

Abstract
Terpenoid synthases catalyze isoprenoid cyclization reactions underlying the generation of more than 80,000 natural products. Such dramatic chemodiversity belies the fact that these enzymes generally consist of only three domain folds designated α, β, and γ. Catalysis by class I terpenoid synthases occurs exclusively in the α domain, which is found with α, αα, αβ, and αβγ domain architectures. Here, we explore the influence of domain architecture on catalysis by taxadiene synthase from Taxus brevifolia (TbTS, αβγ), fusicoccadiene synthase from Phomopsis amygdali (PaFS, (αα)6), and ophiobolin F synthase from Aspergillus clavatus (AcOS, αα). We show that the cyclization fidelity and catalytic efficiency of the α domain of TbTS are severely compromised by deletion of the βγ domains; however, retention of the β domain preserves significant cyclization fidelity. In PaFS, we previously demonstrated that one α domain similarly influences catalysis by the other α domain [Chen, M., Chou, W. K. W., Toyomasu, T., Cane, D. E., Christianson, D. W. (2016) ACS Chem. Biol. 11, 889–899]. Here, we show that the hexameric quaternary structure of PaFS enables cluster channeling. We also show that the α domains of PaFS and AcOS can be swapped so as to make functional chimeric αα synthases. Notably, both cyclization fidelity and catalytic efficiency are altered in all chimeric synthases. Twelve newly formed and uncharacterized C20 diterpene products and three C25 sesterterpene products are generated by these chimeras. Thus, engineered αβγ and αα terpenoid cyclases promise to generate chemodiversity in the greater family of terpenoid natural products.
Graphical abstract
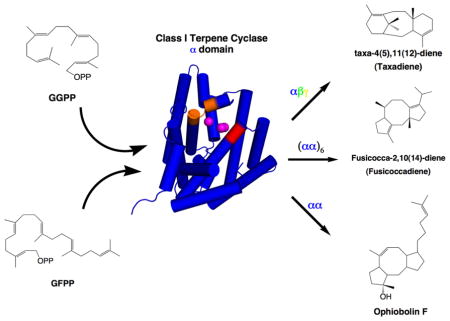
Introduction
More than 80,000 terpenoids have been identified to date, thereby comprising the largest and most diverse family of natural products. Such molecular diversity fosters utility for humankind, since many terpenoids have been widely used as fragrances, insect attractants or repellents, and medicines since times of antiquity.1–5 The molecular complexity of the terpenome belies simple origins in the C5 building blocks isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). Prenyl diphosphate synthases catalyze the condensation of one molecule of DMAPP and one or more molecules of IPP in head-to-tail fashion to form C10, C15, C20, and C25 prenyl diphosphates.6 Terpene cyclases then convert these linear achiral allylic diphosphates into polycyclic terpene hydrocarbons and alcohols through multi-step cyclization cascades.7–13 High-energy carbocation intermediates are hallmarks of these reaction cascades, which cyclases manipulate to direct carbon-carbon bond formation along a wide range of specific pathways. The extraordinary structural diversity of terpenoid natural products arises in large part from the extraordinary functional diversity of terpene cyclases, which are capable of generating myriad carbon skeletons from a single acyclic prenyl diphosphate precursor.
The first crystal structures of terpenoid cyclases were reported twenty years ago.14–16 These structures have guided the exploration of structure-function relationships for both class I cyclases, which initiate carbocation formation through the metal-dependent ionization of the substrate diphosphate group, and class II cyclases, which initiate carbocation formation through the protonation of an isoprenoid carbon-carbon double bond. Diterpene cyclases, which utilize the acyclic C20 substrate geranylgeranyl diphosphate (GGPP), are unique among the greater family of terpenoid cyclases in that both class I and class II enzymes are known. The first crystal structures of class I and class II plant diterpene cyclases revealed a three-domain architecture designated αβγ. Class I cyclization reactions are catalyzed exclusively in the α domain, while class II cyclizations are catalyzed at the interface of the β and γ domains.17–19 Although class I terpene synthases are also found as single α-domain enzymes, such as the bacterial diterpene cyclase cyclooctat-9-en-7-ol synthase (CotB2),20 as well as αβ domain assemblies exemplified by the plant sesquiterpene cyclase 5-epi-aristolochene synthase15 and the plant monoterpene cyclase bornyl diphosphate synthase,21 the occurrence of more complex multi-domain assemblies in terpenoid cyclases raises intriguing questions regarding the relationship between domain architecture and enzyme function. For example, consider taxadiene synthase from Taxus brevifolia (TbTS), a class I diterpene cyclase that catalyzes the first committed step in the biosynthesis of the blockbuster cancer chemotherapeutic drug Taxol.22–24 The crystal structure of TbTS reveals an αβγ domain architecture in which the catalytic function is localized solely in the α domain (Figure 1, Figure S1).17 How, then, does the adjacent β domain, or the distant γ domain, influence catalysis in the α domain of this cyclase?
Figure 1. Domain architecture in αβγ and αα diterpene synthases.

Taxadiene synthase from Taxus brevifolia (TbTS) catalyzes the cyclization of GGPP to form taxa-4(5),11(12)-diene as the major product (henceforth designated simply “taxadiene”). Alternative proton elimination steps yield taxa-4(20),11(12)-diene and taxa-3(4),11(12)-diene as minor products. This cyclization occurs exclusively in the α domain (blue); neither the β domain (green) nor the γ domain (yellow) contain a functional active site. A polypeptide segment connected to the N-terminal helix of the β-domain (magenta) is believed to help cap the active site in the α domain during catalysis. Fusicoccadiene synthase from Phomopsis amygdali (PaFS) is a hexamer of subunits with αα domain architecture. The chain elongation reaction of DMAPP and 3 IPP molecules is catalyzed in the C-terminal GGPP synthase α domain (green), and the GGPP cyclization reaction forming fusicoccadiene is catalyzed in the N-terminal cyclase α domain (blue). Although neither the tertiary structure nor the quaternary structure of ophiobolin F synthase from Aspergillus clavatus (AcOS) are known, this bifunctional sesterterpene synthase also adopts an αα domain architecture in which the chain elongation reaction of DMAPP and 4 IPP molecules is catalyzed in the C-terminal α domain to form GFPP, which then undergoes cyclization in the N-terminal α domain to form ophiobolin F. Detailed catalytic mechanisms for each enzyme are found in Figure S1.
In a contrasting example, consider fusicoccadiene synthase from Phomopsis amygdali (PaFS). This bifunctional diterpene synthase adopts αα domain architecture in which the C-terminal α domain generates GGPP and the N-terminal α domain cyclizes GGPP to form fusicoccadiene.25,26 The tricyclic ring skeleton of fusicoccadiene is critical for the anti-tumor activity of fusicoccin A, a diterpene glycoside that locks 14-3-3 proteins in place with their binding partners.27–29 The recently determined crystal structures of the individual chain elongation and cyclase α domains of PaFS reveal that each utilizes three Mg2+ ions to initiate catalysis.30 Analytical ultracentrifugation and low-angle X-ray scattering experiments indicate that PaFS is a hexamer (Figure 1, Figure S1),30 which raises the possibility of proximity channeling31 between active sites. Proximity channeling may enhance product flux in a biosynthetic pathway even in the absence of a direct tunnel between enzyme active sites, since the product generated in one active site need not completely diffuse into solution and re-diffuse back into the second active site if the two active sites are sufficiently close. The clustering of active sites in such a hexameric assembly may enhance this effect since the product generated in one active site can re-diffuse back into one of multiple second active sites that are nearby as a result of the quaternary structure of the oligomer.32 Thus, multi-domain assembly and oligomerization may enhance the efficiency of cyclic terpene biosynthesis through cluster channeling of the acyclic substrate.13
An αα domain architecture comparable to that of PaFS is found in ophiobolin F synthase from Aspergillus clavatus (AcOS), a bifunctional sesterterpene synthase that first generates C25 geranylfarnesyl diphosphate (GFPP) in the C-terminal α domain and then cyclizes GFPP in the N-terminal α domain to form ophiobolin F.33 This tricyclic sesterterpene is characterized by a 5-8-5 ring skeleton similar to that of fusicoccadiene (Figure 1, Figure S1). Although the crystal structure of AcOS has not yet been determined, PaFS and AcOS share 41% amino acid sequence identity and catalyze GGPP and GFPP cyclization reactions, respectively, that proceed through a common 5–11 ring system intermediate.25,26,33,34 This mechanistic similarity suggests structural similarity between the active site contours of PaFS and AcOS. Pairwise comparisons of active site residues between the two cyclases reveal that while most residues are conserved or isosteric, only two residue pairs substantially differ in size: W225 and V228 in PaFS correspond to L217 and A220 in AcOS, respectively.
Here, we probe the influence of domain architecture on catalytic function in αβγ and αα diterpene cyclases. We demonstrate that the vestigial βγ domains of TbTS influence cyclization fidelity and catalytic efficiency in the α domain of this αβγ cyclase. In PaFS, we demonstrate that αα domain architecture of the wild-type hexamer enables cluster channeling of the acyclic substrate to the cyclase domain. We also show that the α domains of PaFS and AcOS can be swapped to make functional chimeric αα diterpene synthases, while the cyclase domains of PaFS and AcOS can be engineered with just two amino acid substitutions so as to swap substrate specificity for C20 and C25 substrates. Cyclization fidelity and catalytic efficiency are altered in all chimeric synthases studied, however, indicating that domain-domain interactions play a significant role in stabilizing the cyclase domains in catalytically competent conformations.
