

Abstract

Small-molecule inhibitors of the mycobacterial transcriptional repressor EthR have previously been shown to act as boosters of the second-line antituberculosis drug ethionamide. Fragment-based drug discovery approaches have been used in the past to make highly potent EthR inhibitors with ethionamide boosting activity both in vitro and ex vivo. Herein, we report the development of fragment-sized EthR ligands with nanomolar minimum effective concentration values for boosting the ethionamide activity in Mycobacterium tuberculosis whole-cell assays.
Introduction
Twenty years after the World Health Organization declared tuberculosis (TB) disease to be a global health emergency, limited progress has been made on curbing, let alone eradicating, the TB epidemic.1 It has been estimated that one in three people worldwide harbors the Mycobacterium tuberculosis bacillus,2 and over 1.5 million people die from TB each year.1 The first-line antibiotics isoniazid, pyrazinamide, ethambutol, and rifampicin used for the treatment of active drug-susceptible TB3 are complemented by a cohort of second-line drugs such as ethionamide and prothionamide, which are prescribed in cases where there is evidence for multidrug-resistant TB infection. In an attempt to combat the global TB epidemic, there has been extensive research into the development of novel vaccines4 and chemotherapeutics5−7 against TB. In spite of these efforts, a reliable vaccine against the infection has not yet been introduced to the market, and the desperate need for antibiotics with novel mechanisms of action remains.
While there has been a concerted effort to develop new strategies to target TB directly, an alternative strategy involves boosting the effect of existing second-line antituberculars such as ethionamide.8
Ethionamide and isoniazid are pro-drugs, which in their activated form as nicotinamide adenine dinucleotide (NAD) adducts are potent inhibitors of InhA, the 2-trans-enoyl reductase enzyme belonging to the type II fatty acid synthase system of M. tuberculosis (Figure 1).9−11 While activation of isoniazid in M. tuberculosis depends on KatG,9,12 the flavin-dependent monooxygenase enzyme EthA, whose expression is controlled by the transcriptional repressor EthR,13 is responsible for the activation of ethionamide.14,15 It has been previously shown that small-molecule ligands, which abolish the DNA-binding ability and hence the transcriptional repressor function of EthR, can exhibit ethionamide boosting activity both in vitro and ex vivo.8,16,17 Boosting the effect of ethionamide by coadministration with EthR inhibitors is an attractive therapeutic strategy because the reduction of the required daily dose of this second line antibiotic is expected to decrease the toxicity-related side effects and improve patient compliance with the drug.
Figure 1.

Mechanisms of activation of isoniazid (INH) and ethionamide (ETH) and inhibition of InhA by the INH-NAD and ETH-NAD adducts.
Fragment-based approaches have previously been explored for the design and synthesis of potent ethionamide boosters.18−20 We previously carried out a fragment screen against EthR and used a fragment-linking strategy for the design and synthesis of a disulfide-linked EthR ligand capable of boosting ethionamide activity (screened at a concentration of 1 μM).19 Recently we reported a fragment merging approach toward the development of small-molecule inhibitors of M. tuberculosis EthR, which gave us access to molecular probes, potent at inhibiting the interaction between EthR and its DNA operator in vitro but nevertheless unable to boost ethionamide activity in cellular assays.20
In contrast to our merged EthR ligands, an interesting observation was made that fragment 1 (Figure 2) was found to be capable of boosting ethionamide activity against M. tuberculosis.19 The biological activity of 1 could at least in part be due to its small size, which allows it to permeate more effectively through the mycobacterial envelope and/or host cell membrane. Small molecules are also thought to contribute to the sterilizing activity due to their ability to diffuse into caseum within lesions and thus have the potential to shorten the duration of TB treatment.21 In this Article we describe the design, synthesis, and biological evaluation of a set of fragment-sized, highly ligand-efficient derivatives of fragment 1, which exhibit low nanomolar ethionamide boosting ability in a previously described macrophage assay.8,16−18
Figure 2.

X-ray crystal structure of fragment 1 bound to EthR with a 2:1 stoichiometry (PDB code 5F1J), with subdivision of the EthR binding cavity into four distinct subpockets (I, II, III, and IV). The distances between the carbon atoms marked X and Y and residues Asn176 and Thr149 are indicated.
