

Abstract
Intracellular protein-protein interactions (PPIs) are challenging targets for conventional drug modalities, because small molecules generally do not bind to their large, flat binding sites with high affinity, whereas monoclonal antibodies cannot cross the cell membrane to reach the targets. Cyclic peptides in the 700–2000 molecular-weight range have the sufficient size and a balanced conformational flexibility/rigidity for binding to flat PPI interfaces with antibody-like affinity and specificity. Several powerful cyclic peptide library technologies were developed over the past decade to rapidly discover potent, specific cyclic peptide ligands against proteins of interest including those involved in PPIs. Methods are also being developed to enhance the membrane permeability of cyclic peptides through both passive diffusion and active transport mechanisms. Integration of the permeability-enhancing elements into cyclic peptide design has led to an increasing number of cell-permeable and biologically active cyclic peptides against intracellular PPIs. In this account, we review the recent developments in the design and synthesis of cell-permeable cyclic peptides.
Graphical abstract

Introduction
Protein-protein interactions (PPIs) serve as the foundation of essentially all cellular processes by enabling and regulating the function of individual proteins. Molecules that are capable of specifically modulating PPIs are in high demand as molecular probes and therapeutic agents. Unfortunately, PPIs, especially those that occur intracellularly, have proven challenging targets for conventional drug modalities, namely small molecules and biologics. While small molecules excel in targeting proteins containing deep binding pockets, PPIs typically involve large, flat binding sites which are devoid of any major binding pocket. Biologics such as monoclonal antibodies are effective for recognizing PPI interfaces, but cannot cross the cell membrane to reach the intracellular PPIs. Clearly, a general solution to the problem of inhibiting intracellular PPIs requires exploring chemical spaces beyond the conventional drug modalities. Therefore, in recent years, many investigators have turned their attention to macrocycles, particularly cyclic peptides, as potential PPI inhibitors [1–3]. With sizes that are typically 3–5 times larger than conventional small-molecule drugs and a balanced conformational flexibility/rigidity, macrocycles in the 700–2000 molecular-weight range are capable of binding to the flat PPI interfaces with antibody-like affinity and specificity. At the meantime, macrocycles retain many of the drug-like properties of small molecules such as metabolic stability [4,5]. Cyclic peptides are also synthetically accessible and generally less toxic than small molecules. Nevertheless, developing cyclic peptide inhibitors against intracellular PPI targets still faces two significant challenges: 1) how to engineer a macrocyclic structure to engage a target of interest with high affinity and specificity, often in the absence of any structural information; and 2) how to achieve cell permeability for the macrocycle. In this perspective, we will provide an overview of the recent developments toward overcoming both challenges, followed by selected examples of cell-permeable macrocyclic PPI inhibitors.
Generating Cyclic Peptides as PPI Inhibitors
Over the past decade, a variety of structure-based design and combinatorial library approaches have been developed to discover cyclic peptide inhibitors against PPIs (Figure 1). Since these methodologies have been the subject of several recent, exhaustive reviews [6–10], we will provide here only a brief overview of them. The proper method of choice depends on the nature of the PPI in question and the expertise available within a research laboratory. If the PPI is predominantly mediated by a contiguous structural epitope (e.g., an α-helix, a β-turn, or a peptide loop) on one of the binding partners and structural information is available, rational design has been a productive approach for generating potent cyclic peptide PPI inhibitors. One special type of cyclic peptides that have been extensively explored are “stapled peptides”, which are stabilized α-helices by covalently crosslinking their side chains at i and i+4 (or i+7) positions [11–15]. Compared to their linear counterparts, stapled peptides often have greatly improved binding affinity to the intended target, better metabolic stability, and in some cases increased cell permeability. Another common practice is to mimic the β-turn motifs found at PPI interfaces with cyclic peptides. For example, recognizing that the Grb2 SH2 domain binds to a phosphotyrosine-containing peptide ligand in a β-turn conformation, Burke and co-workers rationally designed highly potent cyclic peptide inhibitors against the Grb2 SH2 domain (KD = 75 pM; Figure 2) [16]. Zhou et al. designed a cyclic peptidomimetic inhibitor against menin-mixed lineage leukemia-1 (MLL1), starting from a conserved menin-binding motif (RWRFPARP) [17]. Further SAR studies yielded a highly-potent, cell-permeable cyclic inhibitor with a Ki of 4.7 nM against the MLL1-menin interaction (Figure 2). More recently, the stapling strategy has been applied to generate structurally constrained hairpin loops as PPI inhibitors [18]. These examples demonstrate that naturally occurring epitope sequences at PPI interfaces, when available, can serve as good starting points for rational design of potent cyclic peptide PPI inhibitors.
Figure 1.
