPHX2, ERAP1, and HBB), and “oxygen and reactive oxygen species metabolic process” (CRYAB, AGT, EPHX2, BNIP3, and PARK7).
Figure 2.
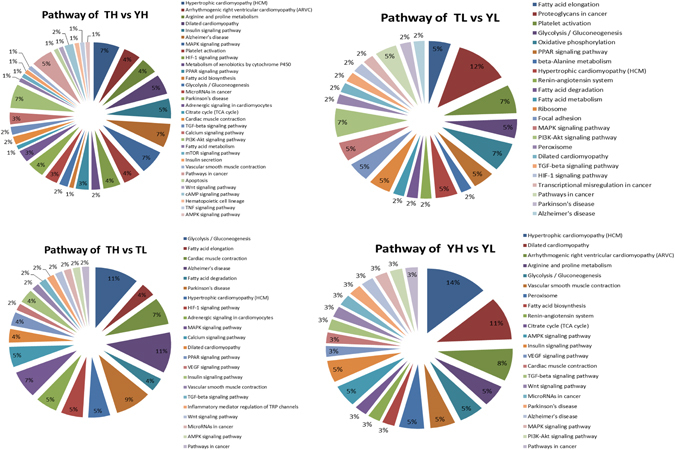
Differentially expressed protein (DEP) pathway analysis. TH, Tibetan highland pig; TL, Tibetan lowland pig; YH, Yorkshire highland pig; YL, Yorkshire lowland pig.
It is worth noting that 2–5% of the DEPs detected in the TH vs. YH, TL vs. YL, and TH vs.TL comparisons belonged to the HIF-1 signaling pathway. Likewise, 3–5% of the DEPs identified in the in TH vs. TL and YH vs. YL comparisons were associated with the vascular endothelial growth factor (VEGF) signaling pathway, which regulates angiogenesis and plays important roles in adaptation to hypoxic conditions. Meanwhile, the MAPK signaling pathway was enriched in 3–7% of the DEPs and pathways in cancer were enriched for 3–5% of the DEPs in the four comparison groups. The MAPK signaling pathway participates in HIF activation, and is involved in various cellular functions, including cell proliferation, differentiation, and migration28, while the proteins in pathways involved in cancer influence sustained angiogenesis, cellular proliferation, genomic damage, and inhibition of differentiation, all of which are closely related to cancer growth and development under hypoxic conditions29. In addition, the insulin signaling pathway, hypertrophic cardiomyopathy (HCM), glycolysis/gluconeogenesis, and dilated cardiomyopathy can be involved in hypoxic adaptation. All of the potential DEPs that were related to hypoxic adaptation in the four groups are shown in Table 2.
Table 2.
Summary of hypoxia-regulated differentially expressed proteins (DEPs).
| Protein | TH/YH | TL/YL | TH/TL | YH/YL | Functional analysis |
|---|---|---|---|---|---|
| MYL7 | 2.05 | 0.79 | 1.60 | 0.62 | muscle contraction, actin cytoskeleton, actin binding, ATPase activity, contractile fiber part |
| PTPMT1 | 1.46 | 0.67 | 1.80 | 0.83 | mitochondrion, inflammatory response, acute inflammatory response |
| PDLIM3 | 1.60 | 1.21 | 1.34 | 1.02 | heart development, actin cytoskeleton, contractile fiber, cardiovascular development |
| A2M | 1.42 | 0.72 | 1.62 | 0.83 | Complement and coagulation cascades, blood microparticle, inflammatory response, regulation of immune effector process, VEGF signaling pathway |
| C6 | 1.29 | 0.78 | 1.28 | 0.77 | inflammatory response, positive regulation of immune system process, acute inflammatory response, positive regulation of immune response, response mediated by circulating, defense response |
| CRYAB | 1.46 | 0.86 | 1.41 | 0.82 | response to hypoxia, mitochondrion, response to reactive oxygen species, oxygen and reactive oxygen species metabolic process, muscle contraction, oxygen species metabolic process |
| PDK4 | 1.10 | 1.02 | 1.45 | 1.34 | mitochondrion, ATP binding, mitochondrion, reactive oxygen species metabolic process, glucose metabolic process |
| ERK2 | 1.20 | 0.98 | 1.15 | 0.94 | HIF-1 signaling pathway, mTOR signaling pathway, TGF-beta signaling pathway, Vascular smooth muscle contraction, VEGF signaling pathway, cardiac muscle contraction |
| ACE | 1.25 | 0.77 | 1.30 | 0.80 | Vasculature development, response to oxygen levels, response to hypoxia, immune system development, regulation of blood pressure, heart contraction, Renin-angiotensin system, blood circulation |
| NAPG | 1.24 | 0.77 | 1.21 | 0.75 | mitochondrion, inflammatory response, actin filament binding, |
| CPS1 | 1.22 | 0.44 | 1.49 | 0.54 | mitochondrion, glucose metabolic process, oxidation of organic compounds |
| HSPE1 | 0.98 | 1.50 | 0.80 | 1.23 | inflammatory response, mitochondrion |
| DECR1 | 0.95 | 1.59 | 0.81 | 1.35 | mitochondrion, 2, 4-dienoyl-CoA reductase (NADPH) activity, NADPH binding, oxidation reduction, iron ion homeostasis, muscle contraction |
| AHSG | 0.72 | 1.20 | 0.78 | 1.30 | inflammatory response, defense response, |
| ENO3 | 0.76 | 1.20 | 0.65 | 1.03 | HIF-1 signaling pathway, glycolysis/gluconeogenesis, glucose metabolic process |
| MRPS26 | 0.63 | 1.40 | 0.68 | 1.52 | mitochondrion, ribosome |
| DCN | 0.50 | 0.80 | 0.80 | 1.29 | TGF-beta signaling pathway |
| COL3A1 | 0.41 | 0.70 | 0.83 | 1.41 | response to radiation, blood vessel development, Platelet activation |
| COL1A2 | 0.40 | 0.80 | 0.83 | 1.65 | blood circulation, regulation of blood pressure, small GTPase mediated signal transduction |
| KRT8 | 0.32 | 0.67 | 0.75 | 1.56 | contractile fiber part |
TH, Tibetan highland pig; TL, Tibetan lowland pig; YH, Yorkshire highland pig; YL, Yorkshire lowland pig.