Fragment 1 was originally identified in a fluorescence-based thermal shift screening campaign of our 1250-member fragment library against EthR.19 The presence of fragment 1 at a concentration of 1 μM was shown to reduce the minimum inhibitory concentration (MIC) of ethionamide from 15 μM to approximately 2 μM under the conditions of the resazurin reduction microplate assay used.19 An X-ray crystal structure of fragment 1 bound to EthR (Figure 2) was obtained that provided a good starting point for the design of a small focused library of compounds containing hydrogen-bond donor or acceptor groups at appropriate positions within the scaffold of compound 1. The distances between the primary amide of Asn176 and position X of 1, and between the hydroxyl oxygen atom of Thr149 and position Y of 1, are shown in Figure 2. The substitution of the carbon atoms of 1 at positions X and Y for hydrogen-bond donor or acceptor functional groups, designed to interact with residues Asn176 and Thr149, is an attractive strategy for developing a structure–activity relationship (SAR) around the ethionamide booster compound 1 (Table 1).
Table 1. Exploration of SAR around Fragment 1: Dissociation Constants (KD) Determined by ITC (n = 1).
MEC = minimum effective concentration. Results are mean ± standard deviation of two independent replicates.
By analogy to the starting fragment 1, derivatives 2–10 were intended to form polar interactions with the amide of Asn179 (via their carbonyl or sulfonamide oxygen atoms). In addition, the urea 3, the amide 4, and the sulfonamide urea 6 were designed to hydrogen-bond to Asn176 via their N–H functionality. The sp3 oxygen atom of carbamate 2 was envisaged to act as a weak hydrogen-bond acceptor from Asn176. Finally, the furan/tetrahydrofuran oxygen atoms of amides 7–10, were designed to form weak hydrogen-bonding interactions with the hydroxyl group of Thr149. Compounds 1–8, however, showed no improvements in activity when tested for binding to EthR by isothermal titration calorimetry (KD values between 6 and 22 μM were measured for derivatives 1–8).
The ability of compounds 1–10 to boost ethionamide activity (in the presence of 1/10 of the MIC of ethionamide) in M. tuberculosis culture grown on 7H9/ADC/Tw media was tested (Table 1). Interestingly, the starting fragment 1 showed the highest ethionamide boosting ability (minimum effective concentration (MEC) = 3.0 ± 1.8 μM; i.e., ligand 1 needed to be present at a concentration of 3 ± 1.8 μM to render ethionamide present at 1/10 of its MIC bactericidal against M. tuberculosis). The carbamate 2 was the only other compound in this series, which showed ethionamide boosting activity in this whole-cell assay (MEC = 19 ± 7.5 μM), while compounds 3–10 were shown to be less active (MEC > 50 μM). Thus, the SAR strategy of introducing hydrogen-bond donor or acceptor functionality in the scaffold of fragment 1 did not afford EthR inhibitors with improved ethionamide boosting ability. The increased polarity and higher number of hydrogen-bond donor and acceptor atoms of molecules 3–10 are likely to be detrimental to the permeability of the inhibitors across the mycobacterial cell envelope and the host cell membrane.
The KD values measured by ITC and the relative ability of the ligands to boost ethionamide in cellular assays do not correlate well. The lack of ethionamide boosting ability of compounds 3–8, which have KD values measured by ITC comparable to those of the active ethionamide boosters 1 and 2, could be due to the low permeability of these compounds across the mycobacterial envelope or host cell membrane. However, a different mode of binding for compound 2 in the cavity of EthR cannot be excluded as this point, which could also explain why this compound was found to be active while 3–8 were not.