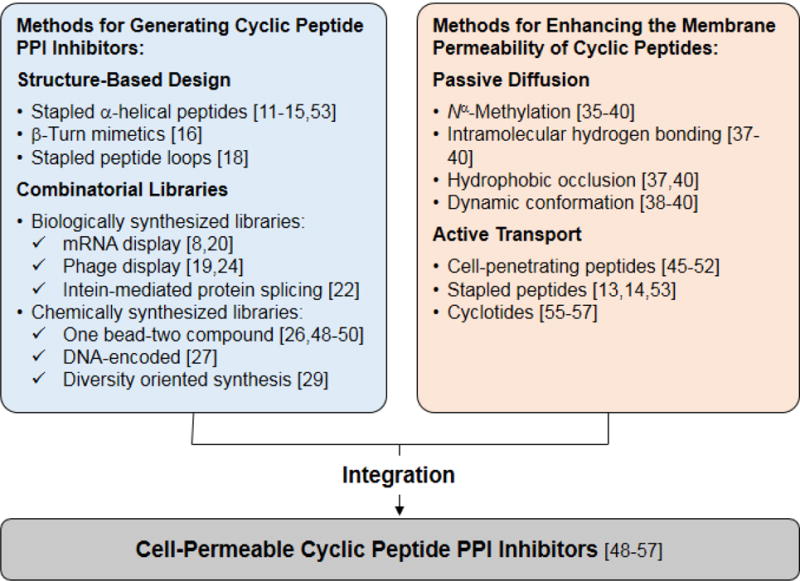
Summary of methods that have been developed to generate cyclic peptide ligands for inhibition of PPI targets and to enhance their membrane permeability.
Figure 2.
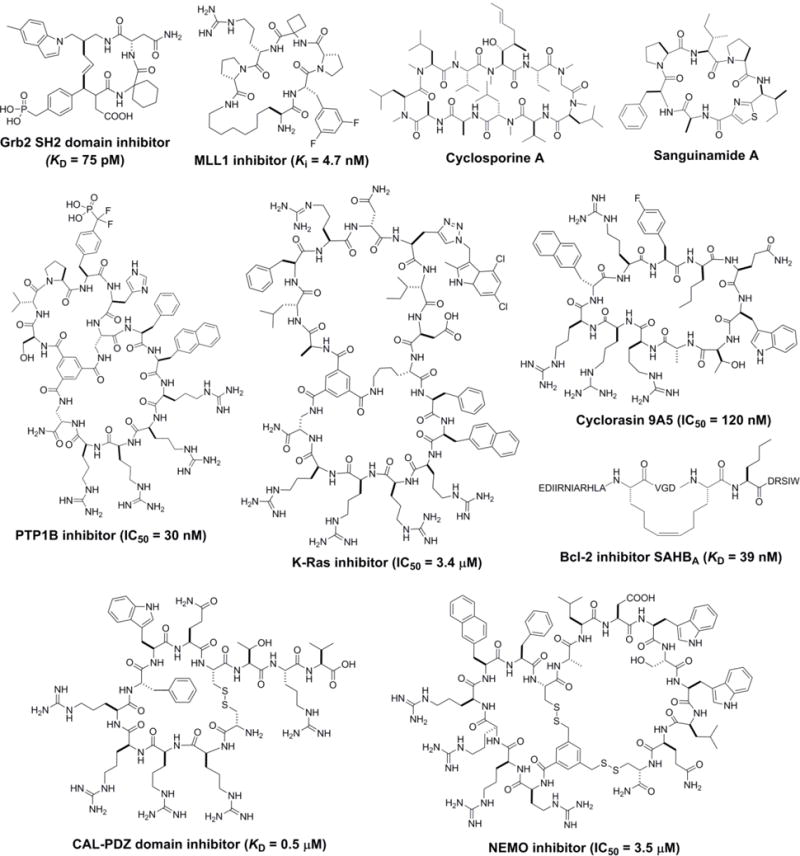
Examples of cell-permeable and biologically active cyclic peptides which were derived from natural sources or rational design and/or combinatorial library screening.