Integrated analysis of DEGs and DEPs and iTRAQ and RNA-seq data
In total, 18,585 genes and 2,578 proteins were identified in the RNA-seq and iTRAQ analyses, of which 10,001 and 2,170 were annotated, respectively. Using these methods, the expression of 1,402 genes was detected at both the mRNA and protein levels (Fig. S9A). The proteome and transcriptome reflect gene expression at two different levels, and one of the goals of the combined analysis was to achieve data complementation and to obtain more complete information regarding the gene expression profiles of each organism at high and low altitudes. We obtained 473, 297, 394, and 297 DEPs in TH vs. YH, TL vs. YL, TH vs. TL, and YH vs. YL, respectively, of which 378, 220, 319, and 226, respectively, were annotated. A total of 26, 8, 23, and 7 genes were identified as DEGs and DEPs in the TH vs. YH, TL vs. YL, TH vs. TL, and YH vs. YL comparisons, respectively (Fig. S9B). The Pearson correlation coefficients obtained from a log2 function for TH vs. YH, TL vs. YL, TH vs. TL, and YH vs. YL were 0.40, 0.47, 0.60, and 0.42, respectively (Fig. S9C), indicating that only partial correlations were found at the mRNA and protein levels of overall gene expression. In summary, based on these results, we suggest that a substantial degree of post-transcriptional regulatory activity occurs during hypoxic adaptation, which has not been described in previous studies that focused solely on analyses of transcriptional and/or translational regulation.
Discussion
The Tibetan pig, which inhabits high-altitude regions on the Qinghai-Tibet Plateau, has evolved genetic adaptations to these extreme elevations via natural and artificial selection3. Previous studies demonstrated that Tibetan pigs have a distinct suite of phenotypic and physiological characteristics, in regard to pulmonary vascular structures and blood flow30, 31. Sequencing of multiple individuals from various pig breeds revealed that certain genomic regions, including genes involved in the hypoxia response, olfaction, energy metabolism, and drug responses, are under selection in the Tibetan pig10. Moreover, miRNA-seq analysis identified several miRNAs that play regulatory roles in the hypoxic adaptation of these animals4. High-altitude adaptation is not controlled by a single gene, but by multiple evolved genetic adaptations acting in concert32. In the current study, we screened for key genes and proteins related to hypoxic adaptation by comparing Tibetan and Yorkshire pigs raised at different altitudes via RNA-seq and iTRAQ protein sequencing analyses. Subsequent functional enrichment analysis of each DEG and DEP identified via these analyses using GO and KEGG software identified 21 DEGs (A2M, AGT, ANXA2, ATF3, CD44, CCL5, COL3A1, CRYAB, CTGF, DUSP1, EGLN3, FGF1, DPP4, HBB, HBA, NPPA, NPPB, DECR1, TGFB2, PDLIM3, and FOS) and 20 DEPs (MYL7, PTPMT1, PDLIM3, A2M, C6, CRYAB, PDK4, ERK2, ACE, NAPG, CPS1, HSPE1, DECR1, AHSG, ENO3, MRPS26, DCN, COL3A1, COL1A2, and KRT8) that appear to be associated with adaptation to hypoxic conditions in the Tibetan pig. In particular, A2M, COL3A1, CRYAB, DECR1, and PDLIM3 were identified as both DEGs and DEPs.