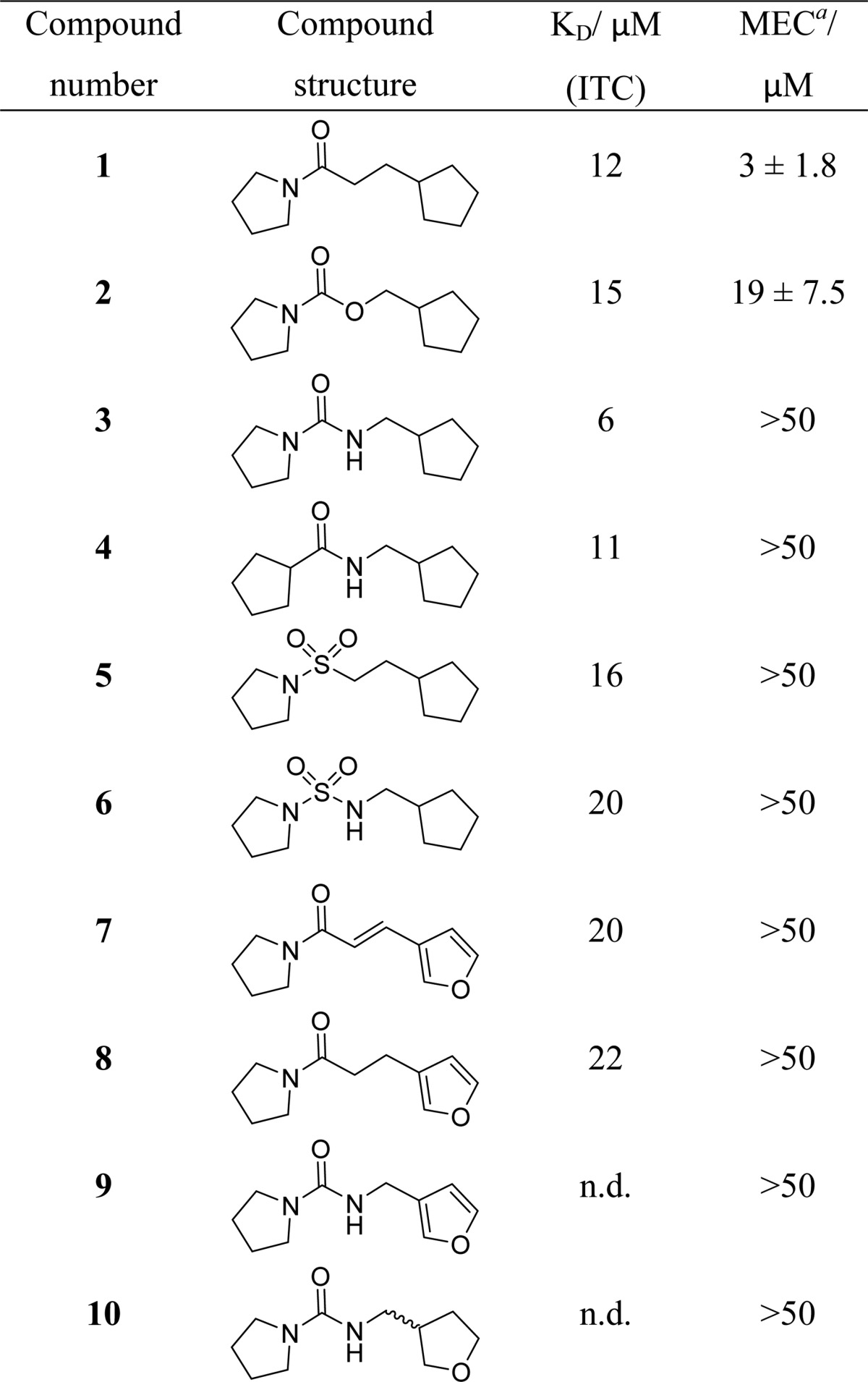
The X-ray crystal structures of compounds 3–10 bound to EthR have been solved to resolutions in the range between 1.7 and 2.0 Å. It was unfortunately not possible to obtain suitable crystals of EthR in complex with compound 2. The X-ray crystal structure of 3 bound to EthR is of particular interest since three molecules of this ligand were found to bind to a single EthR monomer (Figure 3a). The two molecules of 3, which bind inside subpockets II and IV, respectively, recapitulate the binding modes of fragment 1. There is evidence for the formation of a hydrogen-bond between the urea N–H of 3 bound to subpocket II and Asn176 (Figure 3a). The third molecule of 3 binds at the very entrance of the EthR binding cavity (subpocket I).
Figure 3.
(a) X-ray crystal structure of urea 3 (PDB code 5IOY) bound to EthR. Three units of ligand 3 soak inside the binding cavity of the protein, filling subpockets I, II, and IV. (b) X-ray crystal structure of amide 4 (PDB code 5IOZ) bound to subpocket II of EthR.