When structural information is not available or the PPI is mediated by multiple epitopes that are distant in the protein sequence (which is true for most PPIs), rational design of cyclic peptide ligands is very challenging. A more productive approach involves synthesizing and screening large combinatorial libraries of mono- and bicyclic peptides. Several complementary platform technologies have been developed over the past decade to synthesize cyclic peptide libraries of enormous structural diversity (up to 1014 different compounds), either biologically or chemically (Figure 1). The biological methods include phage display [7,19], mRNA display [8,9,20], and peptide splicing mediated by split inteins [21,22], all of which involve ribosomal peptide synthesis. In the case of phage and mRNA display libraries, the peptides are physically linked to their encoding DNA/RNA, which can be readily sequenced to reveal the identity of a peptide ligand. The displayed peptides are converted into mono- or bicyclic peptides posttranslationally by reacting the N-terminal amine and/or side chain nucleophiles (usually Cys and Lys) with a bifunctional (e.g., disuccinimidyl glutarate) [23] or trifunctional crosslinking agent [e.g., tris(bromomethyl)benzene] [24]. In intein-mediated libraries, peptides are produced within living cells and N-to-C cyclized through a protein splicing mechanism, with each cell harbouring a different macrocycle and the corresponding encoding DNA [21]. The most widely practiced chemical methods include one bead-two compound (OBTC) libraries [25,26] and macrocycle libraries derived from DNA-templated synthesis (DTS) [27,28] or diversity-oriented synthesis [29,30]. In OBTC libraries, each library bead displays a unique cyclic or bicyclic peptide on its surface and contains a linear peptide of the same sequence in the interior as an encoding tag, which can be sequenced by Edman degradation or mass spectrometric methods. In DTS libraries, each macrocycle is covalently linked to the DNA template that serves as the “blueprint” for its construction, analogous to that of mRNA display. The advantages of biological libraries include greater diversity (up to 1014 different compounds), compatibility with library amplification (via the polymerase chain reaction) and iterative selection, and straightforward hit identification (by sequencing the encoding DNA/mRNA). However, ribosomal synthesis limits the library building blocks to the 20 proteinogenic amino acids and certain modified amino acids (e.g., Nα-methylated amino acids) [31]. In contrast, both natural and unnatural building blocks (e.g. D-amino acids) can be readily incorporated into chemically synthesized libraries to generate more structurally diverse cyclic peptides. The rational design and combinatorial library approached have also been combined to generate stapled peptide inhibitors of exceptionally high potencies [32,33].
Enhancing the Membrane Permeability of Cyclic Peptides
Cyclic peptides violate Lipinski’s Rule of 5 (molecular weight ≤500, ≤5 hydrogen bond donors, ≤10 hydrogen bond acceptors, and cLog P≤ 5) [34] and are generally impermeable to the cell membrane. However, some naturally-occurring cyclic peptides (e.g., cyclosporine A, Figure 2) possess the unusual ability of crossing the cell membrane by passive diffusion and are orally bioavailable. Investigating the physiochemical and molecular properties of these natural products, with the ultimate goal of designing cell-permeable and orally bioavailable synthetic cyclic peptides, has been an active area of research [4,5,35–39]. In silico modelling of naturally occurring cell-permeable cyclic peptides identified some shared structural features, such as Nα-methylation of the peptide backbone and formation of extensive intramolecular hydrogen bonds [39]. Using model systems, researchers have demonstrated that Nα-methylation, formation of intramolecular hydrogen bonds, and strategic placement of sterically occluding hydrophobic groups are effective for improving the membrane permeability of synthetic cyclic peptides [4,35–38]. These methods decrease the solvent-accessible polar surface area, thus reducing the desolvation energy associated with membrane crossing. Using Sanguinamide A (Figure 2) as a model system, Bockus et al. noted the importance of conformational flexibility when designing cyclic peptides with favourable balance between solubility and permeability [40]. Kihlberg and co-workers performed NMR- and computer-based conformational analysis of orally administered macrocycle drugs (e.g. cyclosporine A) and clinical candidates (e.g. cyclic HCV protease inhibitors) and again demonstrated the importance of conformational dynamics in achieving the proper balance between membrane permeability and aqueous solubility [5]. When in an aqueous environment, the cyclic peptides exist in open conformations, allowing the hydrogen bond donors and acceptors to interact with water or a protein target; whereas in the hydrophobic region of a lipid bilayer, they adopt closed conformations by forming intramolecular hydrogen bonds. The practical challenge inherent in developing passively diffusible cyclic peptides is how to properly balance membrane permeability against tolerating varieties of hydrophilic functional groups that may be required to engage diverse protein targets. So far, essentially all of the designed cell-permeable cyclic peptides have been small model systems consisting of almost exclusively hydrophobic amino acids. None of these hydrophobic cyclic peptides have demonstrated intracellular PPI inhibition or any other biological activity, although they may be useful for targeting hydrophobic PPI interfaces.
A second and likely more general strategy to engineer cell-permeable cyclic peptides is to incorporate cell-penetrating motifs into the cyclic peptide design. Earlier efforts involved covalent attachment of cyclic peptide ligands to short cell-penetrating peptides (CPPs) such as HIV Tat peptide (YGRKKRRQRRR) and nonaarginine (R9) [41–43]. This approach had only limited success, because the linear CPPs have poor cytosolic delivery efficiencies (<5% [44]) and undergo rapid proteolytic degradation in vivo. A major breakthrough in the field was the recent discovery of a family of small amphipathic cyclic peptides as exceptionally active and metabolically stable CPPs [e.g. cyclo(D-Phe-Nal-Arg-D-Arg-Arg-D-Arg-Gln) or CPP9, where Nal is L-2-naphthylalanine], which have cytosolic delivery efficiencies of up to 120% (or 60-fold higher than that of the Tat peptide) [45,46]. The cyclic CPPs (and the CPP-cargo conjugates) bind directly to the plasma membrane phospholipids and enter cells by endocytosis [46]. They then efficiently escape from the early endosome into the cytosol, apparently through budding and subsequent rupture of small vesicles [47]. The cyclic CPPs have provided a general, effective system for intracellular delivery of a wide variety of cargos including peptides and proteins (see below).