Under hypoxic conditions, VEGF-mediated induction of MEF2C and its target gene A2M is strongly reduced, as is the expression of HLX and UNC5B. Together, these data suggest that up-regulation of MEF2C/A2M (and HLX/UNC5B) by VEGF is most strongly pronounced under normoxic conditions, and is inversely correlated with hypoxia33. We found that the expression level of A2M in TH and TL hearts was higher than that in YH and YL hearts, respectively, which could explain why Tibetan pigs adapt better to hypoxic conditions than Yorkshire pigs, regardless of altitude. Meanwhile, TH hearts exhibited 4.68-fold higher levels of CRYAB expression than YH and TL hearts. Given that the siRNA mitochondrial pathway is activated in hypoxic cardiomyocytes in the presence of CRYAB, as evidenced by increased production of cytosolic cytochrome c34, our findings indicate that enhanced expression of this protein could comprise another reason for the superior adaption to hypoxic conditions of TH, compared to YH or TL. PDLIM3 (PDZ and LIM domain protein 3, also known as ALP) is necessary to maintain normal myocardial contractility. GO annotations related to this gene include sion. In summary, based nding and structural constituent of muscle”, and cytoskeletal mutations are known causes of genetically based forms of dilated cardiomyopathy. Moreover, the disruption of the gene encoding ALP is associated with right ventricular chamber dilation and dysfunction, directly implicating α-actinin-associated proteins in the onset of cardiomyopathy35. PDLIM3-specific up-regulation in Tibetan pigs might therefore increase cardiovascular permeability, promote vascular endothelial cell proliferation and migration, and increase the regulation of cardiovascular development to prevent the occurrence of cardiomyopathy.
In this study, we identified a set of associated DEGs (CD44, CTGF, DPP4, CRYAB, EGLN3, FGF1, and FOS) with at least one functional enrichment, such as “angiogenesis”, “binding of endothelial cells”, “development of blood vessel”, MAPK signaling, HCM, and pathways in cancer (Table 1). Of these, CTGF (connective tissue growth factor) and FGF1 (fibroblast growth factor 1), which are involved in blood vessel and vascular development, were expressed at significantly higher levels in TH than in YH, suggesting an important role for the cardiovascular system in hypoxia adaptation. In particular, CTCF restricts upstream enhancers from activating VEGF, thereby inhibiting the induction of VEGF and angiogenesis36. Hypoxia is the principal driver of induction of VEGF during both physiological and pathological angiogenesis37. FGF1 is recognized as an angiogenesis factor in vivo, and angiogenesis is known to be induced by hypoxia38. Our findings therefore indicate that Tibetan pigs must increase CTGF and FGF1 expression and modify their cardiovascular response to hypoxia by increasing blood flow and cardiac pumping.
The expression levels of CD44 and PDLIM3 were also significantly higher in the heart tissues of TH than in those of YH, TL, or YL. CD44 (CD44 molecule) transmembrane glycoproteins are cell adhesion molecules associated with aggressiveness and metastasis37–40, and cycling hypoxia has been shown to increase the population of CD44+/CD24− cells in a metastatic breast cancer cell line41. Therefore, it seems probable that CD44 expression is regulated by hypoxia. The functional effects of the up-regulation of CD44 and its variant isoforms under hypoxic conditions, particularly in the context of hyaluronan levels, should be considered, as cell signaling events that promote anchorage-independent tumor cell growth, survival, migration, and metastasis occur through the binding of hyaluronan with CD4440, 42, 43. In addition, expression of FOS (also known as c-FOS), which was previously reported to be associated with the hypoxia response44, 45, was 17.3-fold higher in TL than in YL. Together, these data suggest that characterization of DEGs that differ between the two pig breeds could provide clues to understanding the distinct hypoxic adaptations of these animals.
According to the GO and pathway analyses, the DEGs CRYAB (crystallin, alpha B), DPP4 (dipeptidyl-peptidase 4), and EGLN3 (egl nine homolog 3) comprise three functional candidate genes that were related to “response to hypoxia”. Indeed, EGLN1 and EGLN3 are strongly induced by hypoxia in most cell types46. his effect is likely important for cellular adaptation to hypoxic conditions, and is responsible for the increased oxygen-mediated HIF-1a degradation observed after long periods of hypoxia47. Indeed, the induction of EGLN3 mRNA expression by hypoxia is particularly significant when compared to that of other hypoxia-responsive genes44, 46, 48, 49. DPP4 is a novel marker of HIF-1 activity in tumors50. In our study, the DPP4 expression level in TH hearts was 0.22-fold higher than that in YH hearts, and 0.31-fold higher than in TL or YL hearts.
In the presence of low atmospheric oxygen, the oxygen levels in highland Yorkshire pigs introduced from the mainland were correspondingly low, and the difference in oxygen levels between capillary blood and mitochondria was small, resulting in hypoxia. A series of genes, such as FLT-1 [fms-related tyrosine kinase 1 (vascular endothelial growth factor/vascular permeability factor receptor)], which was categorized with the GO term “vascular regulation” and HIF-1 signaling pathway in the YH group and is considered the strongest of the VEGF receptors, as well as the HIF-1 signaling pathway, were up-regulated in Yorkshire pigs in responses to hypoxia in the YH group. Hypoxic exercise can result in increased production of VEGF and Flt-1 in muscle tissue; following VEGF protein production, Flt-1 expression at the vascular endothelial cell membrane can be regulated via autocrine or paracrine responses and participate in muscle tissue angiogenesis51. In this study, expression of the Flt-1 gene was the highest in YH and the lowest in TH, possibly because compensatory responses increase vascular permeability in response to a hypoxic environment.