Materials and Methods
Expression, and purification of TbTS variants
We previously described the pET22bTV plasmid encoding a catalytically-active24 pseudomature TbTS truncation variant lacking the N-terminal plastidial targeting sequence, initially designated “M79-TXS” by Köksal and colleagues.17 Here, this variant is referred to as TbTS. This vector was used to prepare all TbTS variants described herein as summarized in Figure 2. The γ domain of TbTS (S136-Y348) is an insertion domain between the first and second helix of the β domain. The γ domain was spliced out of the full-length protein using assembly PCR to yield an αβ construct designated “TbTSαβ” (Figure 2; primers are listed in Table S1). The N-terminal segment M79-I135 includes the first helix of the β domain and remained intact in this construct; this segment is believed to help cap the active site upon substrate binding, as observed in 5-epi-aristolochene synthase and bornyl diphosphate synthase.15,21 Flanked by NdeI and BamHI restriction sites, the gene sequence encoding TbTSαβ was ligated into plasmid pET22bTV and transformed into Escherichia coli DH5α cells. The correct gene sequence was confirmed by DNA sequencing at the University of Pennsylvania Perelman School of Medicine Sequencing Facility. The TbTSαβ protein was expressed but suffered from intractable aggregation problems, so the construct was modified so as to insert a single glycine residue at the splice site. Guided by molecular modeling, it was hypothesized that this additional residue would improve the geometry of the loop connecting the first and second helix of the β domain. This modified construct, designated TbTSαβG, was prepared using site-directed insertion through PCR amplification (Figure 2; primers are listed in Table S1). The resulting PCR product was digested with DpnI for 1 h then transformed into E. coli DH5α cells. The correct gene sequence was confirmed by sequencing.
Figure 2. Primary structure of truncated and chimeric terpenoid synthases.

(a) For the class I diterpene synthase taxadiene synthase (TbTS), four engineered domain constructs successively downsized the αβγ domain architecture, color-coded as follows: α domain, blue; β domain, green; γ domain, yellow; N-terminal segment, magenta. The location of the single inserted glycine residue (G) in TbTSαβG is indicated by an arrow. The TbTSαβ′ is a chimera containing the N-terminal segment (pink) and part of the β domain (β′, red) of isoprene synthase. (b) Engineered bifunctional class I diterpene and sesterterpene synthases with αα domain architecture are color-coded as follows: dark blue, cyclization α domain of fusicoccadiene synthase (PaFS); orange, cyclization α domain of ophiobolin F synthase (AcOS); aquamarine, GFPP synthase α domain of stellatatriene synthase (EvSS); green, GGPP synthase α domain of fusicoccadiene synthase (PaFS).
The native forms of many class I plant terpene synthases are similarly found as αβ domain assemblies. For example, isoprene synthase from Populus x canescens (PcIS) has an active site in the α domain and an N-terminal β domain with a similar N-terminal capping segment.35 It is possible that removal of the γ domain exposes a large hydrophobic patch (the former domain-domain interface) on the β domain of TbTS. To probe the effects of a modified β domain “pre-evolved” to have a hydrophilic solvent-exposed surface in the absence of a γ domain, the α domain (S553-V862) and 120 residues from the β domain (L432-Q552) of TbTS were spliced together with the complementary portion of the β-domain of PcIS (M1-F151) using assembly PCR to result in the 581-residue chimera designated TbTSαβ′ (Figure 2). PCR primers are listed in Table S1.
Finally, a minimalist truncation variant, designated TbTSα (Figure 2), was prepared by completely splicing out the β and γ domains and connecting the N-terminal segment M79-I135 directly to the N-terminus of the first helix of the α domain, starting at N537. This was achieved using assembly PCR (primers are listed in Table S1). Flanked by NdeI and BamHI restriction sites, the gene sequence encoding TbTSα was ligated into plasmid pET22bTV and transformed into E. coli DH5α cells. The correct gene sequence was confirmed by DNA sequencing.
TbTSα, TbTSαβG, and TbTSαβ′ were transformed into E. coli BL21 (DE3) RIL-CodonPlus (Novagen) cells and grown on LB-Agar plates with 50 μg/mL ampicillin and 34 μg/mL chloramphenicol overnight at 37 °C. For the preparation of each construct, individual colonies were transferred to two 15-mL LB broth cultures with 50 μg/mL ampicillin and 34 μg/mL chloramphenicol and allowed to grow at 37 °C with aeration for 12–16 h. A total of 6 × 1 L LB Broth cultures containing 50 μg/mL ampicillin and 34 μg/mL chloramphenicol were inoculated with 5 mL of the cultures grown overnight and allowed to shake at 37 °C until cells reached OD600 = 1.0. Cells were induced with 500 μM isopropyl-β-D-1-thiogalactopyranoside (IPTG) and the temperature was reduced to 16 °C overnight. Cells were pelleted at 5400g for 20 min. The pellet was harvested, flash frozen in liquid nitrogen and stored at −20 °C.
Cells with TbTSαβG were thawed and resuspended in buffer A [20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) (pH 8.2), 300 mM NaCl, 1 mM tris-(2-carboxyethyl)phosphine (TCEP), 10% glycerol]. Cells were lysed by sonication and spun at 37000g for 1 h. Supernatant was loaded onto a His60 Superflow Resin (Clontech) column using buffer A. The column was washed with 30 mM imidazole and then 300 mM imidazole. The 300 mM fractions were pooled and dialyzed overnight at 4 °C in buffer B [50 mM Tris (pH 7.5), 1 mM TCEP, 10% glycerol]. Pooled fractions were loaded onto a 5-mL Hi-Trap Q HP column (GE Healthcare). The enzyme was eluted using a gradient of 0.0–1.0 M NaCl. Fractions were analyzed using SDS-PAGE, and fractions containing protein of the desired molecular weight were again pooled and dialyzed overnight in buffer C [50 mM Tris (pH 7.5), 300 mM NaCl, 1 mM TCEP, 10% glycerol]. TbTSαβ′ was expressed and purified in a similar fashion.
Cells with TbTSα were thawed and resuspended in buffer D [50 mM Tris (pH 7.5), 300 mM NaCl, 1 mM TCEP, 1% Tween20]. Cells were lysed by sonication and then spun at 37000g for 1 h. The supernatant was discarded. The pellet was resuspended in buffer D to a final volume of 50 mL. The pH of the resuspended pellet was gradually increased to 12. The pellet was then allowed to mix at 4 °C for 30 min. The pH was then gradually decreased to 7.5 and allowed to mix at 4 °C for 30 min. This mixture was then spun at 37000g for 1 h. Supernatant was loaded onto a Ni-NTA column using buffer D and purified as described for TbTSαβG. Enzymes were concentrated using Amicon centrifuge tubes with 10-kDa pore size for TbTSα and 30-kDa pore size for TbTSαβG and TbTSαβ′. Enzymes were concentrated to approximately 8 mg/mL as determined by absorbance at 280 nm.
Enzyme assays were performed as described previously for TbTS and other terpenoid cyclases.23,36 Briefly, enzymes were solubilized in buffer E [30 mM HEPES (pH 8.2), 1 mM MgCl2, 1mM TCEP, 10% glycerol]. Assays were performed in triplicate using a range of 0–15 μM [1-3H]GGPP (100 mCi/mmol, American Radiolabeled Chemicals, Inc.) for full-length TbTS, 0–3 μM [1-3H]GGPP (100 mCi/mmol) for TbTSαβG, and 0–25 μM [1-3H]GGPP (100 mCi/mmol) for TbTSα. The final concentration of each construct used for assays was determined to be that at which the dependence of product formation on enzyme concentration was linear and the level of substrate turnover was less than 10%. A 500-μL reaction mixture was prepared and allowed to react for 10 min at room temperature before being quenched with 20 μL of 500 mM ethylenediaminetetraacetic acid (EDTA) at pH 8.0. The extraction of diterpene products was achieved by adding 1 mL of hexane to the reaction mixture and vortexing for 20 s. The hexane layer was removed and passed over a silica gel column (1 cm of silica gel 230–400 mesh, Grade 60, in a Pasteur pipette) into a scintillation vial containing 5 mL of scintillation solution (Ecoscint O, National Diagnostics) and the process was repeated two more times. A Beckman scintillation counter (LS6500) was used to measure total product formation. Data for the rate of product formation as a function of substrate concentration were fit by nonlinear regression using GraphPad Prism software to determine steady-state kinetic parameters kcat and KM based on the known total enzyme concentration.
For product analysis, reaction mixtures were set up with a total volume of 5 mL of buffer E containing 1.7 μM enzyme and 100 μM GGPP. The reaction mixture was overlaid with 3 mL of n-pentane and allowed to react for approximately 20 h at room temperature. The reaction mixture was extracted three times with n-pentane; pentane extracts were dried with anhydrous MgSO4, filtered, and concentrated on an ice/water mixture under reduced pressure until the volume reached 100 μL. Extracts were analyzed using an Agilent 5890 Series II mass spectrometer with a 30 m × 0.25 mm HP5MS capillary column in EI+ mode using a temperature program with an initial hold at 60 °C for 2 min, a 20 °C/min temperature gradient from 60 to 280 °C, a final hold at 280 °C for 2 min, and a solvent delay of 3 min. Analysis of the organic extracts resulting from the incubation of GGPP with the mutant cyclases by gas chromatography and mass spectrometry (GC-MS) revealed the formation of mixtures of diterpene hydrocarbons at m/z 272. Compounds were identified by comparison of their individual mass spectra and chromatographic retention indices with those of authentic compounds using the MassFinder 4.0 Program and Database and the NIST Mass Spectral Library.37
Expression and purification of PaFS and AcOS chimeras
The plasmid for PaFS was prepared as previously described.30 Plasmids for AcOS and stellatatriene synthase (from which the GFPP synthase α domain was utilized) were synthesized and codon-optimized by Genscript. The AcOS gene was on a pET28a vector, using NdeI and BamHI as restriction sites. For cloning chimeric enzymes, a pET His6 TEV LIC cloning vector (a generous gift from Dr. Scott Gradia, Addgene ID: 29653) with an N-terminal hexahistidine tag was used unless otherwise specified. Primers used for cloning and site-directed mutagenesis are listed in Table S1. DNA sequencing was performed at the DNA Sequencing Facility at the Perelman School of Medicine, University of Pennsylvania.