Cell-Permeable Cyclic Peptide PPI Inhibitors
Most of the cell-permeable cyclic peptide PPI inhibitors reported so far enter cells by active transport mechanisms. Cyclic peptides can be rendered cell-permeable by integrating short CPP motifs into their structures, through either rational design or combinatorial approaches (Figure 1). Lian et al. fused membrane impermeable cyclic peptide inhibitors against intracellular enzymes PTP1B and Pin1, which had previously been discovered by screening OBTC libraries, with a cyclic CPP [48]. The resulting bicyclic peptides were readily cell-permeable while retaining the enzyme inhibitory activities, and potently inhibited the signalling pathways downstream of the enzymes in cell culture at nM concentrations (e.g., a PTP1B inhibitor with IC50 = 30 nM; Figure 2). The generality of the bicyclic delivery strategy was later demonstrated by the synthesis of a combinatorial library of 5.7 million cell-permeable bicyclic peptides [49]. Screening of the library against the K-Ras protein identified a bicyclic peptide inhibitor (Figure 2) which blocked the Ras-Raf interaction with an IC50 value of 3.4 μM, inhibited MEK and AKT phosphorylation, and induced apoptosis of lung cancer cells at low μM concentrations. Upadhyaya et al. designed an OBTC cyclic peptide library by integrating a CPP-like pentapeptide motif into 1.5 million different target-binding sequences [50]. Screening of the library against G12V K-Ras followed by medicinal chemistry efforts resulted in a cycloundecapeptide (cyclorasin 9A5; Figure 2) which is cell-permeable and potently blocked the interaction of K-Ras with its downstream effectors (e.g., Raf) with an IC50 value of 120 nM, inhibited signalling pathways downstream of Ras, and induced apoptosis of lung cancer cells (LD50 ~3 μM).
Some PPIs require one of the binding partners in the extended conformation for binding (e.g., PDZ domains); in such cases, cyclization of the binding peptide reduces or abolishes the binding affinity. To render the linear peptide ligands cell-permeable and proteolytically stable, Qian et al. devised reversible cyclization strategies, in which the linear peptide ligands were fused with a short CPP motif (e.g., Arg-Arg-Arg-Arg-Nal-Phe) and cyclized into mono- or bicyclic structures by disulphide bond(s) [51,52]. When outside the cell (e.g., in serum), the cyclic peptides have dramatically improved cellular uptake efficiency and metabolic stability relative to their linear precursors. Upon entering the cytosol, the disulphide bond(s) is reduced by intracellular glutathione to release the biologically active linear peptide for binding to the intended target. As a proof-of-concept, the investigators designed a cyclic peptide inhibitor against the PDZ domain of CFTR-associated ligand (CAL-PDZ; Figure 2), which inhibited the CAL-CFTR interaction and reduced lysosomal degradation of CFTR, thereby increasing CFTR chloride channel activity in lung epithelial cells that harbour a defective CFTR mutant (ΔF508) [51]. More recently, the same group applied the reversible bicyclization strategy to design a peptidyl inhibitor against the PPI between nuclear factor-κB (NF-κB) essential modulator (NEMO) and IKKs. The bicyclic peptide (Figure 2) demonstrated greatly improved serum half-life (10 h vs 15 min for the linear counterpart) and cell permeability, and was ~10-fold more potent for inhibition of NF-κB activation in a cell-based assay relative to the linear peptide (NBD) [52].
It is worth noting that other types of cell-permeable peptidyl PPI inhibitors have also been found to enter cells by endocytosis. Energy-dependent endocytosis was demonstrated to be the major mechanism of uptake of a stapled peptide inhibitor (SAHBA; Figure 2) against the Bcl-2/BH3 interaction, which effectively inhibited the proliferation of a human leukemia xenograft in vivo [53,54]. Likewise, cyclotides, which are a family of highly-stable, disulphide-rich cyclic peptides, were also shown to enter mammalian cells by energy-dependent mechanisms [55,56]. By grafting a known α-helical peptidyl ligand of Hdm2 protein into Momordica cochinchinensis trypsin inhibitor (MCoTI), Camarero and co-workers developed a cell-permeable cyclotide that effectively inhibited the p53/Hdm2 interaction in vitro and in vivo [57].