The specific KEGG enrichments in the Yorkshire pig were primarily concentrated in energy metabolism pathways, such as the glycolysis pathway. This was not surprising as, under hypoxic conditions, myocardial contraction requires increased adenosine triphosphate (ATP) consumption. However, while most differentiated cells employ mitochondrial oxidative phosphorylation to generate ATP to maintain cell processes under normoxic conditions, cells exposed to hypoxia typically produce ATP via glycolysis, which produces lower levels of this energy source than aerobic oxidation. As such, under hypoxic conditions, animals must up-regulate glycolysis to meet the consumption requirements of myocardial cells. In particular, the primary DEGs associated with energy metabolism in Yorkshire pigs were ITGA5, PCK1, and PCK2. Glycolysis produces phosphoenolpyruvate (PEP), and the overexpression of PCK1 can lead to increased PEP production and gluconeogenesis52.
In this study, many genes were found to exhibit consistency in regard to changes in transcript and protein levels; however, there were several cases in which inconsistent results were obtained by RNA-seq and iTRAQ analysis, suggesting that post-transcriptional regulation plays an important role in the adaptive response to hypoxia in Tibetan pigs. Both transcriptomic and proteomic data are important for deciphering the molecular processes involved in this process. Previous studies have shown that the abundance of mRNA transcripts is does not completely correlate with expression level of certain genes53. Indeed, we also found only partial correlations between the mRNA and protein expression levels of certain genes54. Thus, transcriptomic analyses do not fully represent protein expression55. As such, analysis of transcriptomic or proteomic data alone is insufficient for accurate characterization of functional mechanisms; only by combining these approaches can we obtain a more comprehensive understanding of the biological functions of organisms.
In conclusion, we identified important hypoxia-adapted genes and proteins in Tibetan pigs, and by combining RNA-seq and iTRAQ data obtained from pig heart tissues, we elucidated the regulatory relationship between DEGs and DEPs. A combination of transcriptomic and proteomic data revealed several key candidate regulators (A2M, PDLIM3, CRYAB, and ACE) and pathways (HIF-1 signaling pathway, cardiovascular development, VEGF signaling pathway, and pathways in cancer) that might play high-priority roles in the hypoxic adaptation of Tibetan pigs. These results provide new insights into the molecular mechanisms involved in hypoxia-adaptation regulatory networks, and a greater understanding of human hypoxic diseases. However, further studies are required to confirm the DEGs and DEPs involved in regulating hypoxia identified in this work.
Materials and Methods
Sample preparation and extraction
The experimental design comprised four treatment (comparison) groups (n = 8 per group): Tibetan and Yorkshire pigs raised in the highlands [Linzhi, Tibet, 3000 m above mean sea level (AMSL)] and lowlands (Beijing, China, 100 m AMSL). The animals were slaughtered at the age of six months, and heart tissue samples were collected, immediately frozen in liquid nitrogen, and stored at −80 °C (Table S1). The procedures for animal care were approved by the Animal Welfare Committee of the State Key Laboratory for Agro-biotechnology of the China Agricultural University (Approval number XK257), and all experiments were conducted in accordance with approved relevant guidelines and regulations.
RNA isolation, library preparation, and sequencing
Total RNA was isolated using Trizol® reagent (Invitrogen, Waltham, MA, USA)55. The integrity and concentration and purity of each sample was evaluated via 1% agarose gel electrophoresis and using a NanoDrop™ 2000 Biophotometer (Thermo Fisher Scientific, Waltham, MA, USA), respectively. Samples were then reverse transcribed into cDNA using Superscript II reverse transcriptase (Invitrogen) and random hexamer primers. For RNA-seq analyses, RNA pools were created using equal quantities of RNA from four individuals; two biological replicates were included in each group. RNA-seq libraries were constructed according to the manuals provided by Illumina, Inc. (San Diego, CA, USA), and were sequenced using the HiSeq 2000 platform to generate 100-bp paired-end reads. All RNA sequencing data are deposited in the Gene Expression Omnibus under accession number GSE92981.
Mapping and annotation of sequencing reads
Raw RNA-seq reads were arranged using CLC Genomics Workbench 4.8 software (CLC Bio, Aarhus, Denmark). All reads for which the quality of more than half of the bases was less than 10, as well as all reads that contained more than two Ns or were smaller than 20 bp in length, were eliminated from subsequent analyses. After removal of the adapters, the remaining clean reads were aligned to the whole pig genome (Sscrofa10.2.72) (ftp://ftp.ensembl.org/pub/release-72/fasta/sus_scrofa/dna/Sus_scrofa.Sscrofa10.2.72.dna.toplevel.fa.gz) using TopHat (version 2.0.9) software56; two mismatches were allowed per 100-bp read for each alignment. Finally, BAM files generated using SAMtools57 were used for subsequent analysis. Quality control and read statistics were determined using FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
Quantification and differential gene analysis for RNA-seq
FPKM values obtained using Cufflink software (version 2.1.1) were used as values for normalized gene expression58. Differential expression analyses of four comparison groups (TH vs. YH, TH vs.TL, YH vs. YL, and TL vs. YL) were performed using the DESeq R package (1.10.1)59. The resulting P-values were adjusted using the Benjamini-Hochberg method. Results were expressed as the fold change (FC) of the average expression of case groups to that of the respective control group. Among the four comparison groups, DEGs were identified as those genes for which log2 (FC) > 1 and P < 0.01.