For expression of AcOS, E. coli BL21(DE3) cells (Novagen) were transformed with the AcOS plasmid and grown on an LB-Agar plate with 50 μg/mL kanamycin overnight at 37 °C. A single colony was transferred to a 5-mL LB culture with 50 μg/mL kanamycin, which grew at 37 °C with aeration for 8–12 h. A total of 6 × 1 L 2XYT culture containing 50 μg/mL kanamycin was inoculated with 6 × 5mL LB culture, and was shaken at 37 °C until OD600 reached 1.0. Protein expression was induced at 16 °C by adding IPTG to a final concentration of 50 μM. After expression for 18–22 h, cells were pelleted via centrifuge (5400g for 15 min) and stored at −80 °C.
All four chimera protein constructs (Figure 2) were expressed in E. coli BL21(DE3) RIL-CodonPlus strain (Novagen). A total of 6 × 1 L 2XYT medium containing 50 μg/mL kanamycin and 34 μg/mL chloramphenicol was inoculated with 5 mL starting culture and grown at 37 °C with aeration. Cells were induced with 50 μM IPTG at 16 °C when OD600 reached 0.8–1.0. After expression for 18 h, cells were pelleted by centrifugation (5400g for 15 min).
For each construct, the cell pellet was resuspended in Ni-NTA (QIAGEN) wash buffer [50 mM K2HPO4 (pH 7.7), 300 mM NaCl, 10% (v/v) glycerol, 3 mM TCEP]. Cells were lysed by sonication for 10 min. Cell debris was discarded after centrifugation, and supernatant was loaded on a Ni-NTA column (QIAGEN). Protein was eluted with a 0–400 mM imidazole gradient in Ni-NTA wash buffer. Protein fractions were combined, concentrated, exchanged with buffer Q [10 mM Tris (pH 8.0), 10 mM NaCl] and loaded on a 5-mL HiTrap Q HP column (GE Healthcare). Elution was performed with a 0–500 mM NaCl gradient in buffer Q. Fractions were analyzed by SDS-PAGE, combined, concentrated, and loaded onto a 26/60 Superdex 200 size-exclusion column (GE Healthcare) pre-equilibrated with 10 mM Tris (pH 7.9), 200 mM NaCl, 5% (v/v) glycerol, and 3 mM TCEP. Fractions of the expected molecular weight were combined and concentrated.
Diterpene synthase assays on a 100-μL scale were conducted in triplicate, in similar fashion as described above for TbTS assays, using 0.16 nM AcOS, 0.43 nM AcOSα-PaFSα, or 42 nM AcOSα′-PaFSα in assay buffer [25 mM HEPES (pH 8.0), 200 mM NaCl, 5 mM MgCl2]. Concentration ranges of [1-3H]-GGPP were adjusted accordingly. Reactions were initiated by adding enzyme at 30 °C, and quenched with 10 μL 0.5 M EDTA. A 1-mL aliquot of hexane was used to extract hydrocarbon products twice with vigorous vortexing and subsequently loaded onto a 1-mL silica gel column (1 cm of silica gel 230–400 mesh, Grade 60, in a Pasteur pipette). The column was washed with 1 mL hexane and 1 mL ethyl ether. Eluate was collected into a scintillation vial containing 5 mL of scintillation solution (Ecoscint O, National Diagnostics), and radioactivity counts were determined using a Beckman liquid scintillation counter (LS6500). Control experiments were performed to determine background counts, and data were processed with GraphPad Prism software.
Product assays were carried out in duplicate on a 5-mL scale in similar fashion to that described above for TbTS assays. To calibrate peak integrals, nitrobenzene (NBZ) was added to each reaction mixture to serve as internal standard. Reactions were initiated at 30 °C by adding enzyme into assay buffer containing substrate (DMAPP and IPP) overlaid with 5 mL n-pentane. After quenching by EDTA after 16 h of reaction, hydrocarbon products were extracted from the reaction mixture with three 5-mL portions of n-pentane and the extracts were run through anhydrous MgSO4 to eliminate trace amounts of water. Samples were then concentrated under vacuum in an ice water bath using a rotary evaporator and analyzed by GC-MS. Mixtures of diterpene hydrocarbons were detected at m/z 272, some of which were directly identified by comparison of their individual mass spectra and chromatographic retention indices with those of authentic compounds using the MassFinder 4.0 Program and Database.37 Sesterterpene hydrocarbons were detected at m/z 340.
PaFS-TbTS substrate competition assays
PaFS and TbTS were expressed and purified as described above. Monomer concentrations of 1 μM PaFS and 1 μM TbTS in 6 mL of assay buffer [25 mM HEPES pH 8.0, 200 mM NaCl, 5 mM MgCl2] were co-incubated with the following substrates in two separate experiments: (1) 50 μM DMAPP + 150 μM IPP, and (2) 50 μM GGPP. The samples were overlaid with 5 mL HPLC grade n-pentane in a glass test tube and incubated at 30 °C for 16 h. The reaction mixture was extracted four times with 5 mL n-pentane (total 20 mL n-pentane extracts). The pentane extracts were dried with MgSO4, filtered, and concentrated in an ice/water bath under reduced pressure to approximately 100 μL. Reaction mixture extracts were analyzed with an Agilent mass spectrometer with a 30 m × 0.25 mm HP5MS capillary column using a temperature program with an initial 2 min hold at 60 °C, a 20 °C/min temperature gradient from 60 °C to 280 °C, and a final 2 min hold at 280 °C. Analysis of the pentane extracts resulting from co-incubation of PaFS and TbTS with various substrates by GC-MS showed the formation of diterpene products. Compounds were identified by comparison of their mass spectra and GC retention indices with those of authentic compounds using the MassFinder 4.0 Program and Database.37 Since the catalytic fidelities of PaFS and TbTS are comparable for the generation of their major products (PaFS generates 64–72% fusicoccadiene30 and TbTS generates 58–82% taxadiene (Table 1)), the fusicoccadiene:taxadiene ratio measured in the GGPP competition assay provides an accurate reflection of the ratio of steady-state kinetic parameters (kcat/KM)PaFS/(kcat/KM)TbTS for GGPP utilization by individual PaFS and TbTS enzymes, even though these parameters reflect the generation of all diterpene products and not just the major product of each enzyme.
Table 1.
Product arrays generated by TbTS mutantsa
| Retention Index (calc) | Retention Index (ref) | Compound | TbTS run 1 (%) | TbTS run 2 (%) | TbTSαβG (%) | TbTSαβ′ (%) | TbTSα run 1 (%) | TbTSα run 2 (%) |
|---|---|---|---|---|---|---|---|---|
| 1893 | 1922b |
 geranylmyrcene, a.k.a. β-springene |
1.7 | – | 20.3 | 12.6 | – | – |
| 1912 | – |
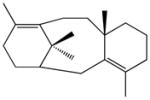 taxa-3(4),11(12)-dienec |
2.7 | – | – | – | – | – |
| 1919 | – | unknown diterpene 1 | – | 16.8 | – | – | – | – |
| 1954 | – | unknown diterpene 2 | 1.5 | – | – | – | – | – |
| 1965 | – | unknown diterpene 3 | 0.3 | 10.0 | 7.1 | 8.6 | 53.9 | 24.2 |
| 1970 | – | unknown diterpene 4 | 3.0 | 5.7 | 1.7 | – | – | |
| 1998 | – |
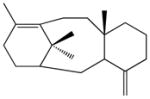 taxa-4(20),11(12)-dienec |
7.2 | 15.0 | 4.3 | 4.9 | – | – |
| 2008 | 2008d |
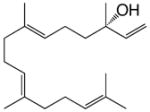 geranyllinalool |
1.4 | 21.0 | 16.0 | 46.1 | 18.3 | |
| 2043 | – |
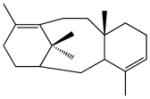 taxa-4(5),11(12)-diene (taxadiene)c |
82.0 | 58.2 | 25.9 | 37.6 | ||
| 2066 | – | unknown diterpene 5 | 0.1 | – | 15.6 | 18.5 | – | 38.9 |
| 2173 | 2167d |
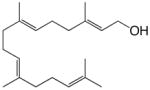 geranylgeraniol |
– | – | – | – | – | 18.7 |
Results
TbTS truncation and chimera variants
Truncated domain constructs were prepared to study the effects of the β and γ domains on the class I GGPP cyclase activity that resides solely in the α domain of full-length αβγ TbTS. The γ domain was spliced out and replaced by a single glycine residue in the di-domain construct TbTSαβG, and both β and γ domains were spliced out to yield the single-domain construct TbTSα. These constructs retained the N-terminal segment M79-I135 of the full-length pseudomature enzyme, since this segment is believed to help cap the active site during catalysis. In TbTSαβ′, approximately half of the β domain of TbTSαβG was replaced by the β domain of PcIS. Since deletion of the γ domain in TbTSαβG exposes nonpolar surface area on the β domain at the former βγ domain interface, the utilization of a related β domain already evolved to be solvent-exposed at this region in TbTSαβ′ might potentially be beneficial. The primary structure of each engineered construct is schematically illustrated in Figure 2.