In contrast to this, the close analogue of urea 3, the amide 4, was shown to bind to EthR only in a 1:1 stoichiometry nevertheless taking advantage of the hydrogen bonding opportunities with both Asn179 and Asn176 within subpocket II (Figure 3b). The markedly different stoichiometry of binding of compounds 1, 3, and 4 to EthR does not result in a significant difference in the dissociation constants (KD by ITC) of these two compounds (Table 1).
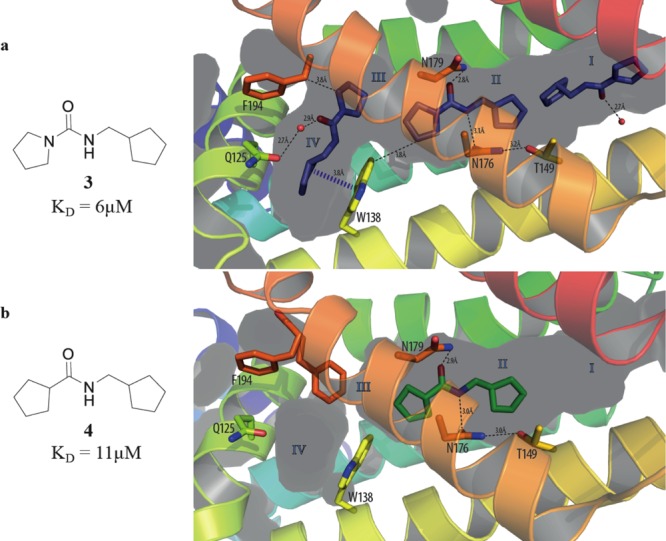
From a crystallographic perspective the sulfonamide group of derivative 5 is a suitable isostere for the amide functionality of the starting fragment 1. Unlike 1, ligand 5 only binds to EthR in a 1:1 stoichiometry (Figure 4a); however, it closely recapitulates the binding mode of 1 inside subpocket II. Both oxygen atoms of the sulfonamide group of 5 are stabilized by polar interactions with the side chain of Asn179 (Figure 4a).
Figure 4.
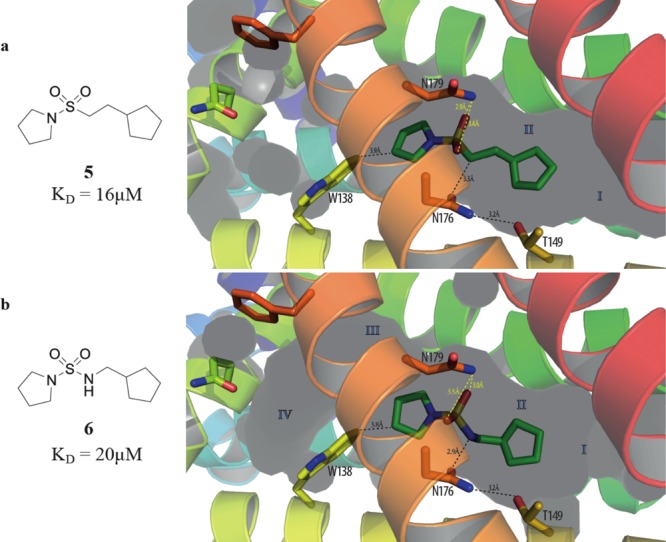
X-ray crystal structures of (a) compound 5 (PDB code 5J3L) and (b) compound 6 bound to subpocket II of EthR. Relevant interatomic distances are indicated.
The sulfonamide urea 6 was found to bind to EthR in an identical position to its close derivative 5 with the two oxygen atoms of 6 interacting with Asn179 (Figure 4b). There is also evidence for the displacement of the sulfonamide urea N–H of 6 relative to the corresponding methylene carbon atom of 5, presumably in order to optimize the available polar interaction with Asn176 (Figure 4).
The electron density in the X-ray crystal structures of 7 and 8 bound to EthR is less well-defined, and for this reason the figures showing the binding modes of these ligands to the protein are given in the Supporting Information (Figures S6 and S7, respectively).