Protein isolation, enzymolysis, and iTRAQ labeling
Heart tissues were ground into powder in liquid nitrogen using lysis buffer (Roche). The resulting samples were then ultrasonically disrupted for extraction of total protein. After centrifugation at 10,000 × g for 30 min at 4ly, supernatants were collected, and protein concentrations were determined using an enhanced BCA (bicinchoninic acid) Protein Assay Kit (P0010; Beyotime Biotechnologies, Ltd., Beijing, China), according to the manufacturer’s instructions. The protein samples (200 μg) were mixed with dl-dithiothreitol and alkylated with iodoacetamide, and then treated with trypsin overnight at a trypsin-to-protein ratio of 1:100.
Protein peptides (15 μg) from each group were labeled using an 8plex iTRAQ reagent multiplex kit (SCIEX, Framingham, MA, USA). The samples were labeled as 113 (TH1), 114 (TH2), 115 (YH1), 116 (YH2), 117 (TL1), 118 (TL2), 119 (YL1), and 121 (YL2). The labeled samples were pooled and further fractionated offline using an ÄKTA Purifier 100 (GE Healthcare) with a strong cation exchange column (PolySULFOETHYL A™; PolyLC Inc., Columbia, MD, USA). The retained peptides were eluted with Buffer A [10 mM KH2PO4 in 25% ACN (acetonitrile), pH 3.0] and Buffer B (10 mM KH2PO4 and 500 mM KCl in 25% ACN, pH 3.0) with a flow rate of 1.0 mL/min.
LC-MS/MS analysis
Eluted fractions were lyophilized using a centrifugal speed vacuum concentrator (CentriVap® Complete Vacuum Concentrator; Labconco, Kansas City, MO, USA) and dissolved in 0.1% formic acid. Equivalent amounts of peptides from each fraction were mixed and then subjected to reversed-phase nanoflow LC-MS/MS analysis using a high-performance liquid chromatography system (EASY-nLC™, Thermo Fisher Scientific) connected to a hybrid quadrupole/time of-flight mass spectrometer equipped with a nano-electrospray ion source. The peptides were separated on a C18 analytical reverse-phase column with mixtures of Solution A (0.1% formic acid in water) and Solution B (0.1% formic acid in ACN). A full MS scan was conducted using a Q Exactive™ mass spectrometer (Thermo Fisher Scientific).
Database search and protein identification and quantification
For peptide identification and quantification, MS/MS data were searched against the “Sus_scrofa_35257_20151120_uniprot.fasta” file using Mascot 2.2 and Proteome Discoverer™ 1.4 software (Thermo Fisher Scientific). A unique protein with at least two unique peptides that had an FDR <0.0160 was used for data analysis. Protein quantification was based on the total intensity of the assigned peptides. The average of eight labeled sample mixes was used as a reference, and was based on the weighted average of the intensity of report ions in each peptide identified. The final protein ratios were normalized to the median average protein content of the 8plex samples. FC > 1.2 or FC < 0.83 was set as the threshold for identifying differentially expressed proteins.
GO and KEGG enrichment analysis of DEGs and DEPs
DEGs and DEPs were classified by GO and KEGG using DAVID online software (https://david.ncifcrf.gov/)61. For these analyses, an official gene symbol for each DEG or DEP was uploaded, and the species with the maximum number of annotations was used. The GO terms used were BP, CC, and MF, and KEGG pathways with corrected P-values < 0.05 were considered significantly enriched. In addition, IPA software (http://www.ingenuity.com/; Ingenuity Systems, Redwood City, CA, USA) was used to compare the DEGs among the four treatment groups. Accession numbers for these genes were imported into IPA, and the “Core Analysis” function was used to analyze the genes in the context of networks, biological functions, and canonical pathways. Detailed information concerning IPA analyses was published previously60, 62, 63. GO annotation and KEGG pathway analysis of the DEPs was also performed using KOBAS 2.0 (http://kobas.cbi.pku.edu.cn/)64 online software.
Verification of RNA-seq and iTRAQ data
RT-qPCR primers were designed to span the exon-exon boundaries of eight genes selected by RNA-seq; the characteristics of the primers are listed in Table S2. RT-PCR analysis was performed using a SYBR® Green I PCR Master Mix Kit (FP204; Tiangen Biotech Co. Ltd., Beijing, China) on a CFX96™ Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA), according to the manufacturer’s instructions.