The double-domain construct TbTSαβG was obtained in very poor yield after purification (0.5–1 mg/L culture, a level approximately 10-fold lower than that of wild-type TbTS), but sufficient quantities of pure protein could be prepared for kinetic and product array assays. Michaelis-Menten kinetics indicated a 2750-fold loss of catalytic efficiency (kcat/KM) for TbTSαβG relative to full-length TbTS (Figure 3). Thus, an intact γ domain is required for 99.9% of the maximum catalytic activity in the α domain of TbTS, even though the γ domain makes no direct contact with the α domain. Comparisons of the diterpene product arrays of full-length TbTS and TbTSαβG using GC-MS are similarly informative. Full-length TbTS generates taxadiene as its major cyclization product (58–82%), along with 9.9–15.0% taxadiene isomers taxa-3(4)11(12)-diene and taxa-4(20),11(12)-diene, and 8.0–26.8% diterpene side products; interestingly, some run-to-run variations are observed (Table 1; mass spectra of diterpene products are reported in Figure S2). TbTSαβG still generates taxadiene as its major product, although cyclization fidelity is somewhat compromised since the major cyclization products are 25.9% taxadiene and 4.3% taxadiene isomers. This di-domain construct also generates a mixture of the linear solvolysis product geranyllinalool (21.0%) and the elimination product geranylmyrcene (20.3%), in addition to small amounts of other unidentified diterpenes. Comparisons of the fragmentation patterns in mass spectra of unidentified diterpenes with those of known cyclic and linear diterpenes suggest that unknown 4 is a cyclic diterpene, whereas unknowns 3 and 5 are linear diterpenes (Figure S2). With the formation of taxadiene thus compromised only 3-fold while the kcat/KM is decreased by 2750-fold, cyclization fidelity appears to be less susceptible to the deletion of the γ domain than is catalytic efficiency in TbTSαβG.
Figure 3. Catalytic activity measurements.

(a) The cyclization of GGPP by full-length TbTS exhibits Michaelis-Menten kinetics with kcat = 0.16 s−1, KM = 2.9 μM, and kcat/KM = 5.5 × 104 M−1s−1. The highest concentrations of GGPP are not included in the final curve due to substrate inhibition. (b) The cyclization of GGPP by TbTSαβG exhibits Michaelis-Menten kinetics with kcat = 0.000019 s−1, KM = 0.96 μM, and kcat/KM = 20 M−1s−1.
To ascertain whether the substitution of part of an alternative β domain for the βγ domains of TbTS would support catalysis, the N-terminal β domain of PcIS was substituted for the β domain of TbTSαβG to generate the chimera TbTSαβ′. This chimera was obtained in slightly better yield (approximately 1–2 mg/L culture) compared with TbTSαβG. TbTSαβ′ retains the ability to generate taxadiene as its major cyclization product (37.6%) and one taxadiene isomer (4.9%). This chimera also generates the linear solvolysis product geranyllinalool (16.0%) and elimination product geranylmyrcene (12.6%), as well as small amounts of additional unidentified diterpenes almost identical in composition to the unknown diterpenes generated by TbTSαβG (Table 1). Again, comparisons of the fragmentation patterns in mass spectra of unidentified diterpenes with those of known cyclic and linear diterpenes suggest that unknown 4 is cyclic and unknowns 3 and 5 are linear diterpenes (Figure S2). The catalytic activity of TbTSαβ′ was sufficiently low that the steady-state kinetic parameters could not be measured.
Finally, deletion of both the β and γ domains yields the single-domain construct TbTSα. In full-length TbTS, a 148-residue helix (N537-F685) connects the α domain to the β domain. This polypeptide segment was retained in TbTSα and connected to the N-terminal segment M79-I135 believed to help cap the active site during catalysis. The yield of TbTSα was comparable to that of TbTSαβ′ (ca. 1–2 mg/L culture), but the expressed protein was insoluble. A basic extraction protocol was performed which rescued much of the insoluble enzyme from the pellet. We were unable to measure Michaelis-Menten kinetics for TbTSα due to very low activity. Product array analysis revealed a complete absence of taxadiene; instead, significant proportions of the acyclic diterpene alcohols geranyllinaool (18.3–46.1%) and geranylgeraniol (0.0–18.7%), as well as 53.9–63.1% of new unidentified diterpenes 3 and/or 5, were generated. Here, too, some run-to-run variations were observed in product arrays (Table 1). The mass spectra of unknown diterpenes 3 and 5 share similar fragmentation patterns with linear diterpenes (Figure S2), suggesting that these are also linear diterpene products. Thus, deletion of both β and γ domains not only compromises catalytic efficiency, but also appears to abolish the ability to cyclize GGPP since only linear diterpene products are observed.
Taken together, these results indicate that, although the β and γ domains do not directly participate in the chemistry of cyclization of GGPP by the α domain, these domains influence catalysis by their direct or indirect interaction with the α domain, presumably by stabilizing the α domain in a catalytically competent conformation. The effects of the β and γ domains on catalytic efficiency are more pronounced than on cyclization fidelity. While retention of the β domain preserves substantial taxadiene formation, complete deletion of the β and γ domains abolishes taxadiene formation with production of other diterpene products.
Cluster channeling in PaFS
We previously reported that PaFS adopts hexameric (αα)6 quaternary structure in solution.30 In addition to a modest two-fold enhancement of product flux due to covalent linkage of the GGPP synthase and GGPP cyclase domains on the same polypeptide chain,30 the hexameric quaternary structure is proposed to enhance the efficiency of fusicoccadiene formation through cluster channeling.13 Before preparing chimeric synthases with PaFS, we decided to evaluate the possibility of cluster channeling in the wild-type enzyme hexamer, since this potential catalytic advantage would likely be compromised in chimeric synthases that do not maintain hexameric quaternary structure.
When exogenous GGPP is utilized as a substrate, the catalytic efficiencies (kcat/KM) for individual full-length PaFS30 and TbTS (see below) are 3.3 × 104 M−1 s−1 and 2.1 × 103 M−1 s−1, respectively. Theoretically, the ratio of the catalytic efficiencies for GGPP cyclization by the two enzymes, (kcat/KM)PaFS/(kcat/KM)TbTS, represents their ability to compete for a common exogenous substrate. In other words, equimolar PaFS and TbTS should yield a fusicoccadiene:taxadiene product ratio of approximately 16:1 when incubated with exogenous GGPP (i.e., in the absence of any possible channeling). Experimentally, using GC-MS we measure a fusicoccadiene:taxadiene product ratio that is slightly lower, 4.3:1, when equimolar PaFS and TbTS are incubated with exogenous GGPP (Figure 4).
Figure 4. Substrate competition between PaFS and TbTS.
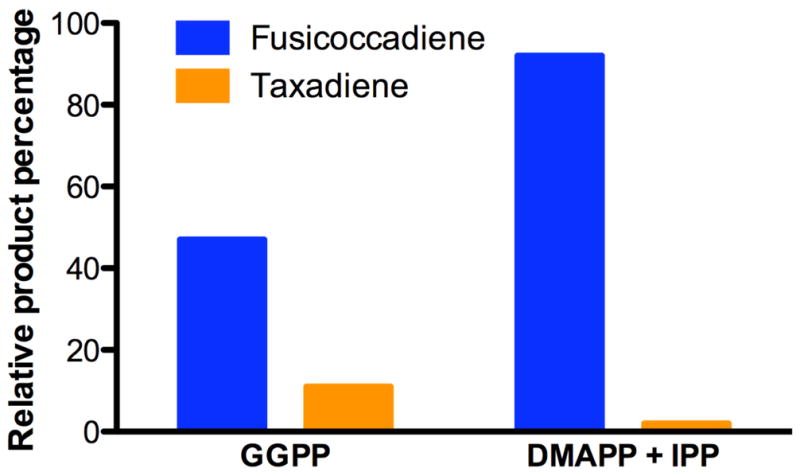
Relative product percentages for the generation of fusicoccadiene and taxadiene in reaction mixtures containing equimolar PaFS and TbTS. When the enzyme mixture is incubated with exogenous GGPP, the fusicoccadiene:taxadiene product ratio is 4.3:1. When the enzyme mixture is incubated with DMAPP and IPP, the only source of cyclization substrate GGPP is that generated by the C-terminal α domain of PaFS and the resulting fusicoccadiene:taxadiene product ratio increases to 46:1. That very little taxadiene is generated in this experiment strongly suggests a cluster channeling model for bifunctional catalysis, in which most of the GGPP generated remains bound in the PaFS hexamer to be utilized for cyclization to fusicoccadiene.
Strikingly, when equimolar PaFS and TbTS are co-incubated with one equivalent of DMAPP and three equivalents of IPP, such that the only source of GGPP is that generated by the GGPP synthase domain of PaFS, the resulting fusicoccadiene:taxadiene product ratio is 46:1 (Figure 4). This excess is much larger than both the theoretical maximum of 16:1 and the experimentally-measured value of 4.3:1 when PaFS and TbTS compete for exogenously supplied GGPP. These results suggest that the GGPP synthesized by the chain elongation domain of PaFS remains mostly associated with the hexamer instead of dissociating to bulk solution and then rebinding to the cyclase domain. If GGPP were instead to dissociate into bulk solution, an equal probability of cyclization by PaFS and by TbTS would result in an increase in taxadiene formation. These results therefore suggest that the (αα)6 hexameric quaternary structure of PaFS enables cluster channeling with a resultant enhancement in fusicoccadiene formation.