Compounds 9 and 10 were also found to bind singly to subpocket II of EthR (Figure 5a), recapitulating the binding mode of the original fragment 1 in this subpocket. While the urea N–H functionalities of both 9 and 10 seem to be stabilized by polar interactions with the side chain of Asn176, the evidence for hydrogen-bonding between the furan/tetrahydrofuran oxygen atoms of 9 and 10, respectively, with Thr149 is less convincing.
Figure 5.
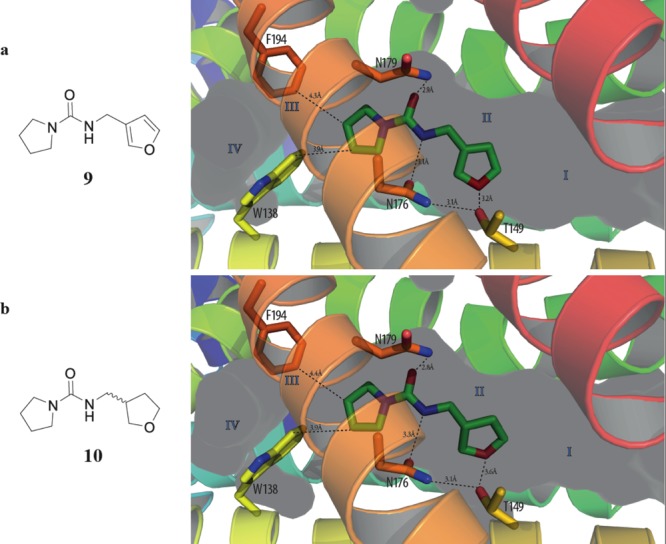
X-ray crystal structures of (a) compound 9 (PDB code 5J1U) and (b) compound 10 (PDB code 5IP6) bound to subpocket II of EthR.
Since amide 1 and carbamate 2 were the only two compounds capable of boosting ethionamide activity in M. tuberculosis culture, further SAR was carried out around these two molecules. A range of compounds were designed by varying the hydrocarbon group attached to the carbonyl atom of pyrrolidine amide 1. Removing the ethylene linker bridging the pyrrolidine amide and the cyclopentyl ring of 1 resulted in loss of ethionamide boosting activity of compound 11 (MEC > 50 μM, see Table 2). Reintroducing a methylene linker in amide 12 (MEC = 19 ± 7.5 μM) restored some of the boosting effect. Increasing the ring size of 1 from cyclopentyl to cyclohexyl resulted in an approximately 4-fold increase in potency (compound 13, MEC = 0.8 ± 0.5 μM). The best ethionamide boosting was achieved by using a propylene linker and a cyclohexyl ring (compound 14, MEC = 0.4 ± 0.2 μM), which gave approximately 7-fold improvement in MEC on the starting fragment 1. Addition of an extra methylene unit to the flexible linker of 14 caused a 4-fold drop in the potency of ligand 15 (MEC = 1.6 ± 0.8 μM). The cyclohexyl ring of 15 proved essential, and its removal resulted in loss of activity (compounds 18 and 19, MEC > 50 μM). Finally, introducing a methylene linker between the pyrrolidine amide and the adamantyl group of the inactive amide 16 (MEC > 50 μM) caused an over 30-fold increase in ethionamide boosting ability of compound 17 (MEC = 1.6 ± 0.8 μM).
Table 2. Exploration of SAR around Fragment 1 by Varying the Hydrocarbon Group Attached to the Carbonyl Carbon Atom of the Pyrrolidine Amide.

MEC = minimum effective concentration. Results are shown as mean ± standard deviation of two independent replicates.
Table 3 shows SAR around carbamate 2. Substitution of the pyrrolidine ring of 2 for a six- or seven-member ring (carbamates 20 and 21, respectively) resulted in loss of ethionamide boosting ability, while decreasing the size of the pyrrolidine ring had the opposite effect in compounds 26 (MEC = 6.3 ± 3.6 μM) and 27 (MEC = 13 ± 7.2 μM). Decreasing the ring size of the cyclopentyl ring of 2 to cyclopropyl or cyclobutyl in compounds 23 (MEC = 3.1 ± 1.8 μM) and 24 (MEC = 3.1 ± 1.8 μM), respectively, appeared beneficial in improving the ethionamide boosting of these compounds. The optimal length of the flexible linker for the carbamate series was shown to consist of three carbon atoms (carbamate 28, MEC = 0.4 ± 0.2 μM). Addition of a fourth methylene group to the flexible linker resulted in a roughly 16-fold decrease in the potency of ligand 29 (MEC = 6.3 ± 3.6 μM). Boc-pyrrolidine 30 (MEC > 50 μM) was shown to be inactive in M. tuberculosis culture.