The results for TH vs. YH obtained by iTRAQ analysis were validated via the label-free technique, as described previously. Label-free quantification is a mass spectrometry method used to determine the relative amounts of proteins of interest in two or more biological samples. Unlike other methods of protein quantification, label-free quantification does not use a stable isotope-containing compound to chemically bind to, and thus, label the protein63, 65, 66.
Electronic supplementary material
Supplementary Figure S1-S9 and Table S1-S3
Acknowledgements
This study was supported by the National Major Special Project on New Varieties Cultivation for Transgenic Organisms (2016ZX08009-003-006), the National Natural Science Foundation of China (31560615), the National High Technology Research and Development Program (2013AA102503-03), and the Program for Changjiang Scholar and Innovation Research Team in University (IRT1191).
Author Contributions
H.Z. conceived and designed the experiments; B.Z. performed the experiments; B.Z., Y.C., P.S., and H.Z. analyzed the data; B.Z., P.S., Z.W., J.M., and L.W. contributed reagents/materials/analysis tools; B.Z. and H.Z. wrote the paper.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-03976-3
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Chaillou T, et al. Effect of hypoxia exposure on the phenotypic adaptation in remodelling skeletal muscle submitted to functional overload. ACTA PHYSIOL. 2013;209:272. doi: 10.1111/apha.12110. [DOI] [PubMed] [Google Scholar]
- 2.Kon M, et al. Effects of systemic hypoxia on human muscular adaptations to resistance exercise training. Physiological reports. 2014;2:e12033. doi: 10.14814/phy2.12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang S, et al. The local origin of the Tibetan pig and additional insights into the origin of Asian pigs. PLOS ONE. 2011;6:e28215. doi: 10.1371/journal.pone.0028215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang B, et al. A Comprehensive MicroRNA Expression Profile Related to Hypoxia Adaptation in the Tibetan Pig. PLOS ONE. 2015;10:e143260. doi: 10.1371/journal.pone.0143260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jia C, et al. Gene Co-Expression Network Analysis Unraveling Transcriptional Regulation of High-Altitude Adaptation of Tibetan Pig. PLOS ONE. 2016;11:e168161. doi: 10.1371/journal.pone.0168161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gou W, et al. Expression pattern of heme oxygenase 1 gene and hypoxic adaptation in chicken embryos. Comparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology. 2014;174:23. doi: 10.1016/j.cbpb.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Simonson TS, et al. Genetic evidence for high-altitude adaptation in Tibet. SCIENCE. 2010;329:72. doi: 10.1126/science.1189406. [DOI] [PubMed] [Google Scholar]
- 8.Yi X, et al. Sequencing of 50 human exomes reveals adaptation to high altitude. SCIENCE. 2010;329:75. doi: 10.1126/science.1190371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bigham A, et al. Identifying signatures of natural selection in Tibetan and Andean populations using dense genome scan data. PLOS GENET. 2010;6:e1001116. doi: 10.1371/journal.pgen.1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li M, et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. NAT GENET. 2013;45:1431. doi: 10.1038/ng.2811. [DOI] [PubMed] [Google Scholar]
- 11.Hakimov HA, et al. Application of iTRAQ to catalogue the skeletal muscle proteome in pigs and assessment of effects of gender and diet dephytinization. PROTEOMICS. 2009;9:4000. doi: 10.1002/pmic.200900049. [DOI] [PubMed] [Google Scholar]
- 12.Ramayo-Caldas Y, et al. Liver transcriptome profile in pigs with extreme phenotypes of intramuscular fatty acid composition. BMC GENOMICS. 2012;13:1. doi: 10.1186/1471-2164-13-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang T, et al. Deep sequencing of the transcriptome reveals inflammatory features of porcine visceral adipose tissue. INT J BIOL SCI. 2013;9:550. doi: 10.7150/ijbs.6257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang C, et al. A group of novel serum diagnostic biomarkers for multidrug-resistant tuberculosis by iTRAQ-2D LC-MS/MS and Solexa sequencing. INT J BIOL SCI. 2016;12:246. doi: 10.7150/ijbs.13805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ream M, Ray AM, Chandra R, Chikaraishi DM. Early fetal hypoxia leads to growth restriction and myocardial thinning. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 2008;295:R583. doi: 10.1152/ajpregu.00771.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tintu A, et al. Hypoxia induces dilated cardiomyopathy in the chick embryo: mechanism, intervention, and long-term consequences. PLOS ONE. 2009;4:e5155. doi: 10.1371/journal.pone.0005155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ai H, et al. Population history and genomic signatures for high-altitude adaptation in Tibetan pigs. BMC GENOMICS. 2014;15:1. doi: 10.1186/1471-2164-15-834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. NAT REV CANCER. 2008;8:851. doi: 10.1038/nrc2501. [DOI] [PubMed] [Google Scholar]