Chimeric αα domain cyclases
Four different chimeric αα bifunctional synthases were constructed using the cyclase α domains of PaFS and AcOS, the GGPP synthase α domain of PaFS, and the GFPP synthase α domain of Emericella variecolor stellatatriene synthase (EvSS) (Figure 2). We used the GFPP synthase α domain of EvSS instead of the GFPP synthase domain of AcOS because the latter generates GGPP as a side product,33 whereas the former is reported to generate solely GFPP when incubated with DMAPP and IPP.38,39 In accord with the αα architecture of the wild-type enzymes, the cyclase domain is the N-terminal α domain and the chain elongation domain is the C-terminal α domain in all chimeras prepared in the current study.
In the first step of converting the C20 diterpene synthase PaFS into a C25 sesterterpene synthase, we swapped the C-terminal GGPP synthase α domain of PaFS with the GFPP synthase α domain of EvSS to ensure the generation of the C25 cyclization substrate GFPP. The resulting chimeric bifunctional αα domain enzyme was designated PaFSα-EvSSα (i.e., the N-terminal cyclase α domain of fusicoccadiene synthase linked to the C-terminal GFPP synthase α domain of EvSS). To complete the conversion into a sesterterpene synthase, we made the double mutant W225L/V228A in the N-terminal cyclase domain of this chimera, designated PaFSα′-EvSSα, to enlarge the active site so as to mimic that of AcOS and thus better accommodate GFPP.
To convert the bifunctional sesterterpene synthase AcOS into a C20 diterpene synthase, we replaced its C-terminal GFPP synthase domain with the GGPP synthase domain of PaFS. The resulting chimeric bifunctional αα domain enzyme was designated AcOSα-PaFSα. Additionally, we made the double mutant L217W/A220V in the N-terminal cyclase domain of this chimera, designated AcOSα′-PaFSα, in order to constrict the active site so as to mimic that of PaFS and better accommodate GGPP.
PaFSα-EvSSα and PaFSα′-EvSSα chimeras
Upon incubation with DMAPP and IPP at a ratio of 1:10, GC-MS analysis indicates that PaFSα-EvSSα generates no cyclic sesterterpenes (Table 2, Figure S3), confirming that the active site of the wild-type PaFS cyclase domain is too small to accommodate GFPP. However, this chimera yields sesquiterpene and diterpene products resulting from the farnesyl diphosphate (FPP) and GGPP intermediates generated by the GFPP synthase domain. Although the GFPP synthase domain of EvSS generates solely GFPP in the wild-type enzyme,38 its detachment and utilization in the PaFSα-EvSSα chimera apparently relaxes the chain elongation specificity and allows for the generation and release of FPP and GGPP intermediates en route to the generation of GFPP. Accordingly, the predominant product generated by the PaFSα-EvSSα chimera is fusicoccadiene (69.1%), but the C15 alcohols farnesol and nerolidol are also formed (total 30.9%). The farnesol:nerolidol ratio of 25.9:5.0 suggests that solvolysis is enzyme-catalyzed, since nonenzymic FPP solvolysis ordinarily yields a 1:3–4 ratio.40,41 While we were unable to measure steady-state kinetics for these chimeras due to the lack of a sesterterpene synthase assay with a C25-radiolabeled substrate, these chimeras are nonetheless interesting due to changes in their product arrays brought about by the mutations.
Table 2.
Product arrays generated by αα synthase chimerasa
| Retention Index (calc) | Retention Index (ref) | Compound | PaFSα-EvSSα (%) | PaFSα′-EvSSα (%) | AcOSα-PaFSα (%) | AcOSα′-PaFSα (%) |
|---|---|---|---|---|---|---|
| 1539 | 1553b | nerolidol | 5.0 | – | 0.1 | 5.0 |
| 1695 | 1694b | farnesol | 25.9 | – | – | – |
| 1843 | – |
 fusicoccadienec |
69.1 | – | – | – |
| 1888 | – | unknown diterpene 6 | – | – | 8.4 | 5.8 |
| 1928 | – | unknown diterpene 7 | – | – | 7.1 | 6.1 |
| 1937 | – | unknown diterpene 8 | – | – | 7.0 | 22.1 |
| 1964 | 1962b |
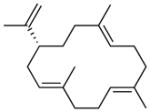 cembrene A |
– | – | – | 9.8 |
| 2002 | 2008b |
geranyllinalool |
– | – | 18.1 | 13.3 |
| 2016 | – | unknown diterpene 9 | – | – | 4.3 | – |
| 2128 | – | unknown diterpene 10 | – | – | 3.0 | – |
| 2140 | – | unknown diterpene 11 | – | – | 5.7 | – |
| 2149 | – | unknown diterpene 12 | – | – | 6.7 | 12.2 |
| 2162 | 2167b |
geranylgeraniol |
– | – | 25.4 | – |
| 2214 | – | unknown diterpene alcohol | – | – | 14.2 | 25.6 |
| 2299 | – | unknown sesterterpene 1 | – | 40.7 | – | – |
| 2338 | – | unknown sesterterpene 2 | – | 14.5 | – | – |
| 2400 | – | unknown sesterterpene 3 | – | 44.8 | – | – |
With its enlarged cyclase active site, the PaFSα′-EvSSα chimera exclusively generates three unknown sesterterpene products (Table 2). The mass spectra of these C25 products indicate molecular weights of 340 and hence 6 degrees of unsaturation (Figure S3); GC-MS analysis further indicates that these unidentified sesterterpenes are distinct from both acyclic geranylfarnesene and tricyclic ophiobolin F (mass spectra of all products generated are reported in Figure S3). These results demonstrate that just two site-specific mutations readily enlarge the active site of the PaFS cyclase domain so as to preferentially accommodate a C25 substrate instead of a C20 substrate.
AcOSα-PaFSα and AcOSα′-PaFSα chimeras
Upon incubation with DMAPP and IPP in a ratio of 1:10, AcOSα-PaFSα and AcOSα′-PaFSα chimeras yield only sesquiterpene and diterpene products (Table 2). Thus, the GGPP synthase domain of PaFS is not functionally compromised by its fusion to the cyclase domain of AcOS in these chimeras.
A total of 11 terpenes are generated by AcOSα-PaFSα (Table 2). Some of these products are conclusively identified on the basis of GC-MS data (Figure S3), but the majority of these products are newly formed and uncharacterized diterpenes. A minor amount of nerolidol is detected (0.1%), indicating release of intermediate FPP in the chain elongation reaction catalyzed by the C-terminal GGPP synthase domain. This broad product array suggests that there is significant flexibility in the binding of GGPP in the active site of the AcOS cyclase domain that normally accommodates the larger substrate GFPP. A more loosely-held substrate can adopt alternative conformations leading to the generation of a wider product array upon initial carbocation formation. However, constriction of the AcOS cyclase domain through the L217W and A220V mutations in AcOSα′-PaFSα reduces the products array to 8 compounds, including newly generated cembrene A (9.8%) (Table 2). While AcOSα-PaFSα generates 43.5% of the linear alcohols geranylgeraniol and geranyllinalool, AcOSα′-PaFSα generates 13.3% geranylgeraniol as the sole linear alcohol. A diterpene alcohol of unknown structure is also generated by AcOSα-PaFSα and AcOSα′-PaFSα (14.2% and 25.6%, respectively). These results suggest that while the constricted active site of AcOSα′-PaFSα cannot enforce a GGPP binding conformation leading to the formation of the core 5-8-5 ring structure of fusicoccadiene, it can enforce an alternative binding conformation leading to a 1,14-cyclization reaction that yields cembrene A, among other diterpene products.
We measured the steady-state kinetics of GGPP cyclization by these chimeric enzymes using a radiolabeled substrate (Table 3, Figure 5). Although the turnover number (kcat) of AcOSα-PaFSα is greater than that of wild-type AcOS, an increase in KM results in equal catalytic efficiencies (kcat/KM) for the generation of diterpenes by AcOSα-PaFSα and wild-type AcOS. For AcOSα′-PaFSα, with its cyclase active site constricted by the L217W and A220V mutations, catalytic efficiency is significantly reduced (Table 3). Interestingly, the utilization of GGPP by AcOSα′-PaFSα did not follow Michaelis-Menten kinetics; while curve-fitting is less than ideal, we interpreted these data as reflecting cooperativity with a Hill coefficient of 1.8. This chimera exhibited a catalytic efficiency approximately 100-fold lower than that of AcOSα-PaFSα. Curiously, the fusion of the PaFS GGPP synthase domain with the AcOS cyclase domain in AcOSα-PaFSα is not sufficient to induce cooperativity – only when the L217W and A220V mutations are introduced is cooperativity observed.
Table 3.
Steady-state kinetics for wild-type and chimeric bifunctional αα terpene synthases with substrate GGPP
| Enzyme | PaFSa | AcOS | AcOSα-PaFSα | AcOSα′-PaFSα |
|---|---|---|---|---|
| kcat (s−1) | 0.021 ± 0.002 | 0.0069 ± 0.001 | 0.011 ± 0.002 | 0.0012 ± 0.0003 |
| KM (μM) | 0.63 ± 0.03 | 0.024 ± 0.01 | 0.038 ± 0.01 | 0.48 ± 0.2 |
| h | 2.2 | 1 | 1 | 1.8 |
| kcat/KM (M−1s−1) | 3.3 × 104 | 2.9 × 105 | 2.9 × 105 | 2.5 × 103 |
From ref. 30.
Figure 5. Steady-state kinetics.