Table 3. Exploration of SAR around Carbamate 2.

MEC = minimum effective concentration. Results are shown as mean ± standard deviation of two independent replicates.
The SAR exploration around molecules 1 and 2 led to the identification of ligands 14 and 28, respectively, which have MEC = 400 nM for the 10-fold boosting of ethionamide, and therefore represent a roughly 7-fold improvement upon the starting fragment 1 (MEC = 3 ± 1.8 μM).
Finally, the ability of a selection of the molecules from Tables 1–3 to boost ethionamide activity in M. tuberculosis infected murine macrophages in a previously described and well-established assay was tested.16,17 Experimental data from the macrophage assay are shown in Figure 6. In summary, compounds 1, 22, 23, and 25 exhibit significant boosting of ethionamide in the macrophage assay. In addition to an ethionamide boosting effect, compounds 1, 22, and 25 have an intrinsic bactericidal activity against M. tuberculosis at concentrations higher than 1 μM. The most potent derivatives of compounds 1 and 2, inhibitors 14 and 28, were also tested in the macrophage assay and were shown to exhibit EC50 = 40 and 50 nM, respectively, in the presence of 1/10 of the MIC of ethionamide. Nevertheless, compounds 14 and 28 also exhibited a significant intrinsic bactericidal effect in the absence of ethionamide with IC50 ≈ 1 μM. Compounds 3–10 from Table 1 were also tested in the macrophage assay; however, they did not boost ethionamide activity in M. tuberculosis infected macrophages to the same degree as boosters 1, 14, 22, 25, and 28. This agrees with the observation that compounds 3–10 were also less effective at boosting ethionamide in M. tuberculosis culture grown on 7H9/ADC/Tw media (MECs > 50 μM, Table 1).
Figure 6.

Inhibition of bacterial replication in macrophages for (a) compound 1, (b) compound 22, (c) compound 25 alone and in the presence of ethionamide (normal MIC/10, experiments have been carried out in triplicate (n = 3)). The parameter used as a read-out was the area of bacteria present per infected macrophage. Normalizations were performed based on the average values obtained for the negative (DMSO 1%) and positive (isoniazid 1 μg/mL) controls. Percentage of inhibition is plotted against the log10 of the compounds concentration, determined in the absence or in the presence of ethionamide at 1/10 of its MIC for the macrophage assay (0.033 μg/mL). Fitting was performed by Prism software using the sigmoidal dose–response (variable slope) model.
Conclusion
We have shown that ligand 1, identified previously in a fragment screen against EthR,19 exhibits an exceptionally strong ethionamide boosting effect in whole-cell M. tuberculosis assays. SAR aimed at introducing hydrogen bond donor or acceptor functionality in the scaffold of 1 did not lead to improvement of the boosting effect but was nevertheless instrumental in identifying carbamate 2 as a second ligand exhibiting an ethionamide boosting effect in whole-cell M. tuberculosis assays.
Subsequent SAR around 1 and 2 led to the identification of the potent ethionamide boosters, the amide 14 (MEC = 0.4 ± 0.2 μM) and the carbamate 28 (MEC = 0.4 ± 0.2 μM), which represent approximately 7-fold improvement in boosting the effect of ethionamide in M. tuberculosis culture compared to the starting fragment 1 (MEC = 3 ± 1.8 μM). Compounds 14 and 28 also exhibited low nanomolar EC50 (40 and 50 nM, respectively) activity in our macrophage assay in the presence of ethionamide at 1/10 of its MIC. Nevertheless, compounds 14 and 28 also showed a significant intrinsic bactericidal effect in the absence of ethionamide with IC50 ≈ 1 μM. The strong bactericidal effect of compounds 14 and 28 in the presence of ethionamide at 1/10 of its MIC for the macrophage experiment is presumably a combination of the antitubercular activity intrinsic to these compounds on their own and an ethionamide boosting effect.