Conclusion
Intracellular PPIs were once considered as undruggable targets. It is now generally accepted that cyclic peptides and other types of macrocycles may provide a general modality for targeting this PPI class. Over the past decade, tremendous progress has been made toward overcoming the two major challenges associated with developing cyclic peptide drugs: target engagement and membrane permeability. The advent of several powerful cyclic peptide library technologies has now made it a relatively routine exercise to discover cyclic peptide ligands against most proteins including those involved in PPIs. Our improved understanding of membrane permeation of cyclic peptides via both passive diffusion and active transport mechanisms has begun to allow rational design of cell-permeable cyclic peptides that specifically target intracellular PPIs. Some of these cyclic peptides have already provided useful molecular probes for chemical biology applications. We are confident that during the next decade, some of these cyclic peptides will advance into clinical applications.
Highlights.
Cyclic peptides are an emerging class of drug modality for PPI inhibition
Rational design and library screening are established to generate lead peptides
Membrane permeability can be enhanced by various techniques
Integration of membrane permeability and binding affinity is essential
Acknowledgments
The work in our laboratory was supported by the National Institutes of Health (GM062820 and GM110208). We thank all of the Pei group members and collaborators for their contributions to the projects.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors declare no conflict of interest.
References
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Driggers EM, Hale SP, Lee J, Terrett NK. The exploration of macrocycles for drug discovery - an underexploited structural class. Nat Rev Drug Discov. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 2.Marsault E, Peterson ML. Macrocycles are great cycles: applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J Med Chem. 2011;54:1961–2004. doi: 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]
- 3.Mallinson J, Collins I. Macrocycles in new drug discovery. Fut Med Chem. 2012;4:1409–1438. doi: 10.4155/fmc.12.93. [DOI] [PubMed] [Google Scholar]
- 4.Hewitt WM, Leung SSF, Pye CR, Ponkey AR, Bednarek M, Jacobson MP, Lokey RS. Cell-permeable cyclic peptides from synthetic libraries inspired by natural products. J Am Chem Soc. 2015;137:715–721. doi: 10.1021/ja508766b. [DOI] [PubMed] [Google Scholar]
- 5 •.Matsson P, Doak BC, Over B, Kihlberg J. Cell permeability beyond the rule of 5. Adv Drug Deliv Rev. 2016;101:42–61. doi: 10.1016/j.addr.2016.03.013. A comprehensive review of the effects of Nα-methylation, intramolecular hydrogen bonds, large hydrophobic side chains, and conformational flexibility on the membrane permeability and oral bioavailability of molecules beyond the Rule of 5. [DOI] [PubMed] [Google Scholar]
- 6.Pelay-Gimeno M, Glas A, Koch O, Grossmann TN. Structure-based design of inhibitors of protein-protein interactions: mimicking peptide binding epitopes. Angew Chem Int Ed. 2015;54:8896–8927. doi: 10.1002/anie.201412070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinis H, Winter G. Encoded libraries of chemically modified peptides. Curr Opin Chem Biol. 2015;26:89–98. doi: 10.1016/j.cbpa.2015.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Morioka T, Loik ND, Hipolito CJ, Goto Y, Suga H. Selection-based discovery of macrocyclic peptides for the next generation therapeutics. Curr Opin Chem Biol. 2015;26:34–41. doi: 10.1016/j.cbpa.2015.01.023. [DOI] [PubMed] [Google Scholar]
- 9.Josephson K, Ricardo A, Szostak JW. mRNA display: from basic principles to macrocycle drug discovery. Drug Disc Today. 2014;19:388–399. doi: 10.1016/j.drudis.2013.10.011. [DOI] [PubMed] [Google Scholar]
- 10.Foster AD, Ingram JD, Leitch EK, Lennard KR, Osher EL, Tavassoli A. Methods for the creation of cyclic peptide libraries for use in lead discovery. J Biomol Screen. 2015;20:563–576. doi: 10.1177/1087057114566803. [DOI] [PubMed] [Google Scholar]
- 11.Blackwell HE, Grubbs RH. Highly efficient synthesis of covalently cross-linked peptide helices by ring-closing metathesis. Angew Chem Int Ed. 1998;37:3281–3284. doi: 10.1002/(SICI)1521-3773(19981217)37:23<3281::AID-ANIE3281>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 12.Schafmeister CE, Po J, Verdine GL. An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J Am Chem Soc. 2000;122:5891–5892. [Google Scholar]