(a) AcOS and AcOSα-PaFSα exhibit Michaelis-Menten kinetics for GGPP utilization. (b) AcOSα′-PaFSα exhibits sigmoidal kinetics for GGPP utilization. (c) Comparison of catalytic efficiencies for AcOS, AcOSα-PaFSα, and AcOSα′-PaFSα.
Discussion
Terpene synthases catalyze the most complex reactions in biology, in which more than half of the substrate carbon atoms typically undergo changes in chemical bonding during the course of a chain elongation or cyclization reaction. Since cyclic terpenes cannot be generated from a linear precursor such as GGPP in the absence of an enzyme, the rate enhancement over the uncatalyzed rate for even the most sluggish terpene cyclase is immeasurably large. A terpenoid cyclase is also unique among enzymes in that its catalytic prowess is represented by its product diversity. Indeed, the fidelity or promiscuity of the cyclization cascade is a defining feature of a terpenoid cyclase.
The active site of a terpenoid cyclase can be altered by mutation of single amino acid residues or the addition/deletion of exonic domains containing hundreds of amino acid residues, and these changes may or may not influence cyclization fidelity. For example, early domain-swapping experiments involving the αβ sesquiterpene cyclases Nicotiana tabacum 5-epi-aristolochene synthase and Hyoscyamus muticus vetispiradiene synthase demonstrated that the β domain of one cyclase could be partially or completely swapped with that of the other while retaining wild-type cyclization fidelity.42 Specific activity measurements indicated that most chimeras retained wild-type levels of catalytic activity, which was probably a reflection of the fact that these enzymes are already related by 77% amino acid sequence identity.43 Structural analysis15 indicates that 75% of αβ interface residues (van der Waals contact or hydrogen bonding) are conserved and 88% are similar between these two enzymes, likely ensuring a complementary fit in domain-swapped αβ chimeras. The preparation of catalytically-active chimeric enzymes is not limited to sesquiterpene synthases, since subsequent examples of domain-swapping include monoterpene synthases from Citrus limon and Salvia officinalis.44,45
In contrast, the β domains of TbTS and PcIS are related by 31% sequence identity, with conservation of 42% of αβ interface residues (53% similarity) as illustrated by the crystal structure of TbTS.17 Thus, to ensure an intact αβ interface in TbTSαβ′, only the first half of the β domain was substituted with that of PcIS (Figure 2). Consequently, with a conserved αβ interface, TbTSαβ′ retains comparable cyclization fidelity to that of TbTSαβG (Table 1), albeit with catalytic activity insufficiently high to permit measurement of steady-state kinetic parameters. A complementary fit between α and β domains in an αβ cyclase is likely to be an important determinant of cyclization fidelity, since deletion of the βγ domains altogether in TbTSα yields a compromised catalyst that appears to generate only linear and not cyclic diterpene products.
Prior domain engineering experiments with bifunctional αβγ cyclases reveal contrasting degrees of interdependence of one domain on the other. Separate α and βγ domain constructs of ent-kaurene synthase from Phaeosphaeria sp. L487 expressed as GST fusion proteins are catalytically active, but exhibit up to 30-fold reductions in catalytic activity.46 In contrast, separate α and βγ domain constructs of abietadiene synthase from Abies grandis (grand fir) are catalytically inactive,47 indicating that the structure and function of the α and βγ domains are much more interdependent than those of ent-kaurene synthase. While the βγ domains of TbTS are not catalytically active, that their deletion results in a severely compromised α domain catalyst unable to generate a cyclic diterpene product is similar to the domain interdependence previously observed for abietadiene synthase.
Bifunctional αα diterpene cyclases make for an interesting contrast with bifunctional and monofunctional αβγ diterpene cyclases in terms of the interdependence of one domain on the other. PaFS serves as a paradigm αα system for probing such interactions. Here, too, catalytic efficiency and cyclization fidelity are substantially altered in all chimeric synthases studied, indicating that domain-domain interactions play a significant role in stabilizing cyclase domains in catalytically competent conformations. Even so, the substitution of a GFPP synthase α domain for the wild-type GGPP synthase α domain in the PaFSα-EvSSα chimera does not result in the utilization of the GFPP by the cyclase domain of PaFS. Additional mutations are required to increase the volume of the PaFS active site to accommodate the larger C25 substrate, as guided by structural homology between the cyclase domains of PaFS and AcOS. Such protein engineering experiments also demonstrate that the active site of a diterpene cyclase can be readily enlarged to favor the utilization of a larger isoprenoid diphosphate substrate and disfavor the utilization of the smaller native substrate to yield new products.
Interestingly, Abe and colleagues have recently described the bifunctional diterpene synthase Emericella variecolor variediene synthase (EvVS) which, similar to PaFS, exhibits double αα domain architecture with a C-terminal GGPP synthase domain and an N-terminal cyclase domain.39 In contrast with PaFS, however, EvVS can utilize both GGPP and GFPP as cyclization substrates. The construction of a chimeric enzyme containing the GFPP synthase domain of EvSS and the cyclase domain of EvVS resulted in the generation of a C25 sesterterpene, (2E)-α-cericerene, as the major product in vivo.39 Notably, minor amounts of variediene were also detected, and the total yield of terpenes was decreased relative to the wild-type system.39 These results indicate, too, that functional α domains can be swapped in bifunctional class I αα terpenoid synthases to engineer product size and diversity.
Geosmin synthase from Streptomyces coelicolor (ScGS) is an example of a bifunctional monomeric αα terpenoid synthase in which both domains catalyze cyclization reactions (Figure 6).48–50 The N-terminal α domain converts FPP into germacradienol, which undergoes further cyclization and fragmentation in the C-terminal α domain to form acetone plus the earthy odorant geosmin.51,52 The N-terminal α domain in isolation can convert FPP to germacradienol, and the C-terminal α domain in isolation can convert germacradienol to geosmin and acetone. Furthermore, co-incubation of separately expressed N-terminal and C-terminal α domains with FPP results in the formation of both germacradienol and geosmin, indicating that the two halves of the protein need not be covalently linked for the enzyme to be catalytically competent. It has also been shown that the germacradienol intermediate does not channel from one active site to the other, but instead dissociates into the medium and re-associates with the active site in the C-terminal α domain to complete the reaction sequence.52 Given that ScGS is an αα monomer in solution as determined by small-angle X-ray scattering,50 there is no possibility of cluster channeling similar to that implicated in (αα)6 PaFS.
Figure 6. Geosmin synthase.

Geosmin synthase is a bifunctional enzyme with αα domain architecture that catalyzes FPP cyclization in the N-terminal domain (blue) to generate germacrene D and germacradienol, the latter of which undergoes a cyclization and fragmentation reaction in the C-terminal domain (green) to yield geosmin and acetone. The structure of the N-terminal domain was determined by X-ray crystallographic methods, and the structure of the C-terminal domain was generated through homology modeling. The domain assembly shown is based on the best fit of small-angle X-ray scattering data (the 41-residue interdomain linker is not shown).50
Product arrays observed for full-length ScGS or an equimolar mixture of individual N-terminal and C-terminal α domains reveal only up to 17% geosmin formation when incubated with FPP, so the cyclization of germacradienol catalyzed by the C-terminal domain appears to be a slower reaction in comparison with the FPP cyclization reaction catalyzed by the N-terminal domain.52 Steady-state kinetics for total hydrocarbon product formation by full-length ScGS and the N-terminal α domain of ScGS indicate a slight loss of catalytic efficiency for the isolated N-terminal α domain, with kcat/KM values of 1.0 × 105 M−1s−1 and 2.8 × 104 M−1s−1, respectively.49 Additionally, mutations in the C-terminal α domain of full-length ScGS suppress geosmin formation.52 Thus, the presence or absence of one α domain modestly influences catalytic efficiency and cyclization fidelity in the other α domain in this monomeric αα terpenoid synthase. However, the degree to which one α domain influences the other in this system is smaller than that observed for PaFS.
Conclusions
In conclusion, domain-domain interactions play a significant role in stabilizing cyclization domains in catalytically competent conformations in class I αβγ, αα, and (αα)6 diterpene cyclases, since cyclization specificity and catalytic efficiency are significantly altered in all chimeric synthases described herein. The mutant cyclases described here also generate 12 newly formed and uncharacterized C20 diterpene products and 3 newly formed and uncharacterized C25 sesterterpene products. Thus, engineered terpenoid cyclases promise to generate new molecular species in an already-diverse family of natural products.
The catalytic efficiency of the utilization of GGPP by the α domain of TbTS within the context of its αβγ domain architecture seems to be more sensitive to truncation or swapping of associated βγ domains than is the specificity of the cyclization itself (Table 1, Figure 3). The crystal structure of TbTS reveals that the β domain partially buttresses the adjacent α domain (Figure 1), so the loss of such stabilizing interactions presumably causes compensatory structural changes in the active site that compromise catalysis. Based on the results with TbTSαβG and TbTSαβ′, such compensatory structural changes can be transmitted across more than one domain-domain interface, since deletion of the γ domain of TbTS influences cyclization fidelity and catalytic efficiency even though the γ domain does not directly contact the α domain.
Interestingly, the catalytic efficiency of GGPP utilization by the α domain of AcOS in wild-type AcOS and the AcOSα-PaFSα chimera is relatively insensitive to substitution of the C-terminal domain (Table 3, Figure 5c). Although GGPP is not the biological substrate of AcOS, its activity nonetheless provides a useful measure of catalytic efficiency. The comparable catalytic efficiencies of AcOS and AcOSα-PaFSα could imply minimal interdomain interactions in these αα assemblies.