- 19.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. CELL. 2012;149:274. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ali SS, et al. Hypoxia-adaptation involves mitochondrial metabolic depression and decreased ROS leakage. PLOS ONE. 2012;7:e36801. doi: 10.1371/journal.pone.0036801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tyberg JV, Yeatman LA, Parmley WW, Urschel CW, Sonnenblick EH. Effects of hypoxia on mechanics of cardiac contraction. American Journal of Physiology–Legacy Content. 1970;218:1780. doi: 10.1152/ajplegacy.1970.218.6.1780. [DOI] [PubMed] [Google Scholar]
- 22.Arjamaa O, Nikinmaa M. Hypoxia regulates the natriuretic peptide system. Int J Physiol Pathophysiol Pharmacol. 2011;3:191. [PMC free article] [PubMed] [Google Scholar]
- 23.Hong L, et al. Atrial natriuretic peptide prevents the mitochondrial permeability transition pore opening by inactivating glycogen synthase kinase 3β via PKG and PI3K in cardiac H9c2 cells. EUR J PHARMACOL. 2012;695:13. doi: 10.1016/j.ejphar.2012.07.053. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y. Atrial natriuretic peptide in hypoxia. PEPTIDES. 2005;26:1068. doi: 10.1016/j.peptides.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 25.Patel VJ, et al. A comparison of labeling and label-free mass spectrometry-based proteomics approaches. J PROTEOME RES. 2009;8:3752. doi: 10.1021/pr900080y. [DOI] [PubMed] [Google Scholar]
- 26.Wang H, Alvarez S, Hicks LM. Comprehensive comparison of iTRAQ and label-free LC-based quantitative proteomics approaches using two Chlamydomonas reinhardtii strains of interest for biofuels engineering. J PROTEOME RES. 2011;11:487. doi: 10.1021/pr2008225. [DOI] [PubMed] [Google Scholar]
- 27.Trinh, H. V. et al. iTRAQ-based and label-free proteomics approaches for studies of human adenovirus infections. International journal of proteomics2013 (2013). [DOI] [PMC free article] [PubMed]
- 28.Sang N, et al. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J BIOL CHEM. 2003;278:14013. doi: 10.1074/jbc.M209702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shen C, et al. Genetic and functional studies implicate HIF1α as a 14q kidney cancer suppressor gene. CANCER DISCOV. 2011;1:222. doi: 10.1158/2159-8290.CD-11-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan PW, et al. Identification of differentially expressed genes in the longissimus dorsi muscle tissue between Duroc and Erhualian pigs by mRNA differential display. ASIAN AUSTRAL J ANIM. 2003;16:1066. doi: 10.5713/ajas.2003.1066. [DOI] [Google Scholar]
- 31.Monge C, Leon-Velarde F. Physiological adaptation to high altitude: oxygen transport in mammals and birds. PHYSIOL REV. 1991;71:1135. doi: 10.1152/physrev.1991.71.4.1135. [DOI] [PubMed] [Google Scholar]
- 32.Simonson TS, McClain DA, Jorde LB, Prchal JT. Genetic determinants of Tibetan high-altitude adaptation. HUM GENET. 2012;131:527. doi: 10.1007/s00439-011-1109-3. [DOI] [PubMed] [Google Scholar]
- 33.Sturtzel, C., Testori, J., Schweighofer, B., Bilban, M. & Hofer, E. The transcription factor MEF2C negatively controls angiogenic sprouting of endothelial cells depending on oxygen. PLOS ONE9, e101521 (2014). [DOI] [PMC free article] [PubMed]
- 34.Mitra A, et al. Role of α-crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. CELL DEATH DIS. 2013;4:e582. doi: 10.1038/cddis.2013.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pashmforoush M, et al. Adult mice deficient in actinin–associated LIM-domain protein reveal a developmental pathway for right ventricular cardiomyopathy. NAT MED. 2001;7:591. doi: 10.1038/87920. [DOI] [PubMed] [Google Scholar]
- 36.Tang M, et al. Restraint of angiogenesis by zinc finger transcription factor CTCF-dependent chromatin insulation. Proceedings of the National Academy of Sciences. 2011;108:15231. doi: 10.1073/pnas.1104662108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirota K, Semenza GL. Regulation of angiogenesis by hypoxia-inducible factor 1. Critical reviews in oncology/hematology. 2006;59:15. doi: 10.1016/j.critrevonc.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 38.Carreira CM, Landriscina M, Bellum S, Prudovsky I, Maciag T. The comparative release of FGF1 by hypoxia and temperature stress. GROWTH FACTORS. 2001;18:277. doi: 10.3109/08977190109029116. [DOI] [PubMed] [Google Scholar]
- 39.Peng S, Su C, Kuo C, Shaw C, Wang H. CD44 crosslinking-mediated matrix metalloproteinase-9 relocation in breast tumor cells leads to enhanced metastasis. INT J ONCOL. 2007;31:1119. [PubMed] [Google Scholar]
- 40.Götte M, Yip GW. Heparanase, hyaluronan, and CD44 in cancers: a breast carcinoma perspective. CANCER RES. 2006;66:10233. doi: 10.1158/0008-5472.CAN-06-1464. [DOI] [PubMed] [Google Scholar]