- 13 •.Chu Q, Moellering RE, Hilinski GJ, Kim Y-W, Grossman TN, Yeh JT, Verdine GL. Towards understanding cell penetration by stapled peptides. Med Chem Commun. 2015;6:111–119. This study analysed >200 stapled peptides and found that cell penetration ability is strongly related to staple type and formal charge (optimal charge of +2–7), whereas other physicochemical parameters do not appear to have a significant effect. It also demonstrated that stapled peptides penetrate cells through endocytosis pathways. [Google Scholar]
- 14 •.Bird GH, Mazzola E, Opoku-Nsiah K, Lammert MA, Godes M, Neuberg DS, Walensky LD. Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices. Nat Chem Biol. 2016;12:845–852. doi: 10.1038/nchembio.2153. This study used an unbiased statistical approach to determine which biophysical parameters dictate the uptake of stapled peptides. The study found that staple placement at the amphipathic boundary combined with optimal hydrophobic and helical content are the key drivers of cellular uptake, whereas excess hydrophobicity and positive charge trigger membrane lysis at elevated peptide dosing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang YS, Graves B, Guerlavais V, Tovar C, Packman K, To K-H, Olson KA, Kesavan K, Gangurde P, Mukherjee A, Baker T, Darlak K, Elkin C, Filipovic Z, Qureshi FZ, Cai H, Berry P, Feyfant E, Shi XE, Horstick J, Annis DA, Manning AM, Fotouhi N, Nash H, Vassilev LT, Sawyer TK. Stapled α-helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci. 2013;110:E3445–E3454. doi: 10.1073/pnas.1303002110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shi Z-D, Wei C-Q, Lee K, Liu H, Zhang M, Araki T, Roberts LR, Worthy KM, Fisher RJ, Neel BG, Kelley JA, Yang D, Burke TR. Macrocyclization in the design of non-phosphorus-containing Grb2 SH2 domain-binding ligands. J Med Chem. 2004;47:2166–2169. doi: 10.1021/jm030510e. [DOI] [PubMed] [Google Scholar]
- 17.Zhou H, Liu L, Huang J, Bernard D, Karatas H, Navarro A, Lei M, Wang S. Structure-based design of high-affinity macrocyclic peptidomimetics to block the menin-mixed lineage leukemia 1 (MLL1) protein-protein interaction. J Med Chem. 2013;56:1113–1123. doi: 10.1021/jm3015298. [DOI] [PubMed] [Google Scholar]
- 18.Siegert TR, Bird MJ, Makwana KM, Kritzer JA. Analysis of loops that mediate protein–protein interactions and translation into submicromolar inhibitors. J Am Chem Soc. 2016;138:12876–12884. doi: 10.1021/jacs.6b05656. [DOI] [PubMed] [Google Scholar]
- 19.Giebel LB, Cass RT, Milligan DL, Young DC, Arze R, Johnson CR. Screening of cyclic peptide phage libraries identifies ligands that bind streptavidin with high affinities. Biochemistry. 1995;34:15430–15435. doi: 10.1021/bi00047a006. [DOI] [PubMed] [Google Scholar]
- 20.Roberts RW, Szostak JW. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci. 1997;94:12297–12302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott CP, Abel-Santos E, Wall M, Wahnon DC, Benkovic SJ. Production of cyclic peptides and proteins in vivo. Proc Nat Acad Sci. 1999;96:13638–13643. doi: 10.1073/pnas.96.24.13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miranda E, Nordgren IK, Male AL, Lawrence CE, Hoakwie F, Cuda F, Court W, Fox KR, Townsend PA, Packham GK, Eccles SA, Tavassoli A. A cyclic peptide inhibitor of HIF-1 heterodimerization that inhibits hypoxia signaling in cancer cells. J Am Chem Soc. 2013;135:10418–10425. doi: 10.1021/ja402993u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Millward SW, Takahashi TT, Roberts RW. A general route for post-translational cyclization of mRNA display libraries. J Am Chem Soc. 2005;127:14142–14143. doi: 10.1021/ja054373h. [DOI] [PubMed] [Google Scholar]
- 24.Heinis C, Rutherford T, Freund S, Winter G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat Chem Biol. 2009;5:502–507. doi: 10.1038/nchembio.184. [DOI] [PubMed] [Google Scholar]
- 25.Liu R, Marik J, Lam KS. A Novel Peptide-Based Encoding System for “One-Bead One-Compound” Peptidomimetic and Small Molecule Combinatorial Libraries. J Am Chem Soc. 2002;124:7678–7680. doi: 10.1021/ja026421t. [DOI] [PubMed] [Google Scholar]
- 26.Joo SH, Xiao Q, Ling Y, Gopishetty B, Pei D. High-throughput sequence determination of cyclic peptide library members by partial Edman degradation/mass spectrometry. J Am Chem Soc. 2006;128:1300–13009. doi: 10.1021/ja063722k. [DOI] [PubMed] [Google Scholar]