Domain-domain and subunit-subunit interactions facilitate cluster channeling in PaFS (Figure 4), in which the bifunctional wild-type enzyme adopts a hexameric quaternary structure, (αα)6.30 Quaternary structure can also be used to regulate the catalytic activity of a class I terpenoid cyclase. A model for such functional regulation is found in Mentha piperita GPP synthase, in which a truncated, catalytically-inert α domain (α′) regulates the function of a catalytically-active α domain in an α2α′2 tetramer.53 It is conceivable that such regulatory strategies could also be engineered in αα or αβγ diterpene cyclases, such that domain-domain or subunit-subunit interactions could direct cyclization fidelity toward one product or another. There are many examples demonstrating that just single-residue substitutions,54–61 or even epistatic pair substitutions,62 can significantly alter product outcome for a diterpene cyclase, so interdomain interactions can be reasonably expected to influence product outcome as well. Future work from our laboratory in this regard will be reported in due course.
Supplementary Material
Acknowledgments
We are grateful to Drs. Ikuro Abe and Takaaki Mitsuhashi for helpful discussions and advice. This work was supported by NIH grant GM56838 to D.W.C. and grant GM30301 to D.E.C.
Footnotes
Supporting Information Available: Table S1, primers used for mutagenesis; Figure S1, detailed cyclization mechanisms of taxadiene synthase, fusicoccadiene synthase, and ophiobolin F synthase; Figure S2, mass spectra of diterpene products generated by TbTS mutants; Figure S3, mass spectra of sesquiterpene, diterpene, and sesterterpene products generated by PaFS and AcOS chimeras. This material is available free of charge via the Internet.
References
- 1.Dolara P, Luceri C, Ghelardini C, Monserrat C, Aiolli S, Luceri F, Lodovici M, Menichetti S, Romanelli MN. Analgesic effects of myrrh. Nature. 1996;379:29. doi: 10.1038/379029a0. [DOI] [PubMed] [Google Scholar]
- 2.Patel T, Ishiuji Y, Yosipovitch G. Menthol: a refreshing look at this ancient compound. J Am Acad Dermatol. 2007;57:873–878. doi: 10.1016/j.jaad.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 3.Gershenzon J, Dudareva N. The function of terpene natural products in the natural world. Nat Chem Biol. 2008;3:408–414. doi: 10.1038/nchembio.2007.5. [DOI] [PubMed] [Google Scholar]
- 4.Baeten J, Deforce K, Challe S, De Vos D, Degryse P. Holy smoke in medieval funerary rites: chemical fingerprints of frankincense in southern Belgian incense burners. PLoS One. 2014;9(11):e113142. doi: 10.1371/journal.pone.0113142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pichersky E, Raguso RA. Why do plants produce so many terpenoid compounds? New Phytol. 2016 doi: 10.1111/nph.14178. in press. [DOI] [PubMed] [Google Scholar]
- 6.Poulter CD, Rilling HC. The prenyl transfer reaction. Enzymic and mechanistic studies of the 1′–4 coupling reaction in the terpene biosynthetic pathway. Acc Chem Res. 1978;11:307–313. [Google Scholar]
- 7.Cane DE. Enzymatic formation of sesquiterpenes. Chem Rev. 1990;90:1089–1103. [Google Scholar]
- 8.Davis EM, Croteau R. Biosynthesis: Aromatic polyketides, isoprenoids, alkaloids. Vol. 209. Berlin: Springer-Verlag Berlin; 2000. Cyclization enzymes in the biosynthesis of monoterpenes, sesquiterpenes, and diterpenes; pp. 53–95. [Google Scholar]
- 9.Tholl D. Terpene synthases and the regulation, diversity and biological roles of terpene metabolism. Curr Opin Plant Biol. 2006;9:297–304. doi: 10.1016/j.pbi.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 10.Christianson DW. Structural biology and chemistry of the terpenoid cyclases. Chem Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 11.Miller DJ, Allemann RK. Sesquiterpene synthases: passive catalysts or active players? Nat Prod Rep. 2012;29:60–71. doi: 10.1039/c1np00060h. [DOI] [PubMed] [Google Scholar]
- 12.Zi J, Mafu S, Peters RJ. To gibberellins and beyond! Surveying the evolution of (di)terpenoid metabolism. Annu Rev Plant Biol. 2014;65:259–286. doi: 10.1146/annurev-arplant-050213-035705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen M, Harris GG, Pemberton TA, Christianson DW. Multi-domain terpenoid cyclase architecture and prospects for proximity in bifunctional catalysis. Curr Op Struct Biol. 2016;41:27–37. doi: 10.1016/j.sbi.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lesburg CA, Zhai G, Cane DE, Christianson DW. Crystal structure of pentalenene synthase: Mechanistic insights on terpenoid cyclization reactions in biology. Science. 1997;277:1820–1824. doi: 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- 15.Starks CM, Back K, Chappell J, Noel JP. Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 16.Wendt KU, Poralla K, Schulz GE. Structure and function of a squalene cyclase. Science. 1997;277:1811–1815. doi: 10.1126/science.277.5333.1811. [DOI] [PubMed] [Google Scholar]
- 17.Köksal M, Jin Y, Coates RM, Croteau R, Christianson DW. Taxadiene synthase structure and evolution of modular architecture in terpene biosynthesis. Nature. 2011;469:116–120. doi: 10.1038/nature09628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Köksal M, Hu H, Coates RM, Peters RJ, Christianson DW. Structure and mechanism of the diterpene cyclase ent-copalyl diphosphate synthase. Nat Chem Biol. 2011;7:431–433. doi: 10.1038/nchembio.578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou K, Gao Y, Hoy JA, Mann FM, Honzatko RB, Peters RJ. Insights into diterpene cyclization from structure of bifunctional abietadiene synthase from Abies grandis. J Biol Chem. 2012;287:6840–6850. doi: 10.1074/jbc.M111.337592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Janke R, Görner C, Hirte M, Brück T, Loll B. The first structure of a bacterial diterpene cyclase: CotB2. Acta Cryst D70. 2014:1528–1537. doi: 10.1107/S1399004714005513. [DOI] [PubMed] [Google Scholar]
- 21.Whittington DA, Wise ML, Urbansky M, Coates RM, Croteau R, Christianson DW. Bornyl diphosphate synthase: Structure and strategy for carbocation manipulation by a terpenoid cyclase. Proc Natl Acad Sci U S A. 2002;99:15375–15380. doi: 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koepp AE, Hezari M, Zajicek J, Vogel BS, LaFever RE, Lewis NG, Croteau R. Cyclization of geranylgeranyl diphosphate to taxa-4(5),11(12)-diene is the committed step of Taxol biosynthesis in Pacific yew. J Biol Chem. 1995;270:8686–8690. doi: 10.1074/jbc.270.15.8686. [DOI] [PubMed] [Google Scholar]
- 23.Hezari M, Lewis NG, Croteau R. Purification and characterization of taxa-4(5),11(12)-diene synthase from Pacific yew (Taxus brevifolia) that catalyzes the first committed step of Taxol biosynthesis. Arch Biochem Biophys. 1995;322:437–444. doi: 10.1006/abbi.1995.1486. [DOI] [PubMed] [Google Scholar]
- 24.Williams DC, Wildung MR, Jin AQ, Dalal D, Oliver JS, Coates RM, Croteau R. Heterologous expression and characterization of a “pseudomature” form of taxadiene synthase involved in Paclitaxel (Taxol) biosynthesis and evaluation of a potential intermediate and inhibitors of the multistep diterpene cyclization reaction. Arch Biochem Biophys. 2000;379:137–146. doi: 10.1006/abbi.2000.1865. [DOI] [PubMed] [Google Scholar]
- 25.Toyomasu T, Tsukahara M, Kaneko A, Niida R, Mitsuhashi W, Dairi T, Kato N, Sassa T. Fusicoccins are biosynthesized by an unusual chimera diterpene synthase in fungi. Proc Natl Acad Sci U S A. 2007;104:3084–3088. doi: 10.1073/pnas.0608426104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toyomasu T, Tsukahara M, Kenmoku H, Anada M, Nitta H, Ohkanda J, Mitsuhashi W, Sassa T, Kato N. Transannular proton transfer in the cyclization of geranylgeranyl diphosphate to fusicoccadiene, a biosynthetic intermediate of fusicoccins. Org Lett. 2009;11:3044–3047. doi: 10.1021/ol901063s. [DOI] [PubMed] [Google Scholar]
- 27.de Vries-van Leeuwen IJ, Kortekaas-Thijssen C, Nzigou Mandouckou JA, Kas S, Evidente A, de Boer AH. Fusicoccin-A selectively induces apoptosis in tumor cells after interferon-α priming. Cancer Lett. 2010;293:198–206. doi: 10.1016/j.canlet.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 28.Bury M, Andolfi A, Rogister B, Cimmino A, Mégalizzi V, Mathieu V, Feron O, Evidente A, Kiss R. Fusicoccin A, a phytotoxic carbotricyclic diterpene glucoside of fungal origin, reduces proliferation and invasion of glioblastoma cells by targeting multiple tyrosine kinases. Transl Oncol. 2013;6:112–123. doi: 10.1593/tlo.12409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wurtele M, Jelich-Ottmann C, Wittinghofer A, Oecking C. Structural view of a fungal toxin acting on a 14-3-3 regulatory complex. EMBO J. 2003;22:987–994. doi: 10.1093/emboj/cdg104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen M, Chou WKW, Toyomasu T, Cane DE, Christianson DW. Structure and function of fusicoccadiene synthase, a hexameric bifunctional diterpene synthase. ACS Chem Biol. 2016;11:889–899. doi: 10.1021/acschembio.5b00960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bauler P, Huber G, Leyh T, McCammon JA. Channeling by proximity: the catalytic advantages of active site colocalization using Brownian dynamics. J Phys Chem Lett. 2010;1:1332–1335. doi: 10.1021/jz1002007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Castellana M, Wilson MZ, Xu Y, Joshi P, Cristea IM, Rabinowitz JD, Gitai Z, Wingreen NS. Enzyme clustering accelerates processing of intermediates through metabolic channeling. Nat Biotechnol. 2014;32:1011–1018. doi: 10.1038/nbt.3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chiba R, Minami A, Gomi K, Oikawa H. Identification of ophiobolin F synthase by a genome mining approach: a sesterterpene synthase from Aspergillus clavatus. Org Lett. 2013;15:594–597. doi: 10.1021/ol303408a. [DOI] [PubMed] [Google Scholar]