- 41.Louie E, et al. Identification of a stem-like cell population by exposing metastatic breast cancer cell lines to repetitive cycles of hypoxia and reoxygenation. BREAST CANCER RES. 2010;12:1. doi: 10.1186/bcr2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bourguignon LY, Wong G, Earle C, Krueger K, Spevak CC. Hyaluronan-CD44 interaction promotes c-Src-mediated twist signaling, microRNA-10b expression, and RhoA/RhoC up-regulation, leading to Rho-kinase-associated cytoskeleton activation and breast tumor cell invasion. J BIOL CHEM. 2010;285:36721. doi: 10.1074/jbc.M110.162305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jothy S. CD44 and its partners in metastasis. CLIN EXP METASTAS. 2003;20:195. doi: 10.1023/A:1022931016285. [DOI] [PubMed] [Google Scholar]
- 44.Rybnikova E, Glushchenko T, Tyulkova E, Baranova K, Samoilov M. Mild hypobaric hypoxia preconditioning up-regulates expression of transcription factors c-Fos and NGFI-A in rat neocortex and hippocampus. NEUROSCI RES. 2009;65:360. doi: 10.1016/j.neures.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 45.Yuen RK, Chen B, Blair JD, Robinson WP, Nelson DM. Hypoxia alters the epigenetic profile in cultured human placental trophoblasts. EPIGENETICS-US. 2013;8:192. doi: 10.4161/epi.23400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Metzen E, et al. Intracellular localisation of human HIF-1α hydroxylases: implications for oxygen sensing. J CELL SCI. 2003;116:1319. doi: 10.1242/jcs.00318. [DOI] [PubMed] [Google Scholar]
- 47.Berra E, Richard DE, Gothié E, Pouysségur J. HIF‐1‐dependent transcriptional activity is required for oxygen‐mediated HIF‐1α degradation. FEBS LETT. 2001;491:85. doi: 10.1016/S0014-5793(01)02159-7. [DOI] [PubMed] [Google Scholar]
- 48.Del Peso L, et al. The von Hippel Lindau/hypoxia-inducible factor (HIF) pathway regulates the transcription of the HIF-proline hydroxylase genes in response to low oxygen. J BIOL CHEM. 2003;278:48690. doi: 10.1074/jbc.M308862200. [DOI] [PubMed] [Google Scholar]
- 49.Naranjo-Suárez S, et al. Down-regulation of hypoxia-inducible factor-2 in PC12 cells by nerve growth factor stimulation. J BIOL CHEM. 2003;278:31895. doi: 10.1074/jbc.M304079200. [DOI] [PubMed] [Google Scholar]
- 50.Dang DT, et al. Hypoxia-inducible factor-1 target genes as indicators of tumor vessel response to vascular endothelial growth factor inhibition. CANCER RES. 2008;68:1872. doi: 10.1158/0008-5472.CAN-07-1589. [DOI] [PubMed] [Google Scholar]
- 51.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. NAT REV MOL CELL BIO. 2006;7:359. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 52.Kleffel S, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. CELL. 2015;162:1242. doi: 10.1016/j.cell.2015.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang B, et al. Proteogenomic characterization of human colon and rectal cancer. NATURE. 2014;513:382. doi: 10.1038/nature13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. NAT REV GENET. 2012;13:227. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chomczynski P, Sacchi N. The single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction: twenty-something years on. NAT PROTOC. 2006;1:581. doi: 10.1038/nprot.2006.83. [DOI] [PubMed] [Google Scholar]
- 56.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. BIOINFORMATICS. 2009;25:1105. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, et al. The sequence alignment/map format and SAMtools. BIOINFORMATICS. 2009;25:2078. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. NAT BIOTECHNOL. 2010;28:511. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anders S, Huber W. Differential expression analysis for sequence count data. GENOME BIOL. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sandberg A, et al. Tumor proteomics by multivariate analysis on individual pathway data for characterization of vulvar cancer phenotypes. MOL CELL PROTEOMICS. 2012;11:M112. doi: 10.1074/mcp.M112.016998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. NAT PROTOC. 2009;4:44. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 62.Hou Y, et al. Genomic regions showing copy number variations associate with resistance or susceptibility to gastrointestinal nematodes in Angus cattle. FUNCT INTEGR GENOMIC. 2012;12:81. doi: 10.1007/s10142-011-0252-1. [DOI] [PubMed] [Google Scholar]
- 63.Hou Y, et al. Genomic characteristics of cattle copy number variations. BMC GENOMICS. 2011;12:1. doi: 10.1186/1471-2164-12-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xie C, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. NUCLEIC ACIDS RES. 2011;39:W316. doi: 10.1093/nar/gkr483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bantscheff M, Lemeer S, Savitski MM, Kuster B. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. ANAL BIOANAL CHEM. 2012;404:939. doi: 10.1007/s00216-012-6203-4. [DOI] [PubMed] [Google Scholar]
- 66.Asara JM, Christofk HR, Freimark LM, Cantley LC. A label‐free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. PROTEOMICS. 2008;8:994. doi: 10.1002/pmic.200700426. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1-S9 and Table S1-S3