- 27.Gartner ZJ, Tse BN, Grubina R, Doyon JB, Snyder TM, Liu DR. DNA-templated organic synthesis and selection of a library of macrocycles. Science. 2004;305:1601–1605. doi: 10.1126/science.1102629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimmermann G, Neri D. DNA-encoded chemical libraries: foundations and applications in lead discovery. Drug Disc Today. 2016;21:1828–1834. doi: 10.1016/j.drudis.2016.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schreiber SL. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science. 2000;287:1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- 30.Collins S, Barlett S, Nie F, Sore HF, Spring DR. Diversity-oriented synthesis of macrocycle libraries for drug discovery and chemical biology. Synthesis. 2016;48:1457–1473. [Google Scholar]
- 31.Hipolito CJ, Suga H. Ribosomal production and in vitro selection of natural product-like peptidomimetics: the FIT and RaPID systems. Curr Opin Chem Biol. 2012;16:196–203. doi: 10.1016/j.cbpa.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 32.Rezaei Araghi R, Ryan JA, Letai A, Keating AE. Rapid optimization of Mcl-1 inhibitors using stapled peptide libraries including non-natural side chains. ACS Chem Biol. 2016;11:1238–1244. doi: 10.1021/acschembio.5b01002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diderich P, Bertoldo D, Dessen P, Khan MM, Pizzitola I, Held W, Huelsken J, Heinis C. Phage selection of chemically stabilized α-helical peptide ligands. ACS Chem Biol. 2016;11:1422–1427. doi: 10.1021/acschembio.5b00963. [DOI] [PubMed] [Google Scholar]
- 34.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2010;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 35.White TR, Renzelman CM, Rand AC, Rezai T, McEwen CM, Gelev VM, Turner RA, Linington RG, Leung SSF, Kalgutkar AS, Bauman JN, Zhang Y, Liras S, Price DA, Mathiowetz AM, Jacobson MP, Lokey RS. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat Chem Biol. 2011;7:810–817. doi: 10.1038/nchembio.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beck JG, Chatterjee J, Laufer B, Kiran MU, Frank AO, Neubauer S, Ovadia O, Greenberg S, Gilon C, Hoffman A, Kessler H. Intestinal permeability of cyclic peptides: common key backbone motifs identified. J Am Chem Soc. 2012;134:12125–12133. doi: 10.1021/ja303200d. [DOI] [PubMed] [Google Scholar]
- 37.Nielsen DS, Hoang HN, Lohman RJ, Hill TA, Lucke AJ, Craik DJ, Edmonds DJ, Griffith DA, Rotter CJ, Ruggeri RB, Price DA, Liras S, Fairlie DP. Improving on nature: making a cyclic heptapeptide orally bioavailable. Angew Chem Int Ed. 2014;53:12059–12063. doi: 10.1002/anie.201405364. [DOI] [PubMed] [Google Scholar]
- 38.Thansandote P, Harris RM, Dexter HL, Simpson GL, Pal S, Upton RJ, Valko K. Improving the passive permeability of macrocyclic peptides: balancing permeability with other physiochemical properties. Bioorg Med Chem. 2014;23:322–327. doi: 10.1016/j.bmc.2014.11.034. [DOI] [PubMed] [Google Scholar]
- 39.Ahlbach CL, Lexa KW, Bockus AT, Chen V, Crews P, Jacobson MP, Lokey RS. Beyond cyclosporine A: conformation-dependent passive membrane permeabilities of cyclic peptide natural products. Fut Med Chem. 2015;7:2121–2130. doi: 10.4155/fmc.15.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bockus AT, Schwochert JA, Pye CR, Townsend CE, Sok V, Bednarek MA, Lokey RS. Going out on a limb: delineating the effects of β-branching, N-methylation, and side chain size on the passive permeability, solubility, and flexibility of Sanguinamide A analogues. J Med Chem. 2015;58:7409–7418. doi: 10.1021/acs.jmedchem.5b00919. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y, Zhou S, Wavreille A-S, DeWille J, Pei D. Cyclic peptidyl inhibitors of Grb2 and tensin SH2 domains identified from combinatorial libraries. J Comb Chem. 2008;10:247–255. doi: 10.1021/cc700185g. [DOI] [PubMed] [Google Scholar]
- 42.Sasaki Y, Minamizawa M, Ambo A, Sugawara S, Ogawa Y, Nitta K. Cell-penetrating peptide-conjugated XIAP-inhibitory cyclic hexapeptides enter into Jurkat cells and inhibit cell proliferation. FEBS Journal. 2008;275:6011–6021. doi: 10.1111/j.1742-4658.2008.06730.x. [DOI] [PubMed] [Google Scholar]
- 43.Desimmie BA, Humbert M, Lescrinier E, Hendrix J, Vets S, Gijsbers R, Ruprecht RM, Dietrich U, Debyser Z, Christ F. Phage display-directed discovery of LEDGF/p75 binding cyclic peptide inhibitors of HIV replication. Mol Ther. 2012;20:2064–2075. doi: 10.1038/mt.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.LaRochelle JR, Cobb GB, Steinauer A, Rhoades E, Schepartz A. Fluorescence correlation spectroscopy reveals highly efficient cytosolic delivery of certain penta-arg proteins and stapled peptides. J Am Chem Soc. 2015;137:2536–2541. doi: 10.1021/ja510391n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qian Z, Liu T, Liu Y, Briesewitz R, Barrios AM, Jhiang SM, Pei D. Efficient delivery of cyclic peptides into mammalian cells with short sequence motifs. ACS Chem Biol. 2013;8:423–431. doi: 10.1021/cb3005275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qian Z, LaRochelle JR, Jiang B, Lian W, Hard RL, Selner NG, Luechapanichkul R, Barrios AM, Pei D. Early endosomal escape of a cyclic cell-penetrating peptide allows effective cytosolic cargo delivery. Biochemistry. 