- 34.Ye Y, Minami A, Mandi A, Liu C, Taniguchi T, Kuzuyama T, Monde K, Gomi K, Oikawa H. Genome mining for sesterterpenes using bifunctional terpene synthases reveals a unified intermediate of di/sesterterpenes. J Am Chem Soc. 2015;137:11846–11853. doi: 10.1021/jacs.5b08319. [DOI] [PubMed] [Google Scholar]
- 35.Köksal M, Zimmer I, Schnitzler J, Christianson DW. Structure of isoprene synthase illuminates the chemical mechanism of teragram atmospheric carbon emission. J Mol Biol. 2010;402:363–373. doi: 10.1016/j.jmb.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aaron JA, Lin X, Cane DE, Christianson DW. Structure of epi-isozizaene synthase from Streptomyces coelicolor A3(2), a platform for new terpenoid cyclization templates. Biochemistry. 2010;49:1787–1797. doi: 10.1021/bi902088z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harangi J. Retention index calculation without n-alkanes – the virtual carbon number. J Chromatogr A. 2003;993:187–195. doi: 10.1016/s0021-9673(03)00320-0. [DOI] [PubMed] [Google Scholar]
- 38.Matsuda Y, Mitsuhashi T, Quan Z, Abe I. Molecular basis for stellatic acid biosynthesis: a genome mining approach for discovery of sesterterpene synthases. Org Lett. 2015;17:4644–4647. doi: 10.1021/acs.orglett.5b02404. [DOI] [PubMed] [Google Scholar]
- 39.Qin B, Matsuda Y, Mori T, Okada M, Quan Z, Mitsuhashi T, Wakimoto T, Abe I. An unusual chimeric diterpene synthase from Emericella variecolor and its functional conversion into a sesterterpene synthase by domain swapping. Angew Chem Int Ed. 2016;55:1658–1661. doi: 10.1002/anie.201509263. [DOI] [PubMed] [Google Scholar]
- 40.Cramer F, Rittersdorf W. Die Hydrolyse von Phosphaten und Pyrophosphaten Einiger Monoterpenalkohole. Tetrahedron. 1967;23:3015–3022. [Google Scholar]
- 41.George-Nascimento C, Pont-Lezica R, Cori O. Nonenzymic formation of nerolidol from farnesyl pyrophosphate in the presence of bivalent cations. Biochem Biophys Res Commun. 1971;45:119–124. doi: 10.1016/0006-291x(71)90058-1. [DOI] [PubMed] [Google Scholar]
- 42.Back K, Chappell J. Identifying functional domains within terpene cyclases using a domain-swapping strategy. Proc Natl Acad Sci U S A. 1996;93:6841–6845. doi: 10.1073/pnas.93.13.6841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Back K, Chappell J. Cloning and bacterial expression of a sesquiterpene cyclase from Hyoscyamus muticus and its molecular comparison to related terpene cyclases. J Biol Chem. 1995;270:7375–7381. doi: 10.1074/jbc.270.13.7375. [DOI] [PubMed] [Google Scholar]
- 44.El Tamer MK, Lücker J, Bosch D, Verhoeven HA, Verstappen FWA, Schwab W, van Tunen AJ, Voragen AGJ, de Maagd R, Bouwmeester HJ. Domain swapping of Citrus limon monoterpene synthases: impact on enzymatic activity and product specificity. Arch Biochem Biophys. 2003;411:196–203. doi: 10.1016/s0003-9861(02)00711-7. [DOI] [PubMed] [Google Scholar]
- 45.Peters RJ, Croteau RB. Alternative termination chemistries utilized by monoterpene cyclases: chimeric analysis of bornyl diphosphate, 1,8-cineole, and sabinene synthases. Arch Biochem Biophys. 2003;417:203–211. doi: 10.1016/s0003-9861(03)00347-3. [DOI] [PubMed] [Google Scholar]
- 46.Kawaide H, Sassa T, Kamiya Y. Functional Analysis of the Two Interacting Cyclase Domains in ent-Kaurene Synthase from the Fungus Phaeosphaeria sp. L487 and a Comparison with Cyclases from Higher Plants. J Biol Chem. 2000;275:2276–2280. doi: 10.1074/jbc.275.4.2276. [DOI] [PubMed] [Google Scholar]
- 47.Peters RJ, Carter OA, Zhang Y, Matthews BW, Croteau RB. Bifunctional Abietadiene Synthase: Mutual Structural Dependence of the Active Sites for Protonation-Initiated and Ionization-Initiated Cyclizations. Biochemistry. 2003;42:2700–2707. doi: 10.1021/bi020492n. [DOI] [PubMed] [Google Scholar]
- 48.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci U S A. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cane DE, Watt RM. Expression and mechanistic analysis of a germacradienol synthase from Streptomyces coelicolor implicated in geosmin biosynthesis. Proc Natl Acad Sci USA. 2003;100:1547–1551. doi: 10.1073/pnas.0337625100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harris GG, Lombardi PM, Pemberton TA, Matsui T, Weiss TM, Cole KE, Köksal M, Murphy FV, IV, Vedula LS, Chou WKW, Cane DE, Christianson DW. Structural studies of geosmin synthase, a bifunctional sesquiterpene synthase with αα domain architecture that catalyzes a unique cyclization-fragmentation reaction sequence. Biochemistry. 2015;54:7142–7155. doi: 10.1021/acs.biochem.5b01143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang J, He X, Cane DE. Geosmin biosynthesis. Streptomyces coelicolor germacradienol/germacrene D synthase converts farnesyl diphosphate to geosmin. J Am Chem Soc. 2006;128:8128–8129. doi: 10.1021/ja062669x. [DOI] [PubMed] [Google Scholar]
- 52.Jiang J, He X, Cane DE. Biosynthesis of the earthy odorant geosmin by a bifunctional Streptomyces coelicolor enzyme. Nat Chem Biol. 2007;3:711–715. doi: 10.1038/nchembio.2007.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chang TH, Hsieh FL, Ko TP, Teng KH, Liang PH, Wang AHJ. Structure of a heterotetrameric geranyl pyrophosphate synthase from mint (Mentha piperita) reveals intersubunit regulation. Plant Cell. 2010;22:454–467. doi: 10.1105/tpc.109.071738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu M, Wilderman PR, Peters RJ. Following evolution’s lead to a single residue switch for diterpene synthase product outcome. Proc Natl Acad Sci U S A. 2007;104:7397–7401. doi: 10.1073/pnas.0611454104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilderman PR, Peters RJ. A single residue switch converts abietadiene synthase into a pimaradiene specific cyclase. J Am Chem Soc. 2007;129:15736–15737. doi: 10.1021/ja074977g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morrone D, Xu M, Fulton DB, Determan MK, Peters RJ. Increasing complexity of a diterpene synthase reaction with a single residue switch. J Am Chem Soc. 2008;130:5400–5401. doi: 10.1021/ja710524w. [DOI] [PubMed] [Google Scholar]
- 57.Keeling CI, Weisshaar S, Lin RPC, Bohlmann J. Functional plasticity of paralogous diterpene synthases involved in conifer defense. Proc Natl Acad Sci USA. 2008;105:1085–1090. doi: 10.1073/pnas.0709466105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zerbe P, Chiang A, Bohlmann J. Mutational analysis of white spruce (Picea glauca) ent-kaurene synthase (PgKS) reveals common and distinct mechanisms of conifer diterpene synthases of general and specialized metabolism. Phytochemistry. 2012;74:30–39. doi: 10.1016/j.phytochem.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 59.Kawaide H, Hayashi K-i, Kawanabe R, Sakigi Y, Matsuo A, Natsume M, Nozaki H. Identification of the single amino acid involved in quenching the ent-kauranyl cation by a water molecule in ent-kaurene synthase of Physcomitrella patens. FEBS J. 2011;278:123–133. doi: 10.1111/j.1742-4658.2010.07938.x. [DOI] [PubMed] [Google Scholar]
- 60.Irmisch S, Müller AT, Schmidt L, Günther J, Gershenzon J, Köllner TG. One amino acid makes the difference: the formation of ent-kaurene and 16α-hydroxy-entkaurane by diterpene synthases in poplar. BMC Plant Biol. 2015;15:262. doi: 10.1186/s12870-015-0647-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jia M, Peters RJ. Extending a single residue switch for abbreviating catalysis in plant ent-kaurene synthases. Front Plant Sci. 2016;7:1765. doi: 10.3389/fpls.2016.01765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jia M, Zhou K, Tufts S, Schulte S, Peters RJ. A pair of residues that interactively affect diterpene synthase product outcome. ACS Chem Biol. 2017;12:862–867. doi: 10.1021/acschembio.6b01075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jin Q, Williams DC, Hezari M, Croteau R, Coates RM. J Org Chem. 2005;70:4667–4675. doi: 10.1021/jo0502091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.