Acknowledgments
We would like to thank A. Coyne for help in the preparation of this manuscript. P.O.N. would like to thank the EPSRC for providing Ph.D. funding. We also thank the Bill and Melinda Gates Foundation and the EU FP7MM4TB Grant No. 260872, the ERC-STG INTRACELLTB Grant No. 260901, the Agence Nationale de la Recherche (ANR-10-EQPX-04-01), the Feder (12001407 (D-AL) Equipex Imaginex BioMed), the Intramural Research Program of the National Institutes of Health/NIAID, and the Région Nord Pas de Calais, France, for providing funding to support this work.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.7b00091.
Experimental procedures and spectral data of new compounds (PDF)
The authors declare no competing financial interest.
Notes
NMR spectra related to this publication are also available at the University of Cambridge data repository (https://www.repository.cam.ac.uk/handle/1810/254768).
Notes
Protein X-ray crystallography structures of compounds 1, 3–5, and 7–10 bound to M. tuberculosis EthR are available via the RCSB Protein Data Bank via PDB codes 5F1J, 5IOY, 5IOZ, 5J3L, 5IPA, 5J1R, 5J1U, and 5IP6, respectively.
Supplementary Material
References
- World Health Organization, Global tuberculosis report, 2015; http://www.who.int/tb/publications/global_report/gtbr2015_executive_summary.pdf.
- World Health Organization, 2011/ 2012 tuberculosis global facts, 2011.
- World Health Organization. Guidelines for treatment of tuberculosis, 4th ed.; WHO Press: Geneva, Switzerland, 2010. [Google Scholar]
- Checkley A. M.; McShane H. (2011) Tuberculosis vaccines: progress and challenges. Trends Pharmacol. Sci. 32, 601–606. 10.1016/j.tips.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Anand N.; Upadhyaya K.; Tripathi R. P. (2015) Drug development pipeline for the treatment of tuberculosis: Needs, challenges, success and opportunities for the future. Chem. Biol. Interface 5, 84–127. [Google Scholar]
- Koul A.; Arnoult E.; Lounis N.; Guillemont N.; Andries K. (2011) The challenge of new drug discovery for tuberculosis. Nature 469, 483–490. 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- Andries K.; Verhasselt P.; Guillemont J.; Goehlmann H. W. H.; Neefs J.-M.; Winkler H.; Van Gestel J.; Timmermann P.; Zhu M.; Lee E.; Williams P.; de Chaffoy D.; Huitric E.; Hoffner S.; Cambau E.; Truffot-Pernot C.; Lounis N.; Jarlier V. (2005) A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307, 223–227. 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- Willand N.; Dirié B.; Carette X.; Bifani P.; Singhal A.; Desroses M.; Leroux F.; Willery E.; Mathys V.; Déprez-Poulain R.; Delcroix G.; Frénois F.; Aumercier M.; Locht C.; Villeret V.; Déprez B.; Baulard A. R. (2009) Synthetic EthR inhibitors boost antituberculous activity of ethionamide. Nat. Med. 15, 537–544. 10.1038/nm.1950. [DOI] [PubMed] [Google Scholar]
- Rozwarski D. A.; Grant G. A.; Barton D. H. R.; Jacobs W. R. Jr.; Sacchettini J. C. (1998) Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 279, 98–102. 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- Wang F.; Langley R.; Gulten G.; Dover L. G.; Besra G. S.; Jacobs W. R. Jr.; Sacchettini J. C. (2007) Mechanism of thioamide drug action against tuberculosis and leprosy. J. Exp. Med. 204, 73–78. 10.1084/jem.20062100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt A.; Molle V.; Besra G. S.; Jacobs W. R. Jr.; Kremer L. (2007) The Mycobacterium tuberculosis FAS-II condensing enzymes: their role in mycolic acid biosynthesis, acid-fastness, pathogenesis and in future drug development. Mol. Microbiol. 64, 1442–1454. 10.1111/j.1365-2958.2007.05761.x. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Heym B.; Allen B.; Young D.; Cole S. (1992) The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 358, 591–593. 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