2014;53:4034–4046. doi: 10.1021/bi5004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47 ••.Qian Z, Martyna A, Hard RL, Wang J, Appiah-Kubi G, Coss C, Phelps MA, Rossman JS, Pei D. Discovery and mechanism of highly efficient cyclic cell-penetrating peptides. Biochemistry. 2016;55:2601–2612. doi: 10.1021/acs.biochem.6b00226. A family of small amphipathic cyclic peptides was found to be highly active CPPs. A method for quantitatively assessing the overall cellular entry, endosomal escape, and cytosolic delivery efficiency of CPPs was reported. The study also reported the first well-defined mechanism for endosomal escape of CPPs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48 ••.Lian W, Jiang B, Qian Z, Pei D. Cell-permeable bicyclic peptide inhibitors against intracellular proteins. J Am Chem Soc. 2014;136:9830–9833. doi: 10.1021/ja503710n. This study showed that cell-impermeable cyclic peptides could be rendered cell-permeable by fusing them with a cyclic CPP, without losing their target-binding activities. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trinh TB, Upadhyaya P, Qian Z, Pei D. Discovery of a direct Ras inhibitor by screening a combinatorial library of cell-permeable bicyclic peptides. ACS Comb Sci. 2016;18:75–85. doi: 10.1021/acscombsci.5b00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50 •.Upadhyaya P, Qian Z, Selner NG, Clippinger SR, Wu Z, Briesewitz R, Pei D. Inhibition of Ras signaling by blocking Ras-effector interactions with cyclic peptides. Angew Chem Int Ed. 2015;54:7602–7606. doi: 10.1002/anie.201502763. This is an example of the endocyclic cargo delivery method by cyclic CPPs, where the peptidyl cargo is integrated with the CPP motif within a monocyclic peptide ring so that the same residues perform dual functions of cell penetration and target engagement. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qian Z, Xu X, Amacher JF, Madden DR, Cormet-Boyaka E, Pei D. Intracellular delivery of peptidyl ligands by reversible cyclization: discovery of a PDZ domain inhibitor that rescues CFTR activity. Angew Chem Int Ed. 2015;54:5874–5878. doi: 10.1002/anie.201411594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52 ••.Qian Z, Rhodes CA, McCroskey LC, Wen J, Appiah-Kubi G, Wang DJ, Guttridge DC, Pei D. Enhancing the cell permeability and metabolic stability of peptidyl drugs by reversible bicyclization. Angew Chem Int Ed. 2017;56:1525–1529. doi: 10.1002/anie.201610888. The cell permeability and metabolic stability of linear peptides can be dramatically and simultaneously improved by fusing them with a hexapeptide CPP motif and cyclizing the peptide fusion into a structurally rigid bicycle through the formation of two disulfide bonds. The linear, active peptide is regenerated upon cytosolic entry. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fischer J, Smith E, Verdine GL, Kormeyer SJ. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C, Fisher JK, Godes M, Pitter K, Kung AL, Walensky LD. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. 2012;122:2018–2031. doi: 10.1172/JCI46231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Greenwood KP, Daly NL, Brown DL, Stow JL, Craik DJ. The cyclic cystine knot miniprotein MCoTI-II is internalized into cells by macropinocytosis. Int J Biochem Cell Biol. 2007;39:2252. doi: 10.1016/j.biocel.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 56.Contreras J, Elnagar AY, Hamm-Alvarez SF, Camarero JA. Cellular uptake of cyclotide MCoTI-I follows multiple endocytic pathways. J Controlled Release. 2011;155:134–143. doi: 10.1016/j.jconrel.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57 •.Ji Y, Majumder S, Millard M, Borra R, Bi T, Elnagar AY, Neamati N, Shekhtman A, Camarero JA. In vivo activation of the p53 tumor suppressor pathway by an engineered cyclotide. J Am Chem Soc. 2013;135:11623–11633. doi: 10.1021/ja405108p. This study demonstrated that a membrane-impermeable α-helical peptide could be rendered cell-permeable by grafting it onto the structure of a cyclotide. The resulting modified cyclotide is metabolically stable, capable of entering mammalian cells and inhibiting intracellular PPIs. [DOI] [PMC free article] [PubMed] [Google Scholar]