- Engohang-Ndong J.; Baillat D.; Aumercier M.; Bellefontaine F.; Besra G. S.; Locht C.; Baulard A. R. (2004) EthR, a repressor of the TetR/CamR family implicated in ethionamide resistance in mycobacteria, octamerizes cooperatively on its operator. Mol. Microbiol. 51, 175–188. 10.1046/j.1365-2958.2003.03809.x. [DOI] [PubMed] [Google Scholar]
- Baulard A. R.; Betts J. C.; Engohang-Ndong J.; Quan S.; McAdam R. A.; Brennan P. J.; Locht C.; Besra G. S. (2000) Activation of the pro-drug ethionamide is regulated in mycobacteria. J. Biol. Chem. 275, 28326–28331. 10.1074/jbc.M003744200. [DOI] [PubMed] [Google Scholar]
- DeBarber A. E.; Mdluli K.; Bosman M.; Bekker L. G.; Barry C. E. 3rd (2000) Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 97, 9677–9682. 10.1073/pnas.97.17.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flipo M.; Desroses M.; Lecat-Guillet N.; Dirié B.; Carette X.; Leroux F.; Piveteau C.; Demirkaya F.; Lens Z.; Rucktooa P.; Villeret V.; Christophe T.; Jeon H. K.; Locht C.; Brodin P.; Déprez B.; Baulard A. R.; Willand N. (2011) Ethionamide boosters: synthesis, biological activity, and structure–activity relationships of a series of 1,2,4-oxadiazole EthR inhibitors. J. Med. Chem. 54, 2994–3010. 10.1021/jm200076a. [DOI] [PubMed] [Google Scholar]
- Flipo M.; Desroses M.; Lecat-Guillet N.; Villemagne B.; Blondiaux N.; Leroux F.; Piveteau C.; Mathys V.; Flament M.-P.; Siepmann J.; Villeret V.; Wohlkönig A.; Wintjens R.; Soror S. H.; Christophe T.; Jeon H. K.; Locht C.; Brodin P.; Déprez B.; Baulard A. R.; Willand N. (2012) Ethionamide Boosters. 2. Combining bioisosteric replacement and structure-based drug design to solve pharmacokinetic issues in a series of potent 1,2,4-oxadiazole EthR inhibitors. J. Med. Chem. 55, 68–83. 10.1021/jm200825u. [DOI] [PubMed] [Google Scholar]
- Villemagne B.; Flipo M.; Blondiaux N.; Crauste C.; Malaquin S.; Leroux F.; Piveteau C.; Villeret V.; Brodin P.; Villoutreix B. O.; Sperandio O.; Soror S. H.; Wohlkönig A.; Wintjens R.; Déprez B.; Baulard A. R.; Willand N. (2014) Ligand efficiency driven design of new inhibitors of Mycobacterium tuberculosis transcriptional repressor EthR using fragment growing, merging, and linking approaches. J. Med. Chem. 57, 4876–4888. 10.1021/jm500422b. [DOI] [PubMed] [Google Scholar]
- Surade S.; Ty N.; Hengrung N.; Lechartier B.; Cole S. T.; Abell C.; Blundell T. L. (2014) A structure-guided fragment-based approach for the discovery of allosteric inhibitors targeting the lipophilic binding site of transcription factor EthR. Biochem. J. 458, 387–394. 10.1042/BJ20131127. [DOI] [PubMed] [Google Scholar]
- Nikiforov P. O.; Surade S.; Blaszczyk M.; Delorme V.; Brodin P.; Baulard A. R.; Blundell T. L.; Abell C. (2016) A fragment merging approach towards the development of small molecule inhibitors of Mycobacterium tuberculosis EthR for use as ethionamide boosters. Org. Biomol. Chem. 14, 2318–2326. 10.1039/C5OB02630J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prideaux B.; Via L. E.; Zimmerman M. D.; Eum S.; Sarathy J.; O’Brien P.; Chen C.; Kaya F.; Weiner D. M.; Chen P.-Y.; Song T.; Lee M.; Shim T. S.; Cho J. S.; Kim W.; Cho S. N.; Olivier K. N.; Barry C. E. III; Dartois V. (2015) The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat. Med. 21, 1223–1227. 10.1038/nm.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.